

ABSTRACT
Malignant hyperthermia manifests as a rapid and sustained rise in temperature in response to pharmacological triggering agents, e.g. inhalational anesthetics and the muscle relaxant suxamethonium. Other clinical signs include an increase in end-tidal CO2, increased O2 consumption, as well as tachycardia, and if untreated a malignant hyperthermia episode can result in death. The metabolic changes are caused by dysregulation of skeletal muscle Ca2+ homeostasis, resulting from a defective ryanodine receptor Ca2+ channel, which resides in the sarcoplasmic reticulum and controls the flux of Ca2+ ions from intracellular stores to the cytoplasm. Most genetic variants associated with susceptibility to malignant hyperthermia occur in the RYR1 gene encoding the ryanodine receptor type 1. While malignant hyperthermia susceptibility can be diagnosed by in vitro contracture testing of skeletal muscle biopsy tissue, it is advantageous to use DNA testing. Currently only 35 of over 400 potential variants in RYR1 have been classed as functionally causative of malignant hyperthermia and thus can be used for DNA diagnostic tests. Here we describe functional analysis of 2 RYR1 variants (c. 7042_7044delCAG, p.ΔGlu2348 and c.641C>T, p.Thr214Met) that occur in the same malignant hyperthermia susceptible family. The p.Glu2348 deletion, causes hypersensitivity to ryanodine receptor agonists using in vitro analysis of cloned human RYR1 cDNA expressed in HEK293T cells, while the Thr214Met substitution, does not appear to significantly alter sensitivity to agonist in the same system. We suggest that the c. 7042_7044delCAG, p.ΔGlu2348 RYR1 variant could be added to the list of diagnostic mutations for susceptibility to malignant hyperthermia.
KEYWORDS: anesthesia, calcium channel, malignant hyperthermia, ryanodine receptor, skeletal muscle
Introduction
Malignant hyperthermia (MH) is an autosomal dominant pharmacogenetic disorder that predisposes susceptible individuals to an acute increase in cytosolic [Ca2+] during general anesthesia using volatile anesthetics or depolarizing muscle relaxants.1,2 In addition, several studies suggest that MH-susceptibility is commonly associated with elevated resting cytosolic [Ca2+].3-5 Acutely elevated cytosolic [Ca2+] results in a hypermetabolic state, characterized by an increase in end-tidal CO2, increased O2 consumption, and tachycardia. Hyperthermia results with core temperatures increasing by 1°C every 5 minutes.6 Uncontrolled hypermetabolism leads to respiratory and metabolic acidosis, rhabdomyolysis, hyperkalemia, widespread organ failure and death if untreated by the drug dantrolene.7
The majority (50–70%) of individuals affected with MH have gain-of-function variants in the RYR1 gene, encoding the skeletal muscle ryanodine receptor, 8,9 while ∼1% have variants in the α1s subunit of the dihydropyridine receptor (DHPR) gene (CACNA1S).10 The DHPR is a multi-subunit voltage-dependent Ca2+ channel, which resides in the T-tubule membrane. Upon response to membrane depolarization it causes the RyR1 tetramer to open due to physical contact between the 2 channels. As a result Ca2+ is released into the cytoplasm from the sarcoplasmic reticulum (SR) stores, triggering muscle contraction as well as a range of other metabolic responses. This process is known as excitation-contraction coupling.11 Ca2+ is returned to the sarcoplasmic reticulum by a Ca2+-ATPase (SERCA).12 This tightly controlled balance of intracellular Ca2+ concentrations is dysregulated in an MH crisis.
Because MH is a life-threatening disorder, susceptibility should ideally be determined prior to general anesthesia, such that non-triggering agents can be used. MH susceptibility in New Zealand is normally determined by in vitro contracture test (IVCT) using skeletal muscle biopsy tissue following the European Malignant Hyperthermia Group (EMHG) protocol.13 DNA-based diagnostic tests were introduced in New Zealand in 1998,14 to augment the IVCT and, while these are now becoming more accepted, 15,16 they do not replace the IVCT for many individuals because of the genetic heterogeneity of MH.17-19 In addition, DNA-based diagnosis for MH susceptibility can be carried out only in families with known mutations that have been functionally characterized.18 This constraint, while appropriate, represents a major hurdle to being able to offer DNA testing on a more general basis. In addition, an MHN diagnosis cannot be made upon a DNA test alone, rather an IVCT is recommended where a DNA test is negative for a familial mutation.20
Here we report functional analysis of 2 RYR1 variants, c.641C>T, p.Thr214Met and c.7042_7044delCAG, p.ΔGlu2348 in the same family, both of which have been linked to MH previously.21,22 We constructed human RYR1 cDNAs with either variant as well as a construct containing both. Functional analysis was carried out in transiently transfected HEK293T cells using 4-CmC as a specific RyR1 agonist and fura-2 as the fluorescent Ca2+ indicator. The c.641C>T, p.Thr214Met RyR1 variant did not appear to affect Ca2+ release in this system, but the c.7042_7044delCAG, p.ΔGlu2348 deletion resulted in a hypersensitive channel suggesting that it is likely to be causative of MH.
Results
In vitro contracture tests
Muscle biopsies and in vitro contracture tests were carried out as described in methods for the proband and his mother (II:1 and I:1 respectively in Fig. 1). The proband was diagnosed MHS and the mother MHN. Thus far the father (I:2, Fig. 1) has declined IVCT testing. The IVCT results are shown in Table 1.
Figure 1.
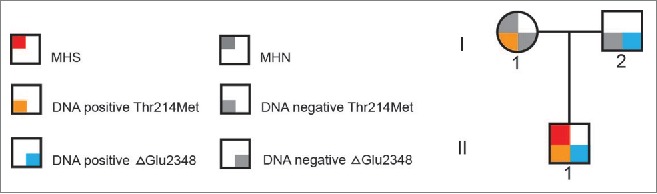
Pedigree diagram pedigree diagram showing inheritance of RyR1 variants from each parent and MH status where known.
Table 1.
