

Abstract
PDK1 (3-Phosphoinositide-dependent kinase 1) is a key member of the AGC protein kinase family. It plays an important role in a variety of cellular functions, leading to the activation of the PI3K signaling pathway, an event often associated with the onset and progression of several human cancers. Numerous recent observations suggest that PDK1 inhibitors may provide novel opportunities for the development of effective classes of therapeutics. On these premises, recent years have witnessed an increased effort by medicinal chemists to develop novel scaffolds to derive potent and selective PDK1 inhibitors. The intent of this review is to update the reader on the recent patent literature covering applications published between June 2008 and September 2011 that report on PDK1 inhibitors.
Introduction
Aberrant activation of the PI3K/AKT signaling pathway has been experimentally validated as one of the most common molecular events toward the initiation and progression of cancer [1–4]. For this reason, many pharmaceutical companies and academic laboratories are developing inhibitors targeting several components of the PI3K signaling cascade, among which PI3K, AKT and mTOR have so far taken center stage [5–9]. Indeed, less attention has been paid to a direct downstream effector of PI3K, the 3-phosphoinositide-dependent protein kinase 1 (PDK1), whose genetic and pharmacological inhibition is supporting its crucial role in cancer progression and metastasis [10]. Unlike PI3K and AKT, only a single PDK1 isoform has been reported in humans. PDK1 is a 556-amino-acid containing enzyme possessing a Pleckstrin Homology (PH) domain and a catalytic domain characterized by the typical bi-lobal kinase fold where the ATP co-factor is sandwiched between an amino-terminal small lobe and a carboxy terminal larger lobe (Figure 1) [11]. PDK1 is constitutively active owing to its auto-phosphorylation at residue Ser241 located at the center of the so called “activation loop”, which mediates an inter-molecular (trans) reaction with its substrates (Figure 1). PDK1 has been defined as the ‘master regulator’ of the AGC protein kinase family due to its ability to phosphorylate and activate at the least 23 AGC kinases including isoforms of AKT (also known as PKB), p70 ribosomal S6 kinase (S6K), serum and glucocorticoid-dependent kinase (SGK), p90 ribosomal S6 kinase (RSK), and atypical protein kinase (PKC) [12–14]. The mutual ability of both AKT and PDK1 to interact with phosphatidylinositol-3,4,5-triphosphate (PtdIns(3,4,5)P3), and phosphatidylinositol-4,5-bisphosphate (PtdIns(3,4)P2), via their PH domains, allows their co-localization at the plasma membrane, hence enabling PDK1 to activate AKT by phosphorylating Thr308 on its activation loop (Figure 2) [15–17]. Differently from AKT, all the other PDK1 substrates lack of the PH domain but possess a hydrophobic motif named PDK1 interacting fragment (PIF), which contains a conserved Phe-Xaa-Xaa-Phe/Tyr-Ser/Thr-Phe/Tyr sequence. Phosphorylation at these specific Ser or Thr residues by distinct upstream kinases promotes the interaction of the PIF fragment with a small phosphate binding groove located in the PDK1 catalytic domain, named PIF pocket (Figures 1 and 2). Such interaction in turn allows PDK1 to phosphorylate the activation loop of its substrates (Figure 2) [18,19].
Figure 1.
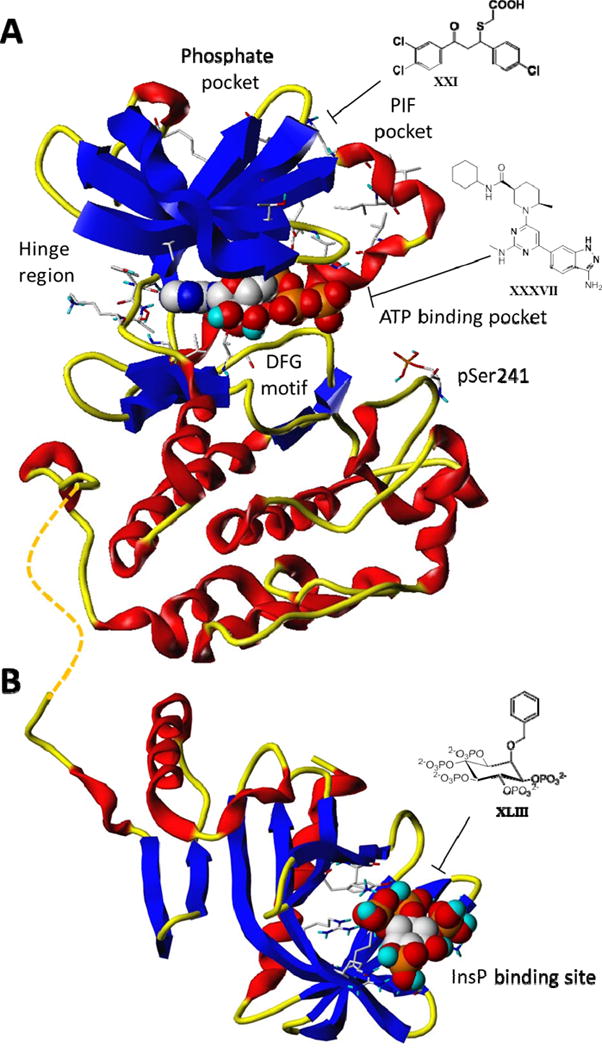
Overview of (A) PDK1 kinase domain (residues 71–232) in complex with ATP (PDB id: 1H1W) and of (B) PDK1 PH-domain (residues 407–550) in complex with inositol (1,3,4,5)-tetrakisphosphate (PDB id: 1W1D). The protein backbone is shown in a ribbon representation generated with MOLCAD [47] and color coded by secondary structures. Key protein residues forming the Hinge region, the PIF pocket, the phosphate pocket, the phosphoserine pSer241 and the Inositol phosphate (InsP) (discussed in the text) are shown as capped sticks while the ligands are depicted as spaced fill model. Chemical structures of representative inhibitors for each site are reported.
Figure 2.
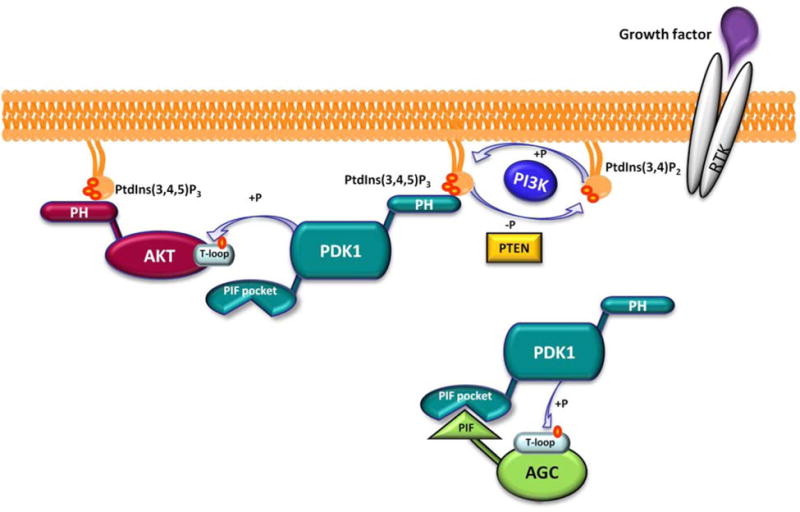
Schematic representation of the PI3K/PDK1/AGC signaling pathway underlying the different mechanism of AKT and other AGC substrates activation by PDK1. AGC substrates, here represented in light green, include the following kinases: S6K, SGK, PKC, RSK, and ROCK1.
