

Abstract
Mouse models of neurodegenerative diseases such as Alzheimer’s disease (AD) are important for understanding how pathological signaling cascades change neural circuitry and in time interrupt cognitive function. Here, we introduce a non-genetic preclinical model for aging and show that it exhibits cleaved tau protein, active caspases and neurofibrillary tangles, hallmarks of AD, causing behavioral deficits measuring cognitive impairment. To our knowledge this is the first report of a non-transgenic, non-interventional mouse model displaying structural, functional and molecular aging deficits associated with AD and other tauopathies in humans with potentially high impact on both new basic research into pathogenic mechanisms and new translational research efforts. Tau aggregation is a hallmark of tauopathies, including AD. Recent studies have indicated that cleavage of tau plays an important role in both tau aggregation and disease. In this study we use wild type mice as a model for normal aging and resulting age-related cognitive impairment. We provide evidence that aged mice have increased levels of activated caspases, which significantly correlates to increased levels of truncated tau and formation of neurofibrillary tangles. In addition, cognitive decline was significantly negatively associated with increased levels of caspase activity and tau truncated by caspase-3. Experimentally induced inhibition of caspases prevented this proteolytic cleavage of tau truncation and the associated formation of neurofibrillary tangles. Our study shows the strength of using a non-transgenic model to study structure, function and molecular mechanisms in aging and age related diseases of the brain.
Keywords: age-related cognitive decline, tauopathy, neurofibrillary tangles, molecular mechanisms in brain aging, preclinical
Introduction
Tauopathies are a group of neurodegenerative disorders that are associated with the pathological aggregation of the tau protein in neurofibrillary tangles; one such disease is Alzheimer’s disease (AD). AD is a neurological disorder that leads to progressive cognitive decline. This includes memory loss, decline in cognitive control and a diminished capacity in visual–spatial skills (Huang and Mucke 2012). A hallmark of AD is the accumulation of senile plaques and neurofibrillary tangles (NFTs), composed of abnormally processed tau (Iqbal et al. 2009).
Tau is a microtubule-associated protein that stabilizes the neuronal cytoskeleton and participates in vesicular transport (Avila et al. 2004). Pathological alterations in tau occur in several neurodegenerative disorders, including AD and Parkinson’s disease (Kosik et al. 1986; Wray and Lewis 2010; Xie et al. 2014).
Several studies have demonstrated the activation of apoptotic machinery within the AD brain (Calissano et al. 2009; Dean 2008). Apoptosis is characterized by plasma membrane blebbing, chromosome condensation and DNA fragmentation and is initiated by caspases.
Caspases are a group of cysteine proteases that cleave after specific Asp residues and play a central role in the cell death pathway (Turk and Stoka 2007). In addition to their established role in apoptosis there is evidence that caspases have non-apoptotic functions (McIlwain et al. 2013; Kuranaga and Miura 2007). Additionally, components of the neuronal cytoskeleton, including tau, are targeted by caspases.
There is evidence for increased caspase activation in AD brain (Rohn et al. 2002). Moreover, the presence of caspase-cleaved tau has also been detected in AD brain. The caspase cleavage of tau plays an important role in the oligomerization and formation of a pathological tau species in AD. This cleavage generates a highly fibrillogenic tau species, which aggregates more freely and to a greater degree than the full length tau and also assists in aggregate formation of the full length tau (Gamblin et al. 2003). Antibodies recognizing truncated tau at Asp421 showed that cleaved tau (Asp421), active caspase-3 and fibrillary tau colocalize in AD brains (Quintanilla et al. 2012). In addition, a mouse model of tauopathies showed that the majority of cells with active caspases also had NFTs (Spires-Jones et al. 2008). Moreover, experiments in tissue culture provide evidence that the Asp421 cleaved tau is toxic to neurons (Chung et al. 2001; Matthews-Roberson et al. 2008; Quintanilla et al. 2009).
Mouse models of age related cognitive decline such as seen in AD and other tauopathies are important for understanding how pathological cascades change neural circuitry and in time interrupt cognitive function (Spires and Hyman 2005). In this study we used an established non-genetic preclinical model for aging (Kaja et al. 2013; 2015) and showed that in cognitively impaired mice, cleaved tau, active caspases and NFTs were present and this correlated significantly to behavioral deficits. To our knowledge this is the first report of a non-transgenic, non-interventional mouse model displaying structural, functional and molecular aging deficits associated with AD and other tauopathies in humans.
Methods
Animals
The animal experiments performed were approved by the local Institutional Animal Care and Use Committee. Young (6 months; analogous to an approximately 34 year old human) and old (24 months; analogous to an approximately 70 year old human) male C57BL/6J mice were obtained from the National Institute on Aging. Individual animals were maintained in the institutional vivarium at ambient temperature (23 ±1 °C), under a 12-hour light/dark cycle starting at 0600 hours. Mice had ad libitum access to food and water, except during testing.