IVCT values.
| Patient | Contracture (g) at 2% halothane | Contracture (g) at 2 mM caffeine | MH Status |
|---|---|---|---|
| I:1 | 0.4 | 0.2 | MHN |
| II:1 | 5.2 | 3.0 | MHS |
DNA sequencing and variant screening
The entire RYR1 gene of the proband (Fig. 1, II:1) and his mother (Fig. 1, I:1) as well as the CACNA1S gene and 48 others with potential roles in skeletal muscle Ca2+ handling were screened using the HaloPlex™ target enrichment system followed by next generation sequencing as described in methods. All exons of the CACNA1S and RYR1 genes were represented in the data set with an average coverage between 30 and 156 with the exception of RYR1 exon 91, which was completed using Sanger sequencing. The proband carries both the c.641C>T, p.Thr214Met and c. 7042_7044delCAG, p.ΔGlu2348 RyR1 variants. Neither variant is listed in the Exome Variant Server. The mother carries the c.641C>T, p. Thr214Met RyR1 variant only. No other rare (minor allele frequency of < 0.1%) variants in any other genes in this dataset were identified for either subject. The c. 7042_7044delCAG, p.ΔGlu2348 RyR1 variant was detected in genomic DNA from the father (Fig. 1, I:2) using kinetic PCR followed by high resolution amplicon melting (HRM).23 Neither variant was detected by HRM screening of a panel representing 150 MHN patients.
Expression of recombinant RyR1 in HEK293T cells
RyR1 expression in transiently transfected HEK293T cells was determined by western blotting to confirm approximately equal amounts of recombinant protein was produced for each construct (Fig. 2). Alpha-tubulin (∼50 kDa) was used as a loading control. No RyR1 protein was detected in the vector-only (pcDNA3.1+) negative control.
Figure 2.
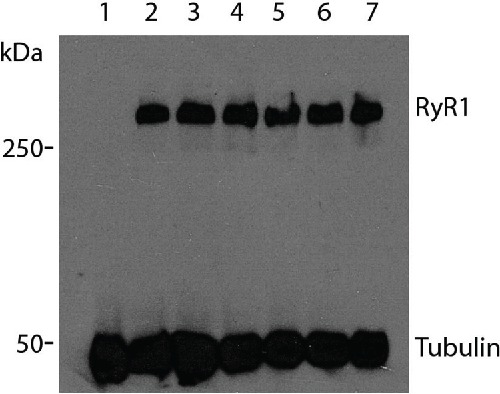
Immunoblot showing expression of RyR1 constructs Immunoblot of HEK293T cells transiently transfected with RYR1 expression plasmids. Total protein (270 µg) extracts were separated on a 7.5% SDS-PAGE gel and transferred onto a PVDF membrane. RyR1, > 250 kDa, and α-tubulin, 50 kDa, were detected using anti-RyR1 (34C) and anti-α-tubulin antibodies respectively. Lane 1, pcDNA3.1+ vector only; lane 2, pcRyR1 wildtype; lane 3, pcRyR1 Gly248Arg; lane 4, pcRyR1 Arg2452Trp; lane 5, pcRyR1 Thr214Met; lane 6, pcRyR1 Thr214Met/ΔGlu2348; lane 7, pcRyR1 ΔGlu2348.
Immunofluorescence was used to confirm correct co-localization of the recombinant RyR1 proteins with the endoplasmic reticulum (ER) marker protein disulfide isomerase, (PDI, Fig. 3). No RyR1 was detected in HEK293T cells transfected with empty pcDNA3.1+ vector, used as a negative control.
Figure 3.

Co-localization of RyR1 with the endoplasmic reticulum. Immunofluorescence of transiently transfected HEK293T cells with RyR1 cDNA. Variants are indicated by their amino acid change. Primary antibodies that specifically recognize RyR1 (34C) and PDI as well as fluorescently-labeled secondary antibodies FITC (green) and TRITC (red) were used to visualize RyR1 and PDI respectively, while nuclei were visualised by staining the cells with DAPI (blue). Cells were examined by confocal fluorescence microscopy at a magnification of 1260 X; the scale bar in each merged image represents a length of 20 microns.
Ca2+ release assays
The activities of variant RyR1 channels expressed in the ER of HEK293T cells were compared with the activity of the wild-type (WT) RyR1 channels using Ca2+ release assays under identical conditions. The increase in cytoplasmic calcium was measured after the addition of incremental doses of the RyR1 agonist 4-CmC to the cells and then calculated as a percentage of total Ca2+ release (Fig. 4). The RyR1 variants c.742G>A, p.Gly248Arg and c.7354C>T, p.Arg24-52Trp were used as positive controls as the former has been validated as an MH-causative mutation by the EMHG (https://emhg.org/genetics/mutations-in-ryr1/) and the latter was shown to be causative using our system.24 Both had significantly increased sensitivities to 4-CmC compared to WT as expected (Table 2). There was no significant difference between the c.641C>T, p. Thr214Met RyR1 variant and WT, with respect to stimulation by 4-CmC, although the concentration-response curve lay between WT and the positive controls (Fig. 4, yellow trace). The p.ΔGlu2348 RyR1 variant was hypersensitive compared to WT when stimulated with 4-CmC as with both agonists the curve was shifted to the left (Fig. 4, dark blue trace). A similar result was observed for the double RyR1 variant p.Thr214Met/ p.ΔGlu2348. EC50 values are presented in Table 2. Resting cytosolic calcium was significantly different to empty vector for the p.Arg2452Trp and p.ΔGlu2348 RyR1 constructs only. All other constructs including the double variant, p.Thr214Met/p.ΔGlu2348 were similar to the vector only control (Table 3).
Figure 4.
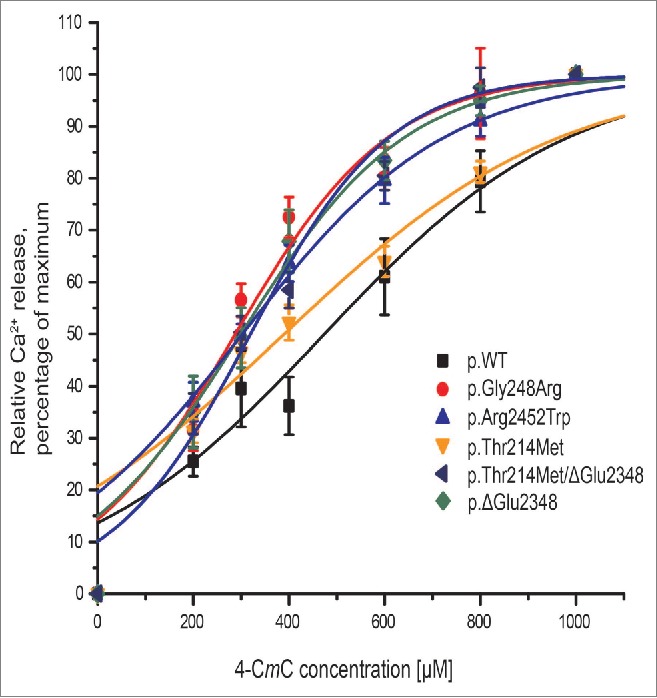
Ca2+ release assays in HEK293T cells. Ca2+-release is illustrated in concentration-response curves for transiently transfected HEK293T cells. Ca2+ released was measured for 4-CmC concentrations between 0 and 1000 μM, values were normalized to Ca2+ released at 1000 μM 4-CmC and represented as mean ± SEM (n ≥ 8). Curves were plotted using OriginLab Origin 8 software.