Interestingly, several PDK1 substrates belong to both the oncogenic PI3K- and RAF-mitogen activated protein kinase (MAPK)-signaling pathways and are themselves oncology targets (i.e. PKB, RSK, PKC and p706K). This increases the attractiveness of direct PDK1 inhibition for drug discovery particularly as it represents an alternative pharmacological strategy to avoid the need of combined treatments targeting both signaling pathways [20].
Furthermore, encouraging recent studies on PDK1 hypomorphic mice expressing ~10% of the normal level of PDK1 showed no obvious harmful phenotype and, when crossed with PTEN+/− mice, showed a reduced tumor development confirming that a selective PDK1 kinase inhibitor might provide effective anticancer activity with an acceptable therapeutic index [21].
A number of drug discovery efforts in the past decade led to the identification of several putative PDK1 inhibitors [22]. A comprehensive list of such small molecules developed between 2005 and 2008 was assembled by Peifer and Alessi, where individual compounds were discussed in terms of their possible mechanism of inhibition [22]. These structures are reiterated here in Table 1 reporting both the general scaffolds and respective representative compounds (series I–XXI). With this manuscript we intend to provide an update on patent literature covering applications published between June 2008 and September 2011 that report on PDK1 inhibitors.
Table 1.
Compounds published before June 2008 grouped by assignee. Of each patent, a general structure and a representative compound are reported and the claimed core is highlighted in red*. Respective Protein Data Bank (PDB) identification codes are reported where available.
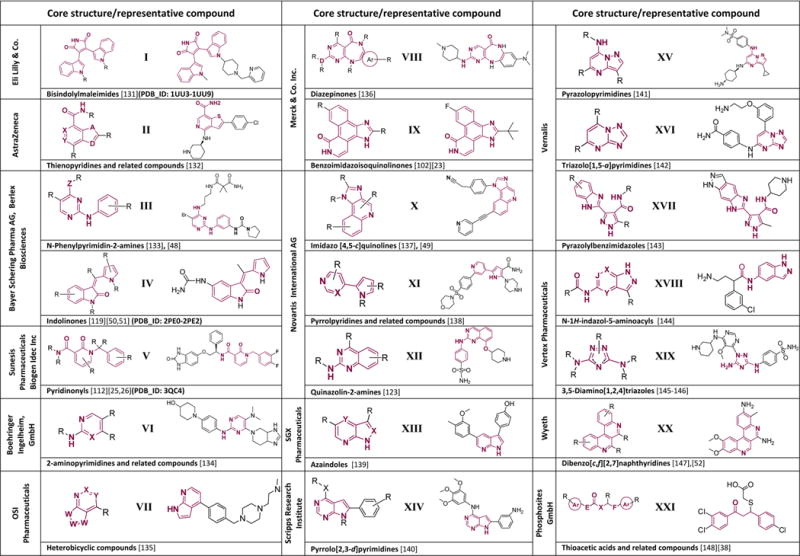
|
In all the reported structures R is any chemical group (hydrogen, aryl, alkyl, halogen etc); A and D are independently N, CH, S, O, NH and derivatives; J, X and Y are independently N and CH; Z is O, (un)substituted NH; W is N, O, S,:C(E1) or C(E1a); Ar is any 5 membered aromatic heterocycle; E is valence bond, CH2, NH, O; F is valence bond, CH2.
The SciFinder database (covering 62 patent authorities worldwide) and the European Patent Office website were used to search, select and download the original document files of the reviewed patents which are grouped and discussed by assignee (Table 1). Due to the numerous publications available in this arena, those patents which refer to general protein kinase inhibitors that do not explicitly report on PDK1 as protein target in either claim I or in the abstract have been excluded from the present discussion.
Patents evaluation: PDK1 inhibitors in recent patent literature
Merck & Co., Inc
A recent analysis of patent literature reveals a 2008 publication from scientist at Merck & Co., Inc. reporting on the synthesis of benzonaphthyridinones (XXII, Table 2) as inhibitors of Janus Kinase (JAK) and PDK1 [101]. These compounds represent an evolution of the previously patented benzoimidazoisoquinolinones, known as pan-JAK inhibitors (IX, Table 1) [23] [102]. The first generation benzonaphthyridinones, including 419 compounds, was still very potent against JAK2 with IC50 values as low as 0.1 nM while very poor inhibition was observed against PDK1 (IC50 ≤ 50 μM). Subsequent structural modifications of this class of tricyclic compounds, aimed to separate the JAK2 from the PDK1 inhibitory activity, led in 2010 to selective and potent PDK1 inhibitors, as the one reported in Table 2 (XXII), (PDK1 IC50 = 9 nM, cell (p-RSK) EC50 = 340 nM) [23].
Table 2.
Compounds claimed in the analyzed patents grouped by assignee. A general structure and a representative compound are reported and the patented core is highlighted in red*. Respective Protein Data Bank (PDB) identification codes are reported where available.
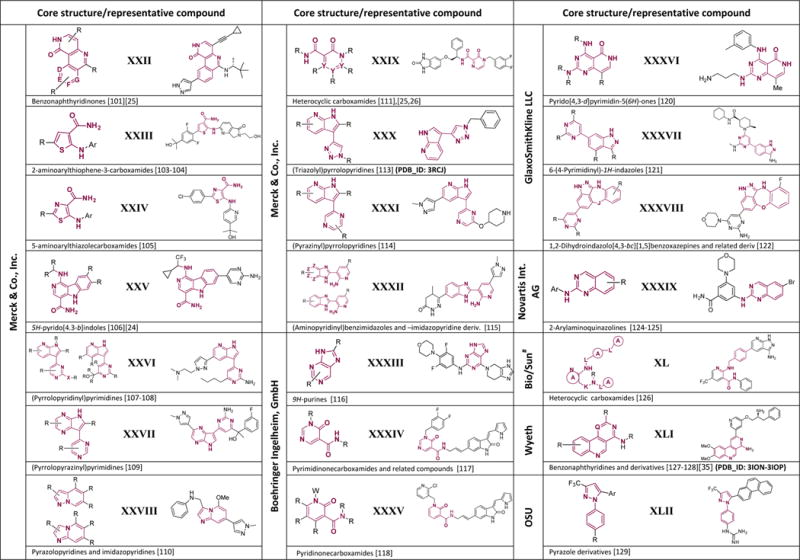
|
In all the reported structures R is any chemical group (aryl, alkyl, halogen etc); D, E, F and G are independently CH, N or NO; X is any heteroatom (NH, O, S etc); Y is independently N and C, provided that at least one Y is N; Z is independently N and C, provided that no more than two Z are N; J is O, S, N-cyclopropyl, NH, NEt, and NMe; W is O, (un)substituted CH2, NH; K C=(O) or SO2; L is a bond, A is any 3–7 membered, substituted or (un)substituted, aliphatic or aromatic ring; Q is CH or N; Ar is any aromatic ring. For a more detailed description of each chemical substituent we recommend to refer to the original patent application.
