

Abstract
Homozygous mutations in SLC30A10 lead to the development of familial manganese-induced parkinsonism. We previously demonstrated that SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its trafficking and efflux activity. Interestingly, other transporters in the SLC30 family mediate zinc efflux. Determining the mechanisms that allow SLC30A10 to transport manganese, which are unclear, is essential to understand its role in parkinsonism. Here, we generated a predicted structure of SLC30A10, based on the structure of the bacterial zinc transporter YiiP, and performed functional studies. In YiiP, side chains of residues Asp-45 and Asp-49 in the second and His-153 and Asp-157 in the fifth transmembrane segments coordinate zinc and are required for transport. In SLC30A10, the corresponding residues are Asn-43 and Asp-47 in the second and His-244 and Asp-248 in the fifth transmembrane segments. Surprisingly, although alanine substitution of Asp-248 abolished manganese efflux, that of Asn-43 and Asp-47 did not. Instead, side chains of charged or polar residues adjacent to Asp-248 in the first (Glu-25) or fourth (Asn-127) transmembrane segments were required. Further analyses revealed that residues His-333 and His-350 in the cytoplasmic C-terminal domain were required for full activity. However, the C-terminal domain failed to transfer manganese transport capability to a related zinc transporter. Overall, our results indicate that residues in the transmembrane and C-terminal domains together confer optimal manganese transport capability to SLC30A10 and suggest that the mechanism of ion coordination in the transmembrane domain of SLC30A10 may be substantially different from that in YiiP/other SLC30 proteins.
Keywords: intracellular trafficking, manganese, metal homeostasis, neurodegenerative disease, structure-function, transporter, zinc
Introduction
The essential metal manganese is required for the activity of numerous cellular enzymes (1). However, elevated levels of manganese are cytotoxic and induce oxidative stress, mitochondrial dysfunction, and apoptosis (2, 3). In humans, when systemic levels of manganese increase, the metal accumulates in the basal ganglia in the brain and leads to the development of an incurable parkinsonian syndrome (1, 4, 5). Manganese toxicity is commonly observed under certain occupational settings where workers are chronically exposed to high levels of manganese (1). Recent evidence suggests that individuals exposed to elevated manganese from environmental sources may also develop aspects of the disease (6). Furthermore, as the primary mode of manganese excretion is by secretion into bile, individuals who fail to excrete manganese due to hepatic dysfunction, secondary to diseases such as cirrhosis, may develop manganese-induced parkinsonism in the absence of exposure to elevated manganese (7). Finally, although manganese-induced parkinsonism is pathologically and clinically distinct from Parkinson disease, prolonged exposure to elevated manganese (>20 years) was reported to increase the risk of developing Parkinson disease (8), suggesting that manganese may be an environmental risk factor for Parkinson disease.
A set of studies over the last few years led to the discovery of the first inherited or familial form of manganese-induced parkinsonism (9–12). Genetic analyses revealed that affected patients had homozygous mutations in SLC30A10 (10, 11). Patients developed parkinsonian symptoms, had large increases in blood manganese levels, and exhibited evidence of manganese deposition in the basal ganglia (10, 11). Importantly, these individuals had no history of exposure to elevated manganese from occupational or environmental sources, suggesting that the disease probably occurred due to a defect in manganese metabolism (10, 11). Our recent work provided insights into the underlying molecular mechanisms. Using assays in HeLa and AF5 cells and primary neuronal cultures as well as in Caenorhabditis elegans, we reported that the wild type (WT) SLC30A10 protein functioned as a cell surface-localized manganese efflux transporter that transported manganese from the cytosol to the cell exterior and protected cells and neurons against manganese toxicity (13). Parkinsonism-causing mutations trapped the transporter in the endoplasmic reticulum and blocked its manganese efflux activity; consequently, cells and neurons expressing these mutants exhibited enhanced sensitivity to manganese toxicity (13).
Although our previous work provided a functional difference between SLC30A10WT and disease-causing mutants, the fundamental question of how SLC30A10 transports manganese is not clear. The SLC30 family has 10 transporters, SLC30A1–A10 (14–16). These transporters are vertebrate members of the cation diffusion facilitator superfamily of metal transporters (14–16). Members of this superfamily are present in all kingdoms of life, and the bacterial transporter YiiP, which mediates zinc efflux, is the archetype (14–16). Similar to YiiP, SLC30A1–A8 as well as ZRC1, the yeast homolog of SLC30A10, are zinc efflux transporters (classification of SLC30A9 as a transporter is likely incorrect; it acts as a nuclear receptor coactivator and has been re-named GAC63) (14–16). In contrast, multiple lines of evidence suggest that the primary cellular function of SLC30A10 is the efflux of manganese, instead of zinc. First, although all patients with SLC30A10 mutations have increased blood manganese levels, increases in plasma or brain zinc levels have not been reported (9–12). Second, in cell culture, expression of SLC30A10WT reduces intracellular manganese levels, increases transport of manganese from the cytosol to the cell exterior, and protects against manganese-induced death, but it does not affect intracellular zinc levels or protect against zinc toxicity (13). Finally, in C. elegans, SLC30A10WT expression protects against manganese but not zinc toxicity (13, 17). In the absence of challenging in vitro transport assays, it is not possible to say whether SLC30A10 completely lacks zinc transport activity or whether the presence of more efficient zinc transporters in the above cell- and organism-based experimental systems mask SLC30A10-mediated zinc transport. However, the ability of SLC30A10 to mediate manganese efflux provides a clear difference between it on the one hand and other SLC30 family proteins on the other hand.