Table 2.
EC50 values 4-CmC.
| construct | EC50 µM 4-CmC | SEM | p-value |
|---|---|---|---|
| WT | 481 | 15.00 | |
| Gly248Arg | 270 | 6.00 | *8.21×10−4 |
| Arg2452Trp | 341 | 7.70 | *6.6×10−4 |
| Thr214Met | 403 | 15.00 | 0.250 |
| Thr214Met/ΔGlu2348 | 219 | 15.22 | *2.08×10−5 |
| ΔGlu2348 | 261 | 20.70 | *1.45×10−4 |
The concentration of 4-CmC required to half maximally activate RyR1 from individual assays was determined using OriginLab Origin 8 software. Results are represented as mean ± SEM. The Student's unpaired T test, followed by a Bonferroni correction, was used to determine the statistical difference between the EC50 value for cells transfected with WT RYR1 cDNA and each variant.
Table 3.
Resting cytosolic Ca2+.
| construct | Ratio 340/380 | SEM | p-value |
|---|---|---|---|
| pcDNA3.1+ vector only | 1.030348 | 0.006029 | |
| WT | 1.038354 | 0.002792 | 0.22291 |
| Gly248Arg | 1.030568 | 0.004414 | 0.13118 |
| Arg2452Trp | 1.061786 | 0.005017 | *2.99 × 10−5 |
| Thr214Met | 1.038319 | 0.003382 | 0.99381 |
| Thr214Met/ΔGlu2348 | 1.049861 | 0.005694 | 0.05691 |
| ΔGlu2348 | 1.088946 | 0.004969 | *7.98 × 10−16 |
Ratio of fluorescence emission at 510 nm using excitation at 340 nm and 380 nm prior to the exposure of RyR1 agonists. Represented is the mean fluorescence emission ratio ± SEM for each construct. The Student's unpaired T test, followed by a Bonferroni correction, was used to determine the statistical difference between cells transfected with the pcDNA3.1+ vector only, and each RyR1 construct.
Discussion
The objective of the current study was to determine the effect of 2 RyR1 variants on the function of the skeletal muscle Ca2+ channel using a heterologous system in vitro. Both variants occur in the proband, having been inherited from each parent (Fig. 1). As positive controls for hypersensitivity, the previously-characterized MH-causative c.742G>A, p.Gly248Arg25 and c.7354C>T, p.Arg2452Trp24 RyR1 mutations were also tested under the same conditions. Under our experimental conditions RyR1 containing the c.641C>T, p. Thr214Met variant cannot be functionally distinguished from the WT RyR1 channel, although the different shaped concentration response curves for 4-CmC suggest some abnormality may exist. While the mother who carries this variant was diagnosed as MHN by IVCT, 3 families in the United Kingdom show concordance between this variant and disease and/or IVCT phenotype.21 The IVCT results for the mother (Fig. 1, I:1 and Table 1) are borderline between MHS and MHN so the MHN diagnosis could be a false negative given that the IVCT has been determined to have a specificity of only 93.6% and sensitivity of 99%.26 False negative IVCT results could be due to the testing laboratory-defined cut-off values27-29 or to variable penetrance of mutations detected.9 In support of the MHN diagnosis however, a retrospective study of 329 patients diagnosed MHN in New Zealand, showed no evidence of any adverse reaction in subsequent anesthesia using triggering agents.28 In addition it is important to note that the transfected HEK293T cells express only exogenous RyR1 with none of the other proteins known to be part of the skeletal muscle triad, the functional unit of excitation-contraction coupling, being present. This variant may therefore have an RyR1 function that cannot be investigated in our system due to the lack of protein-protein interactions with partners of the excitation-contraction system. The c.641C>T, p.Thr214Met residue is located within a β sheet in the β-trefoil B domain in the structurally-characterized N-terminal domain encompassing residues 1-559 of rabbit RyR1. This region forms 3 domains predicted to interact through a hydrophilic interface.30 The Thr214 residue does not appear to be part of the direct domain interaction but is located in a linker region between 2 domains, which is buried in the 3-dimensional structure. Methionine is an aliphatic residue however, as well as being larger than the hydrophilic threonine, which may lead to structural alteration of the folded protein or localized misfolding. Although our functional data suggest the c.641C>T, p. Thr214Met variant is unlikely to be a variant altering Ca2+ release from the SR/ER, the concordance between this variant and MHS in 3 other families, as well as the limitations of the HEK293T assay system indicate that currently we cannot rule out a functional consequence of this variant for RyR1.
On the other hand the c. 7042_7044delCAG, p.ΔGlu2348 variant results in a hypersensitive channel, which suggests that it is likely to cause MH. While the clinical signs of MH for the proband (Fig. 1, II:1) were minor, the IVCT data (Table 1) indicated very strong contractions and a clear MHS diagnosis. DNA testing has shown that the father (Fig. 1, I:2) carries the c. 7042_7044delCAG, p.ΔGlu2348 RyR1, and while MH status has not been confirmed by IVCT, he should be considered at high risk. This RYR1 variant has been associated with MHS in at least one other unrelated family, also associated with very strong contracture tension.31 No predictions however, can be made about any mechanistic effect that this deletion would have on structure or function of the channel. While a near-atomic resolution cryo EM structure predicts this residue to be situated in a helical domain, the resolution was sufficient to trace the backbone structure only.32
It has been suggested that MH can be caused by hyperactive RyR1 channels resulting in a higher than normal Ca2+ release under stimulating conditions causing a crisis event.33 Our results support this conclusion for the c. 7042_7044delCAG, p.ΔGlu2348 variant. As RyR1 with the p.Arg2452Trp and p.ΔGlu2348 variants demonstrated significantly higher resting calcium levels, these variants may result in a functionally “leaky” channel. The p.Arg2452 residue is thought to lie at a domain interface 34 in the cytoplasmic domain of the RyR1 tetramer and perturbation of predicted surface interactions by the introduction of the bulky hydrophobic tryptophan in place of the positively charged arginine residue could have functional consequences for channel stability in the unstimulated state. These observations support the “leaky” channel mechanism suggested as an alternative mechanism leading to an MH crisis.35 They also support previous work with synthetic peptides representing this region, which indicate that interdomain interactions are likely to be responsible for opening and closing the tetrameric channel.36,37
While calcium release was similar for the RyR1 p.ΔGlu2348 and the p.Thr214Met/p.ΔGlu2348 variants, the p.Thr214Met/p.ΔGlu2348 variant did not appear to result in a “leaky” channel. This suggests that the p.Thr214Met variant may in some way compensate for the p.ΔGlu2348 variant in the resting state. It should be noted however, that these 2 variants are in the same RyR1 monomer in this recombinant system, rather than in different monomers as would be the case in the proband. Even though co-transfection of different RYR1 constructs has been performed in previous studies, 33,38 the exact assembly of the tetrameric RyR1 channel with the different monomers cannot be predicted.
