

Abstract
Deep learning is rapidly advancing many areas of science and technology with multiple success stories in image, text, voice and video recognition, robotics and autonomous driving. In this paper we demonstrate how deep neural networks (DNN) trained on large transcriptional response data sets can classify various drugs to therapeutic categories solely based on their transcriptional profiles. We used the perturbation samples of 678 drugs across A549, MCF‐7 and PC‐3 cell lines from the LINCS project and linked those to 12 therapeutic use categories derived from MeSH. To train the DNN, we utilized both gene level transcriptomic data and transcriptomic data processed using a pathway activation scoring algorithm, for a pooled dataset of samples perturbed with different concentrations of the drug for 6 and 24 hours. In both gene and pathway level classification, DNN convincingly outperformed support vector machine (SVM) model on every multiclass classification problem, however, models based on a pathway level classification perform better.
For the first time we demonstrate a deep learning neural net trained on transcriptomic data to recognize pharmacological properties of multiple drugs across different biological systems and conditions. We also propose using deep neural net confusion matrices for drug repositioning. This work is a proof of principle for applying deep learning to drug discovery and development.
Keywords: Deep learning, DNN, predictor, drug repurposing, drug discovery, confusion matrix, deep neural networks
INRODUCTION
Drug discovery and development is complicated and time and resource consuming process and various computational approaches are regularly being developed to improve it. In silico drug discovery1,2 has evolved over the past decade and offers a targeted, efficient approach compared to those of the past, which often relied on either identifying active ingredients in traditional remedies or, in many cases, serendipitous discovery. Modern methods include data mining, structure modeling (homology modeling), traditional Machine Learning3 (ML) and its biologically inspired branch technique, Deep Learning (DL).4
DL4 methods modeling high-level representations of data using Deep Neural Networks (DNNs). DNNs are flexible systems of connected and interacting artificial neurons that perform non-linear data transformations. They have several hidden layers of neurons, which number variation allows adjusting level of data abstraction. DL now play a dominant role in the areas of physics5, speech, signal, image, video and text mining and recognition6, improving state of the art performances by more than 30% where the prior decade struggled to obtain 1–2% improvements. Traditional machine learning approaches have achieved significant levels of classification accuracy, but at the price of manually selected and tuned features. Arguably, feature engineering is the dominating research component in practical applications of ML. In contrast, the power of NNs is in automatic feature learning from massive datasets. Not only does it simplify manual and laborious feature engineering but also allows learning task-optimal features.
Modern biology has entered the era of Big Data, wherein datasets are too large, high-dimensional and complex for classical computational biology methods. The ability learn at the higher levels of abstraction made DL is a promising and effective tool for working with biological and chemical data7. Methods using DL architecture capable to deal with sparse and complex information, which is especially demanded in the analysis of high-dimensional gene expression data. “Curse of dimensionality” is one of the major problems of gene expression data that can be solved by feature selection implementing standard data projections methods as PCA or more biologically relevant as pathway analysis.8 DNNs demonstrate the state-of-art performance extracting features from sparse transcriptomics data (both mRNA and miRNA data)9, in classifying cancer using gene expression data10 and predicting splicing code patterns.11 DL have been effectively applied in biomodeling and structural genomics to predict protein 3-D structure using protein sequence (order or disorder protein (with lack of fixed 3-D structure)12,13 and may become an essential tool for development of new drugs.14 DL approaches were successfully implemented to predict drug-target interactions15, model reaction properties of molecules16 and calculate toxicity of drugs.17 As deep networks incorporate more features from biology18, application breadth and accuracy will likely increase.
Drug repurposing or target extension allows prediction of new potential applications of medications or even new therapeutic classes of drugs using gene expression data before and after treatment (e.g. before and after incubation of a cell line with multiple drugs). There are multiple in silico approaches to drug discovery and classification.19–21 And many attempts were made to predict transcriptional response with functional properties of drugs.22–24 In this study we addressed this problem by classifying various drugs to therapeutic categories with DNN solely based on their transcriptional profiles. We used the perturbation samples of X drugs across A549, MCF-7 and PC-3 cell lines from the LINCS project and linked those to 12 therapeutic use categories derived from MeSH therapeutic use section (Fig. 1). After that we independently used both gene expression level data for “landmark genes” and pathway activation scores to train DNN classifier.