Determining the mechanisms that allow SLC30A10 to transport manganese is important for several reasons. From a translational perspective, prior studies suggest that enhancing efflux may be a useful strategy to protect against the onset or control the progression of familial and non-familial manganese-induced parkinsonism (13, 18). As SLC30A10 is a major manganese efflux transporter, it is a promising candidate for therapeutic intervention. However, drugs that enhance the manganese efflux activity of SLC30A10 cannot be developed without knowledge of the mechanisms by which SLC30A10 mediates manganese transport. Moreover, single nucleotide polymorphisms in SLC30A10 that are associated with altered blood manganese levels were recently identified (19), raising the possibility that polymorphisms in SLC30A10 may alter the risk for developing manganese neurotoxicity in the general population. Information about amino acids in SLC30A10 that are required for manganese transport is essential to provide a mechanistic context to such epidemiological studies and aid in the identification of disease-relevant SLC30A10 polymorphisms. Finally, from a basic science perspective, the means by which ion transporters gain substrate specificity has wide significance. In the cation diffusion facilitator superfamily, compared with zinc, manganese transporters have received less attention, and thus far, in mammals, SLC30A10 is the only known manganese-transporting member. As molecular details of the process by which YiiP and some SLC30 proteins transport zinc are now available (14–16, 20–32), elucidating the mechanisms of manganese transport via SLC30A10 will likely help answer the important question of how cation diffusion facilitators distinguish between metals and thereby improve our understanding of the general principles that guide substrate specificity in ion transporters.
In this study, we used a mutational approach to analyze the manganese efflux function of SLC30A10. Our studies were guided by a structural model of SLC30A10. We assayed the manganese transport activity of SLC30A10 mutants using three different assays, including direct measurements of intracellular manganese, in HeLa cells. Our results show that sequence elements in the transmembrane and C-terminal domains together confer complete manganese transport capability to SLC30A10. Our findings provide novel insights into the biology of SLC30A10 and suggest that, in comparison with YiiP and other SLC30 family proteins, the molecular mechanisms of ion coordination by, and transport through, SLC30A10 are considerably different.
Results
Alanine Substitution of Asp-248, but Not of Asn-43, Asp-47, or His-244, Abolishes the Manganese Efflux Activity of SLC30A10
YiiP is the only cation diffusion facilitator that has a solved crystal structure (23, 24). The structure shows that YiiP has a transmembrane domain with six membrane-spanning helices/segments and a C-terminal domain (Fig. 1A) (23, 24). Side chain oxygen or nitrogen atoms of residues Asp-45 and Asp-49 in the second and His-153 and Asp-157 in the fifth transmembrane segments directly coordinate the zinc ion that is transported (Fig. 1A) (23, 24). This ion coordination site is referred to as site A (23). Phylogenetic and experimental studies suggest that, in other cation diffusion facilitators, residues that correspond to site A of YiiP are crucial for metal coordination and transport and that the identity of these amino acids may play an important role in determining metal specificity (15, 22, 33, 34). Indeed, in most zinc transporters, on the second transmembrane segment, a histidine residue followed by an aspartate residue (the aspartate resides four residues downstream of the histidine) contribute to site A (HXXXD motif) (33). This HXXXD motif is present in zinc-transporting SLC30 proteins and in the yeast zinc transporter ZRC1 (Fig. 1B). However, in most manganese transporters, such as Arabidopsis MTP11 and Stylosanthes hamata MTP8, the corresponding motif is DXXXD (33, 35). Interestingly, although SLC30A10 transports manganese, the motif in its second transmembrane segment differs from zinc or manganese transporters and is NXXXD (Fig. 1B). The presence of the asparagine residue in the putative site A of SLC30A10 (the asparagine residue is at position 43) was intriguing because, although asparagine does not often coordinate with metals, it has a higher propensity to coordinate with manganese than with zinc (36). Furthermore, a recent study on the bacterial manganese transporter MntE revealed that an asparagine residue is present in its putative site A and is required for activity (34). Interestingly, this asparagine residue of MntE corresponds to Asn-43 of SLC30A10, and its requirement provides the precedence for the involvement of an asparagine residue in the putative site A of a cation diffusion facilitator in mediating manganese transport. These observations raised the hypothesis that a single amino acid change in the second transmembrane segment, from histidine in zinc-transporting SLC30 proteins to asparagine in SLC30A10, conferred manganese transport capability to SLC30A10.
FIGURE 1.

Comparison of the solved crystal structure and amino acid sequence of YiiP with the predicted structure and amino acid sequence of SLC30A10. A, crystal structure of YiiP (Protein Data Bank code 3H90) and the predicted structure of SLC30A10 are depicted from the membrane plane (side) and from the periplasmic or extracellular region looking into the proteins (top). Close-up views of site A are also depicted (site A). In this panel, both proteins are shown as monomers; site A residues are shown as red sticks with nitrogen atoms in blue; in YiiP, zinc ions are shown as yellow spheres. B, amino acid sequence alignment of SLC30A10 with YiiP, ZRC1, SLC30A1–A5, and SLC30A7–A8. The site A residues are highlighted in blue; TM stands for transmembrane. Accession numbers of depicted sequences are as follows: YiiP, EIQ66948.1; SLC30A10, NP_061183.2; ZRC1, NP_013970.1; SLC30A1, NP_067017.2; SLC30A2, NP_001004434.1; SLC30A3, NP_003450.2; SLC30A4, NP_037441.2; SLC30A5, NP_075053.2; SLC30A7, NP_001138356.1; SLC30A8, NP_776250.2.