As the c. 7042_7044delCAG, p.ΔGlu2348 RyR1 variant has been identified in at least 2 unrelated families and is a conserved residue, 31 as well as conferring hypersensitivity to a specific RyR1 agonist in a heterologous system, it should be classed as an MH causative mutation. A substitution at this position (c.7043 A>G, p.Glu2348Gly) also segregates with MHS in a UK family, 39 supporting the potential importance of this residue in RyR1 function.
Patients and methods
Case history
A 7 y old, 22 kg child with no family history presented for squint correction in 1999. He had a gas induction with Sevoflurane in oxygen, and was then intubated with 15 mg Rocuronium. Once intubated, he was noted to have a high end-tidal CO2 of 67 mmHg. This resolved with minute ventilation of 2.5 L/min bringing end-tidal CO2 down to 42 mmHg. After induction, the volatile agent was changed to isoflurane for maintenance of anesthesia. Approximately 20 minutes into the case, the end-tidal CO2 again rose from 42 to 58 mmHg, and he developed a mild tachycardia. A rectal temperature was taken which was 37°C and he felt warm to the touch. He was given analgesia with fentanyl 40 µg, and then morphine 2 mg, and the tachycardia resolved. He was given antiemetic prophylaxis with 2 mg of Ondansetron. The anesthetist was concerned that these subtle changes (tachycardia and raised end-tidal CO2) may represent MH, so he stopped administering the volatile agent, went onto high fresh gas flows and maintained anesthesia with a propofol infusion. At the closure of surgery the child was extubated and taken to recovery. Creatine kinase was 221 U/L; the upper limit of normal at the laboratory was 220 U/L. His temperature never rose above 37°C degrees and he was not given dantrolene. He was observed overnight in the intensive care unit, and no other signs of MH were noted. He was referred for in vitro contracture testing at Palmerston North hospital and a diagnosis of MHS was confirmed.
Regulatory authority approvals
This study has been approved by the Central Regional Ethics Committee, Ministry of Health, Wellington, New Zealand (MWH/03/04/018) and the Massey University Genetic Technology Committee (GMO 05/MU/01 and GMO 00/MU/60) acting as an Institutional Biological Safety Committee for the Environmental Protection Authority, Wellington, New Zealand.
In vitro contracture tests
Muscle biopsies were obtained and in vitro contracture testing (IVCT) was performed at Palmerston North hospital using the standard diagnostic procedure for MH-susceptibility according to the European Malignant Hyperthermia group (EMHG) protocol and local laboratory control data.26-28 Briefly this involves exposure of muscle bundles to incremental amounts of halothane and caffeine, while measuring contracture tension in g. A diagnosis of MHS is made if the responses to both halothane and caffeine are above the threshold of 0.4 g using 2% halothane and 0.2 g using 2 mM caffeine. An MHN diagnosis is made if the response is below threshold for both agents. If the response is above threshold for only one of the agents the diagnosis is MHS(h) or MHS(c) for halothane and caffeine respectively. All patients tested MHS, MHS(h) or MHS(c) would be classed as susceptible to MH and be given non-triggering anesthesia.13
DNA sequencing
Genomic DNA was isolated from patient blood samples (3 mL) using the Promega Wizard™ genomic DNA extraction kit according to the manufacturer's instructions. This was followed by quanti-fication using the Quant-iT™ dsDNA high-sensitivity DNA assay kit (Life Technologies, Q-33120) following the manufacturer's instructions and diluted in TE buffer to a final concentration of 5 ng/µL. The HaloPlex™ Target Enrichment system (Agilent Technologies, G9901C) was used to prepare custom libraries from the genomic DNA representing 50 genes with known or potential association with MH. Purified amplified libraries were quantified using the High Sensitivity DNA kit (Agilent Technologies, 5067-4626 ) with the 2100 Bioanalyzer. Libraries were pooled in equimolar (400 pmol) amounts prior to massively parallel sequencing on the Illumina MiSEQ platform by the Next Generation Sequencing Facility, University of Leeds and the Leeds Teaching Hospitals NHS Trust, St James University Hospital Leeds, UK or New Zealand Genomic Limited at Massey University, Palmerston North, New Zealand. Sequences were aligned using Agilent SureCall™ software against hg19 using the default parameters. Exon 91 of RYR1 was amplified in 3 segments using Dream Taq™ DNA polymerase (Fermentas, FMTEP0702), the products purified using ExoSAP-IT® (Affymetrix, 78200) and then 10 ng of each PCR product was mixed with 4 pmol of the appropriate primer and Sanger sequencing was carried out by Macrogen (Korea). The PCR and sequencing primers are available on request. Sequences were aligned to the RYR1 cDNA (NM_000540.2) using the web based tool needle through EMBOSS (http://www.ebi.ac.uk/Tools/psa/emboss_needle/nucleotide.html).
High resolution amplicon melting
Primers (Table 4) for high resolution amplicon melting (HRM) assays were designed for the c.641C>T, p.Thr214Met and p.ΔGlu2348 RyR1 variants using the Light Cycler™ Primer Design software version 2.0 (Roche). cDNA and protein numbering are according to NM_000540.2 and NP_000531.2, respectively. Reactions contained 3 µM MgCl2, 0.3 or 0.2 µM of each primer for the p.Thr214Met and p.ΔGlu2348 variants respectively and, 5 µL of 2x LC480 HRM master mix (Roche, 04909631001) and 25 ng of genomic DNA in a total volume of 10 µL. PCR amplification was carried out on a Roche Light Cycler 480 using a touch-down protocol as follows: 95°C for 10 minutes (ramp rate 4.40°C/sec), followed by 40 cycles of 95°C (ramp rate 4.40°C/sec) for 10 sec, 65°C (ramp rate 2.20°C/sec) with secondary target of 58°C (step size 0.5°C) for 10 sec and 72°C (ramp rate 4.40°C/min) for 4 sec. Single acquisition of data was at the 72°C amplification step. The melt program was as follows: 95°C for 1 min (ramp rate 4.40°C/sec), 40°C for 1 min (ramp rate 1.5°C/sec), 76°C for 1 sec (ramp rate 4.40°C/sec) followed by continuous acquisition at 92°C with a ramp rate of 0.02°C/sec and 25 acquisitions per °C. The final cooling cycle had a 40°C target, 30 sec hold and a ramp rate of 1.5°C/sec. Alleles were called using GeneScan software version 1.5.1.