Figure 1.
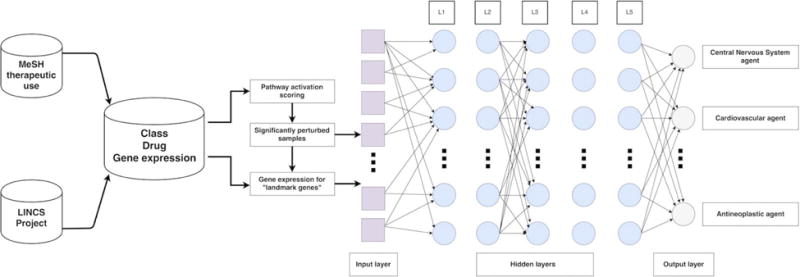
Study design. Gene expression data from LINCS Project was linked to 12 MeSH therapeutic use categories. DNN was trained separately on gene expression level data for “landmark genes” and pathway activation scores for significantly perturbed samples, forming an input layers of 977 and 271 neural nodes, respectively.
RESULTS
The main aim of this study was to apply and estimate the accuracy of DL methods to classify various drugs to therapeutic categories solely based on their transcriptional profiles. In total, we analysed 26,420 drug perturbation samples for three cell lines from Broad LINCS database. All samples were assigned to 12 specific therapeutic use categories according to MeSH classification of particular drug (Supplementary Table 1). Since a number of drugs was present in multiple categories we considered only those drugs that belong only to one category. To increase the number of samples in each of the categories and to make the classification more robust for each given drug we aggregated all samples corresponding to all possible perturbation time, perturbation concentration and cell line parameters (Supplementary Table 2).
When dealing with transcriptional data at the gene level, a common problem is the so called “curse of dimensionality”. Indeed, when we applied DNN on gene level data for whole dataset of 12,797 genes it didn’t perform very well, achieving only 0.24 mean F1 score on 12 classes. So our first step was proper feature selection. Here we investigated two approaches: pathway activation scoring and using “landmark genes” as new features.
Pathway level
For pathway level analysis we used previously established pathway analysis method called OncoFinder.25–30 In contrast with other pathway analysis tools, which implement basic pathway enrichment analysis, OncoFinder performs quantitative estimation of signaling pathway activation strength. It preserves biological function and allows for dimensionality reduction at the same time. All perturbation samples were analysed with this tool and for each sample we calculated pathway activation profiles for 271 signaling pathways. Samples with zero pathway activation score for all of the pathways were considered as insignificantly perturbed and were excluded from further analysis. That resulted in a final dataset containing 308, 454 and 433 drugs for A549, MCF7 and PC3 cell lines, respectively, and totalling 9352 samples (Supplementary table 2).
Using this dataset we built a deep learning classifier based only on pathway activation scores for drug perturbation profiles of 3 cell lines: A549, MCF-7 and PC-3. Making a classifier based on a pooled dataset with different cell lines, drug concentration and perturbation time, we’re able to estimate the classification performance in recognizing complex drug action patterns across different biological conditions. For the 3-class classification problem we chose the most abundant categories: antineoplastic, cardiovascular and central nervous system agents. DNN achieved 10 fold cross-validation mean F1 score of 0.701. We compared the results of DNN to other popular classification algorithm called support vector machine (SVM) trained via nested 3-fold cross validation for several hyperparameters (see Methods). On 3-class classification problem SVM performed with mean F1 score of 0.530.
Addition of gastrointestinal and anti-infective classes decreased the mean F1 score of DNN to 0.596. Mean F1 score for SVM dropped as well, down to 0.417.
When all 12 classes were considered, the classification neural performance decreased in a minor way, with a cross-validation mean F1 score of 0.546. SVM performed with cross-validation mean F1 score of 0.366 on the same 12-class classification problem. The performance comparison of DNN and SVM on investigated classification problems is depicted in Fig. 2a–c. These results indicate that our model performance far exceeds random chance31 and we can conclude that DNN outperformed SVM on every multiclass classification problem.
Figure 2.
