

Summary
Whole exome sequencing (WES) was performed on stem cell transplant donor-recipient (D-R) pairs to determine the extent of potential antigenic variation at a molecular level. In a small cohort of D-R pairs, a high frequency of sequence variation was observed between the donor and recipient exomes independent of human leucocyte antigen (HLA) matching. Nonsynonymous, nonconservative single nucleotide polymorphisms were approximately twice as frequent in HLA-matched unrelated, compared with related D-R pairs. When mapped to individual chromosomes, these polymorphic nucleotides were uniformly distributed across the entire exome. In conclusion, WES reveals extensive nucleotide sequence variation in the exomes of HLA-matched donors and recipients.
Keywords: alloreactivity potential, stem cell transplant, whole exome sequencing, graft-versus-host disease, single nucleotide polymorphism
Donor-recipient (D-R) alloreactivity may result in either graft-versus-host disease (GVHD) or graft rejection in allogeneic stem cell transplantion (SCT) recipients, compromising outcomes. In human leucocyte antigen (HLA) matched SCT, alloreactivity derives, in part, from minor histocompatibility antigen (mHA) differences (Shlomchik, 2007). Incompatibility in mHA, caused by single nucleotide polymorphisms (SNP) in the genome, results in the recognition of recipient oligopeptides as new (non-self) antigenic epitopes by donor T cells, contributing to the development of GVHD (Goulmy et al, 1996). This implies that, because of the unmeasured minor histo-incompatibility between donors and recipients, outcomes in SCT remain probabilistic despite increasingly stringent HLA matching and improvements in SCT technique (Horowitz, 2009). Profiling known mHA is of limited utility in the larger context of population-based donor identification because the immunogenicity of specific mHA depends on the HLA phenotype of the patient, as individual mHA are presented efficiently only on certain HLA molecules and not on others (Spellman et al, 2009; Griffioen et al, 2012). Thus, donor selection algorithms in use at present leave the recipient at risk for GVHD due to the lack of knowledge of the larger antigenic landscape orchestrated by the antigen presenting cells, and as viewed from the frame of reference of the donor immune effector cells such as T and B cells. Hypothetically, such a landscape would incorporate information on the catalogue of mHA ‘visible’ to the donor and recipient T cells, and thus would represent an ‘alloreactivity potential’ for a D-R pair.
Next generation sequencing (NGS) allows a comprehensive examination of genomic variation between SCT donors and recipients (Lind et al, 2010). In this report we focus on whole exome sequencing (WES), which assays only those nucleotides that code for proteins. It is likely that variation in the exome is a major source of alloreactivity because of its influence on mHA, and knowledge of the entire library of antigenic disparity will yield mechanistic insights into the pathobiology of SCT.
Methods
Cryopreserved pre-transplant DNA samples from patients and their donors were acquired for WES after obtaining approval from the Virginia Commonwealth University Institutional Review Board. The D-R pairs were matched at the allele level for HLA-A, B, C and DRB1 (Tables SI and SII). Four matched related donor (MRD) and 5 unrelated donor (MUD) pairs were selected. TruSeq exome enriched libraries were prepared from the DNA samples following standard Illumina protocol. Donor and recipient sequences were compared with each other to identify all the SNPs in the D-R pair (Table I). The annotated SNPs between donor and recipient samples were coded according to functionality as being either synonymous or nonsynonymous (see Glossary in supplementary material), and amongst the latter as either conservative or nonconservative or stop. The direction, or vector, of the SNPs was analysed such that, if the recipient sample contained a polymorphism not present in the donor, the SNP at that position was counted as being in the graft-versus-host (GVH) direction. Reciprocally, if the recipient sample did not contain a polymorphism present in the donor this was counted as being in the host-versus-graft (HVG) direction. The total counts for functional SNPs per pair are reported. To normalize the results based on the acquired WES data, the chromosomal positions common between the D-R pair samples, regardless of presence or absence of polymorphisms, were determined and used to calculate a normalized SNP count per functional group per pair based on the following equation:
Table I.
