

Abstract
Background
As an extracellularly released mediator, high-mobility group box 1 (HMGB1) initiates sterile inflammation following severe trauma. Serum HMGB1 levels correlate well with acute traumatic coagulopathy (ATC) in trauma patients, which is independently associated with higher mortality. We investigated the involvement of HMGB1 in ATC through blocking extracellular HMGB1.
Material/Methods
The ATC model was induced by polytrauma and hemorrhage in male Sprague-Dawley rats, which were randomly assigned to sham, ATC, and ATCH (ATC with HMGB1 blockade) groups. Thrombelastography (TEG) was performed to monitor changes in coagulation function. Serum levels of HMGB1, TNF-α, and IL-6 were measured, as well as lung levels of HMGB1 and nuclear factor (NF)-κB and expression of receptor for advanced glycation end-products (RAGE).
Results
Compared with the sham group, HMGB1 increased the serum levels of TNF-α and IL-6, whereas HMGB1 blockade inhibited the induction of TNF-α and IL-6. HMGB1 also induced elevated serum soluble P-selectin and fibrinolysis markers plasmin-antiplasmin complex, which both were reduced by HMGB1 blockade. Thrombelastography revealed the hypocoagulability status in the ATC group, which was attenuated by anti-HMGB1 antibody. Furthermore, the lung level of NF-κB and expression of RAGE were decreased by anti-HMGB1 antibody, suggesting the role of RAGE/NF-κB pathway in ATC.
Conclusions
HMGB1 blockade can attenuate inflammation and coagulopathy in ATC rats. Anti-HMGB1 antibody might exert protective effects partly through the RAGE/NF-κB pathway. Thus, HMGB1 has potential as a therapeutic target in ATC.
MeSH Keywords: Hemorrhage, HMGB1 Protein, Inflammation
Background
To date, trauma remains one of the leading causes of the worldwide mortality [1]. Although a proportion of traumatic victims survive at the trauma scene, about one-third of these survivors present to the emergency room with early coagulopathic bleeding [2–4] – acute traumatic coagulopathy (ATC). Clinical studies showed that ATC originates from the combination of tissue injury and hypoperfusion and is characterized by hypocoagulability and hyperfibrinolysis [5,6]. Due to further understandings of post-traumatic pathological changes, ATC now has been recognized as a distinct disease entity and is independently associated with more death and higher transfusion requirements [3,7]. Despite progress in the treatment of ATC (e.g., the new concept of “hemostatic resuscitation [8]”), underlying mechanisms responsible for ATC are not yet fully understood.
Studies showed that activation of protein C may function as a major trigger in ATC through inhibition of coagulation factors and activation of fibrinolytic system [4,6]. Moreover, increasing evidence shows that inflammation response and coagulation cascade following trauma are tightly interlinked [8–10]. To some extent, ATC could be partly viewed as the sequelae of maladapted activation of these 2 systems. Tissue injury and hypoperfusion induced by severe trauma could trigger sterile inflammatory response, leading to remote organ damage and multiple-organ failure [9,11,12]. Unlike exogenous pathogens initiating systemic inflammatory response syndrome (SIRS) in sepsis, the innate immune system activation following severe trauma is due to the interaction between pattern recognition receptors (PPRs) and alarmins released by “stressed” cells and necrotic tissue, including heat shock proteins, mitochondrial DNA, hyaluronan, and high-mobility group box 1 (HMGB1) [13,14].
As an endogenous nuclear protein, HMGB1 can be extracellularly released and produce potent proinflammatory effect via the activation of several PPRs, including members of the Toll-like family of receptors (TLRs) and the receptor for advanced glycation end-products (RAGE) [14]. HMGB1 functions as a late inflammatory mediator in sepsis and its plasma concentration raises from 8 to 32 h after endotoxin injection [15]. However, HMGB1 can be released passively into circulation within 30–60 min following sterile injury and trigger inflammatory response similar to those seen during infection [16,17]. Serum HMGB1 levels correlate well with the magnitude of tissue injury and hypoperfusion, as well as the degree of immune system activation [9,13]. Furthermore, animal experiments and clinical studies have demonstrated that HMGB1 could be a predictor of coagulopathy, organ failure, and clinical outcome in severe trauma [12,17].
Most importantly, the plasma concentrations of HMGB1 are not only elevated immediately after extensive tissue injury in severe trauma patients, but also correlate well with their hypocoagulability and hyperfibrinolysis status [12,17]. Given this background, we hypothesized that HMGB1 plays a pivotal role in the development of ATC and HMGB1-targeting therapeutic strategies could be beneficial. To certify this hypothesis, we employed an ATC rat model and used extracellular HMGB1 blockade to investigate the involvement of HMGB1 in ATC, as well as the therapeutic efficacy of HMGB1 neutralizing antibody.