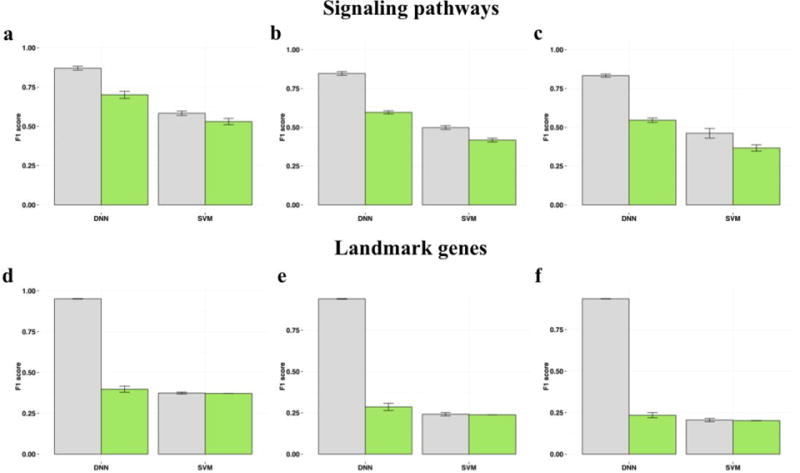
Classification results. Classification performance of DNN and SVM trained on signaling pathways (a, b, c) and landmark genes (d, e, f) for 3, 5 and 12 drug classes, respectively, after 10 fold cross validation. Training and validation set results are shown in gray and green colors, respectively.
Landmark genes level
In our second feature selection approach we used a dataset containing normalized gene expression data for 977 “landmark genes”. According to the authors of LINCS Project they can capture approximately 80% of the information and possess great inferential value. For fair comparison we trained DNN exactly the same way we did on pathway level. We used the same dataset of 9352 significantly perturbed samples and tested the performance of DNN on the same classification problems. DNN trained on “landmark genes” data performed with 10 fold cross-validation mean F1-scores of 0.397, 0.285 and 0.234 for 3-, 5- and 12-class classification tasks, respectively. SVM model showed mean F1-scores of 0.372, 0.238 and 0.202 for respective tasks (Fig. 2d–f).
DNN as drug repurposing tool
Here we tried to dig a bit deeper into classification results on pathway level, since DNN model worked best with pathways as features. To determine which of the 12 therapeutic use categories are the most detectable by DNN, we calculated 10-fold cross-validation classification accuracy of each category. Antineoplastic agents turned out to be the most “recognizable” category by a large margin, with 0.686 accuracy on 12 classes. This was followed by Anti-Infective, Central nervous system and Dermatologic categories, with 0.513, 0.506 and 0.505 accuracy, respectively. The least “recognizable” on the same number of classes was Hematologic agents, with accuracy of 0.23. On 3- and 5-class classification problems, the Antineoplastic drugs category was on top as well, with accuracy of 0.82 and 0.742. Separability of therapeutic categories by DNN can be illustrated with confusion matrices (Fig. 3). Here we observed that the Cardiovascular category drugs was relatively often misclassified as Central nervous system and Antineoplastic agents. In contrast, the level of false positives for the Antineoplastic category was relatively small. If we look even closer into the results, sometimes these misclassified false positive drugs may in fact represent a possibility for drug repurposing. For instance, well known muscarinic receptor antagonist Otenzepad, was misclassified as central nervous system agent, but despite its obvious role in the brain function32,33, according to MeSH therapeutic use section it is only used against cardiac arrhythmia. Another example includes vasodilator Pinacidil, a cyanoguanidine drug that opens ATP-sensitive potassium channels, that was misclassified as central nervous system agent in several cross-validation iterations, although it is used only in cardiovascular conditions. It’s known that potassium channels play important roles in different brain regions34 and pinacidil might influence some of them. Aforementioned cases hint to the fact that imperfect accuracy here might not be a bad thing and DNN model could serve as powerful drug repositioning tool.
Figure 3.