Along the same lines, Merck patented another series of compounds, 2-aminoarylthiophene-3-carboxamides (XXIII, Table 2) as inhibitors of the four known mammalian JAK kinases (JAK1, JAK2, JAK3 and TYK2) and PDK1 [103–104] for the treatment of myeloproliferative disorders and cancer. The claimed compounds showed excellent inhibition against JAK2 with IC50 values in the low nanomolar range and good, but still weaker, inhibition against PDK1 with in vitro IC50 values ranging from 670 nM to 2.5 μM. A structurally similar series of compounds where a thiazole is the central core instead of thiophene (XXIV, Table 2) was patented in 2010 [105]. Herein, Merck claimed 26 compounds, which still showed potent inhibition against JAK2 (IC50 from 10 nM to 1 μM) and only poor inhibition against PDK1 (IC50 > 30 μM). No data on selectivity has been reported.
Similarly, 1-amino-5H-pyrido[4,3-b]indol-4-carboxamides (XXV, Table 2) have been claimed in another patent from Merck [106] as dual JAK-PDK1 inhibitors, even if they are weak PDK1 inhibitors (IC50 ≤ 50 μM) while are very potent as JAK inhibitors (IC50 values from 0.1 nM to 20 μM). Indeed, chemical optimization of the main core structure resulted in a potent, JAK-2 selective, orally active inhibitor whose efficacy translates in in vivo animal models as recently published [24].
Two recent publications [107–108] reported on the synthesis, PDK1 inhibition and cellular activity of pyrrolopyridinylpyrimidines represented by structures XXVI in Table 2. The most potent of these series are reported to inhibit PDK1 in a kinase activity assay with IC50 values in the low nanomolar range. In vitro potency was confirmed in cellular functional assays where they show to inhibit p-AKT Thr308 in PC-3 cells at fairly low concentrations (10 nM to 20 μM). Isosteric replacement of the pyrrolopyridine with the pyrrolopyrazine ring led to the main core structure claimed in a later patent describing pyrrolopyrazinylpyrimidine derivatives (XXVII, Table 2) as new PDK1 inhibitors [109]. The most potent compound of this series (XXVII, Table 2) tested in both PDK1 kinase assay and p-AKT Thr308 cell based assay, exhibited IC50 values of 0.12 μM and 0.8 μM, respectively.
Another patent form Merck discloses the characterization of pyrazolopyridines and imidazopyridines (XXVIII, Table 2) as inhibitors of PDK1 [110]. These compounds, however, inhibited with IC50 values less than 30 μM at least one of the following proteins: Fibroblast Growth Factor Receptor 3 (FGFR3), Neurotrophic Tyrosine Kinase Receptor 3 (NTRK3), Ribosomal Protein S6 Kinase (RP-S6K) and Wee1-like protein kinase (WEE1). Apart from the above kinases which are all potential therapeutic targets in cancer, these classes of compounds inhibit also the Microtubule Affinity Regulating Kinase (MARK) and, for this reason, it has been patented their possible use in the treatment of Alzheimer’s disease as well.
Researchers at Merck in 2010 patented heterocyclic carboxamides (XXIX, Table 2) as PDK1 inhibitors [111], tested in both kinase activity (IC50 values less than 30 μM) and cell based assays (although no inhibition data are reported), looking at the phosphorylation levels in PC-3 cells of the direct PDK1 substrates RSK (p-Ser221), AKT (p-Thr308) and the downstream effector S6RP (p-Ser235/236). From a chemical point of view, these molecules (XXIX, Table 2), are somewhat related to the previously patented and recently published series from Sunesis Pharmaceuticals and Biogen Idec., Inc. (V, Table 1), discovered by tethering two fragments with a flexible linker [25] [112]. Due to their chemical similarity, it could be speculated a similar binding pose, where the urea moiety is hydrogen bonded with the hinge residues Ser160 and Ala162 and the difluorobenzyl ring interacts with hydrophobic residues of the DFG-out pocket; similarly, the heterocyclic keto group could interact with the backbone amide of Asp223 of the DFG motif. The unique allosteric inhibition due to the binding of the inactive (DFG-out) conformation of PDK1 has been extensively studied in cancer cells for the Sunesis lead compound, in a recent paper from the Merck group [26].
Another patent from Merck describes the synthesis of 3-([1,2,3]triazol-4-yl)-pyrrolo[2,3-b ]pyridine derivatives (XXX, Table 2) as PDK1 inhibitors [113]. Using the copper mediated click chemistry approach [27], [3+2] cyclo-addition reaction of alkynes and azides resulted in several triazole containing pyrrolo-pyridine derivatives [28]. They are very potent PDK1 inhibitors, with IC50 values ranging from 0.5 nM to 1 μM in in vitro kinase assays, and IC50 values between 1 μM and 10 μM in cell based evaluation (p-Akt Thr308). Similar potencies have been observed for IKKε, whose inhibition has been validated in cell as well. No biochemical data regarding possible inhibition of other kinases are reported. Interestingly, for a somewhat similar scaffold, having pyrazinylpyrrolopyridine as main core (XXXI, Table 2) [114], the PDK1 inhibition drops dramatically (IC50 < 100 μM) while JAK 2/3 potency is retained (IC50 = 0.1 nM −20μM).
A very recent patent from Merck discloses the preparation of benzimidazoles and imidazopyridines substituted with 2-aminopyridine (XXXII, Table 2) as PDK1 inhibitors for the treatment of cancer and inflammatory diseases [115]. The majority of the patented compounds showed nanomolar potencies (IC50 values range: 0.1 – 100 nM) against PDK1 but also against Interleukin-1 receptor-associated kinase 1 (IRAK-1) and 4 (IRAK-4), with similar or lower potencies (IC50 values range: 0.1 – 10μM).
Boehringer Ingelheim, GmbH
In 2008 investigators at Boehringer Ingelheim, reported on 84 purine derivatives as PDK1 inhibitors (XXXIII, Table 2), [116]. The compounds exhibited good inhibition in PDK1 activity assay with IC50 values less than 1 μM and EC50 values less than 5 μM in PC-3 proliferation tests. There is no report on selectivity for this class of compounds.