Material and Methods
Animals
This study was reviewed and approved by the Animal Care and Use Committee of Nanjing University of Chinese Medicine, and all the procedures performed in experiments were conducted in strict accordance with the National Institutes of Health Guidelines on the use of laboratory animals (NIH Publication No. 80-23). Male Sprague-Dawley rats weighing 280–300 g were obtained from the Animal Research Center, Jinling Hospital, Nanjing, China. Animals were housed at 22°C on a 12-h light/dark cycle with free access to standard rat chow and water. Rats were acclimated to the Animal Research Center for a minimum of 7 days before experiments.
ATC model and experimental protocols
Animals were fasted overnight before the experiments. Rats were anesthetized intraperitoneally with urethane (1.2 g/kg, Sigma Chemical Co., USA), and anesthetic was intermittently added to ensure effective anesthesia. A temperature probe was placed rectally to monitor central body temperature, which was maintained at 37.0°C with a heating pad to avoid influence of hypothermia on coagulation function. Tracheal intubation was performed to preserve the fluent respiratory tract. The left femoral artery and vein both were cannulated with PE-50 tubing for vascular access and arterial pressure monitoring (by blood pressure monitor, Columbus Instruments, USA). After anesthesia and catheterization, rats were left in stabilization for 5 min to obtain baseline parameters (time point 0 h). No resuscitation or antibiotics was given in experiments.
Rats were randomly divided into sham group, ATC group, and ATCH (ATC with HMGB1 blockade) group. Animals in the sham group only received anesthesia, catheterization, and continuous monitoring. The rat model of ATC was developed by polytrauma and hemorrhagic shock with a modified method described previously by Frith [3]. Under an aseptic condition, rats underwent a sterile 6-cm paramedian laparotomy and the abdominal wound was closed using 4-0 interrupted suture. Bilateral femur fractures were produced to generate higher injury severity. Polytrauma procedure was accomplished within 10 min. To simulate hemorrhagic shock, blood was withdrawn through the femoral vein until mean arterial pressure (MAP) decreased to 35–40 mmHg. This target MAP was maintained for the 50-min shock period by withdrawing or returning blood as required. Hence, the modeling of ATC required about 1 h. Immediately after the polytrauma was completed, rats in the ATCH group received intravenous injection of rabbit anti-HMGB1 monoclonal antibody (200 μg/kg, Shino-test, Tokyo, JP). In the sham and ACT groups, rats received the same dosage of non-immune rabbit immunoglobulin G at the same time.
To avoid the effect of anemia, rats (n=10 per group at each time point) were sacrificed at baseline (time point 0 h), and at 0, 1, and 2 h after shock (time points 1, 2, and 3 h, respectively) to obtain blood samples. Lung tissues were collected at 2 h after shock (time point 3 h, n=6 for each group). In addition, separate ATC and ATCH groups (n=20 per group) were observed until death to compare overall mortality between 2 groups.
Blood sample measurements
Thrombelastography (TEG), a viscoelastic method of hemostasis monitoring, uses a cell-based model of coagulation and is more sensitive and comprehensive than the conventional plasma-based coagulation tests [18]. Blood collected from the femoral artery was used for TEG (Haemonetics Corporation, Niles Ill) detection, which was performed according to the technique described previously by Wohlauer et al. [19]. Blood sample was mixed with sodium citrate and inverted 5 times. Then, 340 μl of this sample was pipetted into the TEG cup containing calcium chloride, and analysis was performed at 37°C immediately after blood collection. Standard tracings generated important TEG parameters, including reaction time (R time), coagulation time (K time), α angle, and maximum amplitude (MA).
Levels of hemoglobin and fibrinogen in arterial blood were tested in the Jinling Hospital Core Laboratory. Blood samples were placed in EDTA tubes and centrifuged at 4000 rpm for 15 min. The supernatant plasma was collected and stored at −80°C until analysis. Different serum markers were determined using commercial enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s instructions: HMGB1, tumor necrosis factor- α (TNF-α) and interlukin-6 (IL-6) from R&D Systems Co., MN; activated protein C (aPC) and plasmin-antiplasmin complex (PAP) from Cusabio Biotech Co., China; soluble P-selectin (sP-selectin), and syndecan-1 from Diagnostica Stago Co., NL.