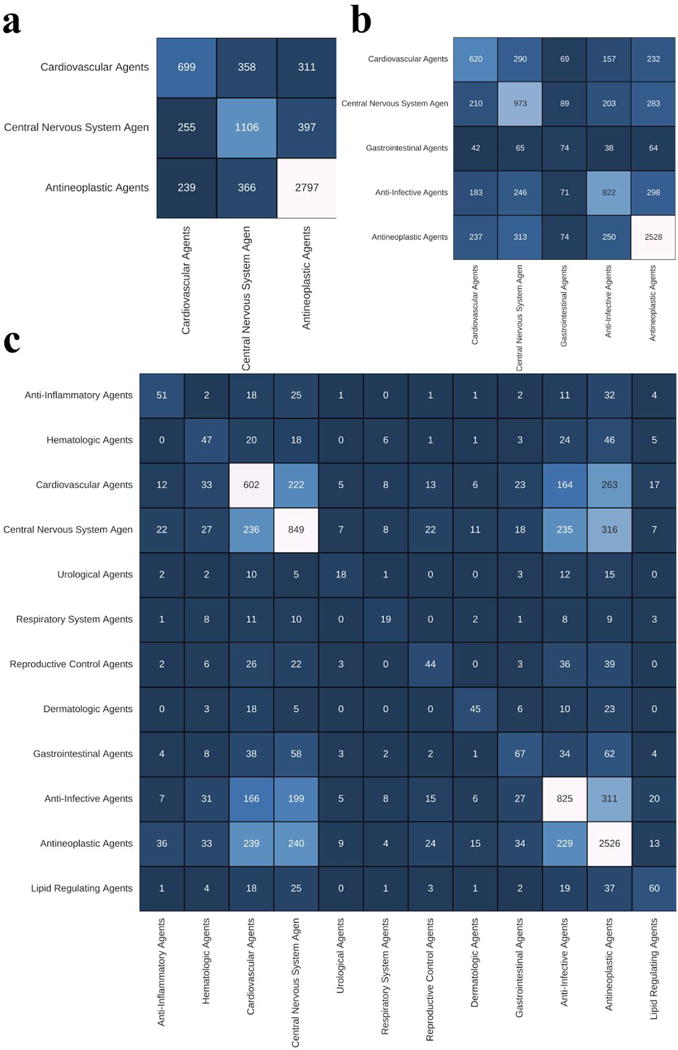
Validation confusion matrix representing deep neural network classification performance over a set of drugs profiled for A549, MCF7 and PC3 cell lines, belonging to 3 (a), 5 (b) and 12 (c) therapeutic classes. C(i,j) element is a sample count of how many times i was the truth and j was predicted.
Discussion
With increasing availability of big data and GPU computing, the entire field of deep learning is experiencing very rapid development, and the breadth of DNN applications goes far beyond text, voice and image recognition problems. In this paper we explored the possibility of using DL to classify various drugs into therapeutic categories solely based on their transcriptomic data. To our knowledge, this is the first DL model to map transcriptomic data onto therapeutical category.
DNN trained on gene level data did not perform very well, achieving only 0.24 F1 score on 12 classes. Thus, as a way to reduce dimensionality and keep biological relevance, we decided to apply pathway activation scoring.27 Translation of perturbation profiles onto the pathway level turned out to be very beneficial. Pathways served as excellent features, and we were able to exclude insignificantly perturbed samples and demonstrate the ability of deep neural network to recognize sophisticated drug action mechanisms on the pathway level. The power of this approach is further highlighted by the fact that it performs with high accuracy on a pooled dataset with samples from cells treated by different drug concentrations and perturbation times. Furthermore, the DNN achieves significant classification accuracy even across different cell lines. Its performance turned out to be better than SVM on every multiclass classification problem. When we used the same set of significantly perturbed samples that we selected with the pathway activation approach, and trained DNN on a dataset with gene expression data for “landmark genes” the results turned out to be significantly worse. Hence, we can conclude that pathway level data is more complementary for DNN and more suitable for classifying drugs into therapeutic use categories. Proper comparison to reference group of samples plays an essential role and merely normalized gene expression data from perturbed samples isn’t sufficient for complex classification tasks.
It is possible to interpret the classification results from different angles. For instance, as it was shown, in confusion matrices (Fig. 3) the “misclassified” samples for a certain drug might in fact be an indication of its potential for novel use, or repurposing, in these exact “incorrectly” assigned conditions. Misclassification, therefore, may lead to unexpected new discoveries. This approach opens a great avenue for application of DL in the drug repurposing field.