Two recent publications from Boehringer Ingelheim report as new PDK1 inhibitors two class of compounds structurally similar: pyrimidinonecarboxamides (XXXIV, Table 2) [117], chosen as example from an even more general structure which claims any saturated or unsaturated C5–10 heterocyclic carboxamide, and pyridinonecarboxamides (XXXV, Table 2) [118]. These molecules could be dissected in two main fragments connected by an unsaturated 3 carbon linker: the heterocyclic carboxamide with N-aryl substituents (as the difluorobenzyl group in the example XXXIV reported in Table 2) and the substituted indolinone moiety. Interestingly both fragments have been independently reported as PDK1 inhibitors: the first one in 2008 by Sunesis Pharmaceuticals and Biogen Idec. Inc. [115] (V, Table 1), (see also above discussion about the Merck patent [111]) and the second one in 2006 by Bayer Schering Pharma AG and Berlex Biosciences (IV, Table 1) [119]. Having the crystal structure for the complexes of both parent molecules with PDK1 kinase domain, we could speculate an allosteric binding mode for this series of molecules in which the indolinone moiety would form hydrogen bonds with the hinge region residues, the heterocyclic keto group would interact with the backbone amide of Asp223 of the DFG motif, and its aromatic substituent would fit in the DFG hydrophobic pocket. All the patented compounds of this series have been first tested in biochemical assays where they showed IC50 values less than 1 μM using both full length PDK1 (aa 52–556) and ΔPH-PDK1 (aa 51–359) enzymes. They have been found to be effective against glioma (U87MG, U373MG), sarcoma (MES-SA, SK-UT-1B), breast (MDA-MB468), colon (HCT116), lung (NCI-H460, NCI-H520), melanoma (MALME-3M, C32), prostate (PC-3, DU-145), ovary (SKOV-3) cells, where the PI3K/AKT/PDK1 signaling pathway is up-regulated. Cell cycle analysis showed that some compounds, for example, induced G1 arrest in PC-3 cells at concentrations of less than 5 μM, and at similar or lower concentrations they induced apoptosis of HCT116 or MALME-3M cells. At the same concentration range they effectively inhibited phosphorylation of the PDK1 substrates RSK (p-Ser221/227), AKT (p-Thr308) and p70S6 kinase (p-Thr229). Because of the importance of the PI3K signaling cascade in regulating vital biological processes, this class of compounds has been proposed for a wide range of applications, not only for cancer but also for infectious, inflammatory, autoimmune, bone, cardiovascular diseases.
GlaxoSmithKline
A patent from GlaxoSmithKline (GSK) disclosed the invention of small molecules PDK1 inhibitors based on a pyrido[4,3-d]pyrimidin-5(6H)-one core structure (XXXVI, Table 2) [120]. In vitro IC50 values of 54 small molecules claimed in this patent varied from 0.003 μM to 10 μM by using both a radiolabeled PDK1 binding assay and a PDK1-AKT coupled activity assay. Compounds were also tested for cell growth/death in several cancer cell lines such as human breast (BT474, HCC1954, T-47D) and prostate (PC-3). There are no reports on in vivo data or selectivity, despite their potent in vitro activity.
Another patent from researchers at GSK [121] describes the invention of pyrimidinyl-indazoles (XXXVII, Table 2) whose synthesis includes as key step a Suzuki coupling of 4-chloropyrimidine intermediate with 4-cyano-3-fluorophenyl boronic ester followed by cyclization with hydrazine. The chemical structure of one of the most potent compound, with nanomolar potency against PDK1, is reported in Table 2 and has been recently published in several papers [29–32]. The aminoindazole scaffold was initially discovered through a Fragment-Based Drug Discovery approach by using Saturation transfer difference (STD) NMR experiments to characterize hits resulted from a biochemical screening of one thousand fragments library [32]. Fragments elaboration using iterative X-ray crystal structures of PDK1 in complex with inhibitors, resulted in the identification of the highly potent and selective GSK2334470 compound [29] (XXXVII, Table 2). Concerning its binding pose in the PDK1 active site, it is the aminoindazole ring to form the key hydrogen bonds with the hinge Ser160 and Ala162 residues; the free amino group of the pyrimidine ring interacts with the catalytic Glu130 while substitutions at the pyrimidine 6-position are used for accessing the hydrophobic pocket along the G-loop. Lead optimization steps helped also to gain selectivity against AurA, AurB, ALK5, ROCK1 kinases which were inhibited with micromolar potency by the parent fragment. PDK1 inhibition was first evaluated in mechanistic cell based assay, looking at the phosphorylation state of Thr308-AKT and Ser221-RSK and subsequently validated for anti-proliferative activity against a panel of leukemia cell lines, finding that only a subset of AML cells were particularly sensitive to PDK1 inhibition. In vivo xenografts using SCID mice bearing OCI-AML2 tumors, however, indicated that GSK2334470 only modestly inhibited tumor growth. This compound, being one of the most potent and highly selective PDK1 inhibitor identified so far, still constitute a useful tool to understand the role of PDK1 in such important signaling pathway, as showed by Dario Alessi’s group and collaborators at GSK [30,31]. Indeed, from extensive cell characterization of GSK2334470 (XXXVII, Table 2) they proved that the potency and the kinetics of inhibition are strictly dependent on the different PDK1 substrates.
In an additional 2010 publication, the GSK group disclosed the synthesis of substituted indazole derivatives as PDK1 inhibitors (XXXVIII, Table 2) [122]. All compounds have been tested by using PDK1 Scintillation Proximity (LEADseeker) and Time Resolved Fluorescence Resonance Energy Transfer (TR-FRET) assays against full length PDK1. They all showed IC50 values ≤ 100 nM, with the most potent ones having single digit nanomolar inhibition, as for compound XXXVIII reported in Table 2 (IC50 = 7.6 nM). They have been characterized in PC-3 cells for their anti-proliferative activity and inhibition of the phosphorylation of AKT and RSK through ELISA’s assays. However, there is no report regarding selectivity profile to date.
Novartis International AG
Novartis International AG patented in 2007 quinazoline derivatives as PDK1, CDK1 and CDK2 inhibitors [123]. Along the same lines, two patents appeared in 2009 [124] and 2010 [125]. The main core structure in both publications is an N-arylquinazolin-2-amine (XXXIX, Table 2) which is decorated with several functional groups (aromatic rings, sulfonamides, piperidines) resulting in over 400 compounds from the first patent (of which a representative compound is shown in Table 2, XXXIX) and 271 compounds from the second one. In a PDK1 Kinase AlphaScreen® assay [33], they resulted as inhibitors with IC50 values less than 5 μM with the most potent compounds in the low nanomolar range (IC50 ≤ 0.05μM). In ovarian A2780 and prostate PC-3 proliferation assays they showed submicromolar EC50 values.
