

Abstract
The current study was aimed at analyzing putative protein sequences of the transition protein-like proteins in 12 Drosophila species based on the reference sequences of transition protein-like protein (Tpl94D) expressed in Drosophila melanogaster sperm nuclei. Transition proteins aid in transforming chromatin from a histone-based nucleosome structure to a protamine-based structure during spermiogenesis - the post-meiotic stage of spermatogenesis. Sequences were obtained from NCBI Ref-Seq database using NCBI ORF-Finder (PSI-BLAST). Sequence alignments and analysis of the amino acid content indicate that orthologs for Tpl94D are present in the melanogaster species subgroup (D. simulans, D. sechellia, D. erecta, and D. yakuba), D. ananassae, and D. pseudoobscura, but absent in D. persmilis, D. willistoni, D. mojavensis, D. virilis, and D. grimshawi. Transcriptome next generation sequence (RNA-Seq) data for testes and ovaries was used to conduct differential gene expression analysis for Tpl94D in D. melanogaster, D. simulans, D. yakuba, D. ananassae, and D. pseudoobscura. The identified Tpl94D orthologs show high expression in the testes as compared to the ovaries. Additionally, 2 isoforms of Tpl94D were detected in D. melanogaster with isoform A being much more highly expressed than isoform B. Functional analyses of the conserved region revealed that the same high mobility group (HMG) box/DNA binding region is conserved for both Drosophila Tpl94D and Drosophila protamine-like proteins (MST35Ba and MST35Bb). Based on the rigorous bioinformatic approach and the conservation of the HMG box reported in this work, we suggest that the Drosophila Tpl94D orthologs should be classified as their own transition protein group.
Keywords: ovary gene expression, Protamine, RNA-Seq, sperm chromatin, testes gene expression, Tpl94D, transition proteins
Introduction
During spermatogenesis in most metazoans, haploid round spermatids undergo a dramatic nuclear transformation where the chromatin is remodeled into a highly compacted, transcriptionally silent form. This transformation is accompanied by the production of sperm-specific proteins that replace histones as the DNA-binding proteins. These sperm-specific proteins include histone H1 linker-like proteins,1,2 true protamines,3 protamine-like proteins,1,2 chromatin insulator proteins,4 and transition proteins.2,4-6 Histone H1 linker-like proteins, true protamines and protamine-like proteins appear to have evolved from histone H1 linker and are collectively referred as the “sperm nuclear basic proteins” (SNBPs).7,8 True protamines are present in the sperm nuclei of higher vertebrates such as mice and humans,9-11 while protamine-like proteins are found in some vertebrates,12 but are predominantly found in invertebrate species such as fruit flies,4,6,13 Atlantic surf clam,13-15 and stalked tunicate.16
Adult male Drosophila fruit flies and mammals have a similar process of spermatogenesis. In Drosophila, spermatogenesis advances from tip of the blind-ended tubular or ellipsoid testes, while in mammals spermatogenesis proceeds within the seminiferous epithelium lining seminiferous tubules in the testes.17 In both flies and mammals, the initiation of spermatogenesis occurs in the stem cell niche region, which is located at the apex of the testes in flies,18,19 and in the basal compartment of the seminiferous epithelium in mammals. The fly testis stem cell niche houses the germline stem cells and cyst progenitor stem cells.20 The gonialblast will go through a mitotic amplification stage, followed by 2 meiotic divisions to generate haploid round spermatids. During the post-meiotic stage of spermatogenesis (spermiogenesis), haploid round spermatids transform into functional sperm. This transformation includes the exchange of histones for protamines and chromatin condensation. In flies, nuclear transformation involves the exchange of somatic histones for SNBPs called protamine-like proteins.21,22 In D. melanogaster, the transition protein Tpl94D facilitates the exchange of histones for protamine-like proteins.4-6 It has also been well documented that mammalian transition proteins (TPs) are involved in binding DNA to facilitate the transition from nucleosome-based chromatin to protamine-based chromatin.3
The D. melanogaster protamine-like proteins are male specific transcripts MST35Ba and MST35Bb.1,2,4,13,23 The purpose of MST35Ba and MST35Bb appears to be to serve as the protector of the compacted DNA in the sperm nucleus against detrimental environmental factors such as X-rays.6 Furthermore, deletion of MST35Ba and MST35Bb does not significantly affect chromatin condensation or fertility as it does in mammals when true protamines are deleted.1,2,24,25
Recent studies showed that during spermiogenesis both transition (Tpl94D) and histone H1 linker-like (male specific transcript - MST77F) proteins play a significant role in remodeling the sperm nucleus in D. melanogaster.4,6 During sperm nuclear remodeling, the ubiquitous chromatin insulator protein CTCF has been postulated to be involved in controlling the areas where chromatin can undergo histone modification.4 These histone modifications include H2A mono-ubiquitination and an increase in H4 acetylation, which cause the histones on the chromatin to be removed and degraded.4 Consequently, an opening within the chromatin allows Tpl94D to act as an intermediate for the transition from a histone bound nucleosome to a protamine bound structure.4 A key component of Tpl94D that allows for chromatin condensation to occur is the N terminal high mobility group (HMG) box.4 This HMG box is rich in arginine, which is a very basic amino acid with high affinity for binding DNA.4,5
Recently, we performed a detailed bioinformatic analysis of protamine-like proteins in 12 species of Drosophila (D. melanogaster. D. simulans, D. sechellia, D. yakuba, D. erecta, D. erecta, D. ananassae, D. persimilis, D. pseudoobcura, D. willistoni, D. virilis, and D. grimshawi).13 The current study focuses on an analysis of transition proteins (TPs) in the same 12 species analyzed in our previous work. Here, we include differential gene expression analysis using available next generation sequencing (NGS) RNA-Seq transcriptome data in addition to the genomic analysis. Additionally, we show that Tpl94D orthologs have a conserved N-terminal DNA binding domain and they are highly expressed in the testes as compared to the ovaries.
Results
BLAST results for Tpl94D nucleic acid sequences
The published genomic and mRNA nucleotide sequences for Tpl94D (GI: 442620556) from D. melanogaster were used to search the genomes of D. simulans, D. sechellia, D. yakuba, D. erecta, D. ananassae, D. pseudoobscura, D. persmilis, D. willistoni, D. mojavensis, D. virilis, and D. grimshawi for sequence matches. The best NCBI ORF sequences for transition protein Tpl94D orthologs within the original 12 sequenced Drosophila species are listed in Table 1. The nucleotide BLAST and protein BLAST did not reveal the same gene loci for all the species outside the melanogaster species subgroup (D. ananassae, D. pseudoobscura, D. persimilis, D. willistoni, D. mojavensis, D. virilis, and D. grimshawi). A forced nucleotide BLAST2 alignment for transcripts and genomic sequences for the protein orthologs for Tpl94D illustrates that the protein BLAST, PSI BLAST, and ORF Finder sequences do not align with the genomic or transcript sequences of the Drosophila species from outside the melanogaster species subgroup to Tpl94D. This is due to the poor E-value scores and the percent query coverage for the species outside the melanogaster species subgroup. The current annotation on Flybase shows D. persimilis (Dper GL26871-Tpl94D) to be a putative ortholog of Tpl94D based on protein sequence predictions made using OrthoDB. Our current investigation, however, does not include Dper GL26871-Tpl94D because the next generation sequence RNA-Seq transcriptome data sets were not available for D. persmilis testes and ovaries and Dper GL26871-Tpl94D was below the NCBI ORF Finder's threshold (Tables 1–2). A summary of the best nucleotide BLAST alignment results are shown in Table 2 with their maximum identity, query coverage and E-value(s).
Table 1.
All NCBI open reading frame (ORF) finder sequence matches for Tpl94D in the original 12 sequenced Drosophila species
| GI Number |
||||
|---|---|---|---|---|
| Drosophila Species | Match Number | Gene Locus | Whole Nucleotide and Transcript Sequences | Protein Sequence |
| D. melanogaster‡,# | Control (Tpl94D) | CG31281 | 24649165 | 24649166 |
| D. simulans*,‡,# | 1–2 | GD20990 | 195573025 | 195573026 |
| D. simulans† | 2–2 | GD21472 | 195574870 | 195574871 |
| D. sechellia*,# | 1–2 | GM26474 | 195331176 | 195331177 |
| D. sechellia† | 2–2 | GM12829 | 195341320 | 195341321 |
| D. yakuba*,‡,# | 1–2 | GE10340 | 195502744 | 195502745 |
| D. yakuba† | 2–2 | GE23890 | 195503141 | 195503142 |
| D. erecta*,# | 1–2 | GG11172 | 194910675 | 194910676 |
| D. erecta† | 2–2 | GG24235 | 194857282 | 194857283 |
| D. ananassae*,‡,# | 1–3 | GF19889 | 194743971 | 194743972 |
| D. ananassae† | 2–3 | GF20096 | 194746963 | 194746964 |
| D. ananassae† | 3–3 | GF15002 | 194758514 | 194758515 |
| D. pseudoobscura*,‡,# | 1–1 | GA22645 | 198471329 | 198471330 |
| D. persmilis† | 1–1 | GL26871 | 195168587 | 195168588 |
| D. willistoni† | 1–2 | GK14607 | 195435142 | 195435143 |
| D. willistoni† | 2–2 | GK12423 | 195461023 | 195461024 |
| D. mojavensis♦ | ‐ | ‐ | ‐ | ‐ |
| D. virilis† | 1–1 | GJ16066 | 195385648 | 195385649 |
| D. grimshawi♦ | ‐ | ‐ | ‐ | ‐ |
Denotes better than threshold
Denotes worse than threshold
Denotes verified through RNA-Seq analysis
Denotes no matches found for D. mojavensis and D. grimshawi
Denotes identified orthologs for Tpl94D
The cut off threshold was query coverage of 40% with maximum identity score of 36 and an E-value of 7 × 10−5
Table 2.