Behavioral Assays
Spatial learning and memory
The behavioral outcome measures were described by us previously (Kaja et al 2013). Spatial learning and memory were determined using a swim maze test as described by us previously (Sumien et al. 2006). Briefly, performance in the swim maze test was assessed using a learning index. The learning index was defined as the relative improvement in performance normalized to the average performance of the young mice using path length. The pretraining phase involved the mouse learning the motor components of swimming and climbing onto the platform, without learning the location of the platform in the tank. Afterwards, the mice were tested for their ability to learn the location of the platform. This involved four acquisition sessions and for each session there were five trials during which the mouse had to swim to the platform from a different starting point in the tank.
Bridge Walking
In short, the latency to fall was determined for each mouse after being placed on one of four bridges (1 cm square, 1 cm round, 2 cm square, 2 cm round) mounted 45 cm above a padded surface. The maximum latency to fall was set at 60 seconds. Mice were placed on each bridge three times and the mean latency to fall was used as a measure of performance for each bridge.
Caspase Activity Assay
Caspase activity was measured using the caspase-3 substrate Ac-DEVD-AFC (Santa Cruz Biotech, sc-311274A) as the caspase substrate. To measure caspase activity in SH-SY5Y, cells were harvested 30 minutes after UV-irradiation and spun down at 2000 × g. The cell pellets were resuspended in 100 uL Caspase Buffer (20 mM HEPES, pH 7.5, 50 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT supplemented with protease inhibitor cocktail) and lysed by 3 cycles of freeze thaw. To detect caspase activity in mouse forebrain and cerebellum, 1ug of lysate was brought up to a volume of 100uL with caspase buffer. The lysates were then incubated for one hour at 37°C. 0.2 μM Ac-DEVD-afc (in DMSO) was added and the reaction mixed. The reactions were then analyzed fluorometrically (excitation 405nm, emission 535nm) and activity was expressed in relative arbitrary fluorescence units.
Antibodies
To examine Tau cleavage, mouse anti-Tau, caspase cleaved (truncated at Asp421) antibody (Millipore, MAB5430) (1:1000) was used. Previous work indicated that caspase-3 targets Tau at Asp421 (Jarero-Basulto et al. 2013). For a loading control mouse anti-Actin (Millipore, MAB1501R) (1:1000) was used.
Immunoblot Analysis
Mice were euthanized using carbon dioxide asphyxiation and brains were then dissected into forebrain, olfactory bulb, and cerebellum and snap frozen using liquid nitrogen. Total protein concentration was determined using the method of Lowry. Twenty-five micrograms of total protein in SDS loading buffer were separated electrophoretically and transferred to nitrocellulose membrane as described previously (Kaja et al. 2013). Protein level quantification was performed by densitometry using ImageJ software (NIH). Density values were corrected for background and TAU expression normalized for endogenous actin expression.
Thioflavin S Stain
To examine NFT formation in SH-SY5Y cells (ATCC, Manassas, VA, USA), cells were stained with Thioflavin S (Sigma, T1892). Thioflavin S was used at a concentration of 0.05% diluted in distilled water. Cells were rinsed in distilled water and incubated in 0.05% Thioflavin S for 5 minutes. After 5 minutes, the cells were washed in 70% Ethanol for 5 minutes and rinsed with distilled water overnight. To examine brain sections for Thioflavin S-positive cells, the same procedure was used except Thioflavin S was used at a concentration of 0.01%.
Brain Immunostaining
For tissue processing and fixation, brains were dissected and separated in two halves by cutting along the midline section with a disposable razorblade. The left hemisphere was used for preparation of tissue lysates, essentially as described previously (Kaja et al, 2013; Kaja et al. 2015). In brief, total protein was extracted from frozen samples using CytoBuster tissue lysis reagent (EMD Millipore, Bellerica, MA) in the presence of protease inhibitors (Roche Applied Science, Indianapolis, IN). The right hemisphere was immersion fixed in freshly prepared 4% PFA in PBS pH7.4 overnight, and cryoprotected by sequential immersion in 10%, 20% and 30% sucrose in PBS pH 7.4 supplemented with 0.01% sodium azide. Tissue was embedded in Tissue-Tek® cryoembedding compound (Takura Finetek USA, Torrance, CA) and 12 um sections were cut on a Vibratome Ultra Pro 5500 cryostat (North Central Instruments, Minneapolis, MN). Mouse forebrain tissue sections were blocked at room temperature for an hour, followed by incubation with anti-cleaved TAU for 3 days at 4 degrees C. After primary incubation tissue sections were washed with Phosphate buffer and incubated with goat anti-mouse Alexa Fluor® 488 (1:2000 dilution; Life Technologies, Carlsbad, CA) secondary antibody for 1 day at 4 degrees C. Tissue was co-labeled with Hoechst 33258 (1:50000; Enzo Life Sciences Inc., Farmingdale, NY) to identify cell nuclei. After secondary incubation tissue sections were washed and mounted using AquaPolymount (Polysciences Inc., Warrington, PA). Images were acquired using a Leica SP5X WLL confocal microscope (Leica Microsystems Inc., Buffalo Grove, IL).