To determine whether the above hypothesis was feasible from a structural perspective, we generated a predicted structure of SLC30A10 using the algorithms of the PHYRE 2.0 server (Fig. 1A) (37). The obtained prediction was based on the solved structure of YiiP and contained residues 3–134 and 227–392 of SLC30A10. Despite the missing parts, the majority of the transmembrane and cytoplasmic domains were present, and it served as a useful hypothesis-generating tool. Comparison of the transmembrane segments of the predicted structure with that of the solved structure of YiiP revealed important differences. In site A of YiiP, the side chains of residues Asp-45 and Asp-49 from the second transmembrane segment point toward the ion-binding pocket, which is formed in the cavity between the second and fifth transmembrane segments (Fig. 1A) (23, 24). In contrast, in SLC30A10, the side chains of the corresponding residues, Asn-43 and Asp-47, appear to point away from the space between the second and fifth transmembrane segments (Fig. 1A). Although our initial hypothesis was that Asn-43 may be critical for activity, review of the predicted structure raised doubts about the importance of the side chain of Asn-43, as well as of Asp-47, in mediating manganese transport.
To obtain experimental evidence, we individually substituted these residues with alanine and generated SLC30A10N43A and SLC30A10D47A. Both mutants were well expressed and, analogous to SLC30A10WT, overlapped with the plasma membrane marker, DPP4, but not with the endoplasmic reticulum marker, calnexin (Fig. 2). For quantitative assessment, we performed colocalization analyses using Pearson's colocalization coefficient, as described by us previously (13). Note that values for Pearson's coefficient for two fluorophores imaged in two channels range from −1 to + 1; values close to +1 indicate overlap or colocalization, and those close to 0 and negative numbers indicate lack of overlap (38). We observed that the Pearson's coefficient for colocalization between SLC30A10WT, SLC30A10N43A, or SLC30A10D47A and DPP4 was >0.8, but between each of the SLC30A10 constructs and calnexin it was <0.1 (Fig. 2), indicating that, similar to the WT protein, both the mutants trafficked to the cell surface. After this, we tested the manganese efflux activity of the mutants. First, we used the GPP130 degradation assay, which we have extensively described previously (18, 39, 40). GPP130 is a predominantly cis-Golgi-localized protein that gets degraded in lysosomes when cells are treated with manganese (18, 39, 40). The manganese-induced degradation of GPP130 occurs in response to a specific increase in intra-Golgi and not just cytosolic manganese (18). Our prior work showed that overexpression of SLC30A10WT protected against the manganese-induced loss of GPP130 by increasing manganese efflux and reducing intracellular manganese levels (13). The protective effect was not seen when transport-inactive mutants of SLC30A10 were expressed (13). For this assay, as positive control, we verified that expression of SLC30A10WT, but not a control construct, robustly protected against the loss of GPP130 after manganese treatment (Fig. 3A). We then observed that in cells transfected with SLC30A10WT, SLC30A10N43A, or SLC30A10D47A, treatment with manganese failed to induce GPP130 degradation (Fig. 3, B and D), suggesting that the mutant proteins acted like SLC30A10WT and retained the ability to mediate manganese efflux. Note that for these and all other mutants analyzed later, results of the GPP130 degradation assay were independently verified by direct measurements of intracellular manganese using inductively coupled plasma mass spectrometry (Fig. 5).
FIGURE 2.

Cell surface localization of SLC30A10WT and transmembrane domain mutants. A, HeLa cells were transfected with various FLAG-tagged SLC30A10 constructs. Two days after transfection, cultures were processed for immunofluorescence. SLC30A10 was detected using a monoclonal antibody against the FLAG epitope, and a polyclonal antibody against calnexin was used to demarcate the endoplasmic reticulum. P represents the Pearson's coefficient for colocalization between SLC30A10 and calnexin (mean ± S.E.; n = 10–15 cells per SLC30A10 construct). Scale bar, 10 μm. B, HeLa cells were cotransfected with FLAG-tagged SLC30A10WT or mutants and HA-tagged DPP4. Two days after transfection, SLC30A10 was detected using a polyclonal antibody against FLAG and DPP4 with a monoclonal antibody against HA. P denotes the Pearson's coefficient for colocalization between SLC30A10 and DPP4 (mean ± S.E.; n = 14–15 cells per SLC30A10 construct). Scale bar, 10 μm.
FIGURE 3.