Whole exome sequencing of donor and recipient DNA, and sequence comparison to generate alloreactivity potential.
| TruSeq exome enriched libraries prepared from de-identified, Donor-Recipient pair DNA samples Illumina protocol | |
|---|---|
| 1 | DNA fragmentation, adapter ligation and amplification performed |
| 2 | Libraries validated on BioAnalyser, quantified using real time (quantitative) polymerase chain reaction (qPCR) and pooled |
| 3 | Exome enrichment. Two hybridizations performed using target-specific biotinylated oligos followed by binding to magnetic streptavidin beads and three washes. PCR amplification of enriched product performed. Validation and sequencing on Illumina HiSeq 2000 with 4–8 samples per lane |
| 4 | The ~100 bp paired end FASTQ reads generated by the sequencer run through the Next-generation Sequencing Quality Control (NGS QC) Toolkit (Patel & Jain, 2011) to select high quality (HQ) reads, i.e., reads where at least 70% of the bases had a quality score of ≥25. An average 20% of reads were excluded due to this HQ filtering |
| 5 | HQ reads aligned to the Human Genome (hg18) using CLC Bio Assembly Cell version 3.22. >91% of the HQ reads aligned with at least 95% of the bases matching over 95% of the read length. The alignments converted to the industry-standard Binary sequence Alignment/ Map (BAM) format. |
| 6 | Sequence Alignment/Map (SAM) tools (Li et al, 2009) used to remove PCR duplicates from the BAM files as these may bias subsequent single nucleotide polymorphism (SNP) calling. All samples with at least 28× average coverage of the entire human exome, ensuring credible and accurate SNP calling |
| 7 | SNP calling performed with preprocessed BAM files using the Broad Institute’s Genome Analysis Toolkit (McKenna et al, 2010) (GATKv1.6). The GATK SNP calling (DePristo et al, 2011) involved three steps; 1. DNA insertion-deletion (INDEL) realignment; 2. Quality score recalibration; 3. SNP discovery and genotyping. The SNP caller generates a multi-sample variant-calls file (VCF) |
| 8 | The multi-sample VCF file filtered to remove chromosomal positions that did not have at least 10× coverage and did not exceed 5009 coverage. Insertion/deletion variants removed using VCFtools software (v.0.1.9.0) (Danecek et al, 2011) |
| 9 | Each sample was separated from the multi-sample VCF file into individual files and positions containing missing genotype data removed. Given that the original VCF file contained multiple samples, every alternate allele that occurred in any of the samples was represented |
| 10 | To annotate the SNPs, the alternate allele and genotype data was transformed into an ANNOVAR-acceptable format primarily consisting of a single alternate allele and a genotype containing only combinations of zero and one |
| 11 | Transformed data samples underwent independent comparison and annotation. (i) Recipient samples were compared to the actual donor and to every other donor sample to generate actual matches (recipient with its human leucocyte antigen [HLA]-matched donor) and simulated donor-recipient matches (recipient with other, HLA-unmatched donor). (ii) For annotation, files were first filtered to remove any positions where the genotype was the same as the reference and then annotated using ANNOVAR (v.2012 Mar 08) (Wang et al, 2010) |
| 12 | The sample-pair comparison files were then combined with the annotation files by comparing the variant alleles in sample 1 and sample 2, then annotating the variant position based on which sample contains the SNP |
Normalization of the data allows direct comparison of different donor-recipient pairs because the number of chromosomal positions sequenced is slightly different for each sample and thus, only those chromosomal positions that were sequenced in both the donor and the recipient were considered.