Best NCBI nucleotide BLAST sequence matches and orthologs for Tpl94D (GI: 24649165)
| Genomic DNA Sequence |
Transcript Sequence |
|||||||
|---|---|---|---|---|---|---|---|---|
| Species Name | Gene Locus | GI Number | Maximum Identity (%) | Query Coverage (%) | E- Value Score | Maximum Identity (%) | Query Coverage (%) | E- Value Score |
| D. simulans***# | GD20990 | 195573025 | 86 | 36 | 0 | 86 | 33 | 1e-170 |
| D. sechellia***# | GM26474 | 195331176 | 86 | 36 | 0 | 86 | 33 | 7e-168 |
| D. yakuba***# | GE10340 | 195502744 | 69 | 35 | 1e-82 | 69 | 32 | 1e-69 |
| D. erecta***# | GG11172 | 194910675 | 67 | 34 | 2e-61 | 71 | 31 | 9e-78 |
| D. simulans† | GD21472 | 195574870 | 100 | 0 | 0.11 | — | — | — |
| D. sechellia† | GM12829 | 195341320 | 100 | 0 | 0.11 | — | — | — |
| D. yakuba†,♦ | GE23890 | 195503141 | — | — | — | — | — | — |
| D. erecta†,♦ | GG24235 | 194857282 | — | — | — | — | — | — |
| D. ananassae*,# | GF22417 | 194766791 | 86 | 2 | 4e-07 | 86 | 2 | 3e-07 |
| D. ananassae† | GF19889 | 194743971 | 100 | 0 | 0.38 | 100 | 0 | 0.32 |
| D. ananassae†,♦ | GF20096 | 194746963 | — | — | — | — | — | — |
| D. ananassae† | GF15002 | 194758514 | 100 | 7 | 0.031 | 100 | 0 | 0.32 |
| D. pseudoobscura*,# | GA22363 | 198467493 | 93 | 4 | 3e-08 | 93 | 5 | 3e-08 |
| D. pseudoobscura†,♦ | GA22645 | 198471329 | — | — | — | — | — | — |
| D. persmilis* | GL18087 | 195175349 | 93 | 1 | 1e-07 | 93 | 1 | 1e-07 |
| D. persmilis†,♦ | GL26871 | 195168587 | — | — | — | — | — | — |
| D. willistoni* | GK19855 | 195432301 | 85 | 6 | 1e-06 | 85 | 3 | 1e-06 |
| D. willistoni†,♦ | GK14607 | 195435142 | — | — | — | — | — | — |
| D. willistoni†,♦ | GK12423 | 195461023 | — | — | — | — | — | — |
| D. mojavensis* | GI13566 | 195128228 | 92 | 3 | 2e-05 | 92 | 2 | 1e-05 |
| D. virilis* | GJ22187 | 195383563 | 100 | 5 | 1e-06 | 85 | 3 | 1e-06 |
| D. virilis† | GJ16066 | 195385648 | 100 | 0 | 0.38 | — | — | — |
| D. grimshawi* | GH21505 | 195027639 | — | — | — | 83 | 2 | 1e-05 |
*Denotes best matches for Drosophila species within the Drosophila melanogaster species subgroup and greater than threshold for NCBI Open Reading Frame Finder
Denotes best match for nucleotide sequence of Tpl94D for Drosophila species outside the Drosophila melanogaster species subgroup
Denotes NCBI Open Reading Frame Finder Match and match not found based on transcript sequence of Tpl94D
Denotes identified orthologs for Tpl94D
No match based on Tpl94D's genomic and transcript sequences
Analysis of transition protein (Tpl94D)
The published protein sequence for Tpl94D (GI: 24649166) for D. melanogaster was used to search the genomes of the Drosophila species listed previously for protein sequence matches. BLAST results with maximum identity, query coverage, and E-value scores are shown in Table 3. Only the best matched protein BLAST sequences are listed for each of the Drosophila species. No sequence matches were found outside the melanogaster species subgroup except for D. ananassae and D. pseudoobscura. The amino acid sequences for D. ananassae (Dana GF19889-Tpl94D) and D. pseudoobscura (Dpse GA22645-Tpl94D) were confirmed by analyzing publically available NGS RNA-Seq transcriptome data sets from NCBI SRA, ModENCODE, Flybase, and NCBI EST (Table S1). All of the orthologs were then confirmed using NCBI ORF Finder, PSI BLAST, and protein BLAST. Figure 1 shows a T-Coffee protein alignment of the Tpl94D orthologs for D. melanogaster, D. simulans (Dsim GD20990-Tpl94D), D. sechellia (Dsec GM26474-Tpl94D), D. yakuba (Dyak GE10340-Tpl94D), D. erecta (Dere GG11172-Tpl94D), D. ananassae (Dana GF19889-Tpl94D), and D. pseudoobscura (Dpse GA22645-Tpl94D) with a consensus score of 87. Figure S1 shows the consensus score increase to 97 with the omission of D. ananassae (Dana GF19889-Tpl94D), and D. pseudoobscura (Dpse GA22645-Tpl94D) amino acid residues from the T-Coffee alignment. Similarly, CLUSTAL Omega (conservative global alignment tool) shows the same N terminal region among the Tpl94D orthologs (Dsim GD20990-Tpl94D, Dsec GM26474-Tpl94D, Dyak GE10340-Tpl94D, Dere GG11172-Tpl94D, Dana GF19889-Tpl94D, and Dpse GA22645-Tpl94D) as being conserved (Fig. 2).
Table 3.
NCBI protein BLAST Tpl94D (GI: 24649166) orthologs
| Species Name | Gene Locus | GI Number | Maximum Identity (%) | Query Coverage (%) | E – Value Score |
|---|---|---|---|---|---|
| D. simulans | GD20990 | 195573026 | 78 | 100 | 1e-91 |
| D. sechellia | GM26474 | 195331177 | 78 | 100 | 7e-92 |
| D. yakuba | GE10340 | 195502745 | 52 | 100 | 7e-56 |
| D. erecta | GG11172 | 194910676 | 54 | 100 | 1e-65 |
| D. ananassae* | GF19889 | 194743972 | 46 | 40 | 3e-18 |
| D. pseudoobscura* | GA22645 | 198471330 | 36 | 65 | 5e-13 |
Matches cannot be retrieved through traditional BLAST means due to best genomic sequences not matching their respective best protein matches.
Figure 1.

T-Coffee alignment of Tpl94D for melanogaster species subgroup, D. ananassae, and D. pseudoobscura. T-Coffee conserved region alignment for Tpl94D. Key on the bottom right shows 87 consensus score for all sequence matches.
Figure 2.
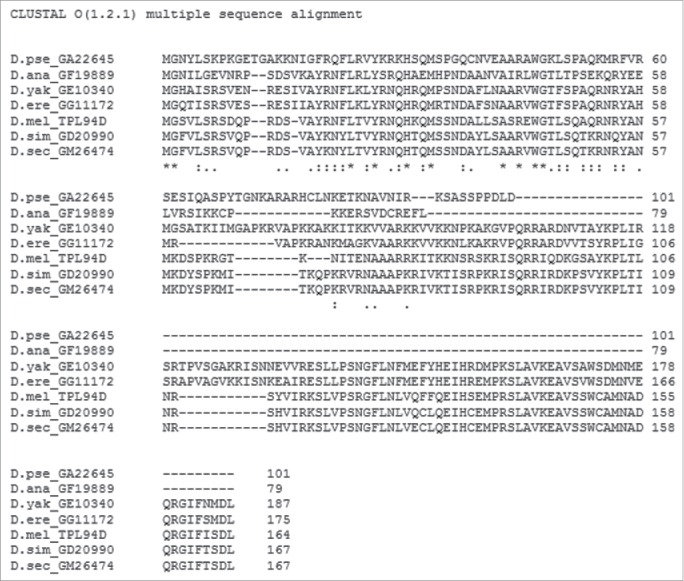
CLUSTAL Omega Alignment of Tpl94D sequence matches CLUSTAL Omega alignment of the best Tpl94D sequence matches in the sequenced 12 Drosophila species.
The Tpl94D protein orthologs were analyzed for their amino acid percentages (Figure S2 and File S1) and total number of amino acids (Figure S3 and File S2). These analyses included published NCBI sequences for D. melanogaster histone H1 linker-like proteins (MST77F), mouse transition proteins, rat transition proteins, protamine-like proteins, and true protamine proteins. These proteins were included to illustrate the change in the percentage of basic amino acids in DNA binding proteins across model and non-model organisms. Previous studies have characterized transition proteins, histone H1 linker-like, protamine-like, and true protamines based on distinct percentage of basic amino acids (lysine and arginine) and other specific amino acids like cysteine, tyrosine, and serine.4,13,15,17,27 Table 4 indicates species that are within the melanogaster species subgroup (Dsim GD20990-Tpl94D, Dsec GM26474-Tpl94D, Dyak GE10340-Tpl94D, and Dere GG11172-Tpl94D) have essentially the same number of amino acid residues as compared to the control Tpl94D found in D. melanogaster. In contrast, Drosophila species found outside the melanogaster species subgroup have greater variance in the number of amino acid residues (79 and 101 amino acids for Dana GF19889-Tpl94D and Dpse GA22645-Tpl94D respectively).
Table 4.
Amino acid analysis for Tpl94D orthologs
| Species | Gene | Cysteine | Lysine | Arginine | Serine |
|---|---|---|---|---|---|
| D. mel | Tpl94D (164) | 0.61 (1) | 7.32 (12) | 11.59 (19) | 13.41 (22) |
| D. sim | GD20990 (167) | 1.8 (3) | 7.78 (13) | 10.18 (17) | 10.78 (18) |
| D. sec | GM26474 (167) | 1.8 (3) | 7.78 (13) | 10.78 (18) | 10.78 (18) |
| D. yak | GE10340 (187) | 0 (0) | 10.16 (19) | 10.16 (19) | 7.49 (14) |
| D. ere | GG11172 (175) | 0 (0) | 8.57 (15) | 12 (21) | 8.57 (15) |
| D. ana* | GF19899 (79) | 2.53 (2) | 7.59 (6) | 11.39 (9) | 7.59 (6) |
| D. pse* | GA22645 (101) | 1.98 (2) | 11.88 (12) | 8.91 (9) | 9.90 (10) |
Percentage (Total number present).
=not confirmed through normal NCBI BLAST / ORF – RNA-Seq only.
Transition proteins are rich in basic amino acids like lysine (K) and arginine (R), serine (S), and low in cysteine (C) amino acid residues.27 All orthologs had a high percentage of the total sum of lysine (K) and arginine (R) amino acids with an average percentage of 19.4 (ranged from 19% to 21%) (Figure S2 and File S1). Overall, there was an equal or larger amount of arginine amino acids for all orthologs with the exception of Dpse GA22645-Tpl94D, which had a higher lysine amino acid percentage of 12% as compared to 9% for arginine amino acids (Figure S2 and File S1). The Drosophila species orthologs closest to the D. melanogaster Tpl94D control (Dsim GD20990-Tpl94D and Dsec GM26474-Tpl94D) had very similar percentages of cysteine, lysine, arginine, and serine (Figure S2 and File S1).
The sum of lysine and arginine amino acids was substantially lower for Tpl94D and its respective orthologs than the sum of both of lysine and arginine amino acids in TP1 and TP2 found in Mus musculus (mouse), Rattus norvegicus (rat), and Bos taurus (bull) (Figure S2 and File S1). In contrast, percentage sum of lysine and arginine amino acids in the Tpl94D orthologs was similar to the percentage sum of lysine and arginine amino acids found in Homo sapiens TP2 (Figure S2 and File S1). A sum percentage average of lysine and arginine amino acids of 19% was obtained when H. sapiens TP2 was included with the Tpl94D orthologs. Cysteine residues are essentially absent from the Tpl94D orthologs, which is similar to TP1 found in M. musculus, R. norvegicus, B. taurus, and Homo sapiens (Figure S2 and File S1).
The whole protein sequences for Tpl94D orthologs in the melanogaster species subgroup are conserved as indicated in Figure S1. The percentage of amino acid residues present among the Tpl94D orthologs are shown in Figure S4 and File S3. Likewise the number of amino acid residues present among the Tpl94D orthologs are shown in Figure S5 and File S4, The lysine and arginine content is slightly lower in the conserved region with an average percentage of 17% (Figure S4 and File S3).