GPP130 degradation assay in cells expressing SLC30A10WT or transmembrane domain mutants. A, HeLa cells were transfected with plasmids coding for GFP-tagged Rab5 (transfection control) or FLAG-tagged SLC30A10WT. Two days after transfection, cultures were treated with or without 500 μm manganese for 4 h and imaged to detect Rab5 (using the GFP signal) or SLC30A10 (using a polyclonal anti-FLAG antibody) and GPP130 (using a monoclonal antibody). For consistency with other panels, the Rab5 signal is depicted in the red channel. Samples that did not receive manganese treatment are denoted as Control. Scale bar, 10 μm. B and C, HeLa cells were transfected with various FLAG-tagged SLC30A10 constructs. Two days after transfection, cells were treated with manganese as described in A and imaged to detect SLC30A10, using a polyclonal antibody against the FLAG epitope, and GPP130, using a monoclonal antibody. Cultures that did not receive manganese treatment are denoted as Control. Scale bars, 10 μm. D, quantification of the amount of GPP130 remaining in cells after manganese treatment from B and C. Untransfected cells did not express any SLC30A10 construct. For each transfection condition, mean GPP130 fluorescence per cell was calculated, with and without manganese treatment, as described under “Experimental Procedures,” and these numbers were used to compute percent GPP130 remaining after manganese (mean ± S.E.; n = 15 cells per manganese treatment and transfection condition; *, p < 0.05 for the difference between WT and other transfection conditions by one-way ANOVA and Dunnett's post hoc test). One-way ANOVA and Dunnett's post hoc test also revealed that there was no difference between untransfected and D248A, E25A, N127A,H244A or D40A groups (p > 0.05); however, there was a significant difference between untransfected and WT, N43A, D47A, H244A, or N127A groups (p < 0.05).
FIGURE 5.
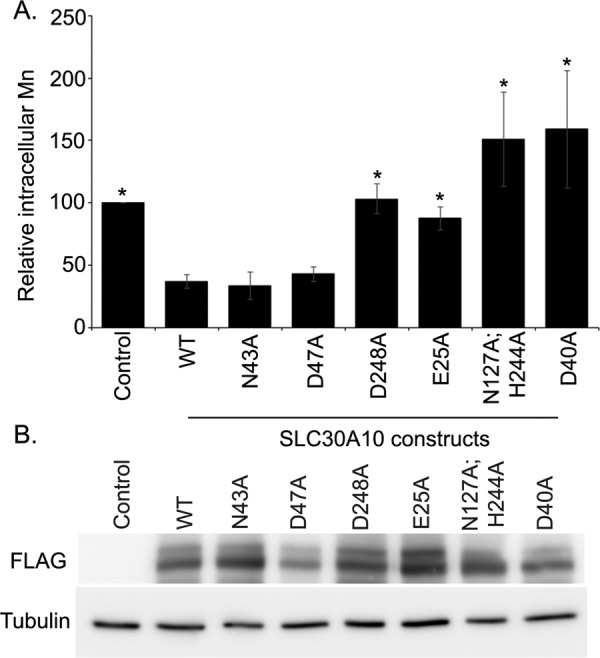
Intracellular manganese measurements in cells expressing SLC30A10WT or transmembrane domain mutants. A, identical numbers of HeLa cells were plated in 35-mm dishes and transfected with a control construct (denoted as Control), SLC30A10WT, or various SLC30A10 mutants. Two days after transfection, cells were treated with 500 μm manganese for 16 h. After this, absolute levels of intracellular manganese were measured using inductively coupled plasma mass spectrometry as described under “Experimental Procedures.” Absolute intracellular manganese in control-transfected cells was normalized to 100 (mean ± S.E.; n ≥ 3 for each construct; *, p < 0.05 for the difference between cells expressing SLC30A10WT and all other transfection conditions by one-way ANOVA and Dunnett's post hoc test). Statistical analyses also revealed that there was no difference between control and D248A, E25A, or N127A,H244A groups (p > 0.05 by one-way ANOVA and Dunnett's post hoc test); however, there was a significant difference between control and WT, N43A, D47A, or D40A groups (p < 0.05 by one-way ANOVA and Dunnett's post hoc test). B, HeLa cells were transfected and treated with 500 μm manganese for 16 h, exactly as described in A. Immunoblot analyses were then performed to detect FLAG or tubulin.
We then tested whether the side chains of residues His-244 and Asp-248 in the fifth transmembrane segment of SLC30A10, which correspond to His-153 and Asp-157 of site A of YiiP (Fig. 1A), were required. For this, we generated SLC30A10H244A and SLC30A10D248A. Colocalization analyses revealed that both these mutants trafficked to the cell surface (Fig. 2). Importantly, in cells transfected with the D248A mutant, GPP130 was robustly degraded after manganese treatment (Fig. 3, B and D), suggesting that it did not have manganese efflux activity. Although the amount of GPP130 remaining in manganese-treated cells expressing SLC30A10H244A was ∼20% less than that of cells expressing SLC30A10WT, there was no statistically significant difference between these two groups (Fig. 3, B and D), suggesting that the H244A mutant retained manganese efflux activity. The above results suggest that the side chains of Asn-43, Asp-47, and His-244 are not required for the manganese efflux activity of SLC30A10, although the side chain of Asp-248 is required. Thus, there is an important difference between SLC30A10 and YiiP/other SLC30 proteins in that, in SLC30A10, of the four residues that correspond to site A of YiiP, the side chain of only one residue, Asp-248, is required for activity.