UV irradiation of cells
SH-SY5Y cells (ATCC® number: CRL-2266) were plated in a 6-well plate (35mm dish) at 1×106 cells per well and allowed to adhere overnight. The next day cells were treated with the pan-caspase inhibitor zVAD-fmk at 100uM concentration. One hour after treatment cells were UV irradiated (UV-C, 254 nm) for 5 minutes using a 3UV transilluminator (BioChemi System, UVP, Upland, CA, USA). One hour after UV treatment, cells were harvested for immunoblot analysis, caspase activity assays or stained for NFT formation using Thioflavin S.
Data Analysis and Statistics
Protein expression levels normalized to mouse ACTB were quantified by densitometry using ImageJ software (NIH). Prism5 software (GraphPad Inc., La Jolla, CA) was used for statistical analysis. Student’s t test was used to compare data and the correlation analyses between protein level and behavioral measurements were performed by calculating a Pearson product-moment correlation coefficient (r) to evaluate the strength of the association (Kaja et al. 2013).
Results
Cleaved tau is present in the forebrain of aged mice
A growing body of evidence highlights the importance of tau truncation in initiation and potentiation of tau aggregation (de Calignon et al. 2010; Novak et al. 1993; Gamblin et al. 2003; Rissman et al. 2004; Ding et al. 2006; Pevalova et al. 2006; Delobel et al. 2008; Zhang et al. 2009). To determine if truncated tau was present we performed quantitative immunoblotting to measure the amount of cleaved tau in the forebrain. We detected significantly greater levels of truncated tau in the aged forebrains compared to the young (Fig. 1A–B) (P=0.0362).
Figure 1. Increased caspase activity and cleaved tau expression in the forebrain of aged mice.

(a) Representative immunoblots from 10 animals for each group (young and aged mice forebrain) showing cleaved tau (CTau) and actin (as a loading control) expression in the forebrain. Each well number represents an individual animal. (b) Quantitative densitometry indicated that cleaved tau expression is significantly higher (P = 0.0362, R2 = 0.3045) in the forebrain of aged mice compared to young. Tau density was normalized to actin. Normalized density is given along the ordinate and the two groups (young vs. aged) are given along the abscissa. (c) DEVD-afc, a substrate used to measure caspase-3 activity indicated that caspase activity is significantly higher (P=0.0064, R2=0.5807) in the forebrain of aged mice compared to young. Caspase activity was normalized (ordinate) to buffer alone. Data are shown as mean+/−SEM. Green squares represent young individuals, blue triangles represent aged. n = 10 mice per group. Blots were confirmed in duplicate.
Tau truncated at amino acid D421 has been detected in AD and other tauopathies (Rissman et al. 2004; Basurto-Islas et al. 2008; Guillozet-Bongaarts et al. 2005). Truncation of tau introduces a conformational change that contributes to aggregation. Caspases are cysteine aspartic proteases typically considered to be activated during apoptosis, but can also be activated without cell death (McLaughlin et al. 2003). In this regard, while caspase activation precedes and promotes tangle formation, tangle-bearing neurons can survive for extended periods (de Calignon et al. 2009; Morsch et al. 1999). Cleavage of tau at D421 is mediated by caspase-3 (Gamblin et al. 2003; Rissman et al. 2004). To determine if there was increased caspase activity we used the caspase 3 substrate DEVD-afc to measure the caspase activity in the forebrain of aged and young mice. Caspase activity was significantly higher in the forebrain of aged mice compared to young (Fig. 1C) (P=0.0064).
Cognitive deficits correlate with increased levels of truncated tau and active caspases in the forebrain of aged mice
Since the forebrain is the region of the CNS that contributes to the generation of spatial learning and memory and motor function (Kaja et al. 2012), we wanted to examine the correlation between truncated tau and active caspases with behavior in the forebrain.
We identified a statistically significant association between truncated tau expression and behavioral performance in both the bridge walking (assessed as the latency of fall, LTF) and swim maze test (assessed by the learning index, LI). Truncated tau expression in the forebrain showed a significant, strong negative association with the learning index (Fig. 2A). In addition, there was a significant negative association with truncated tau expression and the latency of fall (Fig. 2B).
Figure 2. Increased levels of truncated tau expression and caspase activity in the forebrains of aged mice correlates with age-related loss of memory and motor function.

(a–b) Correlation analysis revealed that cleaved tau (CTau) expression in the forebrain was negatively associated with bridge walking performance ((A) Learning Index) (P=0.0008, R2=0.4713) and swim maze performance ((B) Average Latency to Fall) (P=0.468, R2=0.2020). (c) Similarly, increased caspase activity also negatively associated with bridge walking (Learning Index) (P<0.0001, R2=0.7630) and (d) swim maze performance (Average Latency to Fall) (P=0.0049, R2=0.3631). Green squares represent young mice, blue triangles represent aged mice. Linear regressions are shown as solid lines; dotted lines represent 95% confidence intervals.
There was also a significant association between active caspases and behavioral performance in both the bridge walking and swim maze tests. Active caspases in the forebrain showed a strong negative association with the learning index and the latency of fall (Fig. 2C and D).