EXPERIMENTAL SECTION
Data collection
In this study, we performed the analysis of gene expression data produced by the LINCS Project participants (http://www.lincsproject.org/). We utilized the level 3 (Q2NORM) gene expression data for three cell lines: MCF7, A549 and PC3. Q2NORM data contains the expression levels of both directly measured landmark transcripts plus inferred genes, which were normalized using invariant set scaling followed by quantile normalization. Mapping probes onto official HGNC35 gene symbols resulted in a gene expression dataset comprising 12,797 genes total.
Drugs selection
To link the drugs profiled by LINCS project to medical conditions we utilized MeSH (Medical Subject Headings) classification (https://www.nlm.nih.gov/mesh/). We picked only those drugs that had the link to a disease in “therapeutic use” section of classification tree. The drugs belonging to several categories simultaneously were excluded from the analysis.
Gene expression analysis
All of the selected drugs’ samples collected for three cell lines were grouped by the combination of following parameters: cell line, drug, perturbation concentration and perturbation time. This resulted in a total number of 26,420 samples. For gene level analysis we trained our models on both whole dataset of 12,797 genes and a subset of 977 so called “landmark genes” defined in the LINCS Project.
In pathway level analysis, for each given case sample group we generated a reference group consisting of samples perturbed with DMSO that came from the same RNA plate as samples from the case group.
After that, each case sample group was independently analyzed using an algorithm called OncoFinder.27 Taking the preprocessed gene expression data as an input, it allows for cross-platform dataset comparison with low error rate and has the ability to obtain functional features of intracellular regulation using mathematical estimations. For each investigated sample group it performs a case-reference comparison using Student’s t-test, generates the list of significantly differentially expressed genes and calculates the Pathway Activation Strength (PAS), a value which serves as a qualitative measure of pathway activation. Positive and negative PAS values indicate pathway up- and down-regulation, respectively. In this study the genes with FDR-adjusted p-value<0.05 were considered significantly differentially expressed. Samples with zero pathway activation score for all of the pathways were considered as insignificantly perturbed and were excluded from further analysis. The filtered dataset contained 308, 454 and 433 drugs for A549, MCF7 and PC3 cell lines, respectively, and comprised 9352 samples total (Supplementary Tables 1 and 2).
Classification methods
Among the multitude of available classification methods we have employed the two that are highly robust and widely successful in the fields outside drug prediction: SVM36,37 and deep neural network.38 SVMs are a celebrated classification method for their flexibility and ease of use, while deep learning approaches are continuing to break records in many pattern recognition tasks.
Flexibility of SVM, as a maximum margin classifier, is in part reflected in provided ability to select a kernel that fits the data best and choose a soft-margin parameter that allows for best generalization. However, these parameters are not evident for any given dataset. It has been previously shown, that radial basis function (RBF) kernel SVMs perform well on largely different data. In order to be more objective in selection of kernel we have allowed our nested cross validation choice of three. We used grid search for hyperparameters optimization. We have trained the SVM via nested 3-fold cross validation for hyperparameters that include kernel type (linear, RBF, or polynomial) and soft-margin parameter C embedded in the outer 10-fold cross validation loop. For each fold, the algorithm could have selected different kernels and different soft-margin parameters. Nevertheless, RBF was indeed the most preferred.
The deep learning method used in our work was the standard fully connected multilayer perceptron with 977 input nodes for gene expression level data and 271 for pathway activation scores. Similarly to SVM, we used grid search for hyperparameters optimization. We used cross-entropy as cost function and AdaDelta39 as cost function optimizer. We searched for the optimal number of layers, number of hidden units, and the dropout rejection rate in a nested cross validation framework. For the hyper-parameter search, the number of layers varied from 3 to 8, where the number of hidden units per layer was reduced with the depth of the network to half the previous layer. We set the search for the starting hidden layer from 300 to 900 in steps of 150. Each layer was initialized with the Glorot uniform approach.40 The experiment shows that the best combination of parameters was 3 hidden layers with 200 in each with rectified linear activation function. The dropout rejection ratio was tested for 20% and 80% at each layer. We chose an antisymmetric activation function, hyperbolic tangent, as the non-linear function of hidden neurons because the data is normalized to zero mean and unit variance, thus we rely on deviations from the mean to train the network. Also, it has been reported that the hyperbolic tangent speeds convergence up compared to sigmoid functions.39 The number of output nodes was equal to the number of classes to explore in each particular experiment with a softmax activation function. The code that implements the feed-forward neural network used in our experiments is publicly available at https://github.com/alvarouc/mlp (commit: 8b07a1a18b17ca530fdcb482fcec24e26e36b27a).