Side Chains of Residues Glu-25 and Asn-127 Are Required for Manganese Efflux
As the first set of mutations led to the identification of only one residue, Asp-248, whose side chain was required for manganese efflux, we expanded our analyses with the goal of identifying additional transmembrane residues that may be required. We examined the predicted structure of SLC30A10 and attempted to identify charged or polar residues that were located in close proximity to the Asp-248 residue and that had side chains pointing toward the fifth transmembrane segment (Fig. 4). We identified Glu-25 in the first and Asn-127 in the fourth transmembrane segment as potential candidates (Fig. 4), individually substituted these residues with alanine to generate SLC30A10E25A and SLC30A10N127A, and used colocalization analyses to ensure that these new mutants also trafficked to the cell surface (Fig. 2). We then performed the GPP130 degradation assay. Importantly, GPP130 was degraded in manganese-treated cells expressing SLC30A10E25A or SLC30A10N127A (Fig. 3, C and D), suggesting that the side chains of Glu-25 and Asn-127 were required for manganese efflux. Interestingly, however, statistical analyses revealed that, unlike the D248A or E25A mutant, the amount of GPP130 remaining after manganese treatment in cells expressing SLC30A10N127A was greater than that in untransfected cells (Fig. 3D; p < 0.05 for the difference between untransfected cells and SLC30A10N127A-expressing cells by one-way ANOVA2 and Dunnett's post hoc test), raising the possibility that the side chain of the Asn-127 residue may be acting cooperatively with another residue. We had observed that there was a trend toward a decrease in GPP130 levels in manganese-treated cells expressing SLC30A10H244A, and the side chain of His-244 was in proximity to that of Asn-127 (Fig. 4). Therefore, we tested whether the side chain of His-244 cooperated with that of Asn-127. We generated SLC30A10N127A,H244A and validated that the double mutant trafficked to the cell surface using colocalization analyses (Fig. 2). In cells expressing SLC30A10N127A, H244A, after manganese treatment, the level of GPP130 remaining was comparable with untransfected cells (Fig. 3, C and D; p > 0.05 by one-way ANOVA and Dunnett's post hoc test). Thus, the side chain of the His-244 residue likely acts in concert with that of Asn-127 to provide optimal manganese transport capacity to SLC30A10. Finally, as part of these studies, we tested the role of the side chain of residue Asp-40, which is located at the junction of the second transmembrane segment and the adjacent extracellular loop (Fig. 4), and it is aptly positioned to influence ion transport. SLC30A10D40A trafficked to the cell surface (Fig. 2) and did not protect against the manganese-induced loss of GPP130 (Fig. 3, C and D), suggesting that this mutant did not have manganese efflux activity.
FIGURE 4.
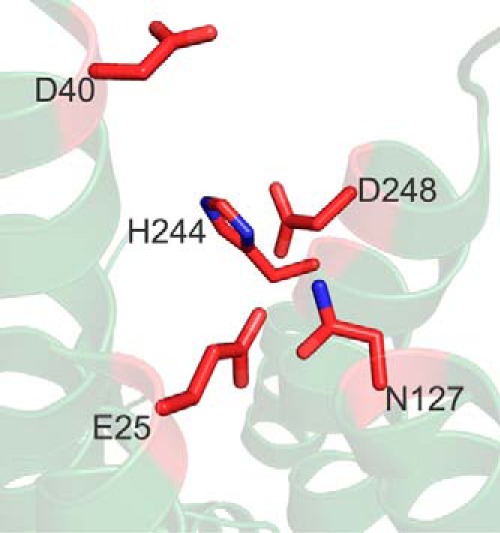
Predicted structure of SLC30A10 depicting residues that are required for its manganese efflux activity. Residues are shown as red sticks with nitrogen atoms in blue. Note that His-244 is not required by itself but acts cooperatively with Asn-127.
Overall, the data in Figs. 2–4 suggest that the side chains of residues Asp-248, Glu-25, Asn-127, and Asp-40 are required for the manganese efflux activity of SLC30A10; the side chains of Asn-43 and Asp-47 are not required; and the side chain of His-244 is not required by itself but plays a direct or indirect role in combination with that of Asn-127.
Validation of GPP130 Degradation Results Using Intracellular Manganese Measurements
The GPP130 degradation assay provides an indirect assessment of intracellular manganese, and it was important to validate the above findings by performing direct manganese measurement studies. We previously showed that intracellular manganese levels, measured using inductively coupled plasma mass spectrometry, were lower in cells expressing SLC30A10WT compared with transfection controls (13). We also demonstrated that the decrease in intracellular manganese was due to an increase in manganese efflux, and it was not seen in cells expressing a transport-inactive mutant of SLC30A10 (13). In this study, we transfected cells with a control construct, SLC30A10WT or various mutants described above and then measured intracellular manganese using inductively coupled plasma mass spectrometry, as described by us previously (13). As expected, intracellular manganese levels in cells expressing SLC30A10WT were less than that of transfection control (Fig. 5A). Importantly, intracellular manganese levels in cells expressing SLC30A10N43A or SLC30A10D47A were also significantly less than that of transfection control and comparable with cells expressing SLC30A10WT (Fig. 5A). In contrast, intracellular manganese levels in cells expressing the D248A, E25A, or N127A,H244A mutant were significantly greater than that of SLC30A10WT and comparable with transfection control (Fig. 5A).