To date, there are no reports on selectivity on these compounds, although it is already reported that 4-anilinoquinazolines are inhibitors of Erb-2 receptor tyrosine kinase and closely related kinases [34]
Biogen, Idec. Inc. and Sunesis Pharmaceuticals, Inc
A patent from Biogen, Idec. Inc. and Sunesis Pharmaceuticals, Inc. discloses the preparation of heterocyclic carboxamides of general formula reported in Table 2, (XL), as small molecules PDK1 inhibitors [126]. In this patent, 443 compounds showed IC50 values ranging from 100 nM to 10 μM in in vitro PDK1 assay, and submicromolar EC50 values using the Meso Scale Discovery (MSD) ELISA kit to detect p-AKT (Thr308) levels in PC-3 cells treated with inhibitors.
Wyeth (now Pfizer Inc.)
Two patents from Wyeth published in 2008 report on the synthesis and biological evaluation of pyrimido[5,4-c]quinoline-2,4-diamines [127] and benzo[c][2,7]naphthyridines [128] as PDK1 inhibitors (XLI, Table 2). These compounds are very effective in inhibiting PDK1 and some exhibited IC50 values in the low nanomolar range, as the one represented in Table 2 (XLI, IC50 = 8 nM). From the available crystal structure of this molecule in complex with the PDK1 kinase domain [35], it is the tricyclic core to anchor the hinge region residues Ser160 and Ala162. The pyridyl moiety forms hydrogen bonds interactions with the catalytic Lys111 while the protonated primary amine of the side chain makes electrostatic interactions with Asp223, critical for the observes increase in potency. Benzonaphthyridines apparently are non-selective against other AGC kinases (PKA, AKT, S6K, and PKC), some of which are inhibited with nanomolar potencies.
Ohio State University
Researchers at Ohio State University (OSU) reported on 77 pyrazole derivatives (XLII, Table 2) as PDK1 inhibitors and as anti-infective agents, due to their ability to induce autophagy and therefore to defend against intracellular pathogens including bacteria, viruses and protozoa [129]. Structurally, all the patented compounds are derivatives of celecoxib which has been optimized to reduce COX-2 inhibition properties and gain in PDK1 potency by increasing bulkiness of the aromatic ring at the pyrazole 5-position (XLII, Table 2). Kinetic and molecular docking data indicated that these compounds act as ATP mimetics anchoring to the hinge residues through a series of heteroatom-rich groups able to form hydrogen bonds such as the sulfonamide, 2-aminoacetamide and guanidine, as for the compound represented in Table 2 (XLII). The planar pyrazole moiety lies perpendicularly to the position occupied by the ATP ribose ring while the phenanthrene moiety forms hydrophobic interactions with a non-polar region formed by residues 88–96. From the PDK1 kinase assay the compound shown in Table 2 is one of the most active ones with an IC50 of 2 μM and, screened against 60 NCI tumor cell lines including ovarian, renal, prostate, leukemia, lung, CNS cancer, melanoma, with a mean GI50 (concentration resulting in 50% growth inhibition) of 1.2 μM.
Allosteric modulators
Most inhibitors of PDK1 described above are ATP competitive and consequently present off-target effects due to the simultaneous interaction with the ATP binding pocket of other protein kinases. Indeed, there has been a growing effort to develop allosteric kinase inhibitors, known as type 3 inhibitors, which targeting outside the ATP pocket are more likely to be selective for a given kinase than type 1 inhibitors. Two alternative allosteric PDK1 binding sites could be explored: the PDK1-Interacting- Fragment, known as PIF pocket (Figures 1 and 2), and the phosphoinositol binding site located on the Pleckstrin Homology (PH) domain (Figure 2). PIF binders include 12–25 residues peptides derived from the Hydrophobic Motif (HM) of different PDK1 substrates known as PIFtides [36,37] and small molecules PIFtides mimics [38–40]. From structural studies, small molecules modulators bind the PIF pocket mainly by hydrophobic interactions between aromatic rings and the hydrophobic pocket and by electrostatic interactions of their carboxylate groups with positively charged residues belonging to the phosphate binding site close to the PIF pocket (XXI, Figure 1 and Table 1). Crystal structure analysis demonstrated that, upon binding, PIF modulators can induce activation or inhibition of the kinase inducing conformational changes at the helix αC, the DFG motif, the activation loop, and therefore at the ATP binding site.
PH domain-targeting inhibitors are often polyphosphate mimics which compete with PtdIns(3,4,5)P3 thus preventing recruitment of the kinase to the plasma membrane and its activation. Phosphatidylinositol ether lipid analogs (PIA) along with the alkylphospholipid perifosine, have been published and developed as selective PH domain inhibitors AKT [41–43]. In a 2011 patent [130], researchers at Universities of London and Bath reported on the identification of inositol phosphate derivatives as therapeutic agents for the treatment of cancer. Among all the claimed analogs, 2-O-benzyl-myo-inositol 1,3,4,5,6-pentakisphosphate (2-O-Bn-InsP5) (XLIII, Figure 1) [44] has been identified as a potent and selective PH domain PDK1 inhibitor. Indeed, from a 60 protein kinases profiling, it inhibited PDK1 with an IC50 value of 26.5 nM and mTOR with an IC50 value of 1.3 μM but none of the AKT and PI3K isoforms or other AGC kinases resulted being inhibited from a selectivity profile screening at 1 μM. Chemically, the compound is a 2-O-benzyl derivative of the previously reported inositol 1,3,4,5,6-pentakisphosphate (InsP5) [45,46] which possesses enhanced pro-apoptotic and antitumor activity compared with the parent molecule. Interestingly, it also inhibited Ser473 and Thr308 Akt phosphorylation in prostate PC3 cells and pancreatic ASPC1 cells which were resistant to InsP5. In vivo prostate cancer xenografts resulted in 52% inhibition of tumor weight when dosed at 50 mg/Kg once daily for 14 days.
Conclusion and Future Perspective
Despite its central position as activator of the PI3K signaling pathway, PDK1 drug discovery efforts have been somehow obscured by the tremendous attention devoted to other more characterized kinases such as PI3K itself, AKT and mTOR [5–9]. Nonetheless, an increasing number of patent applications have reported on putative PDK1 inhibitors since it was discovered in 1998. In this review compounds published in patent literature between June 2008 and September 2011 as putative PDK1 inhibitors are reported. Together with the recent comprehensive review by Peifer and Alessi [22], this article should provide an overview of currently available inhibitors. Hence, the disclosed literature has been summarized in Tables 1 and 2 where the claimed core structures for which PDK1 inhibition data were available are reported together with representative inhibitors. While distinct pharmacophores have been discovered, no direct correlations can be found between a particular scaffold and a selected class of protein kinases that it targets. The majority of these molecules, in fact, act as ATP mimetics utilizing a common general hydrogen bonding and hydrophobic pharmacophore. Hence, most efforts in translating these general scaffolds into potent and selective PDK1 inhibitors required extensive SAR studies, often with the aid of fragment-based and X-ray driven structure-based optimizations. We envision that several additional general scaffolds or those originally developed for other kinases, could likewise be elaborated into novel PDK1 inhibitors in the future. The exploration of binding pockets structurally distinct from the ATP site is becoming a challenging and promising strategy to gain selectivity in PDK1 inhibition.
