

Introduction
Pseudohypoaldosteronism type 1 (PHA1) is a rare disease characterized by congenital resistance to the action of aldosterone on epithelial tissue; PHA1 results in excessive salt wasting despite very high plasma aldosterone and renin levels (1,2,3). There are 3 types of PHA1. The systemic form of PHA1 is inherited in an autosomal recessive manner and manifests as severe life-long salt wasting caused by mineralocorticoid resistance in multiple target tissues (e.g., sweat glands, salivary glands, colonic epithelium, and lung). The renal form of PHA1 (adPHA1) is inherited in an autosomal dominant manner. In this form, mineralocorticoid resistance exists only in the kidney; moreover, salt wasting and other symptoms improve around 1–3 yr of age (1,2,3). The third type of PHA1 is the transient form, which is commonly seen in patients with urinary tract infection or obstructive uropathy. In the transient form, clinical symptoms disappear after treatment.
adPHA1 is caused by a heterozygous mutation in NR3C2, which encodes the mineralocorticoid receptor (MR). Herein, we report the case of a young girl with adPHA1, a novel mutation in NR3C2, hyponatremia, and failure to thrive associated with urinary tract infection. We also describe a genetic analysis of her family.
Patient Report
A 4-mo-old girl was referred to our outpatient clinic because of failure to thrive, dehydration, and vomiting. She was the third child of nonconsanguineous healthy parents; she was born at full term with a body weight of 2820 g (−0.5 SD) after an uneventful pregnancy. There was no significant family history. She had poor weight gain at 1 mo, but subsequent, rapid weight gain was seen without treatment. Therefore, no further evaluation was performed at that time. At the age of 3 mo, she presented with failure to thrive and frequent vomiting once again, and she was admitted to our hospital at the age of 4 mo.
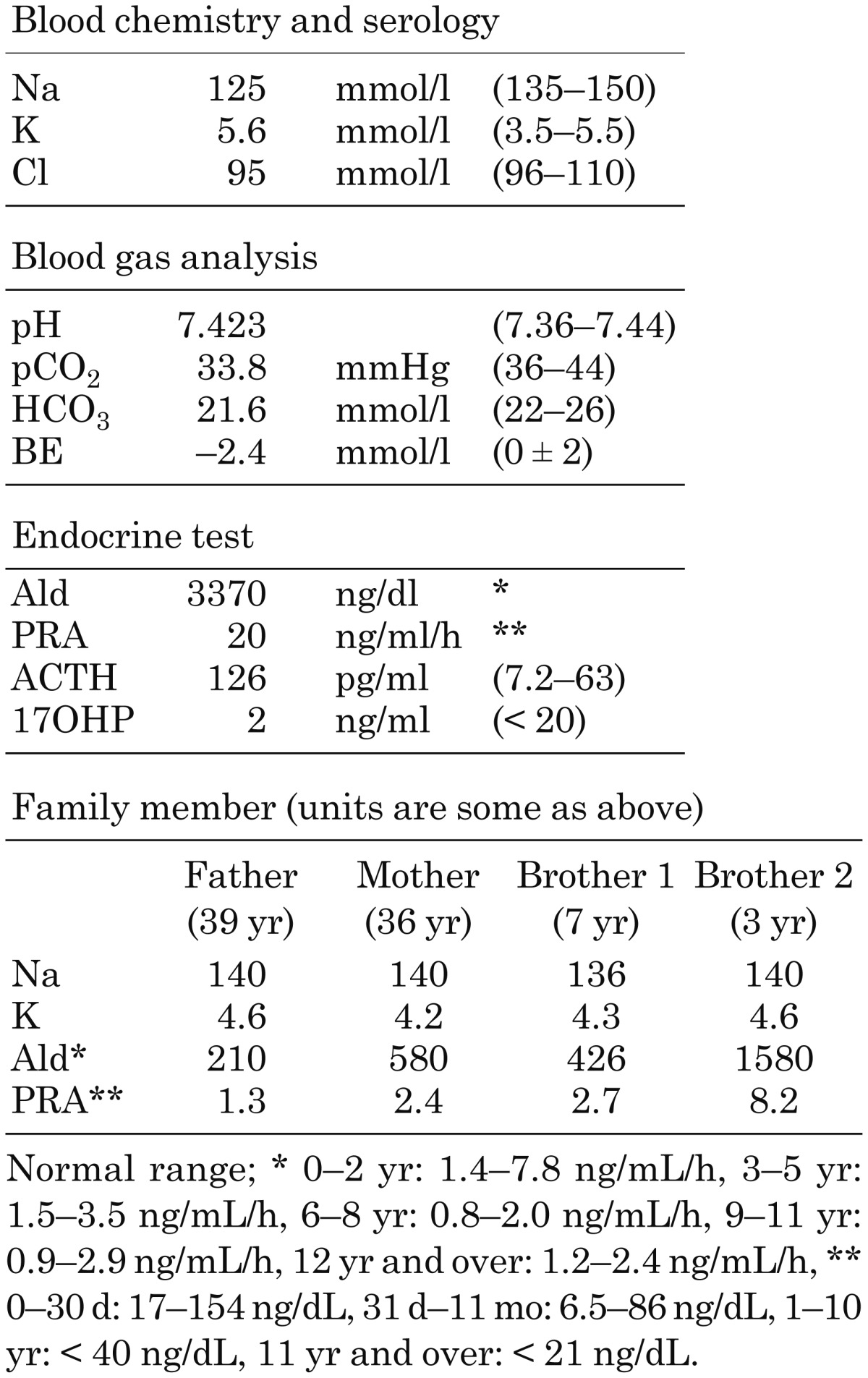