Results
Marked whole exome sequence difference between HLA matched donors and recipients
Whole exome enriched libraries were prepared from the nine D-R pair DNA samples (Table SIII). WES data were considered examining all the differences at the SNP level. The exome sequence of each recipient was compared with their actual donors and also with donors from the other D-R pairs sequenced, in a simulated-matching analysis. The average difference between the whole exome of actual HLA-matched donors and recipients was large, averaging 13 423 SNPs per HLA-matched D-R pair, of which an average 6445 were non-synonymous. There was no substantial difference in the number of SNPs between simulated D-R pairs as compared to actual D-R pairs (Fig S1). This observation implies that sequence variation across the exome in SCT donors and recipients is frequent and independent of HLA matching.
Greater exome variation in HLA-matched unrelated donors
WES differences between actual HLA-matched unrelated and related donors were further characterized to determine the alloreactivity potential that exists in each D-R pair. Nonsynonymous SNP frequency in each D-R pair, when normalized for the number of common bases, varied substantially between unrelated (median 0·18 SNP/Kbp nucleotides sequenced) and related donors (0·11 SNP/Kbp; Mann–Whitney U-test P = 0·016. Table SIV; Fig 1A). When differentiated by nonsynonymous, nonconservative varaints, MUD SCT recipients once again had a higher measure of sequence variation when compared with MRD (0·12 vs. 0·07 SNP/kbp respectively; P = 0·016) (Fig 1B). Further, non-conservative polymorphisms were consistently more frequent in all the pairs sequenced. Thus, in this cohort of patients, greater exome variation and thus, alloreactivity potential, was observed in patients with MUD SCT.
Fig 1.
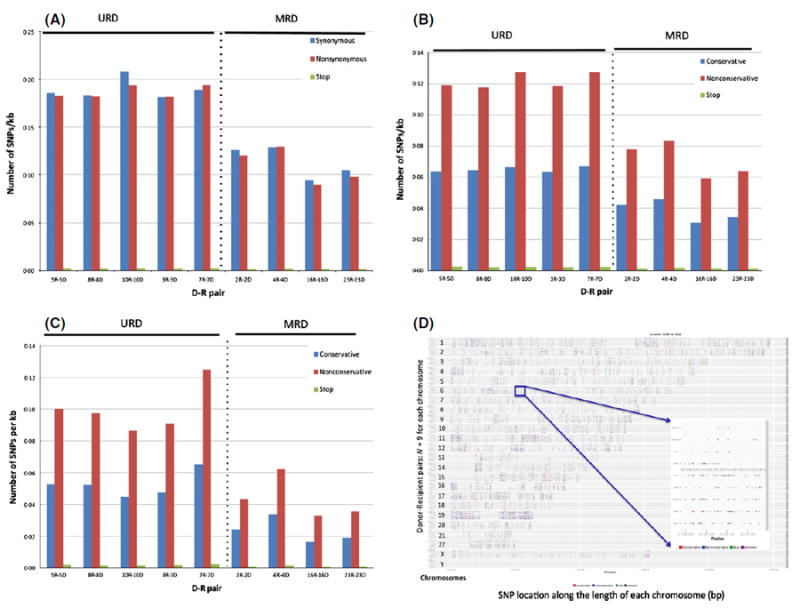
(A–C) Donor-recipient alloreactivity potential. (A) Normalized data from D-R pairs depicting synonymous versus nonsynonymous differences across whole exome. (B) Normalized, nonsynonymous D-R exome variation accounting for conservative, nonconservative substitutions and stop polymorphisms (either, stop-gain or stop-loss). (C) Alloreactivity potential vectors depicting conservative versus nonconservative, nonsynonymous nucleotide variation in D-R pairs across the whole exome in the (C) Graft-versus-host direction, variants present in the recipient and absent in the donor. D-R pairs 3, 5, 7, 8 and 10 underwent MUD SCT; 2, 4, 16 and 23 MRD. D-R pairs 3, 7 and 10 had gender-mismatched SCT (Male/Female) all MUD. (D) Nonsynonymous SNPs mapped on to individual chromosomes in all the D-R pairs, demonstrate the genomic location of polymorphisms along the length of the chromosome in each D-R pair. All nine D-R pairs depicted on the y-axis for each chromosome; SNP coordinates (location) along the length of each chromosome depicted on the x-axis (Data also shown in Fig S2B). Inset shows the major histocompatibity complex (MHC) region on chromosome 6p22. D-R pairs above dotted line (inset) from MRD with less sequence variation compared with URD. Red dots: nonsynonymous, conservative polymorphisms; blue dots: nonsynonymous, nonconservative polymorphisms; green dots: stop polymorphisms. D, donor; R, recipient; MRD, matched related donor; MUD, matched unrelated donor; SCT, stem cell transplantation; SNP, single nucleotide polymorphism.