We confirmed that under these culture, transfection, and manganese treatment conditions, SLC30A10 constructs used in the manganese measurement studies were expressed at comparable levels (Fig. 5B). These results implied that the manganese efflux activities of the N43A and D47A mutants were similar to SLC30A10WT, although the D248A, E25A, and N127A,H244A mutants did not have detectable manganese efflux activity. We also performed manganese measurement assays in cells expressing SLC30A10H244A and observed that although there was a trend toward an increase in levels of intracellular manganese in SLC30A10H244A-expressing cells compared with those expressing SLC30A10WT, consistent with the GPP130 degradation results, the difference between the H244A and WT groups was not statistically significant (p > 0.05; data not shown). Interestingly, we observed that, in cells expressing SLC30A10D40A, levels of intracellular manganese were greater than not only SLC30A10WT-expressing cells but also transfection control (Fig. 5). Although the underlying mechanism for an increase in manganese beyond transfection control in cells expressing the D40A mutant is unclear, for the purpose of this study, the above result meant that SLC30A10D40A also lacked manganese efflux activity. Thus, the results of the manganese measurement assays are consistent with those obtained in the GPP130 degradation studies.
Cell Viability Assays to Confirm GPP130 Degradation and Manganese Measurement Studies
Cell viability assays are commonly used to test whether introduction of a mutation alters transport capacity of a protein. These indirect assays are often difficult to interpret because mutants that sequester, but do not transport, a metal may also confer protection. Nevertheless, as we had the results of the manganese measurement studies in hand, we were in a position to determine whether a mutant that lacked efflux activity (D248A, E25A, N127A, H244A, or D40A) still protected against manganese-induced cell death. Such a result would raise the possibility that the mutant retains manganese binding capacity and could guide future biochemical studies. Moreover, performance of rigorous viability assays would also allow comparison of our results with other viability-based studies performed on SLC30 proteins in the field. Therefore, we used cell viability as a third independent measure of the efflux activity of SLC30A10 mutants generated.
For these assays, we first performed a dose response of manganese. Consistent with our prior work (18, 40), exposure of cells to 500 μm manganese did not affect viability (Fig. 6A), confirming that manganese levels used for the GPP130 degradation and metal measurement studies were non-toxic. In contrast, treatment with 1 or 2 mm manganese led to a significant decrease in viability (Fig. 6A), also consistent with our prior studies (18). Next, we performed a set of important controls. We validated that expression of SLC30A10WT protected against manganese-induced cell death (Fig. 6B). In this assay, we separately expressed the cell surface-localized zinc efflux transporter, SLC30A1/ZnT1 (see Fig. 10) (32, 41); and we observed that expression of ZnT1WT had no effect on manganese-induced cell death (Fig. 6B). Note that SLC30A1 is also called ZnT1, and we will refer to this protein as ZnT1 under “Results” so as to distinguish its zinc transporting property from the manganese transporting activity of SLC30A10. Importantly, however, ZnT1WT, but not SLC30A10WT, protected against zinc-induced cell death (Fig. 6, C and D). These results provide additional evidence, albeit indirect, showing that SLC30A10 transports manganese, but not zinc, although ZnT1 transports zinc, but not manganese. Subsequent GPP130 degradation and manganese measurement studies verified that ZnT1WT lacked manganese efflux activity (see Fig. 10). Finally, we assayed for the ability of the SLC30A10 mutants used in manganese measurement studies to protect against manganese-induced cell death. We discovered that the N43A and D47A mutants strongly protected against manganese-induced cell death (Fig. 6E). Indeed, the viability of manganese-treated cells expressing the N43A or D47A mutant was comparable with those expressing SLC30A10WT and significantly greater than transfection control (Fig. 6E). In contrast, the viability of manganese-treated cells expressing the D248A, E25A, N127A,H244A, or D40A mutant was comparable with transfection control and significantly lower than SLC30A10WT (Fig. 6E). Thus, results of the cell viability assay are consistent with the GPP130 degradation and manganese measurement studies.
FIGURE 6.

Cell viability assays to validate the efflux activities of SLC30A10WT and transmembrane domain mutants. A, untransfected HeLa cells were treated with indicated amounts of manganese for 16 h. Cell viability was then assessed as described under “Experimental Procedures.” Viability at 0 mm manganese was set to 100 and used to calculate percent viability at other manganese concentrations (mean ± S.E.; n = 3; *, p < 0.05 for the difference between viability at 0 mm manganese and other manganese concentrations by one-way ANOVA and Dunnett's post hoc test). B, HeLa cells were transfected with a control construct (indicated as Control), SLC30A10WT, or ZnT1WT. Two days after transfection, cells were treated with 0 or 2 mm manganese for 16 h. Viability was then assessed as described under “Experimental Procedures.” For each transfection condition, viability at 0 mm manganese was set to 100 and used to calculate percent viability at 2 mm manganese (mean ± S.E.; n = 4 for each construct; *, p < 0.05 for the difference between control and other groups using one-way ANOVA and Dunnett's post hoc test). C, untransfected HeLa cells were treated with indicated amounts of zinc for 16 h. Further processing was exactly as described in A. Viability at 0 mm zinc was set to 100 and used to calculate percent viability at other concentrations (mean ± S.E.; n = 3; *, p < 0.05 for the difference between 0 mm zinc and other concentrations by one-way ANOVA and Dunnett's post hoc test). D, HeLa cells were processed as described in B, with the only difference being that instead of treatment with 0 or 2 mm manganese the cultures were treated with 0 or 200 μm zinc. For each transfection condition, viability at 0 μm zinc was set to 100 and used to calculate percent viability at 200 μm zinc (mean ± S.E.; n = 3 for each construct; *, p < 0.05 for the difference between control and other groups using one-way ANOVA and Dunnett's post hoc test). E, HeLa cells were transfected with a control construct (denoted as Control), SLC30A10WT, or various transmembrane domain mutants. After 2 days, cells were treated with 0 or 2 mm manganese for 16 h. Cell viability was then assessed. For each transfection condition, viability at 0 mm manganese was set to 100 and used to calculate percent viability at 2 mm manganese (mean ± S.E.; n ≥ 4 per construct; *, p < 0.05 for the difference between WT and other transfection conditions by one-way ANOVA and Dunnett's post hoc test). One-way ANOVA and Dunnett's post hoc test also revealed that there was no difference between control and D248A, E25A, N127A,H244A, or D40A groups (p > 0.05); however, there was a significant difference between control and WT, N43A, or D47A groups (p < 0.05).