Sequence alignment of Tpl94D orthologs with mammalian transition proteins (TPs)
The orthologs for Tpl94D were compared to TP1 and TP2 from 4 mammalian model organisms: M. musculus, R. norvegicus, B. taurus, and H. sapiens. TP1 for M. musculus, R. norvegicus, B. taurus, and H. sapiens did not show any conservation with Tpl94D orthologs (data not shown). However, there are a small number of amino acid residues at the N terminus of the Tpl94D orthologs that are conserved with the TP2 N terminus for M. musculus, R. norvegicus, B. taurus, and H. sapiens (Fig. 4). This conservation may be attributed to the overall greater sequence and length diversity among TP2s as compared to TP1s.17,27
Figure 4.
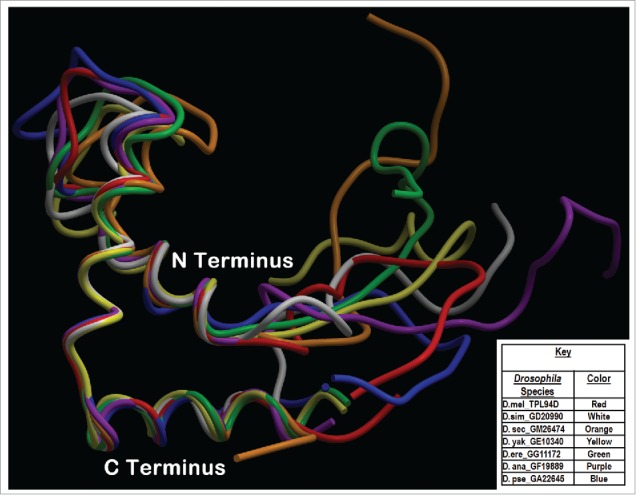
Phyre2 Tpl94D best sequence matches conserved DNA binding region Tertiary structure alignment of a wire frame model for the Tpl94D orthologs. The different colors indicate each of the species indicated on the bottom right.
Functional analysis of the whole protein and conserved region in Tpl94D
Functional analysis of the whole Tpl94D protein orthologs and their respective conserved region was conducted using 3 DNA binding prediction tools: BindN+, DNA-Binder and DP-Bind. All results from DNA binder showed that Tpl94D orthologs and their respective conserved regions were able to bind DNA with average to high confidence (Table S2). Additionally, the conserved regions (Main Data Set) showed a higher affinity to bind DNA as compared to the whole protein (Realistic and Alternative Data sets) (Table S2).
BindN+ was used to predict the actual amino acid residues that will or will not bind to DNA. The whole protein analysis indicates that a minimum of 63% of all amino acids will bind to DNA in all of the orthologs, except for Dana GF19889-Tpl94D with only 57% binding DNA. The conserved N-terminal region in the Tpl94D orthologs illustrates that an increase of DNA binding probability to greater than 71% with the exception of the Dana GF19889-Tpl94D being only 58% (Table S3). Overall, the majority of the putative DNA binding residues were found within the conserved region.
DP-Bind was used to predict DNA binding or non DNA binding amino acid residues in the whole protein orthologs and their respective conserved regions. Overall, a substantial range in the percentages of the Tpl94D orthologs were shown to be DNA binding with the highest percentage found in Dsim GD20990-Tpl94D (53%) and the lowest found in the Dyak GE10340-Tpl94D (29%). The overall decrease in the percentage in Dyak GE10340-Tpl94D and Dere GG11172-Tpl94D is attributed to the larger number of amino acids present as compared to the rest of the orthologs. The conserved regions of Dyak GE10340-Tpl94D and Dere GG11172-Tpl94D have the same number of amino acids shown to be DNA binding as compared to the rest of the melanogaster species subgroup (D. melanogaster Tpl94D, Dsim GD20990-Tpl94D, Dsec GM26474-Tpl94D, Dyak GE10340-Tpl94D, and Dere GG11172-Tpl94D).
The Tpl94D orthologs and their respective conserved regions were further analyzed using Protein homology/analogy recognition engine 2.0 (Phyre 2). A detailed analysis of the conserved regions for Tpl94D is shown in Table 5. All five sample matches (c2e6oA, c2cs1A, d1v64a, d1hmfa, and c2yrqA) have an overlapping region with a protein of unknown function (DUF1074 Family) and high mobility group (HMG) box. Table 6 shows the analysis of the whole protein orthologs for Tpl94D. The DUF1074 protein of unknown function once again overlaps with the HMG box. The Dere GG11172-Tpl94D had N terminal and C-terminal distinct regions matching up for DNA binding and HMG box. This can be attributed to Dere GG11172-Tpl94D being a DNA binding protein as indicated by c2yrqA match, which had residues 2 through 172 covering 97% of the whole protein. Phyre2 was used to generate a tertiary wire frame structure of the conserved regions and Molsoft ICM Browser was used to analyze the alignment of these structures. The conserved regions in Tpl94D orthologs have similar tertiary arrangements of the 3 α helices as shown in Fig. 3.
Table 5.
Detailed analysis of conserved functional groups found in Tpl94D orthologs
| Sample Matches for D. mel Tpl94D | D. mel Tpl94D | D. sim GD20990 Tpl94D | D. sec GM26474 Tpl94D | D. yak GE10340 Tpl94D | D. ere GG11172 Tpl94D | D. ana GF19899 Tpl94D | D. pse GA22645 Tpl94D | |
|---|---|---|---|---|---|---|---|---|
| Model Confidence (Conserved Region) >90% | 94 | 94 | 94 | 100 | 100 | 91 | 89 | |
| c2e6oA | % Confidence | 97.7 | 97.7 | 97.7 | 98.3 | 98.9 | 91.2 | 97.9 |
| % Identity | 15 | 13 | 13 | 15 | 16 | 13 | 13 | |
| Info: a b c d | % Coverage | 71 | 70 | 70 | 81 | 89 | 82 | 92 |
| Residues | 15–60 | 15–60 | 15–60 | 9–61 | 7–64 | 6–59 | 2–62 | |
| c2cs1A | % Confidence | 97.9 | 97.8 | 97.9 | 98.4 | 98.9 | 92 | 98 |
| % Identity | 25 | 27 | 22 | 19 | 17 | 21 | 13 | |
| Info: e f g | % Coverage | 90 | 68 | 68 | 87 | 90 | 89 | 83 |
| Residues | 2–59 | 15–59 | 15–59 | 8–64 | 6–64 | 2–59 | 8–62 | |
| d1v64a | % Confidence | 98 | 98 | 98 | 98.3 | 98.8 | 92.7 | 98.1 |
| % Identity | 20 | 18 | 18 | 12 | 8 | 14 | 12 | |
| Info: h | % Coverage | 69 | 68 | 68 | 76 | 92 | 65 | 86 |
| Residues | 15–59 | 15–59 | 15–59 | 11–60 | 5–64 | 17–59 | 6–62 | |
| d1hmfa | % Confidence | 97.9 | 97.6 | 97.6 | 98.4 | 98.9 | 89.4 | 97.9 |
| % Identity | 19 | 18 | 17 | 14 | 14 | 19 | 20 | |
| Info: h | % Coverage | 85 | 68 | 70 | 85 | 87 | 82 | 87 |
| Residues | 5–59 | 15–59 | 15–60 | 5–60 | 5–61 | 6–59 | 5–62 | |
| c2yrqA | % Confidence | 96.1 | 96.2 | 96.3 | 97.4 | 98.3 | 74.1 | 96.8 |
| % Identity | 20 | 18 | 18 | 16 | 17 | 19 | 18 | |
| Info: e i j | % Coverage | 76 | 75 | 76 | 76 | 96 | 81 | 86 |
| Residues | 11–59 | 11–59 | 11–60 | 11–60 | 2–64 | 7–59 | 6–62 | |
transcription
cell cycle
hmg box-containing protein 1
solution structure of the hmg box domain from human hmg-box2 transcription factor
dna binding protein
pms1 protein homolog 1
solution structure of the hmg domain of human dna mismatch2 repair protein
HMG – box
high mobility group protein b1
solution structure of the tandem hmg box domain from human2 high mobility group protein b1
Table 6.
Detailed analysis of functional groups found in Tpl94D whole protein sequence matches
| Sample Matches for D. mel Tpl94D | D. mel Tpl94D | D. sim GD20990 Tpl94D | D. sec GM26474 Tpl94D | D. yak GE10340 Tpl94D | D. ere GG11172 Tpl94D | D. ana GF19899 Tpl94D | D. pse GA22645 Tpl94D | |
|---|---|---|---|---|---|---|---|---|
| Model Confidence >90% | 98 | 98 | 98 | 82 | 87 | 81 | 56 | |
| c2e6oA | % Confidence | 98.7 | 99.1 | 99.2 | 98.9 | 97.2 | 90.9 | 97.5 |
| % Identity | 18 | 10 | 10 | 15 | 15 | 12 | 12 | |
| Info: a b c d | % Coverage | 40 | 42 | 41 | 27 | 34 | 81 | 55 |
| Residues | 2–69 | 4–75 | 4–74 | 7–59 | 111–172 | 6–70 | 6–62 | |
| c2cs1A | % Confidence | 99 | 99.3 | 99.4 | 98.9 | 97.4 | 91.2 | 97.6 |
| % Identity | 19 | 18 | 20 | 20 | 19 | 21 | 13 | |
| Info: e f g | % Coverage | 51 | 50 | 50 | 31 | 29 | 72 | 43 |
| Residues | 2–86 | 2–87 | 2–87 | 6–64 | 6–57 | 2–59 | 18–62 | |
| d1v64a | % Confidence | 98.9 | 99.3 | 99.4 | 98.8 | 97.4 | 92.3 | 97.8 |
| % Identity | 17 | 16 | 14 | 10 | 10 | 16 | 13 | |
| Info: h | % Coverage | 39 | 53 | 49 | 47 | 33 | 60 | 54 |
| Residues | 9–73 | 4–93 | 9–91 | 11–100 | 114–172 | 17–65 | 7–62 | |
| d1hmfa | % Confidence | 98.8 | 99.2 | 99.3 | 98.9 | 97 | 88.3 | 97.3 |
| % Identity | 19 | 14 | 17 | 15 | 17 | 19 | 20 | |
| Info: h | % Coverage | 41 | 42 | 40 | 28 | 31 | 67 | 56 |
| Residues | 8–76 | 5–76 | 8–76 | 5–59 | 1–56 | 6–59 | 5–62 | |
| c2yrqA | % Confidence | 99.6 | 99.8 | 99.8 | 99.5 | 98.7 | 70.4 | 95.6 |
| % Identity | 16 | 12 | 13 | 26 | 19 | 20 | 18 | |
| Info: e i j | % Coverage | 96 | 97 | 97 | 97 | 97 | 60 | 55 |
| Residues | 2–161 | 2–164 | 2–164 | 2–184 | 2–172 | 11–59 | 6–62 | |
transcription
cell cycle
hmg box-containing protein 1
solution structure of the hmg box domain from human hmg-box2 transcription factor
dna binding protein
pms1 protein homolog 1
solution structure of the hmg domain of human dna mismatch2 repair protein
HMG – box
high mobility group protein b1
solution structure of the tandem hmg box domain from human2 high mobility group protein b1
Figure 3.