Correlation between cognitive deficits and increased caspase activity and truncated tau in the cerebellum
The main role of the cerebellum is to regulate motor function. We quantified truncated tau expression in the cerebellum of aged and young mice by quantitative immunoblotting. In the cerebellum of young mice, truncated tau expression was not detectable (Fig. 3A–B), while truncated tau expression was measured in 30% of aged cerebella (Fig. 3A–B). In addition, the cerebellum of aged mice had detectable caspase activity (Fig. 3C). The caspase activity was not significantly different from aged to young mice. However, the aged mice that had increased caspase activity also had truncated tau expression.
Figure 3. Cleaved tau immunoreactivity and caspase activity in the cerebellum showed no difference between young and aged mice.

(a) Representative immunoblots from 10 animals for each group showing immunoreactivity for cleaved tau (CTau) and actin (loading control) in the cerebellum of young and aged mice. Each well number represents an individual animal. (b) Quantitative densitometry revealed no significant difference in cleaved tau levels in the cerebellum of young and aged mice. Tau density was normalized to actin. (c) Similarly, caspase activity, measured using the substrate DEVD-afc showed no significant difference (P=0.0775, R2=0.3060) in the cerebellum of aged mice compared to young. Caspase activity was normalized to buffer alone. Data are shown as mean+/−SEM. Green squares represent young individuals, blue triangles represent aged. n = 10 mice per group. Blots were confirmed in duplicate.
Consequently, we examined the association between behavioral performance and truncated tau expression and caspase activity in the cerebellum. There was a significant negative association between caspase activity and the latency of fall (Fig. 4D). In addition, although not statistically significant, truncated tau expression did show a weak negative association with the latency of fall (Fig. 4B) In contrast, there were no significant correlations of either caspase activity or truncated tau expression with the learning index (Fig. 4A and C). This is expected since the main role of the cerebellum is to regulate motor function.
Figure 4. Correlation analysis between cleaved tau expression and caspase activity in the cerebellum with behavior.

(a) Correlation analysis revealed that truncated tau (CTau) levels showed no significant association with the learning index (P=0.5434, R2=0.02087) or (b) the latency to fall (P=0.0560, R2=0.1882). (c) Similarly, increased caspase activity showed no significant association with the learning index (P=0.0947, R2=0.0.1474). (d) In contrast increased caspase activity showed a significant negative association with the latency to fall (P=0.0191, R2=0.2690). Square symbols represent young, triangles represent aged. n = 10 mice per group.
A weak learning index correlates to increased caspase activity and tau cleavage – scalability of molecular determinants in the model of cognitive impairment during normal aging
In examining the forebrain of young mice we observed three outliers in which truncated tau and increased caspase activity were observed. Upon further examination, we found a correlation with the learning index. In all three instances the young mice that had a low learning index indicative of cognitive impairment also had increased caspase activity and truncated tau (Fig. S1). Similarly, in aged mice that had no detectable truncated tau or caspase activity there was a higher learning index indicative of normal cognitive performance (Fig. S1).
Higher caspase activity and cleaved tau in the forebrain of aged mice
In examining the caspase activity and truncated tau expression in both the forebrain and cerebellum we observed that the forebrain had a significantly higher amount of both active caspases and truncated tau compared to the cerebellum (Fig. S2A–B). In addition, since we detected both increased caspase activity and truncated tau in the forebrain of aged mice we wanted to determine if there was a correlation between the two. In the forebrain of aged mice, there was a significantly strong positive association between increased caspase activity and cleaved Tau expression (Fig S2C).
Presence of cleaved tau and Thioflavin-S positive NFTs is present in cortical layers of aged mice
To visually observe cleaved tau and NFT formation we performed immunohistochemistry on the forebrains of aged and young mice. We stained forebrains using the caspase cleaved tau antibody and observed cleaved tau staining in aged mice, but not young (Fig. 5A). Interestingly, in young mice that demonstrated behavioral impairment we also observed cleaved tau staining in the forebrain.
Figure 5. The aged cortex of cognitively impaired mice is positive for cleaved tau and NFTs.

(a) Aged and young forebrains were immunostained for cleaved tau (CTAU) (green). Aged mice that showed behavioral impairment also were positive for cleaved tau. Average number of cleaved TAU/area is indicated at top right of CTAU panel. (b) Similarly, the aged behaviorally impaired mice were also positive for NFTs based on Thioflavin S staining (Thio-S) (green). DAPI was used to stain nuclei (blue). Images are representative fields of the outer cortical layers of the forebrain (the region where images were taken is identified by a box in panel Af). The CTAU or Thio-S images were overlaid with the DAPI stain and shown in the merge panels, respectively. (c) Aged and young cerebellum were immunostained for cleaved tau (CTAU) (green). Aged mice showing behavioral impairment were positive for cleaved tau. (d) Similarly, aged behaviorally impaired mice were Thioflavin S positive (Thio-S) (green). Cerebellum sections were counterstained with DAPI (blue) as a nuclear marker. Red arrows indicate CTAU and Thio-S positive cells. The region where images were taken is identified by a box in panel A. Molecular layer: ML; Purkinje cell layer: PCL; Granule cell layer: GCL. Scale bars represent 25μm.