On admission, her body length was 58.5 cm (−1.9 SD), and her body weight was 4534 g (−2.6 SD). Her skin exhibited decreased turgor and her anterior fontanelle was retracted; both signs suggested severe dehydration. As shown in Table 1, laboratory findings showed hyponatremia (125 mmol/l) and hyperkalemia (5.6 mmol/l). Urinary Na and K were within the normal ranges. Endocrinological study revealed elevated plasma aldosterone (3370 ng/dL) and renin (20 ng/mL/h) levels and normal 17-hydroxypregn-4-ene-3,20-dione (2 ng/mL) levels. Furthermore, her urinary white blood cell count was elevated, and Escherichia coli was detected in the urinary culture. Abdominal ultrasonography demonstrated morphologically normal kidneys, and a dimercaptosuccinic acid radionuclide scan was negative. Based on these findings, the transient form of PHA1, associated with urinary tract infection, was suspected. Intravenous administration of NaCl supplementation (2.6 mEq/kg/d) and antibiotics was initiated.
Table 1. Laboratory data on admission/family members.
With the antibiotic treatment, the urinary tract infection improved rapidly, but her serum levels of electrolytes, plasma renin activity and aldosterone concentrations, and body weight gain had not normalized. These findings suggested a diagnosis of adPHA1; therefore, oral NaCl supplementation (18 mEq/kg/d) was started. Thereafter, her serum levels of electrolytes normalized, and her body weight gain improved (Fig. 1).
Fig. 1.

Growth chart (for the case of a Japanese girl aged 0–24 mo). At admission, her height and weight were both under −2.0 SD. After NaCl supplementation, her growth curve improved gradually and returned to the normal range.
Mutational Analysis
After obtaining informed consent, and with the approval of the Ethics Committee of The Jikei University School of Medicine, genomic DNA was extracted from the patient and her family members including her 2 elder brothers. All 10 coding exons and flanking introns of NR3C2 were amplified by PCR using specific primers. PCR products were purified and sequenced directly with an Applied Biosystems 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA). Sequence analysis demonstrated a novel heterozygous single base insertion (c.2724insT) predicted to cause a premature stop codon (p.Lys909fsX1) in exon 8 for our patient (Fig. 2). This mutation was also identified in her mother and 2 elder brothers (Fig. 3). Interestingly, plasma aldosterone and renin levels of her family members carrying the same mutation were all elevated, but they each lacked any history of PHA1 symptoms.