Table 4.
Oligonucleotide primers.
| Primer name | Sequence | Purpose |
|---|---|---|
| Thr214Met F_SDM | GAAGAGGGCTTCGTGATGGGAGGTCACGTCCTC | Site-directed mutagenesis |
| Thr214Met R_SDM | GAGGACGTGACCTCCCATCACGAAGCCCTCTTC | Site-directed mutagenesis |
| ΔGlu2348 F_SDM | GTCAACGGCGAGAGCGTGGAGAACGCCAATGTGGTGGTGCGG | Site-directed mutagenesis |
| ΔGlu2348 R_SDM | CCGCACCACCACATTGGCGTTCTCCTCCACGCTCTCGCCGTTGAC | Site-directed mutagenesis |
| Thr214Met F_HRM | CAACTTCCCTTGCTCCT | HRM |
| Thr214Met R_HRM | CGCTGGTCATCACTGTCA | HRM |
| ΔGlu2348 F_HRM | CCAGGCGAGAGCGTGGAG | HRM |
| ΔGlu2348 R_HRM | CGGATGAGCAGCCGCACC | HRM |
Site directed mutagenesis and cloning
Mutagenesis was carried out by PCR-amplification of ∼70 ng of plasmid DNA containing approximately 2.7 kb of the relevant subclones of human RYR1 cDNA 40 (NM_000540.2) using the 2x Kapa HiFi HotStart Readymix (Kapa Biosystems, KR0370) diluted 4x. Complementary mutagenic primer pairs (Table 4) were sourced from Integrated DNA Technologies and used for QuikChange™ (Stratagene) mutagenesis. Cycling conditions were 95°C for 5 min; 98°C for 2 minutes; 57°C for 30 seconds; 72°C at 1 min/kb; 72°C for 5 minutes; steps 2-3 were repeated 18 times. The PCR products were digested with 20 units of restriction endonuclease DpnI (New England BioLabs, R0176S) and buffer according to the manufacturer's instructions.
The full-length RYR1 cDNA containing the specified variants was generated in 3 sequential cloning steps by standard methods.40 Constructs were confirmed by Sanger DNA sequencing using a capillary ABI3730 Genetic Analyzer with BigDye™ Terminator v3.1 chemistry at the Massey Genome Service, Palmerston North, New Zealand.
Cell culture and transient transfection
HEK293T cells for all applications were grown in DMEM (Dulbecco's Modified Eagle's Medium, Sigma D7777), 10% FBS (fetal bovine serum, Gibco 10091-148), 0.5% penicillin/streptomycin (Life Technologies, 15140-122) at 37°C, 5% CO2 in a humidified atmosphere. For immunoblotting cells were grown to 90% confluence in T25 flasks using 8 mL of medium and for immunofluorescence, to 50% confluence in a 4 chambered slide containing 1 mL medium. For Ca2+ release assays, cells were grown to 80% confluence in UV transparent 96 well plates with each well containing 200 µL medium. Medium was replaced one hour prior to transfection once the correct level of confluence was achieved.
For immunoblotting HEK293T cells were transiently transfected with RYR1 cDNA or empty pcDNA3.1+ (Invitrogen, V790-20) plasmids, using 6 µg plasmid DNA and 24 µL FuGENE HD (Promega, E2311) to a total volume of 300 µL with unsupplemented DMEM, which was added directly to the 8 mL of medium in T25 flasks. After 48 hours, the medium was replaced and growth continued for another 24 hours. Protein extracts were prepared after washing cells with PBS and resuspending in 150 µL of lysis buffer [0.1 M Tris HCl, pH 7.8, 0.5% triton X-100, 20 µL 7x cOmplete Mini EDTA-free protease inhibitor (Roche, 11836170001)]. Insoluble proteins were separated via centrifugation at 13,000 rcf at 4°C. Supernatant was stored at −80°C with limited freeze-thaw cycles.
For immunofluorescence, transient transfection was carried out using 1 µg DNA, 3 µL Fugene 6 (Roche, 11814443001) and DMEM without supplements, to a final volume of 50 µL. Fresh complete medium was replaced after 48 hours and growth continued for another 24 hours prior to processing.
For Ca2+ release assays, HEK293T cells (with 100 µL complete medium per well) were transfected with 100 ng DNA, 1.2 µL Fugene HD in unsupplemented DMEM to a final volume of 14 µL. Cells were incubated overnight prior to the addition of an extra 100 µL complete DMEM. After 24 hours incubation, media was replaced with 200 µL complete DMEM. After a further 24 hours incubation the transfected cells were used in calcium release assays.
Immunoblotting
Protein extracts (∼270 µg) were resolved by SDS-PAGE (4% stacking gel and 7.5% separating gel) for 2 hours at 120 V using a Mini PROTEAN electrophoresis system (Bio-Rad). Proteins were transferred to a PVDF membrane at 70 mA for 20 hours at 4°C. After transfer, the membrane was blocked in 5 mL 5% skim milk in TBST (0.05 M Tris HCl, 0.15 mM NaCl, 0.1% tween 20) for 3 hours at room temperature with gentle agitation. The membrane was cut to separate the 565 kDa RyR1 from the 50 kDa α tubulin loading control. The membrane containing higher molecular weight proteins was incubated overnight at 4°C in 5 mL mouse 34C primary antibody (Sigma, R129) diluted 1:1000 in 2.5% skim milk in TBST with gentle shaking. The membrane containing the lower molecular weight proteins was incubated overnight at 4°C in 5 mL mouse α-tubulin primary antibody (Sigma, T8203) diluted 1:5000 in 2.5% skim milk in TBST with gentle shaking. Both membranes were then incubated at room temperature for 20 minutes with shaking prior to 3 washes in TBST. Both membranes were incubated in 5 mL anti-mouse horse radish peroxidase conjugated secondary antibody (Sigma, A9044) diluted 1:5000 in 2.5% skim milk in TBST for one hour at room temperature. The membranes were washed 3 times in TBST prior to detection. Chemiluminescence blotting substrate (Roche, 11500694001) was prepared by mixing 3 mL luminescence substrate solution A with 30 µL starting solution B. The substrate was applied to the membranes just prior to vizualization of the proteins by the exposure to X-ray film.