Western blotting analysis
In short, HMGB1 and nuclear factor (NF)-κB proteins extracted from lung tissue were resolved by SDS-PAGE and transferred to PVDF membranes. The membranes were incubated with specific primary antibodies (anti-HMGB1 antibody from Invitrogen Co., CA and anti-NF-κB p65 antibody from Thermo Fisher Scientific Co., CA) followed by HRP-conjugated secondary antibody (Jackson ImmunoResearch Co., USA). Beta-actin (1:5000; Thermo Fisher Scientific Co., CA) was applied as endogenous control. Finally, blots were detected by NIH Image J software (NIH, Bethesda, MD, USA).
Immunofluorescent staining
Lung tissues were collected and fixed with 4% formaldehyde, and then were embedded and sectioned. Then sections were immunostained with 1:200 rat anti-RAGE monoclonal antibody (Santa Cruz Biotechnology Inc, USA), followed by incubation with 1:500 goat antirat IgG (Invitrogen Co., CA). Finally, images were acquired under a fluorescent microscope (BZ-9000; Keyence Co., JP).
Statistical analysis
Data are expressed as the mean ± standard deviation (SD). Statistical analyses were performed using SPSS 17.0 software (SPSS Inc., USA). Differences between groups were assessed using ANOVAs and multiple time point data were assessed by two-way ANOVA. Significant results were then analyzed post hoc using Bonferroni test. P value less than 0.05 was considered significant.
Results
Changes in vital signs, hemoglobin, and fibrinogen
Central body temperature and respiratory rate had no differences among the 3 groups, and the mean blood losses were similar between the ATC and ATCH groups during the experiments (9.3±0.7 ml and 9.4±0.6 ml, respectively; P>0.05). As shown in Figure 1, no significant differences in heart rate (HR), MAP, hemoglobin, or fibrinogen were found among the 3 groups. Compared to the sham group, trauma and shock together led to the decrease of HR and MAP in the ATC and ATCH groups (P<0.05, Figure 1A, 1B). However, HR in these 2 groups both had partial restoration after shock, and MAP in these 2 groups increased at 2 h and decreased at 3 h. HMGB1 blockade did not influence HR and MAP in the ATCH group compared with the ATC group. Compared to the sham group, the levels of hemoglobin and fibrinogen in the ATC and ATCH groups both evidently decreased after the shock period (P<0.05, Figure 1C, 1D). No significant differences were found in the levels of hemoglobin or fibrinogen between these 2 groups.
Figure 1.

Changes in (A) HR, (B) MAP, (C) hemoglobin, and (D) fibrinogen in the sham, ATC, and ATCH (ATC with HMGB1 blockade) groups. Data are expressed as mean ±SD. & P<0.05, ATC versus sham; # P<0.05, sham versus ATCH.
Effects of HMGB1 blockade on overall survival
As shown in Figure 2, HMGB1 blockade did not evidently alter the survival rate after shock in the ATCH group compared to the ATC group (P>0.05), and the median time to death after the shock period in the ATC and ATCH groups were 125 min and 150 min, respectively. Although the difference in overall mortality between these 2 groups was not statistically significant, the median survival time was still increased by 20% in the ATCH group compared to the ATC group. Because the sham rats did not receive trauma and hemorrhage procedure, the survival rate of the sham group was not shown.
Figure 2.
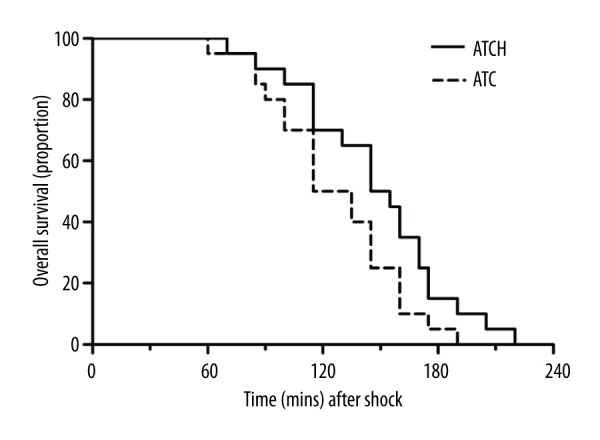
Effects of HMGB1 blockade on overall survival in the ATC and ATCH (ATC with HMGB1 blockade) groups. n=20 per group.