CLUSTAL Omega Alignment of Tpl94D with mammalian TP2 CLUSTAL Omega alignment of the orthologs for Tpl94D and transition protein 2 from M. musculus, R. norvegicus, B. taurus, and H. sapiens.
Ovaries and testes transcriptome RNA-Seq and isoform analysis of Tpl94D in D. melanogaster, D. simulans, D. yakuba, and D. pseudoobscura
File S5 and Table 7 shows a summary of the RNA-Seq analysis using Cuffdiff 2.0.2 with a false discovery rate (FDR) of 0.01 for all transition protein Tpl94D orthologs across D. melanogaster (control), D. simulans, D. yakuba, D. ananassae, and D. pseudoobscura. For these species, Tpl94D was highly expressed in the testes as compared to ovaries. D. melanogaster expressed 2 isoforms for Tpl94D: Tpl94D_A: FBtr0084339 and Tpl94D_B: FBtr0310110 - with higher expression found for Tpl94D_A (Figure S7 and S8). The Fragments Per Kilobase of exon model per Million mapped fragments (FPKM) for the testes samples in D. melanogaster Tpl94D_A showed a high expression (123.52) as compared D. melanogaster ovaries samples (FPKM = 0). A positive log2 fold change of 13.8006 was seen with a p value of 0.0622531 and a q value of 0.222648 for Tpl94D_A isoform. The Tpl94D_B isoform has a lower expression for the testes (19.079 FPKM) as compared to Tpl94D_A isoform for the testes. The ovaries expression for the Tpl94D_B isoform was 0. The relationship of these 2 isoforms for D. melanogaster Tpl94D (FBgn0051281) was analyzed using NCBI Isoform Usage Two-step Analysis (IUTA). IUTA showed that Tpl94D_A isoform (FBtr0084339) is the dominant isoform of the Tpl94D gene with 91% expression as compared to only 9% expression of Tpl94D_B isoform (FBtr0310110) in D. melanogaster testes (Figure S8).28 No other isoforms were detected for any other species (D. simulans, D. yakuba, D. ananassae, and pseudoobscura) based on ENSEMBL GTF files.29
Table 7.
Ovaries vs. testes transcriptome Cuffdiff 2.0.2 RNA-Seq analysis summary
| Species - Gene ID | Ovaries (FPKM)* | Testes (FPKM)* | Log2 (Ovaries/ Testes)* | P value* | Q Value* |
|---|---|---|---|---|---|
| D. mel – TPL94D – Iso A | 0.0087 | 123.524 | 13.8006 | 0.0623 | 0.2226 |
| D. mel – TPL94D – Iso B | 0 | 19.0749 | 1.7977e+308 | 0.1929 | 0.4164 |
| D. sim - GD20990 | 0 | 266.525 | 1.7977e+308 | 0.0001 | 0.0011 |
| D. yak - GE10340 | 0.0120 | 506.227 | 15.3673 | 0.0070 | 0.0105 |
| D. ana - GF19889 | 0.0116 | 78.6323 | 12.7224 | 0 | 0 |
| D. pse - GA22645 | 0.0548 | 232.614 | 12.0528 | 0.0001 | 0.0016 |
Values were rounded to the 10-thousandths decimal point as compared to File S5.
The expression for Dsim GD20990-Tpl94D was 266.525 FPKM in the testes with 0 FPKM found in the D. simulans ovaries. This also resulted in an exponential positive log 2-fold change of 1.79769×10308 with a p value of 0.000117315 and a q value of 0.00109149. The Dyak GE10340-Tpl94D had similar high expression in the testes (506.227 FPKM) and close to 0 FPKM for the ovaries (positive log 2-fold change of 15.3673 with a p value of 0.00698236 and q value of 0.0104932). The Dana GF19889-Tpl94D had testes expression of 78.6323 FPKM while the ovaries were close zero to (0.0116349 FPKM). The decreased expression of the Dana GF19889-Tpl94D is attributed to the sequence length of Dana GF19889-Tpl94D being the smallest among all the orthologs. The p and q values for Dana GF19889-Tpl94D were both 0 with a log 2-fold change of 12.7224. Lastly, the Dpse GA22645-Tpl94D had testes expression of 232.614 FPKM and ovaries expression of 0.054751 FPKM with a p value of 0.000115653 and q value of 0.00159621 (log 2-fold change of 12.0528). The log 2-fold change was approximately the same across all orthologs with the exception of Dsim GD20990-Tpl94D and D. melanogaster Tpl94D_B due to 0 expression being found for respective sequences in ovaries. The gene orthologs for Tpl94D had high expression in the testes as compared to the ovaries.
To confirm the differential expression analysis for testes and ovaries in D. melanogaster, D. simulans, D. yakuba, and D. pseudoobscura, we compared the results to published data in ModENCODE,30 Flybase,31 NCBI EST32,33 and NCBI (File S5).34 A better consensus on the differential expression for the testes and ovaries RNA-Seq datasets for D. ananassae was established through the use of 2 additional approaches because there is only one known RNA-Seq testes and ovaries data set for D. ananassae.34
Tpl94D orthologs alignments and resulting phylogenetic analysis
The results of the sensitivity analysis for the Tpl94D orthologs are shown in Figure S9.35 A stable alignment was found to exist when the gap open penalty (GOP) value varied from 5 to 50 while the gap extension penalty (GEP) was constant at a value of 10. Positional correspondence for amino acids across all the species required gaps to be inserted into the Tpl94D orthologs resulting in an overall length of 189 amino acids. The highest number of gaps were inserted into the D. ananassae (Dana GF19889- Tpl94D) and D. pseudoobscura sequences due to their shorter length relative to the other Tpl94D orthologs. For some sites the primary homology could not be confirmed, therefore, they are designated as ambiguous sites and were eliminated from the character matrix.36-38
The phylogenetic analysis used the portions of the protein alignment from the sensitivity analysis that were unambiguous (a total of 144 characters from character positions 1, 18– 61 and 91–189) (Figure S9). This yielded 2 most equally parsimonious trees (length = 137 steps, consistency index = 0.97 and retention index = 0.94) (Figures S9 and S10). Figure S10 shows that Tree A and Tree B differ in the placements of D. yakuba and D. erecta within the melanogaster species group. Tree A has D. yakuba as sister to the melanogaster species complex and D. erecta as sister to the clade comprised of D. yakuba and the melanogaster species complex. The topology of Tree B shows that D. yakuba and D. erecta form a clade that is sister to the melanogaster species complex.
Discussion
Genomic and transcript sequences among the 12 Drosophila species
Our results show that the best protein sequences (Table 3), genomic DNA and nucleotide transcript sequences (Table 2) have the same gene loci within a species for Tpl94D orthologs for representatives of the melanogaster species subgroup. The diversity in length for Dana GF19889-Tpl94D and Dpse GA22645-Tpl94D prevented the sequences from being found using a typical BLAST search. This meant that there was no gene loci consensus for D. ananassae and D. pseudoobscura across NCBI ORF Finder (Table 1), nucleotide BLAST (Table 2), and protein BLAST (Table 3). We were able to refine the genomic DNA and nucleotide transcript sequences through our rigorous DNA binding predictions and RNA-Seq analysis to establish Dana GF19889-Tpl94D and Dpse GA22645-Tpl94D as orthologs for Tpl94D. The other representative species of the subgenus Sophophora (D. persmilis, and D. willistoni) and representatives of the subgenus Drosophila (D. mojavensis, D. virilis, and D. grimshawi) did not have any gene loci matches within the established threshold of NCBI's ORF Finder (Table 1) for Tpl94D. All conserved regions that were found among the analyzed Drosophila species were based on one open reading frame in Tpl94D that was located at the 5′ end of each transcript sequence. This same conserved region was found at the same locus for the N-terminal HMG group box described by Rathke and colleagues4 for Tpl94D. The N terminal HMG box region is important for the replacement of histones and for the deposition of protamine-like proteins (MST35Ba and MST35Bb) and histone H1 linker-like (MST77F).4,5
Amino acid analysis for Tpl94D and conserved region
Several studies have focused on the number and the percentages of amino acids present in TPs39-41 and SNBPs.3,13,42,43 The Tpl94D orthologs found in the 12 Drosophila species analyzed in the current work are less rich in basic amino acids when compared to their mammalian counterparts, but they still share specific characteristics that classify them as TPs.6,13,27 For example, Tpl94D and mammalian TPs cause a disruption of the histone nucleosome organization to facilitate the sperm chromatin transition to a protamine bound structure.4-6,27 Jeanteur27 summarized the concentration of basic amino acids lysine (K) and arginine (R), serine (S), proline (P), cysteine (C), and tyrosine (Y) in TP1 and TP2 for H. sapiens, B. taurus, R. norvegicus, Sus scrofa (boar), Ovis aries (ram), and M. musculus. That analysis indicated that TP1 and TP2 appeared to have evolved separately from each other, and mammalian TP1 is more conserved when compared to mammalian TP2.27,40,41,44
The TPs are different from the SNBPs in that they have large variations in size and the percentages of specific amino acids.17,27 TPs are more basic than histones, but are less basic than protamines.27 This is probably due to the cascade of evolution of the SNBPs from histone H1 linker protein (H1→H1 like→ protamine-like→ true protamine).21,42
The putative Tpl94D protein orthologs found across the sequenced species of Drosophila described in the current work vary significantly in length, with the largest found in Dyak GE10340-Tpl94D (187 amino acids) and the smallest found in Dana GF19889-Tpl94D (79 amino acids) (Figure S3). Our analysis of the DNA binding domain in the Tpl94D orthologs indicates that the same 26 amino acid DNA binding region is conserved within the melanogaster species subgroup (Dsim GD20990-Tpl94D, Dsec GM26474-Tpl94D, Dyak GE10340-Tpl94D, and Dere GG11172-Tpl94D) (File S6A-G and Table S4). The species outside the melanogaster species subgroup (Dana GF19889-Tpl94D and Dpse GA22645-Tpl94D) had greater variation in number of potential DNA binding residues. This may be attributed to a decrease in the protein sequence length in those respective species.
Dana GF19889-Tpl94D had only 39 predicted DNA binding amino acid residues with 29 of those residues being predicted to be DNA binding within the conserved region (N-terminal HMG box/DNA binding). Dana GF19889-Tpl94D is a small protein with a sequence length of 79 amino acids and a high concentration of DNA binding amino acid resides in the conserved region. In contrast, the Dpse GA22645-Tpl94D conserved region had approximately the same percentage of amino acid residues predicted to bind DNA compared to the whole protein (48%). Overall, the putative DNA binding regions were found mainly within their respective conserved regions (File S6A-G and Table S4). All Tpl94D orthologs had low numbers of cysteine amino acid residues, which is similar to mammalian TP1 and TP2 (Figure S3 and File S2). Disulfide bonding occurs between cysteine amino acids in mammalian protamines which increases the compactness of the sperm chromatin.45,46
Interestingly, a similarity between the mammalian TPs and the Tpl94D orthologs is the concentration of tyrosine in the conserved region. Among the Tpl94D protein orthologs, the tyrosine concentration averages 3% (Figure S2 and File S1) in the whole protein. In contrast, in the conserved region the tyrosine concentration averages 6% (Figure S4 and File S3). The average tyrosine concentration within the conserved region for Tpl94D orthologs is 2% greater than the average tyrosine concentration found within the 12 sequenced Drosophila male specific transcript (MST) 35 Ba/Bb orthologs.13 The concentration of tyrosine amino acid residues appears to be important in destabilizing the chromatin compactness thus allowing the histone-bound nucleosome to become protamine-bound.27
The Tpl94D orthologs are rich in arginine (R) amino acid residues as compared to lysine (K) for all the orthologs except for Dpse GA22645-Tpl94D (Table 4). The increased number of arginine (R) residues probably increases protein affinity for DNA binding during chromatin condensation.8,22 Also arginine (R) has higher hydrogen bond potential as compared to lysine (K).8 This allows chromatin to be more protected from DNA damaging sources.8 These Drosophila TPs are less basic than both histone H1 linker-like and protamine-like proteins (Table 4; Figure S2; File S1).8 This is unlike their mammalian counterparts.