Supplementary Material
Supplementary table 1. MeSH category stratification binary matrix of 678 significantly perturbed drugs. Every drug belongs only to single category.
Supplementary table 2. Number of drugs selected for A549, MCF7 and PC3 cell lines according to MeSH therapeutic use section. Number of samples is shown in brackets.
Acknowledgments
We would like to thank Dr. Leslie C. Jellen of Insilico Medicine for editing this manuscript for language and style. Authors thank Mark Berger and NVIDIA Corporation for providing valuable advice and high performance GPU equipment for deep learning applications.
References
- 1.Loging W, William L, Lee H, Bryn W-J. High-Throughput Electronic Biology: Mining Information for Drug Discovery. Nat Rev Drug Discov. 2007;6(6) doi: 10.1038/nrd2265. [DOI] [PubMed] [Google Scholar]
- 2.Kirchmair J, Johannes K, Göller AH, Dieter L, Jens K, Bernard T, Wilson ID, Glen RC, Gisbert S. Predicting Drug Metabolism: Experiment And/or Computation? Nat Rev Drug Discov. 2015 doi: 10.1038/nrd4581. [DOI] [PubMed] [Google Scholar]
- 3.Schirle M, Markus S, Jenkins JL. Identifying Compound Efficacy Targets in Phenotypic Drug Discovery. Drug Discov Today. 2015 doi: 10.1016/j.drudis.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 4.LeCun Y, Yann L, Yoshua B, Geoffrey H. Deep Learning. Nature. 2015;521(7553):436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 5.Baldi P, Sadowski P, Whiteson D. Searching for Exotic Particles in High-Energy Physics with Deep Learning. Nat Commun. 2014;5:4308. doi: 10.1038/ncomms5308. [DOI] [PubMed] [Google Scholar]
- 6.Schmidhuber J, Jürgen S. Deep Learning in Neural Networks: An Overview. Neural Netw. 2015;61:85–117. doi: 10.1016/j.neunet.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Mamoshina P, Vieira A, Putin E, Zhavoronkov A. Applications of Deep Learning in Biomedicine. Mol Pharm. 2016;13(5):1445–1454. doi: 10.1021/acs.molpharmaceut.5b00982. [DOI] [PubMed] [Google Scholar]
- 8.Hira ZM, Gillies DF. A Review of Feature Selection and Feature Extraction Methods Applied on Microarray Data. Adv Bioinformatics. 2015;2015:198363. doi: 10.1155/2015/198363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ibrahim R, Rania I, Yousri NA, Ismail MA, El-Makky NM. Multi-Level gene/MiRNA Feature Selection Using Deep Belief Nets and Active Learning. 2014 36th Annual International Conference of the IEEE Engineering in Medicine and Biology Society. 2014 doi: 10.1109/EMBC.2014.6944490. [DOI] [PubMed] [Google Scholar]
- 10.Fakoor R, Ladhak F, Nazi A, Huber M. Using Deep Learning to Enhance Cancer Diagnosis and Classification. Proceedings of the International Conference on Machine Learning. 2013 [Google Scholar]
- 11.Leung MKK, Xiong HY, Lee L J, Frey BJ. Deep Learning of the Tissue-Regulated Splicing Code. Bioinformatics. 2014;30(12):i121–i129. doi: 10.1093/bioinformatics/btu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyons J, James L, Abdollah D, Rhys H, Alok S, Kuldip P, Abdul S, Yaoqi Z, Yuedong Y. Predicting Backbone Cα Angles and Dihedrals from Protein Sequences by Stacked Sparse Auto-Encoder Deep Neural Network. J Comput Chem. 2014;35(28):2040–2046. doi: 10.1002/jcc.23718. [DOI] [PubMed] [Google Scholar]