Executive Summary.
PDK1 inhibitors hold great promise for the development of novel anti-cancer therapies targeting the PI3K pathway. Several pharmaceutical companies and some academic laboratories have devoted significant resources to these efforts. These are briefly summarized below by institution.
Merck & Co., Inc
-
■
From June 2008 to September 2011 the Merck group published 13 patents applications, of which several report on dual JAK-PDK1 inhibitors, with higher potency against JAK2/JAK3 versus PDK1.
-
■
Benzonaphthyridinones (XXII, Table 2), pyrrolopyridinyl- (XXVI, Table 2) and pyrrolopyrazinylpyrimidines (XXVII, Table 2), triazolyl-pyrrolopyridines (XXX, Table 2) along with selected heterocyclic carboxamides (XXIX, Table 2) are low nanomolar PDK1 inhibitors and effective in cell based assay (p-Akt Thr308; p-RSK Ser221) as well.
Boehringer Ingelheim, GmbH
-
■
Two publications in 2010 report on pyrimidinonecarboxamides (XXXIV, Table 2) and pyridinonecarboxamides (XXXV, Table 2) with IC50 values less than 1 μM in both PDK1 binding and activity assays. They showed to be effective against a set of cancer cell lines. We speculated an allosteric mechanism of inhibition for this series of compounds due to their possible binding to the inactive (DFG-out) conformation of PDK1.
GlaxoSmithKline
-
■
Pyridopyrimidin-5(6H)-ones (XXXVI, Table 2) and substituted indazole derivatives (XXXVIII, Table 2) exhibited nanomolar IC50 values against PDK1 and have been characterized in several cancer cell lines for both anti-proliferative and inhibition of PDK1 substrates phosphorylation. There are no reports on in vivo data or selectivity, despite their potent in vitro activity.
-
■
Pyrimidinyl-indazole derivatives optimization resulted in the identification of one of the most potent and selective PDK1 inhibitor identified so far: GSK2334470 (XXXVII, Table 2). X-ray crystal structures of analogs in complex with PDK1 have been solved. Only a subset of AML cells was particularly sensitive to its PDK1 inhibition. In vivo xenografts indicated that GSK2334470 only modestly inhibited tumor growth.
Novartis International AG
-
■
Two patents published in 2008 and 2009 report on N-arylquinazolin-2-amines (XXXIX, Table 2) which exhibited IC50 values as low as 50 nM in PDK1 assay and submicromolar EC50 values in ovarian A2780 and prostate PC-3 proliferation assays. There are no reports on selectivity.
Biogen, Idec. Inc. and Sunesis Pharmaceuticals, Inc
-
■
443 disclosed heterocyclic carboxamides (XL, Table 2), showed IC50 values ranging from 100 nM to 10 μM in in vitro PDK1 assay and submicromolar EC50 values detecting p-AKT (Thr308) levels in PC-3 cells.
Wyeth (now Pfizer Inc.)
-
■
Pyrimido[5,4-c]quinoline-2,4-diamines and benzo[c][2,7]naphthyridines (XLI, Table 2) are low nanomolar PDK1 inhibitors. X-ray structures of two selected inhibitors in complex with PDK1 kinase domain are available. Apparently they are non-selective against other AGC kinases (PKA, AKT, S6K, and PKC).
Ohio State University
-
■
In a 2009 publication, celecoxib derivatives have been optimized to reduce COX-2 inhibition properties and gain in PDK1 potency by increasing bulkiness of the aromatic ring at the pyrazole 5-position (XLII, Table 2). Low micromolar inhibition observed from both PDK1 kinase assay and 60 NCI tumor cell lines proliferation assays for this series of compounds.
University of London and University of Bath
-
■
A PH domain-targeting inhibitor, 2-O-benzyl-myo-inositol 1,3,4,5,6-pentakisphosphate (2-O-Bn-InsP5) (XLIII, Figure 1) has been developed from the parent inositol 1,3,4,5,6-pentakisphosphate (InsP5) and identified as a potent (IC50 = 26.5 nM) and selective PDK1 inhibitor. The efficacy translated in in vivo prostate cancer xenografts when dosed at 50 mg/Kg once daily for 14 days.
Acknowledgments
We thank NIH grant P01CA128814 (to MP) for financial support.
Defined key terms
- PDK1
3-phosphoinositide-dependent protein kinase 1
- PH domain
Pleckstrin Homology is a domain of approximately 120 residues that bind to phosphatidylinositol phosphates and occurs in a wide range of proteins involved in intracellular signaling, including Akt and PDK1
- PIF pocket
The kinase domain of PDK1 possesses three distinct ligand binding sites: the ATP binding site, the substrate binding site, and a docking site known as PIF pocket
- DGF motif
A loop region present in PDK1 and other related kinases whose conformation dictates the activation status of the protein. The related terms DFG-in and DFG-out refer to the conformations of this loop in the active and inactive forms of the kinase, respectively
- Hinge region
a loop region in PDK1 and other related kinases located between the two structural lobes of the catalytic domain. This loop region contains the amino acids that make specific interactions with the adenine ring of the ATP and with most type I and type II kinase inhibitors
- Type I inhibitor
A kinase inhibitor that is competitive with ATP, binding to the active form of the kinase, also known as the DFG-in form
- Type II inhibitor
A kinase inhibitor that is competitive with ATP, binding to the inactive form of the kinase, also known as the DFG-out form
- Type III inhibitor
A kinase inhibitor that is not a direct ATP competitor, binding to other sites on the protein surface
Footnotes
Patents
| 101. | Merck WO112217 (2008). |
| 102. | Merc k & Co. Inc. WO145957 (2007). |
| 103. | Merck WO155156 (2009). |
| 104. | Merck WO156726 (2008). |
| 105. | Merck WO011375 (2010). |
| 106. | Merck WO075830 (2009). |
| 107. | Merck WO 2010000364 (2010) |
| 108. | Merck WO076316 (2011). |
| 109. | Merck WO076327 (2011). |
| 110. | Merck WO017047 (2010). |
| 111. | Merck WO065384 (2010). |
| 112. | Sunesis Pharmaceuticals WO005457 (2008). |
| 113. | Merck WO127754 (2010). |
| 114. | Merck & Co. WO054941 (2009). |
| 115. | Merck WO006567 (2011). |
| 116. | Boehringer Ingelheim WO107444 (2008). |
| 117. | Boehringer Ingelheim WO007114 (2010). |
| 118. | Boehringer Ingelheim WO007116 (2010). |
| 119. | Schering AG US07105563 (2006). |
| 120. | GlaxoSmithKline WO019637 (2010). |