Conserved functional domains in Tpl94D
The functional domains shown in Table 5 and Table 6 are present in the protein orthologs and their respective N terminal conserved regions. Rathke and co-workers4,5 found a high mobility group (HMG) box that spanned from amino acid residue 4 through 84 in Tpl94D. The functional domains listed in Table 5 and Table 6 illustrate that HMG proteins are highly conserved chromosomal proteins that have DNA binding properties47 and are often involved in transcription.48 The conserved HMG box in Tpl94D has been postulated to be involved in the disruption of nucleosomal structure during the histone to protamine transition in Drosophila.4,49
A consensus of InterProScan 5, Phyre2, and HMMER found a large overlap of an HMG box within the conserved region described in the current work. The HMG box partially overlapped with the DUF1074 family of proteins. The functionality of DUF1074 family of proteins is currently unknown, although DUF1074 is part of the HMG box-like super family that includes 6 other protein families. These six protein families are CHDNT, DUF1014, DUF1073, DUF1898, HMG Box and YABBY, which have been annotated by the Sanger Institute.50 The secondary and tertiary 3D model wire frame structures of the conserved regions for the putative Tpl94D orthologs found in the current work appear to be nearly identical to each other. Furthermore, these secondary and tertiary wire frame structures are similar to known HMG boxes and DNA binding proteins (Fig. 3). The HMG structure is known for its 3 α helices, which appear to be similar to the DNA-binding motif found in histone H1 linker-like proteins.12,14 The conserved Tpl94D region aligns with the secondary and tertiary 3D models of the conserved region found in Drosophila protamine-like proteins (Fig. 5).13 A T-Coffee alignment of the Tpl94D orthologs and Drosophila protamine-like proteins indicates conservation (Fig. 6). In this alignment, the first translated exon for D. pseudoobscura GA18970 (Dpse GA18970/GA31252-MST35Ba/MST35Bb) was used because the length of the protein is 569 amino acids. A recent annotation update to Flybase has indicated that the first exon for D. pseudoobscura GA18970 is a separate gene called GA31252, but other annotation sites such as ENSEMBL still refer to this exon as part of GA18970.29,31 Additionally, the first translated exon for Dpse GA18970/GA31252 -MST35Ba/MST35Bb contains the conserved region found among the rest of the protamine-like and Tpl94D orthologs.13 When the whole protein sequence of Dpse GA18970/GA31252 -MST35Ba/MST35Bb is used, the same conserved region is found when aligned with rest of MST35Ba/MST35Bb and Tpl94D orthologs (Figure S6).
Figure 5.

Phyre2 Tpl94D orthologs, MST35Ba, MST35Bb orthologs, Dpse GA18970 Exon 1 (GA31252) best sequence matches conserved DNA binding region Tertiary structure alignment of a wire frame model for the Tpl94D orthologs. The different colors indicate each of the species shown on the bottom right.
Figure 6.

T-Coffee Alignment of Tpl94D orthologs with MST35Ba and MS35Bb orthologs. A T-Coffee alignment of the whole protein Tpl94D conserved region and the whole proteins of the Drosophila protamine-like proteins found in the 12 sequenced Drosophila species shows high conservation of the conserved regions with a T-Coffee consensus score of 72. D. pseudoobscura GA18970s first exon (GA31252) was used due to size length of the whole protein being 569 amino acids.
Dana GF19889-Tpl94D and Dpse GA22645-Tpl94D are conserved at the N-terminal HMG box-DNA binding region when aligned with both MST35Ba/MST35Bb and Tpl94D orthologs. In contrast, the N terminal HMG box-DNA binding region of the Drosophila protamine-like protein orthologs is conserved with the C-terminal end of Tpl94D within the melanogaster species subgroup (Dsim GD20990-Tpl94D, Dsec GM26474-Tpl94D, Dyak GE10340-Tpl94D, and Dere GG11172-Tpl94D) (Fig. 6). The melanogaster species subgroup contains a conserved sequence identified as c2yrqA in the Protein Databank (PDB), which spans from the N to the C terminus (Table 6). C2yrqA is known to be involved in DNA binding and contains a HMG box (Table 6). Dere GG11172-Tpl94D aligns 2 PDB proteins (c2e6oA and d1v64a) that span from the middle of the protein sequence to the C terminus. PDB proteins (c2e6oA, c2cs1A, d1v64a, d1hmfa, and c2yrqA) indicated in Table 6 are present in the conserved region in the Tpl94D orthologs (Table 5). The variation in the protein alignments of the Drosophila protamine-like protein (MST35Ba and MST35Bb) orthologs and Tpl94D orthologs can be attributed to vast sequence length differences.13
The conserved regions in Tpl94D protein orthologs and Drosophila protamine-like protein orthologs appear to have the same primary function of binding DNA during Drosophila spermatogenesis as reflected by the T-Coffee alignment (consensus score = 93; Fig. 7). Hence, both conserved regions have a similar function of binding DNA through their respective highly basic HMG box during spermiogenesis.
Figure 7.

T-Coffee Alignment of Tpl94D and Drosophila protamine-like protein (MST35Ba and MST35Bb) conserved regions. T-Coffee alignment of the DNA binding-HMG box conserved regions in Drosophila Tpl94D and Drosophila protamine-like proteins (MS35Ba and MST35Bb) orthologs. The area in red indicates strong conservation. Consensus score equals 93. Also D. pseudoobscura GA18970s first exon (GA31252) contains the conserved region for the D. pseudoobscura MST35Ba/Bb ortholog.
RNA-Seq transcriptome and isoform analysis of Tpl94D
Collectively, the results (File S5) of the transcriptome RNA-Seq analysis of D. melanogaster, D. simulans, D. yakuba, D. ananassae, and D. pseudoobscura reveal that all protein orthologs for Tpl94D are highly expressed in the testes. Dyak GE10340-Tpl94D was reconfirmed to be testes specific by NCBI expressed sequence tag (EST) MEGABLAST.32,33 The testes and ovaries expression results in Cuffdiff2 for Dpse GA22645-Tpl94D yielded similar expression results as published by Van Kuren and Vibranovski.34 Our FPKM differential expression result for Dana GF19889-Tpl94D was lower when compared to Van Kuren and Vibranovski.34 This may be attributed to the different approach for mapping the reads to the reference genome and the quality assessment during the pre-processing stage. Regardless, our RNA-Seq differential expression results and Van Kuren and Vibranovski34 showed high expression for Dana GF19889-Tpl94D in the testes as compared to the ovaries. Overall, Dana GF19889-Tpl94D had comparable log fold change values in EdgeR (File S5).51 Additionally, DESeq was utilized to further test the differential expression of D. ananassae RNA-Seq testes and ovaries data (File S5).52 DESeq revealed high expression in the testes as compared to the ovaries for Dana GF19889-Tpl94D. D. melanogaster Tpl94D and Dsim GD20990-Tpl94D were verified to be highly expressed in the testes by analyzing the gene loci locations in the genome browser in ModENCODE.30 Likewise, the expression of Dpse GA22645-Tpl94D in the testes was verified by analyzing the gene loci location using Flybase and ModENCODE. These Tpl94D orthologs have small p and q values, which signifies confidence in differential expression FPKM values from Cuffdiff2.53 Heatmaps were generated using CummeRbund in R Studio to show the high expression of Tpl94D orthologs in the testes as compared to ovaries (Figure S7).54 This analysis showed that Tpl94D_A isoform (FBtr0084339) is more highly expressed than Tpl94D_B isoform (FBtr0310110) in D. melanogaster testes. Additionally, NCBI IUTA analysis shows that Tpl94D_A isoform (FBtr0084339) is the dominant isoform of the Tpl94D (FBgn0051281) gene as compared to Tpl94D_B isoform (FBtr0310110) in D. melanogaster testes (Figure S8). Our RNA-Seq transcriptome expression results across the available sequenced Drosophila species show that Tpl94D orthologs are highly expressed in the testes and have a similar role to Tpl94D in D. melanogaster during spermatogenesis.
Some RNA-Seq data sets presented in this study contained testes and ovaries with tracts30,31 and without tracts.34 There was minimal differential expression difference for Tpl94D orthologs between whole reproductive organs with tracts versus organs without tracts. Additionally, our differential expression results for Tpl94D and its orthologs in D. melanogaster (control), D. simulans, D. ananassae, D. yakuba, and D. pseudoobscura were very similar to the genome-wide studies conducted by ModENCODE;30 Flybase;31 Begun et al.;33 Begun et al.;32 and Van Kuren and Vibranovski.34
Phylogenetic distribution and features of Tpl94D orthologs among drosophild flies
All of these Tpl94D orthologs exhibit the characteristic HMG box at the N-terminus and a high degree of DNA binding amino acids. A sensitivity analysis of the amino acid sequence alignment was another approach corroborating that the N-terminus HMG box is more conserved (unambigious) across species (Figure S9). Because sequence alignments establish characters used to build evolutionary trees they are also sensitive to species sampling.37 Thus, in the future, when additional Tpl94D sequences are available, we anticipate that there will be fewer gaps and unambiguous sites in the sequence alignments, and that the features of Tpl94D orthologs will be better understood.
As one progresses to hierarchical levels in the phylogeny further from D. melanogaster (Figure S10), the variation in the amino acid length of Tpl94D increases. In fact, the D. ananassae (Dana GF19889-Tpl94D) and D. pseudoobscura (Dpse GA22645-Tpl94D) orthologs required further corroboration through RNA-Seq analysis of their testes and ovaries transcriptome datasets.
The current work does not identify putative transition protein-like proteins in the other Drosophila species, however, they may exist. Our inability to identify Tpl94D orthologs in those species might be due to greater variation in sequence from the D. melanogaster Tpl94D reference sequence. Currently, there are no available testis or ovary transcriptome data sets for D. sechellia, D. erecta, D. persmilis, D.willistoni, D. mojavensis, D. virilis, and D. grimshawi (Table 1).