One pathological hallmark of tauopathies is aggregation of the microtubule-associated protein tau. To examine this we stained aged and young forebrains with the histological dye, Thioflavin-S Thioflavin S stains tau-based neurofibrillary tangles/paired helical filaments (Sun et al. 2002; Liu et al. 2012; Santa-Maria at al. 2007) as well as amyloid β plaques (Bussiere et al. 2004; Ly et al. 2011; Urbanc et al. 2002). In aged, behaviorally impaired mice we detected Thioflavin S-positive forebrains, while in young behaviorally-nonimpaired mice there was no detectable Thioflavin S staining (Fig. 5B). In addition, young behaviorally impaired mice also stained Thioflavin S-positive. The same was observed in the cerebellum (Fig. 5C–D).
Tau cleavage is caspase-3 - dependent and leads to the formation of neurofibrillary tangles
To investigate the upstream and downstream mechanisms of tau cleavage, we tested whether caspases can cleave tau and whether truncated tau can form NFTs in a cellular in vitro model, the human neuroblastoma cell line, SH-SY5Y. Caspase activity was induced by UV irradiation. One hour after UV treatment, cells were harvested for immunoblot analysis. SH-SY5Y cells that were UV irradiated showed cleaved Tau present along with activated caspases (Fig. 6A–C). However, when cells were treated with the broad caspase inhibitor zVAD-fmk prior to UV treatment, caspase activation along with cleaved Tau was not detected. To determine if neurofibrillary tangles were being formed, cells were stained with Thioflavin S and imaged. Cells that were UV irradiated showed Thioflavin S-positive staining and this was inhibited by pretreatment with zVAD-fmk (Fig. 6D).
Figure 6. Active caspases cleave tau in the human neuroblastoma SH-SY5Y cell line.
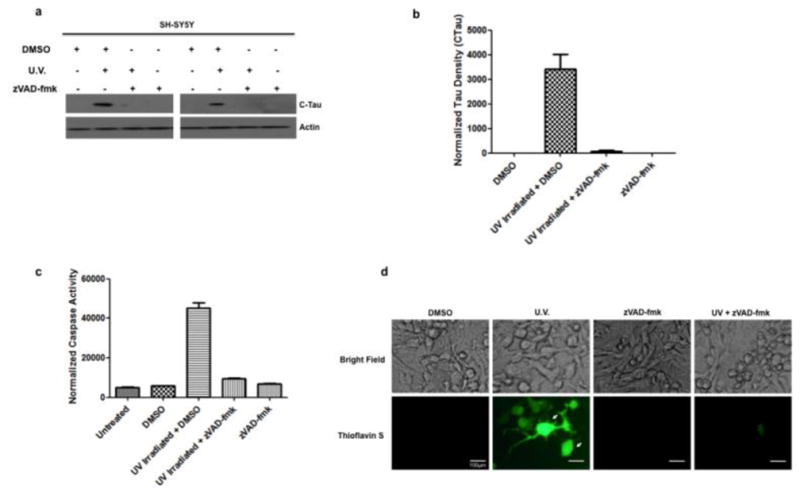
(a) SH-SY5Y cells were UV irradiated in the presence of the general caspase inhibitor zVAD-fmk (100 μM) and immunoblotted for cleaved tau (CTau). Cells that were UV irradiated had detectable cleaved tau but this was blocked by the caspase inhibitor zVAD-fmk. Actin served as a loading control. (b) Quantitative densitometry indicates SH-SY5Y cells show a higher amount of cleaved tau (normalized to actin) compared to control and zVAD-fmk treated cells. (c) SH-SY5Y cells that have been UV irradiated show increased levels of caspase activity (normalized to buffer), measured using the caspase substrate DEVD-afc and this is inhibited by pretreatment with zVAD-fmk. (d) SH-SY5Y cells that have been UV irradiated stain Thioflavin S-positive (green) and this is inhibited by treatment with zVAD-fmk. Arrows indicate Thioflavin S-positive cells (green). Scale bars represent 100μm.
Discussion
The present study provides the first evidence for a non-transgenic mammalian model of tauopathies during normal aging. Normally aged mice that are not genetically modified and have not been subjected to neuronal damage show behavioral cognitive impairment and a high level of active caspase compared to young animals of the same strain. In addition, we detected truncated tau in aged mice, which was most prominent in the forebrain. This increased level of caspase activity and cleaved tau correlated to cognitive deficits associated with aging. This was similar to what was reported in a fly model of AD. In that model aged wildtype flies showed increased caspase activity and tau truncation and showed behavioral impairment (Means et al. 2015). One of the hallmark characteristics of tauopathies is NFT formation. Thioflavin S is used to stain tau-containing neurofibrillary tangles/paired helical filaments (Sun et al. 2002; Liu et al. 2012; Santa-Maria et al. 2007) as well as amyloid β plaques (Bussiere et al. 2004; Ly et al. 2011; Urbanc et al. 2002). We detected NFTs using Thioflavin S in all cognitively-impaired mice.
Activated caspases cleave tau, the main component of NFTs. When cleaved in vitro by caspases, tau “seeds” filamentous aggregates (Cotman et al. 2005). In addition, tau adopts the MC1 conformation when cleaved, one of the earliest pathologic events in tangle formation (Cotman et al. 2005). Truncated tau occurs early in the development of tangles within AD brains and in a transgenic mouse model of AD. This truncated form of tau is therefore likely to initiate or accelerate NFT development.