FIGURE 10.

Cytoplasmic domain of SLC30A10 is not sufficient to confer manganese efflux activity to SLC30A1/ZnT1. A, HeLa cells were transfected with FLAG-tagged ZnT1WT or a FLAG-tagged construct that had the transmembrane domain of ZnT1 followed by the C-terminal domain of SLC30A10 (ZnT1N-termSLC30A10C-term). Two days later, cultures were processed for immunofluorescence exactly as described in Fig. 2A. P denotes the Pearson's coefficient for colocalization between the FLAG constructs and calnexin (mean ± S.E.; n = 15 cells for each construct). Scale bar, 10 μm. B, HeLa cells were cotransfected with FLAG-tagged ZnT1WT or ZnT1N-termSLC30A10C-term and HA-tagged DPP4. Further processing was exactly as described in Fig. 2B. P denotes the Pearson's coefficient for colocalization between the FLAG constructs and DPP4 (mean ± S.E.; n = 10–15 cells for each construct). Scale bar, 10 μm. C, HeLa cells were transfected with FLAG-tagged SLC30A10WT, ZnT1WT, or ZnT1N-termSLC30A10C-term. Two days after transfection, the GPP130 degradation assay was performed as described in Fig. 3. Scale bar, 10 μm. D, quantification of the amount of GPP130 remaining after manganese treatment from C. Untransfected cells did not express any of the constructs. Percent GPP130 remaining after manganese was calculated as described in Fig. 3 (mean ± S.E.; n = 15 cells per manganese treatment and transfection condition; *, p < 0.05 for the difference between SLC30A10WT and other transfection conditions). Note that there was no difference between untransfected and ZnT1WT or ZnT1N-termSLC30A10C-term groups (p > 0.05 by one-way ANOVA and Dunnett's post hoc test); however, there was a significant difference between untransfected and SLC30A10WT groups (p < 0.05 by one-way ANOVA and Dunnett's post hoc test). E, intracellular manganese measurements to validate results of D above. Identical numbers of HeLa cells were plated in 35-mm dishes and transfected with a control construct (denoted as Control), SLC30A10WT, ZnT1WT, or ZnT1N-termSLC30A10C-term. After this, cultures were used to assay for intracellular manganese using inductively coupled plasma mass spectrometry exactly as described in Fig. 5. Absolute amount of intracellular manganese in control-transfected cells was normalized to 100 (mean ± S.E.; n = 3 for each construct; *, p < 0.05 for the difference between SLC30A10WT and other transfection conditions by one-way ANOVA and Dunnett's post hoc test). Additional analyses showed that there was no difference between control-transfected cells and cells expressing ZnT1WT or ZnT1N-termSLC30A10C-term (p > 0.05 by one-way ANOVA and Dunnett's post hoc test); however, there was a significant difference between control and SLC30A10WT groups (p < 0.05 by one-way ANOVA and Dunnett's post hoc test). F and G, cell viability was assessed in HeLa cells transfected with control (denoted as Control), SLC30A10WT, or ZnT1N-termSLC30A10C-term constructs and treated with 0 or 2 mm manganese (F) or transfected with control (denoted as Control), ZnT1WT, or ZnT1N-termSLC30A10C-term constructs and treated with 0 or 200 μm zinc (G). Calculation of percent viability for each panel was exactly as described in Fig. 6 (mean ± S.E.; n = 4 per construct for F and 3 per construct for G; *, p < 0.05 for the difference between cells expressing SLC30A10WT (F) or ZnT1WT (G) and other transfection conditions by one-way ANOVA and Dunnett's post hoc test). Statistical analyses also revealed that there was no difference between control transfected cells and cells expressing ZnT1N-termSLC30A10C-term in either panel (p > 0.05 by one-way ANOVA and Dunnett's post hoc test).