Effects of HMGB1 blockade on serum levels of HMGB1, TNF-α and IL-6
As indicated in Figure 3, basal serum levels of HMGB1, TNF-α, and IL-6 were similar among the 3 groups. After the shock period, tissue trauma and sustained hypoperfusion contributed to evidently increased serum HMGB1 in the ATC and ATCH groups, compared to the sham group (P<0.05, Figure 3A). In the ATCH group, the serum levels of HMGB1 level peaked at 3 h after baseline and then slightly decreased. However, the serum level of HMGB1 in the ATCH group was clearly lower than that in the ATC group after shock (P<0.05, Figure 3A). Compared to the sham group, serum levels of TNF-α and IL-6 in the ATC and ATCH groups were consistently elevated after shock (P<0.05, Figure 3B, 3C). The serum TNF-α level was apparently lower in the ATCH rats compared with ATC rats after shock, and the difference was statistically evident at 2 and 3 h (P<0.05, Figure 3B). Moreover, the anti-HMGB1 treatment also significantly suppressed the serum IL-6 after shock in the ATCH group compared to the ATC group (P<0.05, Figure 3C).
Figure 3.

Effects of HMGB1 blockade on serum levels of (A) high-mobility group box 1 (HMGB1), (B) tumor necrosis factor-α (TNF-α), and (C) interlukine-6 (IL-6) in the sham, ATC and ATCH (ATC with HMGB1 blockade) groups. Data are expressed as mean ±SD. * P<0.05, ATC versus ATCH; & P<0.05, ATC versus sham; # P<0.05, sham versus ATCH.
Effects of HMGB1 blockade on clotting capacity revealed by TEG analysis
In order to determine clotting abnormalities following trauma and shock in our experiments, we applied TEG examination in 3 groups at 1 h after the end of shock period (time point 2 h). The influence of HMGB1 blockade on TEG parameters were indicated in Figure 4. Compared to the sham group, the ATC and ATCH groups had clearly higher R time and K Time, and obviously lower α angle and MA, confirming the early and systemic hypocoagulability status induced by trauma and shock in rats (Figure 4, P<0.05). Compared to the ATC rats, the anti-HMGB1 antibody treatment could markedly attenuate the elevation of K time and the reduction of MA in the ATCH rats (P<0.05, Figure 4B, 4D). However, no significant differences were found in R time and α angle between the ATC and ATCH groups (P>0.05, Figure 4A, 4C).
Figure 4.

Effects of HMGB1 blockade on thrombelastography (TEG) parameters including (A) R time, (B) K time, (C) α angle, and (D) maximal amplitude (MA) in the sham, ATC, and ATCH (ATC with HMGB1 blockade) groups at 2 h after baseline. Data are expressed as mean ±SD. * P<0.05, ATC versus ATCH; & P<0.05, ATC versus sham; # P<0.05, sham versus ATCH.
Effects of HMGB1 blockade on serum markers of anticoagulation, hyperfibrinolysis, glycocalyx shedding, and platelet activation
To determine the effects of HMGB1 blockade on the anticoagulant and fibrinolytic systems as well as endothelial and platelet functions, we detected the levels of aPC, PAP, syndecan-1, and sP-selectin in plasma (Figure 5). These parameters at baseline were similar in the 3 groups. After the end of the shock period, the ATC and ATCH groups had obviously higher levels of these markers compared with those in the sham group. Compared to the sham group, serum levels of aPC and syndecan-1 in the ATC and ATCH groups were continuously elevated, but HMGB1 blockade evidently inhibited the increased serum aPC and syndecan-1in the ATCH group after shock compared with those in the ATC group (P<0.05, Figure 5A, 5C). Moreover, the serum PAP and sP-selectin levels in the ATC and ATCH groups also showed sustained increases after shock, which both were effectively suppressed by the anti-HMGB1 antibody treatment at 2 and 3 h after baseline (P<0.05, Figure 5B, 5D).
Figure 5.

Effects of HMGB1 blockade on (A) activated protein C (aPC), (B) plasmin anti-plasmin (PAP) complex, (C) syndecan-1 and (D) soluble P-selectin (sP-selectin) in the sham, ATC and ATCH (ATC with HMGB1 blockade) groups. Data are expressed as mean ±SD. * P<0.05, ATC versus ATCH; & P<0.05, ATC versus sham; # P<0.05, sham versus ATCH.
Effects of HMGB1 blockade on NF-κB and HMGB1 levels in lung tissue
To investigate the mechanism by which the HMGB1 blockade alleviated systemic inflammation response following trauma and hemorrhage, the NF-κB and HMGB1 protein levels in lung tissue both were detected by Western blot analysis. As shown in Figure 6, Western blot analysis showed the obvious increases in NF-κB and HMGB1 levels in the ATC and ATCH groups in comparison with the sham group (P<0.05). Nevertheless, HMGB1 blockade significantly inhibited the increased NF-κB level in the ATCH group compared to the ATC group (P<0.05, Figure 6A). Additionally, the increasing tendency of HMGB1 level in ATCH group was evidently suppressed by the treatment of anti-HMGB1 antibody in comparison with the ATC group (P<0.05, Figure 6B).