- 13.Wang S, Weng S, Ma J, Tang Q. DeepCNF-D: Predicting Protein Order/Disorder Regions by Weighted Deep Convolutional Neural Fields. Int J Mol Sci. 2015;16(8):17315–17330. doi: 10.3390/ijms160817315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lusci A, Pollastri G, Baldi P. Deep Architectures and Deep Learning in Chemoinformatics: The Prediction of Aqueous Solubility for Drug-like Molecules. J Chem Inf Model. 2013;53(7):1563–1575. doi: 10.1021/ci400187y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang C, Caihua W, Juan L, Fei L, Yafang T, Zixin D, Qian-Nan H. Pairwise Input Neural Network for Target-Ligand Interaction Prediction. 2014 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 2014 [Google Scholar]
- 16.Hughes TB, Miller GP, Joshua Swamidass S. Modeling Epoxidation of Drug-like Molecules with a Deep Machine Learning Network. ACS Central Science. 2015;1(4):168–180. doi: 10.1021/acscentsci.5b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Y, Youjun X, Ziwei D, Fangjin C, Shuaishi G, Jianfeng P, Luhua L. Deep Learning for Drug-Induced Liver Injury. J Chem Inf Model. 2015;55(10):2085–2093. doi: 10.1021/acs.jcim.5b00238. [DOI] [PubMed] [Google Scholar]
- 18.Solovyeva KP, Karandashev IM, Zhavoronkov A, Dunin-Barkowski WL. MODELS OF INNATE NEURAL ATTRACTORS AND THEIR APPLICATIONS FOR NEURAL INFORMATION PROCESSING. Front Syst Neurosci. 2015;9 doi: 10.3389/fnsys.2015.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newby D, Freitas AA, Ghafourian T. Comparing Multilabel Classification Methods for Provisional Biopharmaceutics Class Prediction. Mol Pharm. 2015;12(1):87–102. doi: 10.1021/mp500457t. [DOI] [PubMed] [Google Scholar]
- 20.Wenlock MC, Barton P. In Silico Physicochemical Parameter Predictions. Mol Pharm. 2013;10(4):1224–1235. doi: 10.1021/mp300537k. [DOI] [PubMed] [Google Scholar]
- 21.Broccatelli F, Cruciani G, Benet LZ, Oprea TI. BDDCS Class Prediction for New Molecular Entities. Mol Pharm. 2012;9(3):570–580. doi: 10.1021/mp2004302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herrera-Ruiz D, Faria TN, Bhardwaj RK, Timoszyk J, Gudmundsson OS, Moench P, Wall DA, Smith RL, Knipp GT. A Novel hPepT1 Stably Transfected Cell Line: Establishing a Correlation between Expression and Function. Mol Pharm. 2004;1(2):136–144. doi: 10.1021/mp034011l. [DOI] [PubMed] [Google Scholar]
- 23.Iskar M, Zeller G, Blattmann P, Campillos M, Kuhn M, Kaminska KH, Runz H, Gavin A-C, Pepperkok R, van Noort V, Bork P. Characterization of Drug-Induced Transcriptional Modules: Towards Drug Repositioning and Functional Understanding. Mol Syst Biol. 2013;9:662. doi: 10.1038/msb.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kutalik Z, Beckmann JS, Bergmann S. A Modular Approach for Integrative Analysis of Large-Scale Gene-Expression and Drug-Response Data. Nat Biotechnol. 2008;26(5):531–539. doi: 10.1038/nbt1397. [DOI] [PubMed] [Google Scholar]
- 25.Spirin PV, Lebedev TD, Orlova NN, Gornostaeva AS, Prokofjeva MM, Nikitenko NA, Dmitriev SE, Buzdin AA, Borisov NM, Aliper AM, Garazha AV, Rubtsov PM, Stocking C, Prassolov VS. Silencing AML1-ETO Gene Expression Leads to Simultaneous Activation of Both pro-Apoptotic and Proliferation Signaling. Leukemia. 2014;28(11):2222–2228. doi: 10.1038/leu.2014.130. [DOI] [PubMed] [Google Scholar]