Tau aggregation is a hallmark of several neurodegenerative diseases, including AD (Schraen-Maschke et al. 2010). The mechanism underlying tau aggregation is still unclear. Recent reports have shown that tau cleavage plays an important role in tau aggregation and neurodegeneration (Hanger and Wray 2010). Truncation of tau may generate tau fragments that initiate tau aggregation, which can lead to toxicity or result in tau fragments, which induce neurodegeneration through mechanisms not yet known, independent of aggregation.
In addition, SH-SY5Y cells transfected with a tau mutant are vulnerable to apoptosis (Katsutoshi et al. 2000). It is possible that the C-terminal truncation of Tau eliminates an as yet unidentified motif that functions to modulate cell death.
It is interesting to note that the inclusions that form in cells containing cleaved tau stain for Thioflavin S. This indicates that the aggregates have some ordered structure, which based on previous reports is likely filamentous.
We propose a model on the role of caspases in the cleavage of tau in tangle development. Following the activation of caspases by a stimulus, tau is cleaved predominantly at Asp421 (Gamblin et al. 2003). The truncated tau undergoes a conformational change, leading to increased filament formation and tau aggregation on the microtubule. To compensate for this, tau is phosphorylated, which leads to tau being removed from the microtubules. The hyperphosphorylated tau then leads to paired helical filament (PHF) formation and microtubule destabilization (Grundke-Iqbal et al. 1986a, 1986b). Although microtubule destabilization may be acting as a neuroprotective mechanism or is an attempt of the neuron to initiate neuroprotective signaling, the disruption of intracellular trafficking leads to apoptosis. Based on the role of caspase mediated cleavage of tau in promoting NFT formation in AD, blocking the truncation of tau could provide a promising therapeutic approach for the treatment of AD or other tauopathies.
In the mice we examined we identified outliers from both the aged and young groups. The aged outliers showed no significant caspase activity or tau cleavage. Interestingly, these outliers had fewer cognitive deficits based on the learning index. Conversely, the young outliers that showed significant caspase activity and tau cleavage also had impaired cognitive function suggesting that the same mechanism is at play regardless of age. These outliers are expected in a system that has not been genetically or chemically modified. As with many neurodegenerative disorders such as AD, the older population is more prone to disease onset, as age represents the primary predisposing condition. However, the younger population can also be susceptible, although at a lower rate and to a smaller extent. We consider this finding as highly relevant as it not only contributes to a statistically highly significant finding, but also models the human clinical situation of AD very accurately.
Transgenic mouse models of AD have allowed researchers to examine potential mechanisms involved in the development of disease based on human mutations in the genes for presenilins (PSEN1 and PSEN2) and amyloid beta precursor protein (APP). Some of these models also use mutations in MAPT, the gene for the cytoskeletal protein tau linked to tauopathies such as frontotemporal dementia (Kar et al. 2005). However, because of the large discrepancy in the behavioral findings observed across the AD mouse models, a question that arises is whether we are really any closer today to determining what these mechanisms are. In addition, another argument that complicates the use of animal models based solely on APP and/or tau mutations is that other mechanisms may be at play.
In using a non-genetic aging model to examine AD-like pathologies we remove the variability that is associated with transgenic animals. For instance, transgene integration is apparently random. Also, experiments reveal that the genetic surrounding of the inserted transgenic construct is modulating the expression pattern of the transgene itself both quantitatively and qualitatively. In our system we allow for normal aging and disease onset to occur instead of forcing the system to a disease state.
Supplementary Material
Acknowledgments
Research reported in this publication was supported in part by grants AG022550, AG027956 (NS, PK), AG010485 from NIH/NIA, RR022570 and RR027093 from NIH/NCRR and EY022774 from NIH/NEI (PK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional support by the Felix and Carmen Sabates Missouri Endowed Chair in Vision Research, the Vision Research Foundation of Kansas City and a departmental challenge grant by Research to Prevent Blindness (PK) is gratefully acknowledged. The authors thank Michael J. Forster, Margaret, Richard and Sara Koulen for generous support and encouragement.