| 121. | GlaxoSmithKline WO059658 (2010). |
| 122. | GlaxoSmithKline WO120854 (2010). |
| 123. | Novartis Vaccines & Diagnostics Inc. WO117607 (2007). |
| 124. | Novartis WO153313 (2009). |
| 125. | Novartis WO079988 (2008). |
| 126. | Sunesis & Biogen WO044157 (2011). |
| 127. | Wyeth WO109599 (2008). |
| 128. | Wyeth WO109613 (2008). |
| 129. | The Ohio State University Research Foundation US0111799 (2009). |
| 130. | Universities of London and Bath WO064559 (2011). |
| 131. | Eli Lilly and Co. 041953 (2005). |
| 132. | AstraZeneca UK Ltd. WO106326 (2006). |
| 133. | Schering AG US0186118A1 (2004). |
| 134. | Boehringer Ingelheim Intl. GmbH WO003766 (2008). |
| 135. | OSI Pharmaceuticals Inc. WO084667 (2007). |
| 136. | Merck Patent GmbH WO079826 (2007). |
| 137. | Novartis AG-054238 (2008). |
| 138. | Novartis AG WO034600 (2008). |
| 139. | SGX Pharmaceuticals Inc. WO015124 (2006). |
| 140. | IRM LLC, The Scripps Research Institute WO093812 (2004). |
| 141. | Vernalis Cambridge Ltd. WO087707 (2004). |
| 142. | Vernalis Cambridge Ltd. WO108136 (2004). |
| 143. | Vernalis R&D Ltd. WO134318 (2006). |
| 144. | Vertex Pharmaceuticals Inc. WO064397 (2003). |
| 145. | Vertex Pharmaceuticals Inc. WO046120 (2004). |
| 146. | Vertex Pharmaceuticals WO050249 (2006). |
| 147. | Wyeth US0135429 (2007). |
| 148. | Phosphosites GmbH EP-1486488-A1 (2004). |
Bibliography
Papers of special note have been highlighted as
■ of interest
■■ of considerable interest
- 1.Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008;8(3):187–198. doi: 10.2174/156800908784293659. [DOI] [PubMed] [Google Scholar]
- 2.Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007;13(10):433–442. doi: 10.1016/j.molmed.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Cicenas J. The potential role of Akt phosphorylation in human cancers. Int J Biol Markers. 2008;23(1):1–9. doi: 10.1177/172460080802300101. [DOI] [PubMed] [Google Scholar]
- 4.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brachmann S, Fritsch C, Maira SM, Garcia-Echeverria C. PI3K and mTOR inhibitors: a new generation of targeted anticancer agents. Curr Opin Cell Biol. 2009;21(2):194–198. doi: 10.1016/j.ceb.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Fasolo A, Sessa C. mTOR inhibitors in the treatment of cancer. Expert Opin Investig Drugs. 2008;17(11):1717–1734. doi: 10.1517/13543784.17.11.1717. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008;27(41):5511–5526. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]
- 8.Lindsley CW, Barnett SF, Layton ME, Bilodeau MT. The PI3K/Akt pathway: recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Curr Cancer Drug Targets. 2008;8(1):7–18. doi: 10.2174/156800908783497096. [DOI] [PubMed] [Google Scholar]
- 9.Maira SM, Stauffer F, Brueggen J, et al. Identification and characterization of NVPBEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 10.Raimondi C, Falasca M. Targeting PDK1 in cancer. Curr Med Chem. 2011;18(18):2763–2769. doi: 10.2174/092986711796011238. [DOI] [PubMed] [Google Scholar]
- 11.Biondi RM, Komander D, Thomas CC, et al. High resolution crystal structure of the human PDK1 catalytic domain defines the regulatory phosphopeptide docking site. EMBO J. 2002;21(16):4219–4228. doi: 10.1093/emboj/cdf437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bayascas JR. PDK1: the major transducer of PI 3-kinase actions. Curr Top Microbiol Immunol. 2010;346:9–29. doi: 10.1007/82_2010_43. [DOI] [PubMed] [Google Scholar]
- 13.Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15(2):161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 14.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11(1):9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 15.Alessi DR, James SR, Downes CP, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7(4):261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 16.Filippa N, Sable CL, Hemmings BA, Van Obberghen E. Effect of phosphoinositide-dependent kinase 1 on protein kinase B translocation and its subsequent activation. Mol Cell Biol. 2000;20(15):5712–5721. doi: 10.1128/mcb.20.15.5712-5721.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milburn CC, Deak M, Kelly SM, Price NC, Alessi DR, Van Aalten DM. Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem J. 2003;375(Pt 3):531–538. doi: 10.1042/BJ20031229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 2001;20(16):4380–4390. doi: 10.1093/emboj/20.16.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casamayor A, Torrance PD, Kobayashi T, Thorner J, Alessi DR. Functional counterparts of mammalian protein kinases PDK1 and SGK in budding yeast. Curr Biol. 1999;9(4):186–197. doi: 10.1016/s0960-9822(99)80088-8. [DOI] [PubMed] [Google Scholar]
- 20.Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bayascas JR, Leslie NR, Parsons R, Fleming S, Alessi DR. Hypomorphic mutation of PDK1 suppresses tumorigenesis in PTEN(+/−) mice. Curr Biol. 2005;15(20):1839–1846. doi: 10.1016/j.cub.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 22.Peifer C, Alessi DR. Small-molecule inhibitors of PDK1. ChemMedChem. 2008;3(12):1810–1838. doi: 10.1002/cmdc.200800195. [DOI] [PubMed] [Google Scholar]
- 23.Bobkova EV, Weber MJ, Xu Z, et al. Discovery of PDK1 kinase inhibitors with a novel mechanism of action by ultrahigh throughput screening. J Biol Chem. 2010;285(24):18838–18846. doi: 10.1074/jbc.M109.089946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim J, Taoka B, Otte RD, et al. Discovery of 1-amino-5H-pyrido[4,3-b]indol-4-carboxamide inhibitors of janus kinase 2 (JAK2) for the treatment of myeloproliferative disorders. J Med Chem. 2011;54(20):7334–7349. doi: 10.1021/jm200909u. [DOI] [PubMed] [Google Scholar]
- 25.Erlanson DA, Arndt JW, Cancilla MT, et al. Discovery of a potent and highly selective PDK1 inhibitor via fragment-based drug discovery. Bioorg Med Chem Lett. 2011;21(10):3078–3083. doi: 10.1016/j.bmcl.2011.03.032. [DOI] [PubMed] [Google Scholar]