Immunofluorescence
After an initial washing step in 500 µL of PBS (0.14 M NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 pH 7.2) cells were fixed in 500 µL 2% paraformaldehyde in PBS for 15 minutes at room temperature and washed 3 times in PBS and then permeabilised using 0.1% triton X-100 in PBS for 5 minutes. After washing 3 times in PBS, the cells were blocked in 1 mL 5% bovine serum albumin, 0.5% Tween-20 in PBS with gentle shaking at room temperature for 5 minutes, then incubated overnight at 4°C with gentle shaking in 1 mL primary antibody solution [mouse 34C diluted 1:1000 or rabbit anti protein disulphide isomerase, PDI (Sigma, p7496) diluted 1:1000 in PBS]. The cells were then incubated at room temperature for 20 minutes, and washed 3 times in PBS. Finally, the cells were incubated at room temperature with gentle shaking for 1 hour in 500 µL secondary antibody solution [fluorescein isothiocyanate (FITC) conjugated goat anti-mouse secondary antibody (Jackson immuno research, 15095003) diluted 1:200 in 500 µL PBS, for RyR1 or tetramethylrhodamine (TRITC) conjugated goat anti-rabbit secondary antibody (Jackson immuno research, 11025003) diluted 1:200 in 500 µL for PDI and then washed 3 times in PBS. A cover slip was mounted using 7 µL ProLong Gold AntiFade mounting solution containing DAPI (Invitrogen, p-36931) to stain the nucleus. The slides were incubated over night before being visualised using a Leica SP5 DM6000B Scanning Confocal Microscope at 1260 × magnification at the Massey Microscopy and Imaging Center.
Measurement of Ca2+ release
Transiently-transfected HEK293T cells were incubated in 100 µL balanced salt solution, BSS (140 mM NaCl, 2.8 mM KCl, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES, pH 7.25) containing 2 mM CaCl2, 2 µM fura2-AM (Molecular Probes, F-1221) and 0.01% pluronic F-127 (Sigma P2443) per well for one hour at 37°C in the dark. The cells were washed once with 100 µL BSS plus Ca2+ and once in 100 µL Ca2+-free BSS buffer (containing 2 mM EGTA). Activation of the RyR1 using 4-CmC (Sigma, C55402) as the agonist was measured by the change in the fluorescence emission ratio at 510 nm, when excited at 340 nm and 380 nm, using an Olympus XI81 fluorescence microscope. Each well was assayed after establishing a fluorescence ratio baseline for using 100 µL Ca2+-free balanced salt solution before adding 100 µL Ca2+-free balanced salt solution containing 4-CmC. The baseline of the fluorescence ratio was used to estimate integrity of the ER membrane and potential leakage due the presence of RyR1 variant proteins. The final concentrations of 4-CmC used were: 200, 300, 400, 600, 800 and 1000 µM.
Statistical analysis
The amount of Ca2+ release at each concentration of 4-CmC was normalized to account for any differences in cell density for each assay and calculated as a percentage of the total Ca2+ released with 1000 µM 4-CmC. A minimum of 8 analytical replicates were carried out for each construct, the results were pooled and a sigmoidal curve fitted for each data set using OriginLab Origin 8 software. Results are presented as the mean ± standard error of the mean (SEM) for each value of 4-CmC used. The concentration of agonist required for half maximal fluorescence change (EC50) was calculated from curves fitted to individual replicates and represented as mean ± SEM. The statistical significance of each EC50 value was determined using the unpaired Student's t-test with OriginLab Origin 8 software and represented as a p-value with respect to WT and a Bonferroni correction was carried out to confirm statistical significance.
Abbreviations
- BSS
Balanced salt solution
- CACNA1S
Gene encoding the α1s subunit of the dihydropyridine receptor
- cDNA
complementary DNA
- 4-CmC
4-chloro-m-cresol
- DAPI
4′,6-diamidino-2-phenylindole
- DHPR
Dihydropyridine receptor
- DMEM
Dulbecco's Modified Eagle's Medium
- EC50
Half maximal effective concentration
- EDTA
Ethylenediamine tetraacetic acid
- EGTA
Ethylene glycol tetraacetic acid
- EM
Electron Microscopy
- EMBOSS
European Molecular Biology Open Software Suite
- EMHG
European Malignant Hyperthermia Group
- ER
Endoplasmic Reticulum
- FBS
Fetal Bovine serum
- FITC
Fluoroscein isothiocyanate
- HRM
High Resolution Amplicon Melting
- IVCT
In vitro contracture test
- MH
Malignant hyperthermia
- MHN
Malignant hyperthermia negative
- MHS(h)
Malignant hyperthermia susceptible with abnormal response to Halothane
- MHS(c)
Malignant hyperthermia susceptible with abnormal response to caffeine
- MHS
Malignant hyperthermia susceptible
- PBS
Phosphate buffered saline
- PDI
Protein disulphide isomerase
- PVDF
polyvinylidene fluoride
- RyR1
Ryanodine receptor protein type 1
- RYR1
Gene encoding ryanodine receptor protein type 1
- SDS-PAGE
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SERCA
Sarco/endoplasmic reticulum ATPase
- SEM
Standard error of the mean
- SR
Sarcoplasmic reticulum
- TBST
Tris buffered saline tween-20
- TE
Tris-EDTA buffer
- TRITC
Tetramethylrhodamine
- UV
Ultraviolet
- WT
Wildtype.
Disclosure of potential conflicts of interest
No potential conflicts of interest are disclosed.
Acknowledgments
We would like to thank Professor Phil Hopkins (Leeds MH Unit, St. James University Hospital) for the targeted capture panel, as well as the HaloPlex™ enrichment and next generation sequencing that was carried out while the corresponding author was a visitor to his laboratory and Mrs Lili Rhodes (Institute of Fundamental Sciences, Massey University) for technical support.
Funding
This study was supported by the Massey University Research Fund for consumables and the Australian and New Zealand College of Anesthetists (grant number 15/011) for salary for AS.