References
- Avila J, Lucas JJ, Perez M, Hernandez F. Role of tau protein in both physiological and pathological conditions. Physiol Rev. 2004;84(2):361–384. doi: 10.1152/physrev.00024.2003. [DOI] [PubMed] [Google Scholar]
- Basurto-Islas G, Luna-Munoz J, Guillozet-Bongaarts AL, Binder LI, Mena R, Garcia-Sierra F. Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol. 2008;67(5):470–483. doi: 10.1097/NEN.0b013e31817275c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussière T, Bard F, Barbour R, Grajeda H, Guido T, Khan K, Schenk D, Games D, Seubert P, Buttini M. Morphological Characterization of Thioflavin-S-Positive Amyloid Plaques in Transgenic Alzheimer Mice and Effect of Passive Aβ Immunotherapy on Their Clearance. Am J Pathol. 2004;165(3):987–995. doi: 10.1016/s0002-9440(10)63360-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calissano P, Matrone C, Amadoro G. Apoptosis and in vitro Alzheimer disease neuronal models. Commun Integr Biol. 2009;2(2):163–169. doi: 10.4161/cib.7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CW, Song YH, Kim IK, Yoon WJ, Ryu BR, Jo DG, Woo HN, Kwon YK, Kim HH, Gwang BJ, Mook-Jung IH, Jung YK. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis. 2001;8:162–172. doi: 10.1006/nbdi.2000.0335. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Poon WW, Rissman RA, Blurton-Jones M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol. 2005;64(2):104–12. doi: 10.1093/jnen/64.2.104. [DOI] [PubMed] [Google Scholar]
- De Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT. Caspase activation precedes and leads to tangles. Nature. 2010;464(7292):1201–1204. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Calignon A, Spires-Jones TL, Pitstick R, Carlson GA, Hyman BT. Tangle-bearing neurons survive despite disruption of membrane integrity in a mouse model of tauopathy. J Neuropathol Exp Neurol. 2009;68(7):757–761. doi: 10.1097/NEN.0b013e3181a9fc66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean E. Apoptosis in neurodegeneration: Programmed cell death and its role in alzheimer’s and huntington’s diseases. Eukaryon. 2008;4:42–47. [Google Scholar]
- Delobel P, Lavenir I, Fraser G, Ingram E, Holzer M, Ghetti B, Spillantini MG, Crowther RA, Goedert M. Analysis of tau phosphorylation and truncation in a mouse model of human tauopathy. American Journal of Pathology. 2008;172(1):123–131. doi: 10.2353/ajpath.2008.070627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar A, Kuo D, He RH, Zhou J, Wu JY. Tau alternative splicing and frontotemporal dementia. Alzheimer Dis Assoc Disord. 2005;19:S29–S36. doi: 10.1097/01.wad.0000183082.76820.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Matthews TA, Johnson GVW. Site-specific phosphorylation and caspase cleavage differentially impact tau-microtubule interactions and tau aggregation. J of Biol Chem. 2006;281(28):19107–19114. doi: 10.1074/jbc.M511697200. [DOI] [PubMed] [Google Scholar]
- Furukawa K, D’Souza I, Crudder CH, Onodera H, Itoyama Y, Poorkaj P, Bird TD, Schellenberg GD. Pro-apoptotic effects of tau mutations in chromosome 17 frontotemporal dementia and parkinsonism. Ageing. 2000;11(1):57–59. doi: 10.1097/00001756-200001170-00011. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, Miller R, Berry RW, Binder LI, Cryns VL. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. PNAS. 2003;100(17):10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of alzheimer paired helical filaments. J of Biol Chem. 1986a;261:6084–6089. [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986b;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillozet-Bongaarts AL, Garcia-Sierra F, Reynolds MR, Horowitz PM, Fu Y, Wang T, Cahill ME, Bigio EH, Berry RW, Binder LI. Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol Aging. 2005;26(7):1015–1022. doi: 10.1016/j.neurobiolaging.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Wray S. Tau cleavage and tau aggregation in neurodegenerative disease. Biochem Soc Trans. 2010;38(4):1016–1020. doi: 10.1042/BST0381016. [DOI] [PubMed] [Google Scholar]
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148(6):1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong CX, del Alonso AC, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009;118(1):53–69. doi: 10.1007/s00401-009-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarero-Basulto JJ, Luna-Munoz J, Mena R, Kristofikova Z, Ripova D, Perry G, Binder LI, Garcia-Sierra F. Proteolytic cleavage of polymeric tau protein by caspase-3: implications for Alzheimer’s disease. J Neuropathol Exp Neurol. 2013;72(12):1145–1161. doi: 10.1097/NEN.0000000000000013. [DOI] [PubMed] [Google Scholar]
- Kaja S, Sumien N, Borden PK, Khullar N, Iqbal M, Collins JL, Forster MJ, Koulen P. Homer-1a immediate early gene expression correlates with better cognitive performance in aging. Age (Dordr) 2013;35(5):1799–1808. doi: 10.1007/s11357-012-9479-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaja S, Sumien N, Shah VV, Puthawala I, Maynard AN, Khullar N, Payne AJ, Forster MJ, Koulen P. Loss of Spatial Memory, Learning, and Motor Function During Normal Aging Is Accompanied by Changes in Brain Presenilin 1 and 2 Expression Levels. Mol Neurobiol. 2015;52(1):545–554. doi: 10.1007/s12035-014-8877-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci USA. 1986;83:4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuranaga E, Miura M. Nonapoptotic functions of caspases: caspases as regulatory molecules for immunity and cell-fate determination. Trends in Cell Biol. 2007;17:135–144. doi: 10.1016/j.tcb.