Equal alloreactivity potential vectors in the GVH and HVG directions
Polymorphisms present in the recipient and absent in the donor are more likely to result in GVHD because the donor T cells would lack tolerance to the mHA in the recipient tissue. As with total allo-reactivity potential, the nonconservative variants in the GVH direction were more prevalent in the exomes of MUD SCT recipients (P = 0·016; Fig 1C, Table SIV). Importantly, the alloreactivity potential vectors (frequency of polymoprhisms in either the GVH or HVG direction) were of a similar magnitude in both the GVH and HVG directions for both the whole exome as well as the major histocompatibility complex (MHC) locus (Fig S2A, Table SIV).
Uniform distribution of SNPs across the exome
To determine whether these polymorphisms were concentated in certain regions within the exome they were mapped to the individual chromosomes in each D-R pair. A composite figure depicting the findings demonstrates that the polymorphisms are distributed over the entire genome, and in these HLA-matched individuals appear to be uniformly distributed, even when the MHC region is considered (Fig 1D, Fig S2B). This is true for both the GVH and HVG directions (Fig S2C, Tables SV and SVI).
Discussion
Stringent HLA matching criteria for donor selection has diminished the relative-risk of GVHD in SCT; nevertheless, the incidence of this complication is still substantial despite intensive immunosuppression following SCT (Lee et al, 2007). This is so because the risk for GVHD in both its acute and chronic forms is affected by the level of overall genetic disparity between donors and recipients serving as a trigger for immune response. However, despite the many mHA recognized (>30), identifying unique mHA relevant in individual, HLA-matched D-R pairs remains challenging. This leads to the need for exploring techniques, such as WES, to give an accurate estimate of such variation. In this paper we report WES results on a small cohort of patients, demonstrating a large magnitude of variation across the exomes of SCT donors and recipients. Although the size of our study cohort precludes comparison of WES with established genetic variables, such as the known mHA (e.g. HA-1, HA-2, H-Y antigens) (Miklos et al, 2005; Spellman et al, 2009), a major advantage of WES is that any variation between the donor and recipient will be accounted for, providing a quantitative understanding of transplant immuno-biology. As an example, in our data set, greater variation was consistently observed in MUD compared with MRD D-R pairs, providing a biological basis for greater alloreactivity in MUD SCT. It is to be noted that the oligopeptides resulting from exome variation may not all have equal immunogenicity because of factors like variable binding affinity of the ‘non-self’ oligopeptides for the HLA molecules in a unique D-R pair, or variable expression of the SNP-bearing genes. However, the more numerous variant SNPs are in a given D-R pair, logically, the greater the probability of variant peptides being presented to donor T cells, with alloreactivity developing in the recipient following SCT. These findings imply that in HLA-matched donor recipient pairs, there exists an extensive ‘library’ of potentially immunogenic sequence differences, not accounted for by conventional histocompatibility testing techniques. Indeed, our study raises the important question of why all patients undergoing SCT do not develop alloreactivity?