Fig. 2.

Mutation of NR3C2. The patient, the mother, and two elder brothers each had a heterozygous single base insertion (c.2724insT). This insertion created a premature stop codon (p.Lys909fsX1) in exon 8. In each sequence panel, double bands are present after the insertion site, which is always indicated with an arrow.
Fig. 3.

Pedigree diagram. Her 2 elder brothers and mother had the same mutation but asymptomatic.
Discussion
Human NR3C2 mutation was first identified by Geller et al. in 1998 (1), and since then, more than 50 different adPHA1-causing mutations have been identified in this gene (1,2,3, 5). These mutations are distributed throughout the gene, but pathogenesis of adPHA1 still remains to be elucidated. NR3C2 is located on chromosome 4q31.1, comprises 10 exons, and encodes a protein of 984 amino acids. The mature MR protein can be functionally subdivided into 3 domains: the N-terminal activation domain, the centrally located DNA-binding domain, and the C-terminal ligand-binding domain (LBD).
In the present study, we identified a novel mutation (c.2724insT) of NR3C2 in a female patient and 3 of her family members. This mutation in exon 8 creating premature stop codon is localized in the C-terminal LBD of MR. Several different mutations in the LBD of MR have been reported in patients with adPHA1 (2, 3). Couette et al. (4) demonstrated in vitro with artificial MR that deletion of the last 4 amino acids of MR completely abolished its affinity to aldosterone. Therefore, it is plausible that the p.Lys909fsX1 mutation may affect the ligand-binding capacity because the MR encoded by the gene with this mutation lacks 76 amino acids in the C-terminal region. Alternatively, this mutation may induce nonsense-mediated mRNA decay, resulting in haploinsufficiency. In addition, it is of interest that her mother and 2 elder brothers, who each carried the same c.2724insT mutation, had no electrolyte disturbances despite elevated plasma aldosterone and renin levels (Table 1). These findings indicated that this familial NR3C2 mutation could cause very heterogeneous phenotypic expression within a single family (5).
Finally, we studied an adPHA1 patient and her family members who shared an identical mutation, and confirmed the importance of a genetic diagnosis. Each individual with a confirmed NR3C2 mutation should be regularly monitored to assess sodium homeostasis.
References
- 1.Geller DS, Zhang J, Zennaro MC, Vallo-Boado A, Rodriguez-Soriano J, Furu L, et al. Autosomal dominant pseudohypoaldosteronism type 1: mechanisms, evidence for neonatal lethality, and phenotypic expression in adults. J Am Soc Nephrol 2006;17: 1429–36. doi: 10.1681/ASN.2005111188 [DOI] [PubMed] [Google Scholar]
- 2.Viengchareun S, Le Menuet D, Martinerie L, Munier M, Pascual-Le Tallec L, Lombès M. The mineralocorticoid receptor: insights into its molecular and (patho)physiological biology. Nucl Recept Signal 2007;5: e012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geller DS. Mineralocorticoid resistance. Clin Endocrinol (Oxf) 2005;62: 513–20. doi: 10.1111/j.1365-2265.2005.02229.x [DOI] [PubMed] [Google Scholar]
- 4.Couette B, Jalaguier S, Hellal-Levy C, Lupo B, Fagart J, Auzou G, et al. Folding requirements of the ligand-binding domain of the human mineralocorticoid receptor. Mol Endocrinol 1998;12: 855–63. doi: 10.1210/mend.12.6.0119 [DOI] [PubMed] [Google Scholar]
- 5.Riepe FG. Clinical and molecular features of type 1 pseudohypoaldosteronism. Horm Res 2009;72: 1–9. doi: 10.1159/000224334 [DOI] [PubMed] [Google Scholar]