References
- [1].MacLennan DH, Chen SR. The role of the calcium release channel of skeletal muscle sarcoplasmic reticulum in malignant hyperthermia. Ann N Y Acad Sci 1993; 707:294-304; PMID:9137560; http://dx.doi.org/ 10.1111/j.1749-6632.1993.tb38060.x [DOI] [PubMed] [Google Scholar]
- [2].Hopkins PM. Malignant hyperthermia: pharmacology of triggering. Br J Anaesth 2011; 107:48-56; PMID:21624965; http://dx.doi.org/ 10.1093/bja/aer132 [DOI] [PubMed] [Google Scholar]
- [3].Vukcevic M, Broman M, Islander G, Bodelsson M, Ranklev-Twetman E, Muller CR, Treves S. Functional properties of RYR1 mutations identified in Swedish patients with malignant hyperthermia and central core disease. Anesth Analg 2010:111:185-90; PMID:20142353 [DOI] [PubMed] [Google Scholar]
- [4].Yang T, Esteve E, Pessah IN, Molinski TF, Allen PD, Lopez JR. Elevated resting [Ca2+]i in myotubes expressing malignant hyperthermia RyR1 cDNAs is partially restored by modulation of passive calcium leak from the SR. Am J Physiol Cell Physiol 2007:292:C1591-8 Epub 2006 Dec 20; PMID:17182726; http://dx.doi.org/ 10.1152/ajpcell.00133.2006 [DOI] [PubMed] [Google Scholar]
- [5].Eltit JM, Bannister RA, Moua O, Altamirano F, Hopkins PM, Pessah IN, Molinski TF, Lopez JR, Beam KG, Allen PD. Malignant hyperthermia susceptibility arising from altered resting coupling between the skeletal muscle L-type Ca2+ channel and the type 1 ryanodine receptor. Proc Natl Acad Sci U S A 2012; 109:7923-8; PMID:22547813; http://dx.doi.org/ 10.1073/pnas.1119207109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wappler F. Malignant hyperthermia. Eur J Anaesthesiol 2001; 18:632-52; PMID:11553240; http://dx.doi.org/ 10.1097/00003643-200110000-00002 [DOI] [PubMed] [Google Scholar]
- [7].Lopez JR, Allen P, Alamo L, Ryan JF, Jones DE, Sreter F. Dantrolene prevents the malignant hyperthermic syndrome by reducing free intracellular calcium concentration in skeletal muscle of susceptible swine. Cell Calcium 1987; 8:385-96; PMID:3427616; http://dx.doi.org/ 10.1016/0143-4160(87)90013-3 [DOI] [PubMed] [Google Scholar]
- [8].Broman M, Gehrig A, Islander G, Bodelsson M, Ranklev-Twetman E, Ruffert H, Muller CR. Mutation screening of the RYR1-cDNA from peripheral B-lymphocytes in 15 Swedish malignant hyperthermia index cases. Br J Anaesth 2009; 102:642-9; PMID:19346234; http://dx.doi.org/ 10.1093/bja/aep061 [DOI] [PubMed] [Google Scholar]
- [9].Robinson R, Carpenter D, Shaw MA, Halsall J, Hopkins P. Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat 2006: 27:977-89; PMID:16917943; http://dx.doi.org/ 10.1002/humu.20356 [DOI] [PubMed] [Google Scholar]
- [10].Stewart SL, Hogan K, Rosenberg H, Fletcher JE. Identification of the Arg1086His mutation in the alpha subunit of the voltage-dependent calcium channel (CACNA1S) in a North American family with malignant hyperthermia. Clin Genet 2001; 59:178-84; PMID:11260227; http://dx.doi.org/ 10.1034/j.1399-0004.2001.590306.x [DOI] [PubMed] [Google Scholar]
- [11].Rebbeck RT, Willemse H, Groom L, Casarotto MG, Board PG, Beard NA, Dirksen RT, Dulhunty AF. Regions of ryanodine receptors that influence activation by the dihydropyridine receptor beta1a subunit. Skelet Muscle 2015: 5:23; PMID:26203350; http://dx.doi.org/ 10.1186/s13395-015-0049-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gilchrist JS, Palahniuk C, Abrenica B, Rampersad P, Mutawe M, Cook T. RyR1/SERCA1 cross-talk regulation of calcium transport in heavy sarcoplasmic reticulum vesicles. Can J Physiol Pharmacol 2003: 81:220-33; PMID:12733821; http://dx.doi.org/ 10.1139/y03-035 [DOI] [PubMed] [Google Scholar]
- [13].Hopkins PM, Ruffert H, Snoeck MM, Girard T, Glahn KP, Ellis FR, Muller CR, Urwyler A. European Malignant Hyperthermia Group guidelines for investigation of malignant hyperthermia susceptibility. Br J Anaesth 2015: 115:531-539; PMID:26188342; http://dx.doi.org/ 10.1093/bja/aev225 [DOI] [PubMed] [Google Scholar]
- [14].Stowell KM, Brown R, James D, Couchman K, Hodges M, Pollock N. Malignant Hyperthermia in New Zealand. NZ BioScience 1999. 7:12-17. [Google Scholar]
- [15].Litman RS, Rosenberg H. Malignant hyperthermia: update on susceptibility testing. Jama 2005; 293:2918-24; PMID:15956637; http://dx.doi.org/ 10.1001/jama.293.23.2918 [DOI] [PubMed] [Google Scholar]
- [16].Rosenberg H, Rueffert H. Clinical utility gene card for: malignant hyperthermia. Eur J Hum Genet 2011; 19; PMID:21248738; http://dx.doi.org/ 10.1038/ejhg.2010.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Robinson RL, Anetseder MJ, Brancadoro V, van Broekhoven C, Carsana A, Censier K, Fortunato G, Girard T, Heytens L, Hopkins PM, et al.. Recent advances in the diagnosis of malignant hyperthermia susceptibility: how confident can we be of genetic testing? Eur J Hum Genet 2003; 11:342-8; PMID:12700608; http://dx.doi.org/ 10.1038/sj.ejhg.5200964 [DOI] [PubMed] [Google Scholar]
- [18].Urwyler A, Deufel T, McCarthy T, West S. Guidelines for molecular genetic detection of susceptibility to malignant hyperthermia. Br J Anaesth 2001; 86:283-7; PMID:11573677; http://dx.doi.org/ 10.1093/bja/86.2.283 [DOI] [PubMed] [Google Scholar]
- [19].Robinson R, Curran JL, Hall WJ, Halsall PJ, Hopkins PM, Markham AF, Stewart AD, West SP, Ellis FR. Genetic heterogeneity and HOMOG analysis in British malignant hyperthermia families. J Med Genet 1998; 35:196-201; PMID:9541102; http://dx.doi.org/ 10.1136/jmg.35.3.196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Girard T, Treves S, Voronkov E, Siegemund M, Urwyler A. Molecular genetic testing for malignant hyperthermia susceptibility. Anesthesiology 2004; 100:1076-80; PMID:15114203; http://dx.doi.org/ 10.1097/00000542-200405000-00008 [DOI] [PubMed] [Google Scholar]