Given the small cohort of patients examined in this study, the clinical value of WES in donor selection algorithms remains to be determined; however, we posit that, if determined in large cohorts of patients, measuring D-R alloreactivity potential by WES may lead to an improved understanding of GVHD pathogenesis. This may in the future help optimize immunosuppressive therapy following transplantation and maximize treatment benefit.
Supplementary Material
Data S1. Materials.
Fig S1. WES to quantify SNPs between SCT D-R pairs.
Fig S2. (A) Alloreactivity potential vectors depicting conservative vs. nonconservative, nonsynonymous nucleotide variation in D-R pairs across the whole exome in the host versus graft direction, variants absent in the recipient and present in the donor. (B) Nonsynonymous SNPs in the graft versus host direction mapped on to individual chromosomes in all the D-R pairs demonstrate the genomic location of polymorphisms along the length of the chromosome in each D-R pair. All nine D-R pairs depicted on the y-axis for each chromosome; SNP coordinates (location) along the length of each chromosome depicted on the x-axis. (C) Nonsynonymous SNPs in the host versus graft direction mapped onto each chromosomes (data presented in Fig 1D).
Table SI. Donor-Recipient characteristics and patient outcomes observed in patients transplanted using ATG + 450 cGy TBI.
Table SII. HLA type of the donor-recipient pairs.
Table SIII. Sample statistics for the nine D-R pairs sequenced. The numbers in the parentheses denote percentage with respect to the number of HQ reads.
Table SIV. Median normalized SNP/Kbp for whole exome and for the 6p22·1–21·2 (MHC) locus in the graft versus host, host versus graft and direction independent SNPs.
Table SV. Total SNP counts/functional polymorphism for each D-R pair over the entire exome.
Table SVI. Total SNP counts/functional polymorphism for each D-R pair for the 6p22·1–21·2 (MHC) locus.
Acknowledgments
The authors gratefully acknowledge Ms. Cheryl Jacocks-Terrell for her help in manuscript preparation. This research was supported by grant funding from Virginia’s Commonwealth Health Research Board and by the VCU, Massey Cancer Center pilot project grant. Sequencing was performed in the Nucleic Acids Research Facilities, analysis was provided by the Bioinformatics Computational Core Laboratories, and performed in the computational environment of the Center for High Performance Computing, all at Virginia Commonwealth University. Dr. Neale and Mr. Sheth are supported by Commonwealth Health Research Board grant #236-11-13.
Conflict of interest
Dr. Toor and Dr. Manjili have received research support from Sanofi-Aventis, manufacturers of Thymoglobulin.
Footnotes
Authorship
JS, performed research, wrote the paper; NS, performed research, wrote the paper; VK, performed research, wrote the paper; AS, performed research, wrote the paper; CR, performed research, wrote the paper; MJL, performed research; MS, performed research; VL, performed research; AFG, performed research; MM, designed research, wrote the paper; GB, designed research, wrote the paper; MN, designed research, wrote the paper; AT, proposed and designed research, wrote the paper.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
References
- Danecek P, Auton A, Abecasis G, Albers A, Banks E, DePristo A, Handsaker E, Lunter G, Marth T, Sherry T, McVean G, Durbin R 1000 Genomes Project Analysis Group. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulmy E, Schipper R, Pool J, Blokland E, Falkenburg JH, Vossen J, Gratwohl A, Vogelsang GB, van Houwelingen HC, van Rood JJ. Mismatches of minor histocompatibility antigens between HLA-identical donors and recipients and the development of graft-versus-host disease after bone marrow transplantation. The New England Journal of Medicine. 1996;334:281–285. doi: 10.1056/NEJM199602013340501. [DOI] [PubMed] [Google Scholar]