The phylogenetic analysis yields 2 (Tree A and Tree B) most equally most parsimonious trees (Figure S10). The topology of Tree A in Figure S10 more accurately reflects the taxonomic groupings and well-established phylogeny when all 9 species within the melanogaster species subgroup are included in analyses.31,55,56 The topology of Tree B in Figure S10 depicts an anomalous sister relationship between D. yakuba and D. erecta forming a clade that is sister to the melanogaster species complex. This topology has been seen previously by 12 Drosophila Consortium and Flybase.31,56 Phylogenetic analyses are sensitive to species sampling; therefore, this anomaly is most likely due to the reduced number of species represented within the melanogaster species subgroup in the phylogenetic analyses.
Summary
The work presented here indicates that the orthologs for Tpl94D are present in the sequenced Drosophila species of the melanogaster species subgroup (D. simulans, D. sechellia, D. erecta, and D. yakuba), D. ananassae, and D. pseudoobscura. The RNA-Seq differential expression data for D. melanogaster, D. simulans, D. yakuba, D. ananassae, and D. pseudoobscura indicates a high expression of Tpl94D and its respective orthologs in the testes as compared to the ovaries. Additionally, Drosophila Tpl94D orthologs share a conserved DNA-binding region with Drosophila protamine-like proteins. The conserved HMG box among all the Tpl94D orthologs has been postulated to be involved in the disruption of nucleosomal structure, which facilitates the transition from histone-bound nucleosome chromatin to a protamine-bound chromatin structure in Drosophila.4,49 In addition, the rigorous bioinformatic methodology used in the work reported here can be used to annotate Tpl94D orthologs in any newly sequenced Drosophila species found within the melanogaster species group. We suggest that the Drosophila Tpl94D orthologs should be classified as their own transition protein group.
Materials and Methods
Nucleotide BLAST and protein BLAST on transition protein (Tpl94D)
The reference genomic, transcript, and protein sequences for D. melanogaster transition protein Tpl94D were acquired from NCBI and Flybase. A nucleotide BLAST, protein BLAST, and Position-Specific Iterated (PSI)-BLAST were conducted on the original 12 sequenced Drosophila genomes:56 D. melanogaster, D. simulans, D. sechellia, D. erecta, D. yakuba, D. ananassae, D. pseudoobscura, D. persmilis, D.willistoni, D. mojavensis, D. virilis, and D. grimshawi. Potential orthologs were identified for transition protein Tpl94D using BLASTX and NCBI open reading frame finder (ORF finder). The cut off threshold for Tpl94D open reading frame orthologs was query coverage of 40% with maximum identity score of 36% and an E-value of 7 × 10−5. The best protein matches for Tpl94D were analyzed for conserved domains by the local alignment tool T-Coffee (http://tcoffee.crg.cat/apps/tcoffee/).57
Functional analysis (DNA Binder, BindN+, and DP-Bind) in Tpl94D
The DNA binding bioinformatic tools DNA Binder, BindN+, and DP- Bind, were used to analyze each of the best protein matches for Tpl94D and their respective conserved domains for prospective DNA binding regions. DNA Binder uses a regression based algorithm through support vector machines (SVM) models to determine whether a protein sequence is involved in DNA binding (http://www.imtech.res.in/raghava/dnabinder/).58 Three defined datasets called realistic, alternative, and main set parameters are used to determine whether the user defined protein sequence is DNA binding. The realistic data sets contain 146 DNA binding proteins and 1500 non DNA binding proteins with the analysis parameters set to 47.95% for sensitivity, 93.33% for specificity, and 89.31% accuracy. The alternative dataset is the largest of the 3 data sets with 1153 DNA binding proteins and 1153 non-DNA binding protein chains. The main dataset is the smallest of the 3 types of data sets provided in DNA Binder and is primarily used in the identification of DNA binding regions and domains within a large protein sequence. The main dataset contains 146 DNA bind proteins and 250 non-DNA binding proteins with the analysis parameters set to 78.11% for sensitivity, 80.80% for specificity, and 79.80% for accuracy. The provided sequence is considered as DNA binding if the score is close or above 1 in DNA Binder. In contrast, a non-DNA binding score will be closer to −1 or less. In the case of a score is in between −1 and 1 and is close to zero then the provided protein sequence may or may not be a DNA binding domain.58
The BindN+ uses 2 data sets (PDNA-62 and PRINR25) from the Protein Data Bank (PDB) to analyze user defined amino acid sequences in FASTA format for potential to bind to DNA. The evolutionary information in regards to the user defined amino acid sequence is acquired in BindN+ by searching through UniPortKB and PDB (PDNA-62 and PRINR25) databases. The analysis in BindN+ was conducted using the recommended settings of 79% for the specificity. The results in BindN+ are given a score of positive or negative with confidence score under each amino acid ranging from one to 9 with one being the least confident and 9 being the most confident.59
Lastly, DP-Bind was also used to analyze the probability of the user defined the amino acid sequences to bind to DNA. DP-Bind returns highly sensitive and conservative results as compared to BindN and BindN+.60,61 DP-Bind determines a user defined amino acid sequence based on 3 different approaches:56 support vector machines (SVM),56 kernel logistic regression (KLR), and62 penalized logistic regression (PLR). The three approaches in DP-Bind use non-redundant datasets of 62 experimentally determined structures of proteins that have been shown to bind to double-stranded DNA. These three algorithms are combined with position-specific scoring matrix (PSSM) in PSI-BLAST that are used to generate a score of one (DNA binding) or zero (not DNA binding) for each amino acid in the user defined sequence. The combined PSSM-SVM had the following analysis parameters: 76% +/− 9.1 for accuracy, 76.7% +/− 18.6 for sensitivity, and 74.8% +/− 12.5 specificity. The combined PSSM-KLR had the following analysis parameters: 77.2% +/− 9.3 for accuracy, 76.4% +/− 18.5 for sensitivity, and 76.6% +/− 11.2 specificity. The combined PSSM-PLR had the following analysis parameters: 73% +/− 8.8 for accuracy, 73.3% +/− 18.4 for sensitivity, and 71.8% +/− 12.8 specificity. A probability score ranging from one (high probability) to zero (low probability) states the likelihood of the amino acid residue to bind to DNA. DP-Bind contained 2 additional tests called majority consensus and strict consensus. These two consensus tests summarized the results from PSSM-PLR, PSSM-KLR, and PSSM-SVM with a score of zero (not DNA binding), one (DNA binding), and not assigned (NA – cannot be determined). The majority consensus had the following set analysis parameters: 76% +/− 9.0 for accuracy, 76.9% +/− 18.6 for sensitivity, and 75.3% +/− 12.0 specificity. Likewise the strict consensus had the following set analysis parameters 80% +/− 9.4 for accuracy, 79.1% +/− 19.4 for sensitivity, and 78.6% +/− 12.7 specificity. We used the recommended approach by DP-Bind to seek a consensus of all 5 results (PSSM-SVM, PSSM-KLR, PSSM-PLR, majority consensus, and strict consensus) to determine whether each amino acid in a sequence was DNA binding or not DNA-binding.
Amino acid content analysis in Tpl94D
Sequence Manipulation Suite 2 - Protein Statistics (http://www.bioinformatics.org/sms2/protein_stats.html) was used to analyze the amino acid content for each of the NCBI Open Reading Frame (ORF) Finder, protein BLAST, Position-Specific Iterated (PSI)-BLAST, and BLASTX and conserved sequence regions in Tpl94D matches. The following published sequences were added to the comparison: Mus musculus histone H1 linker-like protein (GI: 9055232), Rattus norvegicus histone linker-like H1 domain, spermatid-specific 1, (GI: 157818369), Mus musculus spermatid nuclear TP1 (GI: 6678395), Mus musculus nuclear TP2 (GI: 31981239), Rattus norvegicus spermatid nuclear TP1 (GI: 8394472), and Rattus norvegicus nuclear TP2 (GI: 51036639).
Functional domains and tertiary models for Tpl94D
The respective NCBI ORF Finder, protein BLAST, PSI-BLAST, and BLASTX and conserved sequence regions in Tpl94D matches were analyzed for functional domains through EMBL-EBI's Interpro Scan 5 (http://www.ebi.ac.uk/interpro/),63 HMMER (http://hmmer.org/),64 and Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2).65 The functional groups were identified using Phyre2, Interpro Scan 5, and HMMER. The putative 3D secondary and tertiary models for each conserved regions for Tpl94D matches were modeled using Phyre2. The 3D models were then analyzed using Molsoft ICM Browser (http://www.molsoft.com/).
RNA-Seq and isoform data analysis for Tpl94D in D. melanogaster, D. simulans, D. yakuba, D. ananassae, and D. pseudoobscura
Testes and ovaries transcriptome Illumina RNA-Seq FastQ data files were acquired from publicly available EMBL-EBI-SRA based on their corresponding NCBI SRA identification codes for D. melanogaster, D. simulans, D. yakuba, D. ananassae, and D. pseudoobscura. The NCBI SRA identifications for these publicly available data sets are listed in Table S1. Quality assessment and trimming of the FastQ files was done using FastQC 0.10.1 (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc) and Trimmomatic 0.32,66 respectively. The trimmed and quality assessed FastQ files were then uploaded onto iPlant Collaborative's Discovery Environment for differential expression assessment.67 The genomic sequences and general transfer formats (GTF) for D. melanogaster 5.21, D. simulans 1.21, D. yakuba 1.3, D. ananassae 1.21, and D. pseudoobscura 2.21 were uploaded to iPlant Collaborative's Discovery Environment67 from ENSEMBL.29 All reads were then mapped using Tophat 2.0.9 with Bowtie 2.1.0 with the settings of -g 1 with species appropriate reference GTF and reference genomic sequence.53,68 The settings for Tophat 2 were acquired from Flybase (http://flybase.org). The – g 1 setting instructed Tophat 2.0.9 with Bowtie 2.1.0 to allow only 1 alignment to the provided reference genome for a given read. This was done so to have a conservative approach in mapping the reads to reference genome as the default setting is 40. All paired-end datasets were aligned with the inner mate distance of -r 150 as stated on Flybase (http://flybase.org). The rest of the parameters for Tophat 2.0.9 were left as default.