- 26.Zhu Q, Qingsong Z, Evgeny I, Aliper AM, Evgeny M, Keren P, Buzdin AA, Zhavoronkov AA, David S. Pathway Activation Strength Is a Novel Independent Prognostic Biomarker for Cetuximab Sensitivity in Colorectal Cancer Patients. Human Genome Variation. 2015;2:15009. doi: 10.1038/hgv.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buzdin AA, Zhavoronkov AA, Korzinkin MB, Venkova L S, Zenin AA, Smirnov PY, Borisov NM. Oncofinder, a New Method for the Analysis of Intracellular Signaling Pathway Activation Using Transcriptomic Data. Front Genet. 2014;5:55. doi: 10.3389/fgene.2014.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Artemov A, Aliper A, Korzinkin M, Lezhnina K, Jellen L, Zhukov N, Roumiantsev S, Gaifullin N, Zhavoronkov A, Borisov N, Buzdin A. A Method for Predicting Target Drug Efficiency in Cancer Based on the Analysis of Signaling Pathway Activation. Oncotarget. 2015;6(30):29347–29356. doi: 10.18632/oncotarget.5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venkova L, Aliper A, Suntsova M, Kholodenko R, Shepelin D, Borisov N, Malakhova G, Vasilov R, Roumiantsev S, Zhavoronkov A, Buzdin A. Combinatorial High-Throughput Experimental and Bioinformatic Approach Identifies Molecular Pathways Linked with the Sensitivity to Anticancer Target Drugs. Oncotarget. 2015;6(29):27227–27238. doi: 10.18632/oncotarget.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makarev E, Cantor C, Zhavoronkov A, Buzdin A, Aliper A, Csoka AB. Pathway Activation Profiling Reveals New Insights into Age-Related Macular Degeneration and Provides Avenues for Therapeutic Interventions. Aging. 2014;6(12):1064–1075. doi: 10.18632/aging.100711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Combrisson E, Jerbi K. Exceeding Chance Level by Chance: The Caveat of Theoretical Chance Levels in Brain Signal Classification and Statistical Assessment of Decoding Accuracy. J Neurosci Methods. 2015;250:126–136. doi: 10.1016/j.jneumeth.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 32.Langmead CJ, Jeannette W, Charlie R. Muscarinic Acetylcholine Receptors as CNS Drug Targets. Pharmacol Ther. 2008;117(2):232–243. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 33.Volpicelli LA, Levey AI. Muscarinic Acetylcholine Receptor Subtypes in Cerebral Cortex and Hippocampus. Progress in Brain Research. 2004:59–66. doi: 10.1016/S0079-6123(03)45003-6. [DOI] [PubMed] [Google Scholar]
- 34.Trimmer JS. Subcellular Localization of K+ Channels in Mammalian Brain Neurons: Remarkable Precision in the Midst of Extraordinary Complexity. Neuron. 2015;85(2):238–256. doi: 10.1016/j.neuron.2014.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gray KA, Yates B, Seal RL, Wright MW, Bruford EA. Genenames.org: The HGNC Resources in 2015. Nucleic Acids Res. 2015;43:D1079–D1085. doi: 10.1093/nar/gku1071. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boser BE, Guyon IM, Vapnik VN. A Training Algorithm for Optimal Margin Classifiers. Proceedings of the fifth annual workshop on Computational learning theory – COLT ’92. 1992 [Google Scholar]
- 37.Cortes C, Corinna C, Vladimir V. Support-Vector Networks. Mach Learn. 1995;20(3):273–297. [Google Scholar]
- 38.Hinton GE, Salakhutdinov RR. Reducing the Dimensionality of Data with Neural Networks. Science. 2006;313(5786):504–507. doi: 10.1126/science.1127647. [DOI] [PubMed] [Google Scholar]
- 39.Zeiler MD. ADADELTA: An Adaptive Learning Rate Method. arXiv. 2012:6. [Google Scholar]
- 40.Glorot Xavier, Bengio Yoshua. Understanding the difficulty of training deep feedforward neural networks. International conference on artificial intelligence and statistics. 2010 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1. MeSH category stratification binary matrix of 678 significantly perturbed drugs. Every drug belongs only to single category.
Supplementary table 2. Number of drugs selected for A549, MCF7 and PC3 cell lines according to MeSH therapeutic use section. Number of samples is shown in brackets.