- 26.Nagashima K, Shumway SD, Sathyanarayanan S, et al. Genetic and pharmacological inhibition of PDK1 in cancer cells: characterization of a selective allosteric kinase inhibitor. J Biol Chem. 2011;286(8):6433–6448. doi: 10.1074/jbc.M110.156463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew Chem Int Ed Engl. 2001;40(11):2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 28.Merkul E, Klukas F, Dorsch D, Gradler U, Greiner HE, Muller TJ. Rapid preparation of triazolyl substituted NH-heterocyclic kinase inhibitors via one-pot Sonogashira coupling-TMS-deprotection-CuAAC sequence. Org Biomol Chem. 2011;9(14):5129–5136. doi: 10.1039/c1ob05586k. [DOI] [PubMed] [Google Scholar]
- 29.Medina JR, Becker CJ, Blackledge CW, et al. Structure-based design of potent and selective 3-phosphoinositide-dependent kinase-1 (PDK1) inhibitors. J Med Chem. 2011;54(6):1871–1895. doi: 10.1021/jm101527u. [DOI] [PubMed] [Google Scholar]
- 30.Najafov A, Sommer EM, Axten JM, Deyoung MP, Alessi DR. Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem J. 2011;433(2):357–369. doi: 10.1042/BJ20101732. [DOI] [PubMed] [Google Scholar]
- 31.Knight ZA. For a PDK1 inhibitor, the substrate matters. Biochem J. 2011;433(2):e1–2. doi: 10.1042/BJ20102038. [DOI] [PubMed] [Google Scholar]
- 32.Medina JR, Blackledge CW, Heerding PA, et al. Aminoindazole PDK1 Inhibitors: A Case Study in Fragment-Based Drug Discovery. Acs Med Chem Lett. 2010;1(8):439–442. doi: 10.1021/ml100136n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Z, Nagashima K, Sun D, et al. Development of high-throughput TR-FRET and AlphaScreen assays for identification of potent inhibitors of PDK1. J Biomol Screen. 2009;14(10):1257–1262. doi: 10.1177/1087057109349356. [DOI] [PubMed] [Google Scholar]
- 34.Barlaam B, Ballard P, Bradbury RH, et al. A new series of neutral 5-substituted 4-anilinoquinazolines as potent, orally active inhibitors of erbB2 receptor tyrosine kinase. Bioorg Med Chem Lett. 2008;18(2):674–678. doi: 10.1016/j.bmcl.2007.11.052. [DOI] [PubMed] [Google Scholar]
- 35.Nittoli T, Dushin RG, Ingalls C, et al. The identification of 8,9-dimethoxy-5-(2-aminoalkoxy-pyridin-3-yl)-benzo[c][2,7]naphthyridin-4-y lamines as potent inhibitors of 3-phosphoinositide-dependent kinase-1 (PDK-1) Eur J Med Chem. 2010;45(4):1379–1386. doi: 10.1016/j.ejmech.2009.12.036. [DOI] [PubMed] [Google Scholar]
- 36.Biondi RM, Cheung PC, Casamayor A, Deak M, Currie RA, Alessi DR. Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J. 2000;19(5):979–988. doi: 10.1093/emboj/19.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frodin M, Jensen CJ, Merienne K, Gammeltoft S. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J. 2000;19(12):2924–2934. doi: 10.1093/emboj/19.12.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hindie V, Stroba A, Zhang H, et al. Structure and allosteric effects of low-molecular-weight activators on the protein kinase PDK1. Nat Chem Biol. 2009;5(10):758–764. doi: 10.1038/nchembio.208. [DOI] [PubMed] [Google Scholar]
- 39.Sadowsky JD, Burlingame MA, Wolan DW, McClendon CL, Jacobson MP, Wells JA. Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc Natl Acad Sci U S A. 2011;108(15):6056–6061. doi: 10.1073/pnas.1102376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stockman BJ, Kothe M, Kohls D, et al. Identification of allosteric PIF-pocket ligands for PDK1 using NMR-based fragment screening and 1H-15N TROSY experiments. Chem Biol Drug Des. 2009;73(2):179–188. doi: 10.1111/j.1747-0285.2008.00768.x. [DOI] [PubMed] [Google Scholar]
- 41.Kozikowski AP, Sun H, Brognard J, Dennis PA. Novel PI analogues selectively block activation of the pro-survival serine/threonine kinase Akt. J Am Chem Soc. 2003;125(5):1144–1145. doi: 10.1021/ja0285159. [DOI] [PubMed] [Google Scholar]
- 42.Gills JJ, Holbeck S, Hollingshead M, Hewitt SM, Kozikowski AP, Dennis PA. Spectrum of activity and molecular correlates of response to phosphatidylinositol ether lipid analogues, novel lipid-based inhibitors of Akt. Mol Cancer Ther. 2006;5(3):713–722. doi: 10.1158/1535-7163.MCT-05-0484. [DOI] [PubMed] [Google Scholar]
- 43.Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther. 2003;2(11):1093–1103. [PubMed] [Google Scholar]
- 44.Falasca M, Chiozzotto D, Godage HY, et al. A novel inhibitor of the PI3K/Akt pathway based on the structure of inositol 1,3,4,5,6-pentakisphosphate. Br J Cancer. 2010;102(1):104–114. doi: 10.1038/sj.bjc.6605408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Razzini G, Berrie CP, Vignati S, et al. Novel functional PI 3-kinase antagonists inhibit cell growth and tumorigenicity in human cancer cell lines. FASEB J. 2000;14(9):1179–1187. doi: 10.1096/fasebj.14.9.1179. [DOI] [PubMed] [Google Scholar]
- 46.Piccolo E, Vignati S, Maffucci T, et al. Inositol pentakisphosphate promotes apoptosis through the PI 3-K/Akt pathway. Oncogene. 2004;23(9):1754–1765. doi: 10.1038/sj.onc.1207296. [DOI] [PubMed] [Google Scholar]
- 47.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11(5):425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 48.Bain J, Plater L, Elliott M, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408(3):297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stauffer F, Maira S-M, Furet P, Garcia-Echeverria C. Imidazo[4,5-c]quinolines as inhibitors of the PI3K/PKB-pathway. Bioorg Med Chem Lett. 2008;18:1027–1030. doi: 10.1016/j.bmcl.2007.12.018. 18248814. [DOI] [PubMed] [Google Scholar]
- 50.Islam I, Brown G, Bryant J, et al. Indolinone based phosphoinositide-dependent kinase-1 (PDK1) inhibitors. Part 2: optimization of BX-517. Bioorg Med Chem Lett. 2007;17(14):3819–3825. doi: 10.1016/j.bmcl.2007.05.060. [DOI] [PubMed] [Google Scholar]
- 51.Islam I, Bryant J, Chou YL, et al. Indolinone based phosphoinositide-dependent kinase-1 (PDK1) inhibitors. Part 1: design, synthesis and biological activity. Bioorg Med Chem Lett. 2007;17(14):3814–3818. doi: 10.1016/j.bmcl.2007.04.071. [DOI] [PubMed] [Google Scholar]
- 52.Gopalsamy A, Shi M, Boschelli DH, et al. Discovery of dibenzo[c,f][2,7]naphthyridines as potent and selective 3-phosphoinositide-dependent kinase-1 inhibitors. J Med Chem. 2007;50:5547–5549. doi: 10.1021/jm070851i. 17941624. [DOI] [PubMed] [Google Scholar]