Cufflinks 2.0.2 was then used to assemble the reads with species appropriate reference GTF and reference genomic sequence. The reference genomic sequences were provided through –b/-frag-bias-correct < reference_genome.fa > setting in Cufflinks 2.0.2, which improved the accuracy of the transcript abundance by running new bias detection and by using a built-in correction algorithm.53 Multi-read correction option, –u/-multi-read-correct, was enabled during Cufflinks 2.0.2 to improve the accuracy of the reads mapped to multiple locations in the reference genome. Cuffmerge 2.0.2 was then used to merge all the GTF output files from Cufflinks 2.0.2 in a species-specific manner with the species-specific reference annotation (-g/-ref-gtf ENSEMBL GTFs) and all isoforms were discarded with abundance below 0.1. This was done to merge all novel isoforms and known isoforms to obtain maximum assembly quality.53,69 The merged output GTF from Cuffmerge 2.0.2 and the species and tissue sample appropriate output from Tophat 2.0.9 were used in Cuffdiff 2.0.2 to evaluate the differential expression between the ovaries and the testes for D. melanogaster Tpl94D orthologs in D. simulans, D. yakuba, D. ananassae, and D. pseudoobscura. In Cuffdiff 2.0.2, the default setting of 10 was used for the minimum number of counts (-c/-min-alignment-count), which signified the minimum number of alignments to be present to test the significance in change between the ovaries and testes at samples for any gene loci.53,69 The accuracy of the transcript abundance was improved by enabling fragment bias correction with species-specific genome (b/-frag-bias-correct < reference_genome.fa >) and multi-read correction (–u/-multi-read-correct) in Cuffdiff 2.0.2. Also the default false discovery rate (-FDR) of 0.05 was changed to 0.01 in Cuffdiff 2.0.2.53 The remaining conditions for Cuffdiff 2.0.2 were left as default. Heatmaps were generated using cummeRbund for the Tpl94D orthologs and isoforms in D. melanogaster, D. simulans, D. yakuba, D. ananassae, and D. pseudoobscura.54
A count-based differential expression approach was used to conduct the 2 additional RNA-Seq approaches. The Tophat 2.0.9 alignment for D. ananassae was converted to counts file by using HT-Seq counts70 with the D. ananassae 1.21 GTF from ENSEMBL.29 Then EdgeR51 and DeSeq52 was used at default settings with false discovery rate (FDR) set to 0.01 to analyze the differential expression between ovaries and testes data sets for D. ananassae. EdgeR and DeSeq were conducted on iPlant Collaborative's Discovery Environment.67
Isoforms for Tpl94D orthologs in D. melanogaster, D. simulans, D. yakuba, D. ananassae, and D. pseudoobscura were analyzed using NCBI Isoform Usage Two-step Analysis (IUTA) in R Studio with R 3.2.1.28 We created 2 array variables in IUTA that contained all the ovaries (bam.list.1) and the testes (bam.list.2) paired-end Tophat 2.0.9 alignments for each specific species. A third variable was created called “transcript.info” that indicated the species specific GTF from ENSEMBL.29 These variables were created in accordance with IUTA's manual. IUTA was run independently for each species with fragment length distribution (FLD) setting set to empirical and 3 statistical tests called SKK, CQ, and KY enabled.26,28,71,72 IUTA recommended the empirical settings to be used for the fragment length distribution for each sample group (ovaries vs. testes) per species. Pie charts were generated using IUTA to illustrate the percentage of each isoform present in the testes and ovaries for D. melanogaster, D. simulans, D. yakuba, D. ananassae, and pseudoobscura.
Phylogenetic analysis and identification of conserved regions
From NCBI Ref-Seq protein sequence for Tpl94D orthologs were identified using D. melanogaster isoform A and isoform B as the reference sequences for BLAST searches (Tables 2–3). The length of the Tpl94D orthologs varies across species. Therefore, a sensitivity analysis was run to create an unbiased approach for placement of gaps and identification of characters by position.35 Multiple alignments were performed using the ClustalW method within the program MEGA6 under a Gonnet weight table for amino acid change where the gap extension penalty (GEP) was held constant while the gap opening penalty (GOP) varied.38,73,74 A stable alignment was found to exist when amino acid sites considered to be ambiguous were eliminated.38 Therefore, the character matrix for the phylogenetic analysis only contained unambiguous positions for the Tpl94D orthologs. An exhaustive search under a maximum parsimony criterion was run on PAUP* version 4.0a14.75 The gaps were treated as missing and the tree was rooted with the outgroup, D. pseudoobscura.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank the anonymous reviewers for suggestions that significantly improved the manuscript. We are grateful to Jennifer Hillman Jackson (Pennsylvania State University and Galaxy), Dr. Roger Barthelson (Iplant Collaborative), Dr. Sheldon McKay (Iplant Collaborative), Nirav Merchant (Iplant Collaborative), Andy Edmonds (Iplant Collaborative) and other members of the Iplant Collaborative team for their support during the transcriptome and isoform analysis. We also would like to thank Michael Campbell (Utah University) and Dr. Chris Childer (USDA) for introducing us to the Iplant Collaborative.
Supplemental Material
Supplemental Material may be downloaded here: publisher's website
References
- 1.Jayaramaiah Raja S, Renkawitz-Pohl R. Replacement by Drosophila melanogaster protamines and Mst77F of histones during chromatin condensation in late spermatids and role of sesame in the removal of these proteins from the male pronucleus. Mol Cell Biol 2005; 25:6165-77; PMID:15988027; http://dx.doi.org/ 10.1128/MCB.25.14.6165-6177.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tirmarche S, Kimura S, Sapey-Triomphe L, Sullivan W, Landmann F, Loppin B. Drosophila protamine-like Mst35Ba and Mst35Bb are required for proper sperm nuclear morphology but are dispensable for male fertility. G3 (Bethesda) 2014; 4:2241-5; PMID:25236732; http://dx.doi.org/full_text [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balhorn R. The protamine family of sperm nuclear proteins. Genome Biol 2007; 8:227; PMID:17903313; http://dx.doi.org/ 10.1186/gb-2007-8-9-227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rathke C, Baarends WM, Jayaramaiah-Raja S, Bartkuhn M, Renkawitz R, Renkawitz-Pohl R. Transition from a nucleosome-based to a protamine-based chromatin configuration during spermiogenesis in Drosophila. J Cell Sci 2007; 120:1689-700; PMID:17452629; http://dx.doi.org/ 10.1242/jcs.004663 [DOI] [PubMed] [Google Scholar]
- 5.Rathke C, Baarends WM, Awe S, Renkawitz-Pohl R. Chromatin dynamics during spermiogenesis. Biochim Biophys Acta 2014; 1839:155-68; PMID:24091090; http://dx.doi.org/ 10.1016/j.bbagrm.2013.08.004 [DOI] [PubMed] [Google Scholar]
- 6.Rathke C, Barckmann B, Burkhard S, Jayaramaiah-Raja S, Roote J, Renkawitz-Pohl R. Distinct functions of Mst77F and protamines in nuclear shaping and chromatin condensation during Drosophila spermiogenesis. Eur J Cell Biol 2010; 89:326-38; PMID:20138392; http://dx.doi.org/ 10.1016/j.ejcb.2009.09.001 [DOI] [PubMed] [Google Scholar]
- 7.Ausio J. Histone H1 and evolution of sperm nuclear basic proteins. J Biol Chem 1999; 274:31115-8; PMID:10531297; http://dx.doi.org/ 10.1074/jbc.274.44.31115 [DOI] [PubMed] [Google Scholar]
- 8.Kasinsky HE, Eirin-Lopez JM, Ausio J. Protamines: structural complexity, evolution and chromatin patterning. Protein Pept Lett 2011; 18:755-71; PMID:21443489; http://dx.doi.org/ 10.2174/092986611795713989 [DOI] [PubMed] [Google Scholar]
- 9.Yan W, Ma L, Burns KH, Matzuk MM. HILS1 is a spermatid-specific linker histone H1-like protein implicated in chromatin remodeling during mammalian spermiogenesis. Proc Natl Acad Sci U S A 2003; 100:10546-51; PMID:12920187; http://dx.doi.org/ 10.1073/pnas.1837812100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bianchi F, Rousseaux-Prevost R, Bailly C, Rousseaux J. Interaction of human P1 and P2 protamines with DNA. Biochem Biophys Res Commun 1994; 201:1197-204; PMID:8024562; http://dx.doi.org/ 10.1006/bbrc.1994.1832 [DOI] [PubMed] [Google Scholar]
- 11.Kanippayoor RLA JH, Moehring AJ. Protamines and spermatogenesis in Drosophila and Homo sapiens: a comparative analysis. Spermatogenesis 2013; 1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saperas N, Chiva M, Casas MT, Campos JL, Eirin-Lopez JM, Frehlick LJ, Prieto C, Subirana JA, Ausio J. A unique vertebrate histone H1-related protamine-like protein results in an unusual sperm chromatin organization. FEBS J 2006; 273:4548-61; PMID:16965539; http://dx.doi.org/ 10.1111/j.1742-4658.2006.05461.x [DOI] [PubMed] [Google Scholar]
- 13.Alvi ZA, Chu TC, Schawaroch V, Klaus AV. Protamine-like proteins in 12 sequenced species of Drosophila. Protein Pept Lett 2013; 20:17-35; PMID:22789106; http://dx.doi.org/ 10.2174/092986613804096847 [DOI] [PubMed] [Google Scholar]
- 14.Lewis JD, Ausio J. Protamine-like proteins: evidence for a novel chromatin structure. Biochem Cell Biol 2002; 80:353-61; PMID:12123288; http://dx.doi.org/ 10.1139/o02-083 [DOI] [PubMed] [Google Scholar]
- 15.Zhang F, Lewis JD, Ausio J. Cysteine-containing histone H1-like (PL-I) proteins of sperm. Mol Reprod Dev 1999; 54:402-9; PMID:10542381; http://dx.doi.org/ 10.1002/(SICI)1098-2795(199912)54:4%3c402::AID-MRD11%3e3.0.CO;2-X [DOI] [PubMed] [Google Scholar]
- 16.Lewis JD, Saperas N, Song Y, Zamora MJ, Chiva M, Ausio J. Histone H1 and the origin of protamines. Proc Natl Acad Sci U S A 2004; 101:4148-52; PMID:15024099; http://dx.doi.org/ 10.1073/pnas.0308721101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zini AA. A. Sperm Chromatin Biological and Clinical Applications in Male Infertility and Assisted Reproduction. New York: Springer, 2011. [Google Scholar]
- 18.White-Cooper H. Studying how flies make sperm–investigating gene function in Drosophila testes. Molecular and Cellular Endocrinology 2009; 306:66-74; PMID:19101606; http://dx.doi.org/ 10.1016/j.mce.2008.11.026 [DOI] [PubMed] [Google Scholar]
- 19.Ricketts PG, Minimair M, Yates RW, Klaus AV. The effects of glutathione, insulin and oxidative stress on cultured spermatogenic cysts. Spermatogenesis 2011; 1:159-71; PMID:22319665; http://dx.doi.org/ 10.4161/spmg.1.2.17031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Decotto E, Spradling AC. The Drosophila ovarian and testis stem cell niches: similar somatic stem cells and signals. Dev Cell 2005; 9:501-10; PMID:16198292; http://dx.doi.org/ 10.1016/j.devcel.2005.08.012 [DOI] [PubMed] [Google Scholar]
- 21.Eirin-Lopez JM, Frehlick LJ, Ausio J. Protamines, in the footsteps of linker histone evolution. J Biol Chem 2006; 281:1-4; PMID:16243843; http://dx.doi.org/ 10.1074/jbc.R500018200 [DOI] [PubMed] [Google Scholar]
- 22.Eirin-Lopez JM, Lewis JD, Howe A, Ausio J. Common phylogenetic origin of protamine-like (PL) proteins and histone H1: Evidence from bivalve PL genes. Mol Biol Evol 2006; 23:1304-17; PMID:16613862; http://dx.doi.org/ 10.1093/molbev/msk021 [DOI] [PubMed] [Google Scholar]