Figure 6.

Effects of HMGB1 blockade on (A) nuclear factor (NF)-κB and (B) HMGB1 expressions in lung tissue detected by Western blot assays in the sham, ATC and ATCH (ATC with HMGB1 blockade) groups. Data are expressed as mean ±SD. * P<0.05, ATC versus ATCH; & P<0.05, ATC versus sham; # P<0.05, sham versus ATCH.
Effects of HMGB1 blockade on the RAGE expression in the lung
Considering the important mediating role of RAGE in the sterile inflammatory response induced by HMGB1, lung sections were obtained and immunostained with anti-RAGE monoclonal antibody (Figure 7). In comparison with the sham rats, the expression of RAGE was obviously enhanced in ATC rats following tissue injury and hypoperfusion (Figure 7A, 7B). However, an evident inhibitive effect of HMGB1 blockade on the increased RAGE expression was found in the ATCH group compared with the ATC group (Figure 7B, 7C).
Figure 7.

Effects of HMGB1 blockade on immunofluorescent expression of the receptor for advanced glycation end- products (RAGE) in lung tissue (original magnification ×200). Red signal shows the expression of RAGE. (A) sham group, (B) ATC group, (C) ATCH (ATC with HMGB1 blockade) group.
Discussion
As an evolutionarily conserved chromosome-binding protein, HMGB1 is a normal cell constituent and can be extracellularly released into circulation following infectious diseases, cellular stress, or tissue damage [16,17]. Subsequently, HMGB1 could act as a damage-associated molecular pattern (DAMP) molecule and provoke inflammatory responses in both infectious and non-infectious inflammatory conditions [15,20,21]. Furthermore, clinical research showed that HMGB1 was closely associated with the main driving factors of ATC – tissue injury and hypoperfusion [17]. HMGB1 might function as a key mediator of inflammation and coagulopathy in ATC in analogy to its important role in sepsis. Hence, identifying the exact role of HMGB1 in ATC is crucial for elucidating the molecular mechanisms of ATC, as well as developing novel therapeutic strategies against post-traumatic coagulopathy. In this study, we showed that HMGB1 facilitated the imbalanced inflammatory response and abnormal hemostasis function in a rat model of ATC, which was attenuated by administration of anti-HMGB1 antibody. Furthermore, extracellularly released HMGB1 contributed to the enhancement of NF-κB and RAGE expressions, which both were clearly suppressed by HMGB1 blockade. These results indicate the pivotal role of HMGB1 in the pathogenesis of ATC.
Remote organ dysfunction following trauma and shock usually originates from overwhelming inflammatory responses [9]. Studies have shown that HMGB1 has potent proinflammatory effects in disparate conditions, whereas neutralization of extracellular HMGB1 reduces the augmentation of proinflammatory cytokines [5,11,20,22–25]. In the current study, trauma and hemorrhage procedures together led to uncontrolled production of extracellular HMGB1 and serum proinflammatory cytokines such as TNF-α and IL-6, which were evidently suppressed by HMGB1 blockade. Evidence from research demonstrated that HMGB1 acted as a late mediator of sepsis and increased from 8 to 32 h after endotoxin exposure in mice [15]. Moreover, a clinical study showed that serum HMGB1 levels remained high in septic patients more than 1 week following admission [26]. In contrast, HMGB1 can function as early mediator of sterile damage following trauma or hemorrhage. Indeed, HMGB1 is promptly released into circulation in less than 1 h and peaks at 2 to 6 h after injury in severe traumatic patients [16,17,27]. In the present study, serum levels of HMGB1 peaked at 2 h after baseline and then began to decline in the ATC group. Additionally, serum levels of TNF-α and IL-6 did not appear to peak, and increased continuously during the observation period. This phenomenon further shows the proinflammatory effects of HMGB1 following tissue injury and hypoperfusion. Research proved that the interaction between HMGB1and RAGE promotes chemotaxis, cell growth, and differentiation, as well as the activation of NF-κB and subsequent production and release of proinflammatory cytokines [28–30]. In our study, rats with ATC showed significantly higher levels of NF-κB and enhanced expression of RAGE in lung tissue, which were inhibited by extracellular HMGB1 blockade. The anti-inflammatory effect of HMGB1 blockade in ATC might be produced, at least in part, through the RAGE/NF-κB signaling pathway.