- [21].Fiszer D, Shaw MA, Fisher NA, Carr IM, Gupta PK, Watkins EJ, Roiz de Sa D, Kim JH, Hopkins PM. Next-generation Sequencing of RYR1 and CACNA1S in Malignant Hyperthermia and Exertional Heat Illness. Anesthesiology 2015; 122:1033-46; PMID:25658027; http://dx.doi.org/ 10.1097/ALN.0000000000000610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sambuughin N, McWilliams S, de Bantel A, Sivakumar K, Nelson TE. Single-amino-acid deletion in the RYR1 gene, associated with malignant hyperthermia susceptibility and unusual contraction phenotype. Am J Hum Genet 2001; 69:204-8; PMID:11389482; http://dx.doi.org/ 10.1086/321270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Grievink H, Stowell KM. Identification of ryanodine receptor 1 single-nucleotide polymorphisms by high-resolution melting using the LightCycler 480 System. Anal Biochem 2008; 374:396-404 Epub 2007 Nov 21; PMID:18082125; http://dx.doi.org/ 10.1016/j.ab.2007.11.019 [DOI] [PubMed] [Google Scholar]
- [24].Roesl C, Sato K, Schiemann AH, Pollock N, Stowell KM. Functional characterisation of the R2452W ryanodine receptor variant associated with malignant hyperthermia susceptibility. Cell Calcium 2014; 56:1950201; PMID:NOT_FOUND; http://dx.doi.org/ 10.1016/j.ceca.2014.07.004 [DOI] [PubMed] [Google Scholar]
- [25].Tong J, Oyamada H, Demaurex N, Grinstein S, McCarthy TV, MacLennan DH. Caffeine and halothane sensitivity of intracellular Ca2+ release is altered by 15 calcium release channel (ryanodine receptor) mutations associated with malignant hyperthermia and/or central core disease. J Biol Chem 1997; 272:26332-26339; PMID:9334205; http://dx.doi.org/ 10.1074/jbc.272.42.26332 [DOI] [PubMed] [Google Scholar]
- [26].Ording H, Brancadoro V, Cozzolino S, Ellis FR, Glauber V, Gonano EF, Halsall PJ, Hartung E, Heffron JJ, Heytens L, et al. In vitro contracture test for diagnosis of malignant hyperthermia following the protocol of the European MH Group: results of testing patients surviving fulminant MH and unrelated low-risk subjects. The European Malignant Hyperthermia Group. Acta Anaesthesiol Scand 1997; 41:955-66; PMID:9311391; http://dx.doi.org/ 10.1111/j.1399-6576.1997.tb04820.x [DOI] [PubMed] [Google Scholar]
- [27].Ording H. Diagnosis of susceptibility to malignant hyperthermia in man. Br J Anaesth 1988; 60:287-302; PMID:3279989; http://dx.doi.org/ 10.1093/bja/60.3.287 [DOI] [PubMed] [Google Scholar]
- [28].Pollock N, Langton EE, Stowell KM, Bulger TF. Safety of exposure of malignant hyperthermia non-susceptible patients and their relatives to anaesthetic triggering agents. Anaesth Intensive Care 2011; 39:887-94; PMID:21970134 [DOI] [PubMed] [Google Scholar]
- [29].Ording H, Islander G, Bendixen D, Ranklev-Twetman E. Between-center variability of results of the in vitro contracture test for malignant hyperthermia susceptibility. Anesth Analg 2000; 91:452-7; PMID:10910867; http://dx.doi.org/ 10.1213/00000539-200008000-00042 [DOI] [PubMed] [Google Scholar]
- [30].Tung CC, Lobo PA, Kimlicka L, Van Petegem F. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature 2010; 468:585-8; PMID:21048710; http://dx.doi.org/ 10.1038/nature09471 [DOI] [PubMed] [Google Scholar]
- [31].Sambuughin N, McWilliams S, de Bantel A, Sivakumar K, Nelson TE. Single-amino-acid deletion in the RYR1 gene, associated with malignant hyperthermia susceptibility and unusual contraction phenotype. Am J Hum Genet 2001; 69:204-8 Epub 2001 May 29; PMID:11389482; http://dx.doi.org/ 10.1086/321270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yan Z, Bai XC, Yan C, Wu J, Li Z, Xie T, Peng W, Yin CC, Li X, Scheres SH, et al.. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 2015; 517:50-5; PMID:25517095; http://dx.doi.org/ 10.1038/nature14063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tong J, McCarthy TV, MacLennan DH. Measurement of resting cytosolic Ca2+ concentrations and Ca2+ store size in HEK-293 cells transfected with malignant hyperthermia or central core disease mutant Ca2+ release channels. J Biol Chem 1999; 274:693-702; PMID:9873004; http://dx.doi.org/ 10.1074/jbc.274.2.693 [DOI] [PubMed] [Google Scholar]
- [34].Ikemoto N, Yamamoto T. Regulation of calcium release by interdomain interaction within ryanodine receptors. Front Biosci 2002; 7:d671-83; PMID:11861212; http://dx.doi.org/ 10.2741/ikemoto [DOI] [PubMed] [Google Scholar]
- [35].Avila G, Dirksen RT. Functional effects of central core disease mutations in the cytoplasmic region of the skeletal muscle ryanodine receptor. J Gen Physiol 2001; 118:277-90; PMID:11524458; http://dx.doi.org/ 10.1085/jgp.118.3.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Murayama T, Oba T, Hara H, Wakebe K, Ikemoto N, Ogawa Y. Postulated role of interdomain interaction between regions 1 and 2 within type 1 ryanodine receptor in the pathogenesis of porcine malignant hyperthermia. Biochem J 2007; 402:349-57; PMID:17107340; http://dx.doi.org/ 10.1042/BJ20061040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yamamoto T, El-Hayek R, Ikemoto N. Postulated role of interdomain interaction within the ryanodine receptor in Ca2+ channel regulation. J Biol Chem 2000; 275:11618-25; PMID:10766778; http://dx.doi.org/ 10.1074/jbc.275.16.11618 [DOI] [PubMed] [Google Scholar]
- [38].Kraeva N, Heytens L, Jungbluth H, Treves S, Voermans N, Kamsteeg E, Ceuterick-de Groote C, Baets J, Riazi S. Compound RYR1 heterozygosity resulting in a complex phenotype of malignant hyperthermia susceptibility and a core myopathy. Neuromuscul Disord 2015; 25:567-76; PMID:25958340; http://dx.doi.org/ 10.1016/j.nmd.2015.04.007 [DOI] [PubMed] [Google Scholar]
- [39].Shepherd S, Ellis F, Halsall J, Hopkins P, Robinson R. RYR1 mutations in UK central core disease patients: more than just the C-terminal transmembrane region of the RYR1 gene. J Med Genet 2004; 41:e33; PMID:14985404; http://dx.doi.org/ 10.1136/jmg.2003.014274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sato K, Pollock N, Stowell KM. Functional studies of RYR1 mutations in the skeletal muscle ryanodine receptor using human RYR1 complementary DNA. Anesthesiology 2010; 112:1350-4; PMID:20461000; http://dx.doi.org/ 10.1097/ALN.0b013e3181d69283 [DOI] [PubMed] [Google Scholar]