- 23.Barckmann B, Chen X, Kaiser S, Jayaramaiah-Raja S, Rathke C, Dottermusch-Heidel C, Fuller MT, Renkawitz-Pohl R. Three levels of regulation lead to protamine and Mst77F expression in Drosophila. Dev Biol 2013; 377:33-45; PMID:23466740; http://dx.doi.org/ 10.1016/j.ydbio.2013.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho C, Willis WD, Goulding EH, Jung-Ha H, Choi YC, Hecht NB, Eddy EM. Haploinsufficiency of protamine-1 or -2 causes infertility in mice. Nat Genet 2001; 28:82-6; PMID:11326282 [DOI] [PubMed] [Google Scholar]
- 25.Dorus S, Freeman ZN, Parker ER, Heath BD, Karr TL. Recent origins of sperm genes in Drosophila. Mol Biol Evol 2008; 25:2157-66; PMID:18653731; http://dx.doi.org/ 10.1093/molbev/msn162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu KKaJ. Modified Nel and Van der Merwe test for the multivariate Behrens–Fisher problem. Statistics & Probability Letters 2004; 66:161-9; http://dx.doi.org/ 10.1016/j.spl.2003.10.012 [DOI] [Google Scholar]
- 27.Jeanteur P. Epigenetics and Chromatin. Berlin: Springer, 2005. [Google Scholar]
- 28.Niu L, Huang W, Umbach DM, Li L. IUTA: a tool for effectively detecting differential isoform usage from RNA-Seq data. BMC Genomics 2014; 15:862; PMID:25283306; http://dx.doi.org/ 10.1186/1471-2164-15-862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al.. Ensembl 2014. Nucleic Acids Res 2014; 42:D749-55; PMID:24316576; http://dx.doi.org/ 10.1093/nar/gkt1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.mod EC, Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, et al.. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 2010; 330:1787-97; PMID:21177974; http://dx.doi.org/ 10.1126/science.1198374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.St Pierre SE, Ponting L, Stefancsik R, McQuilton P, FlyBase C. FlyBase 102–advanced approaches to interrogating FlyBase. Nucleic Acids Res 2014; 42:D780-8; PMID:24234449; http://dx.doi.org/ 10.1093/nar/gkt1-092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Begun DJ, Lindfors HA, Kern AD, Jones CD. Evidence for de novo evolution of testis-expressed genes in the Drosophila yakuba/Drosophila erecta clade. Genetics 2007; 176:1131-7; PMID:17435230; http://dx.doi.org/ 10.1534/genetics.106.069245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Begun DJ, Lindfors HA, Thompson ME, Holloway AK. Recently evolved genes identified from Drosophila yakuba and D. erecta accessory gland expressed sequence tags. Genetics 2006; 172:1675-81; PMID:16361246; http://dx.doi.org/ 10.1534/genetics.105.050336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.VanKuren NW, Vibranovski MD. A novel dataset for identifying sex-biased genes in Drosophila. J Genomics 2014; 2:64-7; PMID:25031657; http://dx.doi.org/ 10.7150/jgen.7955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wheeler WC. Sequence alignment, parameter sensitivity, and the phylogenetic analysis of molecular data. Systematic Biology 1995; 44:321-31; http://dx.doi.org/ 10.1093/sysbio/44.3.321 [DOI] [Google Scholar]
- 36.de Pinna MCC. Concepts and tests of homology in the cladistic paradigm. Cladistics 1991; 7:367-94; http://dx.doi.org/ 10.1111/j.1096-0031.1991.tb00045.x [DOI] [Google Scholar]
- 37.Brower AVZ, Schawaroch V. Three steps of homology assesment. Cladistics 1996; 12:265-72. [DOI] [PubMed] [Google Scholar]
- 38.Gatsey J, DeSalle R, Wheeler WC. Alignment ambiguous nucleotide sites and the exclusion of systematic data. Molecular Phylogenetics and Evolution 1994; 2:152-7; http://dx.doi.org/ 10.1006/mpev.1993.1015 [DOI] [PubMed] [Google Scholar]
- 39.Akama K, Oka S, Tobita T, Hayashi H. The amino acid sequence of a boar transition protein 3. J Biochem 1994; 115:58-65; PMID:8188637 [DOI] [PubMed] [Google Scholar]
- 40.Grimes SR Jr., Platz RD, Meistrich ML, Hnilica LS. Partial characterization of a new basic nuclear protein from rat testis elongated spermatids. Biochem Biophys Res Commun 1975; 67:182-9; PMID:1201018; http://dx.doi.org/ 10.1016/0006-291X(75)90300-9 [DOI] [PubMed] [Google Scholar]
- 41.Singh J, Rao MR. Interaction of rat testis protein, TP, with nucleic acids in vitro. Fluorescence quenching, UV absorption, and thermal denaturation studies. J Biol Chem 1987; 262:734-40; PMID:3805005 [PubMed] [Google Scholar]
- 42.Eirin-Lopez JM, Ausio J. Origin and evolution of chromosomal sperm proteins. Bioessays 2009; 31:1062-70; PMID:19708021; http://dx.doi.org/ 10.1002/bies.200900050 [DOI] [PubMed] [Google Scholar]
- 43.Birkhead TRH DJ, Pitnick S. Sperm Biology: An Evolutionary Perspective. Amsterdam: Elsevier/Academic, 2009. [Google Scholar]
- 44.Meistrich ML, Mohapatra B, Shirley CR, Zhao M. Roles of transition nuclear proteins in spermiogenesis. Chromosoma 2003; 111:483-8; PMID:12743712; http://dx.doi.org/ 10.1007/s00412-002-0227-z [DOI] [PubMed] [Google Scholar]
- 45.Cheng WM, An L, Wu ZH, Zhu YB, Liu JH, Gao HM, Li XH, Zheng SJ, Chen DB, Tian JH. Effects of disulfide bond reducing agents on sperm chromatin structural integrity and developmental competence of in vitro matured oocytes after intracytoplasmic sperm injection in pigs. Reproduction 2009; 137:633-43; PMID:19155332; http://dx.doi.org/ 10.1530/REP-08-0143 [DOI] [PubMed] [Google Scholar]
- 46.McBride AA, Klausner RD, Howley PM. Conserved cysteine residue in the DNA-binding domain of the bovine papillomavirus type 1 E2 protein confers redox regulation of the DNA-binding activity in vitro. Proc Natl Acad Sci U S A 1992; 89:7531-5; PMID:1323841; http://dx.doi.org/ 10.1073/pnas.89.16.7531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wagner CR, Hamana K, Elgin SC. A high-mobility-group protein and its cDNAs from Drosophila melanogaster. Mol Cell Biol 1992; 12:1915-23; PMID:1373803; http://dx.doi.org/ 10.1128/MCB.12.5.1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qin J, Kang W, Leung B, McLeod M. Ste11p, a high-mobility-group box DNA-binding protein, undergoes pheromone- and nutrient-regulated nuclear-cytoplasmic shuttling. Mol Cell Biol 2003; 23:3253-64; PMID:12697825; http://dx.doi.org/ 10.1128/MCB.23.9.3253-3264.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Travers AA. Priming the nucleosome: a role for HMGB proteins? EMBO Rep 2003; 4:131-6; PMID:12612600; http://dx.doi.org/ 10.1038/sj.embor.embor741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, et al.. The Pfam protein families database. Nucleic Acids Res 2004; 32:D138-41; PMID:14681378; http://dx.doi.org/ 10.1093/nar/gkh121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26:139-40; PMID:19910308; http://dx.doi.org/ 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010; 11:R106; PMID:20979621; http://dx.doi.org/ 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012; 7:562-78; PMID:22383036; http://dx.doi.org/ 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goff LTR, Kelley D. cummeRbund: Analysis, exploration, manipulation, and visualization of cufflinks high-throughput sequencing data. 2013. [Google Scholar]
- 55.Ashburner M, Golic KG, Hawley RS. Drosophila: a laboratory handbook, 2nd Edition. Cold Spring Harbor Laboratory. 2005. pp. 1123-1283. [Google Scholar]
- 56.Drosophila 12 Genomes C, Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W, et al.. Evolution of genes and genomes on the Drosophila phylogeny. Nature 2007; 450:203-18; PMID:17994087; http://dx.doi.org/ 10.1038/nature06341 [DOI] [PubMed] [Google Scholar]
- 57.Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang JM, Taly JF, Notredame C. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res 2011; 39:W13-7; PMID:21558174; http://dx.doi.org/ 10.1093/nar/gkr245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar M, Gromiha MM, Raghava GP. Identification of DNA-binding proteins using support vector machines and evolutionary profiles. BMC Bioinformatics 2007; 8:463; PMID:18042272; http://dx.doi.org/ 10.1186/1471-2105-8-463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang L, Huang C, Yang MQ, Yang JY. BindN+ for accurate prediction of DNA and RNA-binding residues from protein sequence features. BMC Syst Biol 2010; 4Suppl 1:S3; PMID:20522253; http://dx.doi.org/ 10.1186/1752-0509-4-S1-S3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu X, Ericksen SS, Mitchell JC. DBSI: DNA-binding site identifier. Nucleic Acids Res 2013; 41:e160; PMID:23873960; http://dx.doi.org/ 10.1093/nar/gkt617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang S, Gou Z, Kuznetsov IB. DP-Bind: a web server for sequence-based prediction of DNA-binding residues in DNA-binding proteins. Bioinformatics 2007; 23:634-6; PMID:17237068; http://dx.doi.org/ 10.1093/bioinformatics/btl672 [DOI] [PubMed] [Google Scholar]
- 62.Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al.. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 2010; 330:1775-87; PMID:21177976; http://dx.doi.org/ 10.1126/science.1196914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R. InterProScan: protein domains identifier. Nucleic Acids Res 2005; 33:W116-20; PMID:15980438; http://dx.doi.org/ 10.1093/nar/gki442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 2011; 39:W29-37; PMID:21593126; http://dx.doi.org/ 10.1093/nar/gkr367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 2009; 4:363-71; PMID:19247286; http://dx.doi.org/ 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- 66.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30:2114-20; PMID:24695404; http://dx.doi.org/ 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goff SA, Vaughn M, McKay S, Lyons E, Stapleton AE, Gessler D, Matasci N, Wang L, Hanlon M, Lenards A, et al.. The iPlant Collaborative: Cyberinfrastructure for Plant Biology. Front Plant Sci 2011; 2:34; PMID:22645531; http://dx.doi.org/ 10.3389/fpls.2011.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013; 14:R36; PMID:23618408; http://dx.doi.org/ 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 2011; 27:2325-9; PMID:21697122; http://dx.doi.org/ 10.1093/bioinformatics/btr355 [DOI] [PubMed] [Google Scholar]
- 70.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 2015; 31:166-9; PMID:25260700; http://dx.doi.org/ 10.1093/bio-informatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Muni S, Srivastava SK, Yutaka Kano. A two sample test in high dimensional data. Journal of Multivariate Analysis 2013; 114:349-58; http://dx.doi.org/ 10.1016/j.jmva.2012.08.014 [DOI] [Google Scholar]
- 72.Qin SXCaY-L. A two-sample test for high-dimensional data with applications to gene-set testing. The Annals of Statistics 2010; 38:808-35; http://dx.doi.org/ 10.1214/09-AOS716 [DOI] [Google Scholar]
- 73.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 2013; 30:2725-9; PMID:24132122; http://dx.doi.org/ 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics 2002; Chapter 2:Unit 2 3; PMID:18792934 [DOI] [PubMed] [Google Scholar]
- 75.Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (*and other methods), version 4.0a147. 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.