In order to mimic the clinical characteristics of ATC, an ATC rat model was successfully established by the combination of tissue trauma and hemorrhagic shock, evidenced by systemic hypocoagulability and hyperfibrinolysis in rats [3,31]. Davenport et al. considered that TEG analysis is superior to conventional coagulation test as a diagnostic tool for ATC, and the characteristic reduction in clot strength could be used as diagnostic criteria for ATC [7]. In our study, rats with ATC showed obviously higher K time and lower MA after shock, which were evidently attenuated by HMGB1 blockade. These results confirmed the promotion of hypocoagulability induced by extracellular HMGB1, suggesting the therapeutic potential of HMGB1 blockade against ATC. Interestingly, although HMGB1 did not affect clotting times by itself, Ito et al. considered that HMGB1 has a procoagulant role in sepsis [32]. On the one hand, our study showed that extracellular HMGB1 contributed to platelet dysfunction, endothelial damage and associated glycocalyx degradation, evidenced by increased sP-selectin and syndecan-1 in the ATC group. Studies have proven that platelet dysfunction inevitably contribute to reduction of clot strength [33,34], and glycocalyx degradation will lead to endogenous heparinization in severe traumatic patients with ATC [35]. On the other hand, treatment of anti-HMGB1 antibody could suppress the high serum levels of aPC in rats with ATC, indicating the activation of aPC induced by extracellular HMGB1. In view of the anti-inflammatory and cytoprotective effects of aPC, the activation of aPC could be viewed as physiological response to released inflammatory mediators, such as HMGB1. Simultaneously, aPC could generate anticoagulant effects by suppressing coagulation factors and derepressing fibrinolytic system [6,31]. Hence, the hypocoagulability status in ATC might be the synergistic effects of HMGB1 and aPC on the hemostasis system following severe trauma.
Numerous studies showed that ATC is characterized by the over-activation of fibrinolytic system – hyperfibrinolysis – which is independently associated with transfusion requirements and mortality [36]. Clinical study also demonstrated that excessive fibrinolytic activation could be sensitively diagnosed by serum PAP levels rather than thromboelastometry analysis [37]. We observed evidently increased serum PAP levels in rats with ATC, which was obviously suppressed by HMGB1 blockade. Roussel et al. recently demonstrated that HMGB1 could promote fibrinolysis of blood clots by facilitate the release of tPA from vascular endothelium [38]. In contrast, our study showed that HMGB1 blockade could reduce the concentration of PAP and suppress fibrinolytic activity, suggesting the anti-fibrinolysis effect of HMGB1 blockade. Cohen et al. proved that soluble RAGE is released into circulation early after severe trauma and is associated with coagulopathy and endothelial damage [39]. Moreover, studies have identified the mediating role of RAGE in the endothelial damage induced by HMGB1 [30,40]. In our study, rats with ATC showed enhanced expression of RAGE in lung tissue, which was inhibited by HMGB1 blockade. These results further indicate that the anti-fibrinolysis effect of HMGB1 blockade might work through the RAGE pathway.
However, several limitations exist in our study. First, studies demonstrated that the activation of TLRs such as TLR4, TLR2, and TLR9 also play a part in organ injury induced by HMGB1. Nevertheless, the effect of HMGB1 on the expression of TLRs in the lung was not determined in our study, and further research is needed to identify the role of TLRs in ATC. Additionally, we applied HMGB1 blockade in an ATC rat model without resuscitation. Hence, further study is necessary to clarify the exact effects of HMGB1 blockade in animals experiencing tissue trauma, hemorrhagic shock, and subsequent resuscitation.
Conclusions
Our study demonstrated in an ATC rat model that HMGB1 blockade could attenuate systemic inflammation and hemostasis abnormality, partly through the RAGE/NF-κB pathway. This study not only shows the role of HMGB1 in the pathogenesis of ATC, but also revealed the therapeutic potential of HMGB1 blockade against coagulopathic bleeding following severe trauma. Hence, the exact role of HMGB1 in ATC deserves further study.
Footnotes
Declaration of interest
The authors report no conflicts of interest.