- Griffioen M, Honders M, van der Meijden E, van Luxemburg-Heijs SA, Lurvink EG, Kester MG, van Bergen CA, Falkenburg JH. Identification of 4 novel HLA-B*40:01 restricted minor histocompatibility antigens and their potential as targets for graft-versus-leukemia reactivity. Haematologica. 2012;97:1196–1204. doi: 10.3324/haematol.2011.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz M. High-resolution typing for unrelated donor transplantation: how far do we go? Best Practice & Research Clinical Hematology. 2009;22:537–541. doi: 10.1016/j.beha.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Klein J, Haagenson M, Baxter-Lowe LA, Confer DL, Eapen M, Fernandez-Vina M, Flomenberg N, Horowitz M, Hurley CK, Noreen H, Oudshoorn M, Petersdorf E, Setterholm M, Spellman S, Weisdorf D, Williams TM, Anasetti C. High-resolution donor-recipient HLA matching contributes to the success of unrelated donor marrow transplantation. Blood. 2007;110:4576–4583. doi: 10.1182/blood-2007-06-097386. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 1000 Genome Project Data Processing Subgroup. The sequence alignment/MAP format and SAM tools. Bioinformatics. 2009;25:2078–2089. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind C, Ferriola D, Mackiewicz K, Heron S, Rogers M, Slavich L, Walker R, Hsiao T, McLaughlin L, D’Arcy M, Gai X, Goodridge D, Sayer D, Monos D. Next-generation sequencing: the solution for high-resolution, unambiguous human leukocyte antigen typing. Human Immunology. 2010;71:1033–1042. doi: 10.1016/j.humimm.2010.06.016. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MAP reduce framework for analysing next-generation DNA sequencing data. Genome Research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklos DB, Kim HT, Miller KH, Guo L, Zorn E, Lee SJ, Hochberg EP, Wu CJ, Alyea EP, Cutler C, Ho V, Soiffer RJ, Antin JH, Ritz J. Antibody responses to HY minor histocompatibility antigens correlate with chronic graft-versus-host disease and disease remission. Blood. 2005;105:2973–2978. doi: 10.1182/blood-2004-09-3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE. 2011;7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomchik WD. Graft-versus-host disease. Nature Reviews in Immunology. 2007;7:340–352. doi: 10.1038/nri2000. [DOI] [PubMed] [Google Scholar]
- Spellman S, Warden MB, Haagenson M, Pietz BC, Goulmy E, Warren EH, Wang T, Ellis TM. Effects of mismatching for minor histocompatibility antigens on clinical outcomes in HLA-matched, unrelated hematopoietic stem cell transplants. Biology of Blood and Marrow Transplantation. 2009;15:856–863. doi: 10.1016/j.bbmt.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANN-OVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Materials.
Fig S1. WES to quantify SNPs between SCT D-R pairs.
Fig S2. (A) Alloreactivity potential vectors depicting conservative vs. nonconservative, nonsynonymous nucleotide variation in D-R pairs across the whole exome in the host versus graft direction, variants absent in the recipient and present in the donor. (B) Nonsynonymous SNPs in the graft versus host direction mapped on to individual chromosomes in all the D-R pairs demonstrate the genomic location of polymorphisms along the length of the chromosome in each D-R pair. All nine D-R pairs depicted on the y-axis for each chromosome; SNP coordinates (location) along the length of each chromosome depicted on the x-axis. (C) Nonsynonymous SNPs in the host versus graft direction mapped onto each chromosomes (data presented in Fig 1D).
Table SI. Donor-Recipient characteristics and patient outcomes observed in patients transplanted using ATG + 450 cGy TBI.
Table SII. HLA type of the donor-recipient pairs.
Table SIII. Sample statistics for the nine D-R pairs sequenced. The numbers in the parentheses denote percentage with respect to the number of HQ reads.
Table SIV. Median normalized SNP/Kbp for whole exome and for the 6p22·1–21·2 (MHC) locus in the graft versus host, host versus graft and direction independent SNPs.
Table SV. Total SNP counts/functional polymorphism for each D-R pair over the entire exome.
Table SVI. Total SNP counts/functional polymorphism for each D-R pair for the 6p22·1–21·2 (MHC) locus.