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-Synaptic Spread of Tau Pathology In Vivo. PLoS ONE. 2012;7(2):e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly PT, Cai F, Weihong &, Song W. Detection of Neuritic Plaques in Alzheimer’s Disease Mouse Model. J Vis Exp. 2011;53:2831. doi: 10.3791/2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews-Roberson TA, Quintanilla RA, Ding H, Johnson GV. Immortalized cortical neurons expressing caspase-cleaved tau are sensitized to endoplasmic reticulum stress induced cell death. Brain Res. 2008;1234:206–212. doi: 10.1016/j.brainres.2008.07.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain DR, Berger T, Mak TW. Caspase Functions in Cell Death and Disease. Cold Spring Harb Perspect Biol. 2013;5:a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in preconditioning. PNAS. 2003;100(2):715–720. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means JC, Venkatesan A, Gerdes B, Fan JY, Bjes ES, Price JL. Drosophila spaghetti and doubletime link the circadian clock and light to caspases, apoptosis and tauopathy. PLoS Genet. 2015;7(11):e1005171. doi: 10.1371/journal.pgen.1005171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J of Neuropathol Exp Neurol. 1999;58(2):188–197. doi: 10.1097/00005072-199902000-00008. [DOI] [PubMed] [Google Scholar]
- Novak M, Kabat J, Wischik CM. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J. 1993;12(1):365–370. doi: 10.1002/j.1460-2075.1993.tb05665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Dolan PJ, Jin YN, Johnson GV. Truncated tau and Aβ cooperatively impair mitochondria in primary neurons. Neurobiol Aging. 2012;33:619e25–619e35. doi: 10.1016/j.neurobiolaging.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GVW. Caspase-cleaved tau expression induces mitochondrial dysfunction in cortical neurons. Implications for the pathogenesis of Alzheimer’s disease. J Biol Chem. 2009;284:18754–18766. doi: 10.1074/jbc.M808908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevalova M, Filipcik P, Novak M, Avila J, Iqbal K. Post-translational modifications of tau protein. Bratislavské Lekárske Listy. 2006;107(9–10):346–353. [PubMed] [Google Scholar]
- Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT, Cotman CW. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J of Clinical Invest. 2004;114(1):121–130. doi: 10.1172/JCI20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT, Rissman RA, Head E, Cotman CW. Caspase activation in the alzheimer’s disease brain: tortuous and torturous. Drug News Perspect. 2002;15(9):549–557. doi: 10.1358/dnp.2002.15.9.740233. [DOI] [PubMed] [Google Scholar]
- Santa-Maria I, Hernández F, Del Rio D, Moreno FJ, Avila J. Tramiprosate, a Drug of Potential Interest for the Treatment of Alzheimer’s Disease, Promotes an Abnormal Aggregation of Tau. Mol Neurodegener. 2007;2(1):17. doi: 10.1186/1750-1326-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraen-Maschke S, Sergeant N, Dhaenens C-M, Bombois S, Deramecourt V, Caillet-Boudin M-L, Pasquier F, Maurage C-A, Sablonniere B, Vanmechelen E, Buee L. Tau as a biomarker of neurodegenerative diseases. Biomark Med. 2008;2(4):363–384. doi: 10.2217/17520363.2.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Hyman BT. Trangenic models of alzheimer’s disease: learning from animals. Neuro Rx. 2005;2(3):423–437. doi: 10.1602/neurorx.2.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, de Calignon A, Matsui T, Zehr C, Pitstick R, Wu HY, Osetek JD, Jones PB, Bacskai BJ, Feany MB, Carlson GA, Ashe KH, Lewis J, Hyman BT. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J Neurosci. 2008;28:862–867. doi: 10.1523/JNEUROSCI.3072-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumien N, Sims MN, Taylor HJ, Forster MJ. Profiling psychomotor and cognitive aging in four-way cross mice. Age. 2006;28:265–282. doi: 10.1007/s11357-006-9015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun A, Nguyen XV, Bing G. Comparative Analysis of an Improved Thioflavin-S Stain, Gallyas Silver Stain, and Immunohistochemistry for Neurofibrillary Tangle Demonstration on the Same Sections. J Histochem Cytochem. 2002;50(4):463–472. doi: 10.1177/002215540205000403. [DOI] [PubMed] [Google Scholar]
- Turk B, Stoka V. Protease signaling in cell death: caspases versus cysteine cathepsins. FEBS Letters. 2007;581:2761–2767. doi: 10.1016/j.febslet.2007.05.038. [DOI] [PubMed] [Google Scholar]
- Urbanc B, Cruz L, Le R, Sanders J, Ashe KH, Duff K, Stanley HE, Irizarry MC, Hyman BT. Neurotoxic Effects of Thioflavin S-positive Amyloid Deposits in Transgenic Mice and Alzheimer’s Disease. Proc Natl Acad Sci USA. 2002;99(22):13990–13995. doi: 10.1073/pnas.222433299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray S, Lewis PA. A Tangled Web – Tau and Sporadic Parkinson’s Disease. Front Psychiatry. 2010;1:150. doi: 10.3389/fpsyt.2010.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Gao J, Xu L, Meng D. Shared Mechanisms of Neurodegeneration in Alzheimer’s Disease and Parkinson’s Disease. Bio Med Research International. 2014;2014:8. doi: 10.1155/2014/648740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhang X, Chen J, Miao Y, Sun A. Role of caspase-3 in tau truncation at D421 is restricted in transgenic mouse models for tauopathies. Journal of Neurochemistry. 2009;109(2):476–484. doi: 10.1111/j.1471-4159.2009.05959.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.