Source of support: National Natural Science Foundation of China (nos. 81270884), the 12th five-year plan major project of PLA (nos. WS12J001) and the Jiangsu Province’s Key Medical Talent Program of China (nos. RC2011128)
References
- 1.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Floccard B, Rugeri L, Faure A, et al. Early coagulopathy in trauma patients: an on-scene and hospital admission study. Injury. 2012;43:26–32. doi: 10.1016/j.injury.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Frith D, Goslings JC, Gaarder C, et al. Definition and drivers of acute traumatic coagulopathy: clinical and experimental investigations. J Thromb Haemost. 2010;8:1919–25. doi: 10.1111/j.1538-7836.2010.03945.x. [DOI] [PubMed] [Google Scholar]
- 4.Cohen MJ, Kutcher M, Redick B, et al. Clinical and mechanistic drivers of acute traumatic coagulopathy. J Trauma Acute Care Surg. 2013;75:S40–47. doi: 10.1097/TA.0b013e31828fa43d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu K, Mori S, Takahashi HK, et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007;21:3904–16. doi: 10.1096/fj.07-8770com. [DOI] [PubMed] [Google Scholar]
- 6.Brohi K, Cohen MJ, Ganter MT, et al. Acute traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C pathway. Ann Surg. 2007;245:812–18. doi: 10.1097/01.sla.0000256862.79374.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davenport R, Manson J, De’Ath H, et al. Functional definition and characterization of acute traumatic coagulopathy. Crit Care Med. 2011;39:2652–58. doi: 10.1097/CCM.0b013e3182281af5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holcomb JB, Wade CE, Michalek JE, et al. Increased plasma and platelet to red blood cell ratios improves outcome in 466 massively transfused civilian trauma patients. Ann Surg. 2008;248:447–58. doi: 10.1097/SLA.0b013e318185a9ad. [DOI] [PubMed] [Google Scholar]
- 9.Pierce A, Pittet JF. Inflammatory response to trauma: Implications for coagulation and resuscitation. Curr Opin Anaesthesiol. 2014;27(2):246–52. doi: 10.1097/ACO.0000000000000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johansson PI, Stensballe J, Rasmussen LS, Ostrowski SR. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann Surg. 2011;254:194–200. doi: 10.1097/SLA.0b013e318226113d. [DOI] [PubMed] [Google Scholar]
- 11.Lu B, Wang C, Wang M, et al. Molecular mechanism and therapeutic modulation of high mobility group box 1 release and action: An updated review. Expert Rev Clin Immunol. 2014;10:713–27. doi: 10.1586/1744666X.2014.909730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong YC, Lai YY, Tan MH, et al. Potential biomarker panel for predicting organ dysfunction and acute coagulopathy in a polytrauma porcine model. Shock. 2015;43:157–65. doi: 10.1097/SHK.0000000000000279. [DOI] [PubMed] [Google Scholar]
- 13.Manson J, Thiemermann C, Brohi K. Trauma alarmins as activators of damage-induced inflammation. Br J Surg. 2012;99(Suppl 1):12–20. doi: 10.1002/bjs.7717. [DOI] [PubMed] [Google Scholar]
- 14.Oppenheim JJ, Yang D. Alarmins: Chemotactic activators of immune responses. Curr Opin Immunol. 2005;17:359–65. doi: 10.1016/j.coi.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 16.Sun Q, Wu W, Hu YC, et al. Early release of high-mobility group box 1 (HMGB1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J Neuroinflammation. 2014;11:106. doi: 10.1186/1742-2094-11-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen MJ, Brohi K, Calfee CS, et al. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: Role of injury severity and tissue hypoperfusion. Crit Care. 2009;13:R174. doi: 10.1186/cc8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoffman M. A cell-based model of coagulation and the role of factor VIIa. Blood Rev. 2003;17(Suppl 1):S1–5. doi: 10.1016/s0268-960x(03)90000-2. [DOI] [PubMed] [Google Scholar]
- 19.Wohlauer MV, Moore EE, Harr J, et al. A standardized technique for performing thromboelastography in rodents. Shock. 2011;36:524–26. doi: 10.1097/SHK.0b013e31822dc518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Wang LK, Wang LW, et al. Blockade of high-mobility group box-1 ameliorates acute on chronic liver failure in rats. Inflamm Res. 2013;62:703–9. doi: 10.1007/s00011-013-0624-1. [DOI] [PubMed] [Google Scholar]
- 21.Eskici ZM, Acikgoz S, Piskin N, et al. High mobility group B1 levels in sepsis and Disseminated Intravascular Coagulation. Acta Biochim Pol. 2012;59:561–66. [PubMed] [Google Scholar]
- 22.Yang R, Harada T, Mollen KP, et al. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med. 2006;12:105–14. doi: 10.2119/2006-00010.Yang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagiwara S, Iwasaka H, Matsumoto S, Noguchi T. High dose antithrombin III inhibits HMGB1 and improves endotoxin-induced acute lung injury in rats. Intensive Care Med. 2008;34:361–67. doi: 10.1007/s00134-007-0887-5. [DOI] [PubMed] [Google Scholar]
- 24.Robinson AP, Caldis MW, Harp CT, et al. High-mobility group box 1 protein (HMGB1) neutralization ameliorates experimental autoimmune encephalomyelitis. J Autoimmun. 2013;43:32–43. doi: 10.1016/j.jaut.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen KB, Uchida K, Nakajima H, et al. High-mobility group box-1 and its receptors contribute to proinflammatory response in the acute phase of spinal cord injury in rats. Spine (Phila Pa 1976) 2011;36:2122–29. doi: 10.1097/BRS.0b013e318203941c. [DOI] [PubMed] [Google Scholar]
- 26.Sunden-Cullberg J, Norrby-Teglund A, Rouhiainen A, et al. Persistent elevation of high mobility group box-1 protein (HMGB1) in patients with severe sepsis and septic shock. Crit Care Med. 2005;33:564–73. doi: 10.1097/01.ccm.0000155991.88802.4d. [DOI] [PubMed] [Google Scholar]
- 27.Manganelli V, Signore M, Pacini I, et al. Increased HMGB1 expression and release by mononuclear cells following surgical/anesthesia trauma. Crit Care. 2010;14:R197. doi: 10.1186/cc9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park JS, Arcaroli J, Yum HK, et al. Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am J Physiol Cell Physiol. 2003;284:C870–79. doi: 10.1152/ajpcell.00322.2002. [DOI] [PubMed] [Google Scholar]
- 29.Palumbo R, Galvez BG, Pusterla T, et al. Cells migrating to sites of tissue damage in response to the danger signal HMGB1 require NF-kappaB activation. J Cell Biol. 2007;179:33–40. doi: 10.1083/jcb.200704015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang W, Liu Y, Li L, et al. HMGB1 increases permeability of the endothelial cell monolayer via RAGE and Src family tyrosine kinase pathways. Inflammation. 2012;35:350–62. doi: 10.1007/s10753-011-9325-5. [DOI] [PubMed] [Google Scholar]
- 31.Chesebro BB, Rahn P, Carles M, et al. Increase in activated protein C mediates acute traumatic coagulopathy in mice. Shock. 2009;32:659–65. doi: 10.1097/SHK.0b013e3181a5a632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ito T, Kawahara K, Nakamura T, et al. High-mobility group box 1 protein promotes development of microvascular thrombosis in rats. J Thromb Haemost. 2007;5:109–16. doi: 10.1111/j.1538-7836.2006.02255.x. [DOI] [PubMed] [Google Scholar]
- 33.Donahue DL, Beck J, Fritz B, et al. Early platelet dysfunction in a rodent model of blunt traumatic brain injury reflects the acute traumatic coagulopathy found in humans. J Neurotrauma. 2014;31:404–10. doi: 10.1089/neu.2013.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wohlauer MV, Moore EE, Thomas S, et al. Early platelet dysfunction: An unrecognized role in the acute coagulopathy of trauma. J Am Coll Surg. 2012;214:739–46. doi: 10.1016/j.jamcollsurg.2012.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ostrowski SR, Johansson PI. Endothelial glycocalyx degradation induces endogenous heparinization in patients with severe injury and early traumatic coagulopathy. J Trauma Acute Care Surg. 2012;73:60–66. doi: 10.1097/TA.0b013e31825b5c10. [DOI] [PubMed] [Google Scholar]
- 36.Kashuk JL, Moore EE, Sawyer M, et al. Primary fibrinolysis is integral in the pathogenesis of the acute coagulopathy of trauma. Ann Surg. 2010;252:434–42. doi: 10.1097/SLA.0b013e3181f09191. discussion 443–44. [DOI] [PubMed] [Google Scholar]
- 37.Raza I, Davenport R, Rourke C, et al. The incidence and magnitude of fibrinolytic activation in trauma patients. J Thromb Haemost. 2013;11:307–14. doi: 10.1111/jth.12078. [DOI] [PubMed] [Google Scholar]
- 38.Roussel BD, Mysiorek C, Rouhiainen A, et al. HMGB-1 promotes fibrinolysis and reduces neurotoxicity mediated by tissue plasminogen activator. J Cell Sci. 2011;124:2070–76. doi: 10.1242/jcs.084392. [DOI] [PubMed] [Google Scholar]
- 39.Cohen MJ, Carles M, Brohi K, et al. Early release of soluble receptor for advanced glycationendproducts after severe trauma in humans. J Trauma. 2010;68:1273–78. doi: 10.1097/TA.0b013e3181db323e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolfson RK, Chiang ET, Garcia JG. HMGB1 induces human lung endothelial cell cytoskeletal rearrangement and barrier disruption. Microvasc Res. 2011;81:189–97. doi: 10.1016/j.mvr.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]