SLC30A10N43H Retains Manganese Efflux Activity
An important conclusion from data in Figs. 2–6 is that the side chain of Asn-43 is not required for the manganese efflux activity of SLC30A10. However, as described earlier, substitution of Asn-43 with histidine is an important difference between SLC30A10 and zinc-transporting SLC30 proteins (Fig. 1). It was possible that, although the side chain of Asn-43 was not required for manganese efflux, the presence of a histidine residue at this position may interfere with a manganese-binding site created by side chains of other required residues (e.g. Asp-248, Asn-127, and Glu-25). In such a scenario, the Asn-43 residue would still be important for manganese efflux as it would prevent interference in ion binding caused by a histidine side chain. To test this possibility, we generated SLC30A10N43H. This mutant also trafficked to the cell surface and was separate from the endoplasmic reticulum (Fig. 7, A and B). Importantly, intracellular manganese levels in cells expressing SLC30A10N43H were comparable with SLC30A10WT and significantly lower than transfection control (Fig. 7C). Moreover, expression of SLC30A10N43H robustly protected against manganese-induced cell death, and there was no difference in the level of protection conferred by SLC30A10N43H or SLC30A10WT (Fig. 7D). These results suggested that SLC30A10N43H retained manganese efflux activity. Thus, the histidine-to-asparagine substitution in the second transmembrane domain of SLC30A10 does not appear to directly contribute to the ability of the transporter to mediate manganese efflux, at least in our cell culture assays.
FIGURE 7.

SLC30A10N43H retains manganese efflux activity. A, HeLa cells were transfected with FLAG-tagged SLC30A10N43H. Two days after transfection, cultures were processed for immunofluorescence. SLC30A10 was detected using a monoclonal antibody against the FLAG epitope, and a polyclonal antibody against calnexin was used to demarcate the endoplasmic reticulum. P represents the Pearson's coefficient for colocalization between SLC30A10 and calnexin (mean ± S.E.; n = 13 cells). Scale bar, 10 μm. B, HeLa cells were cotransfected with FLAG-tagged SLC30A10N43H and HA-tagged DPP4. Two days after transfection, cultures were processed for immunofluorescence. SLC30A10 was detected using a polyclonal antibody against the FLAG tag. A monoclonal antibody against HA was used to detect DPP4. P represents the Pearson's coefficient for colocalization between SLC30A10 and DPP4 (mean ± S.E.; n = 15 cells). Scale bar, 10 μm. C, intracellular manganese levels were measured in HeLa cells transfected with control (denoted as Control), SLC30A10WT (WT), or SLC30A10N43H (N43H) constructs, exactly as described in Fig. 5 (mean ± S.E.; n = 3 for each construct; *, p < 0.05 for the difference between cells expressing SLC30A10WT and all other transfection conditions by one-way ANOVA and Dunnett's post hoc test; n.s. stands for “not significant” and denotes that the difference between WT and N43H groups was not statistically significant (p > 0.05)). D, cell viability was assessed in HeLa cells transfected with control (denoted as Control), SLC30A10WT (WT), or SLC30A10N43H (N43H) constructs after treatment with 0 or 2 mm manganese, exactly as described in Fig. 6E. For each transfection condition, viability at 0 mm manganese was set to 100 and used to calculate percent viability at 2 mm manganese (mean ± S.E.; n ≥ 5 for each construct; *, p < 0.05 for the difference between SLC30A10WT and all other transfection conditions by one-way ANOVA and Dunnett's post hoc test; n.s. stands for not significant and denotes that the difference between WT and N43H groups was not statistically significant (p > 0.05)).
Identification of Residues in the Cytoplasmic Domain of SLC30A10 That Are Required for Optimal Manganese Efflux
YiiP has a cytoplasmic C-terminal domain, which assumes a metallochaperone-like fold and is involved in homodimerization (23, 24). This domain has two zinc-binding sites. One of these sites (site B) is at the interface of the transmembrane and cytoplasmic domains and is formed by residues Asp-68, His-71, His-75, and a water molecule (Fig. 8) (23, 24). The other site (site C) exhibits binuclear zinc coordination; the coordinating residues are Asp-285, His-232, His-248, and His-283 from one monomer, and His-261 from the neighboring subunit of the dimer (Fig. 8) (23, 24). Residue Asp-285 forms two individual monodentate coordinate bonds with two zinc ions, whereas the other residues form single coordinate bonds (Fig. 8) (23, 24). The molecular details of the role played by the C-terminal domain in regulating transport are not completely understood as yet, but available evidence suggests that zinc binding to this domain may allosterically regulate transport through site A (16, 23). Indeed, in YiiP, a single point mutation in site C (H232A) reduces the kinetics of zinc transport (23). Similarly, in the bacterial cation diffusion facilitators ZitB and CzcD, mutants that lack the C-terminal domains exhibit decreased activity (42). In SLC30A10, the C-terminal domain is predicted to begin at residue Gln-308. Interestingly, a parkinsonism-causing mutation, Q308Stop, leads to the deletion of the entire C-terminal domain in humans (11). We previously showed that SLC30A10Q308Stop was trapped in the endoplasmic reticulum and was incapable of mediating manganese efflux (13). However, we felt that it was important to determine whether the C-terminal domain played a role in mediating manganese transport separate from its requirement for intracellular trafficking.
FIGURE 8.

Comparison of site B and site C of YiiP with that of SLC30A10. The solved crystal structure of YiiP (Protein Data Bank code 3H90) is depicted as a homodimer (green and gray colors denote individual monomers). The predicted structure of SLC30A10 is shown as a monomer. For both proteins, a side view from the plane of the membrane is shown on the left (Side). Boxes in the side view denote site B (solid box) or site C (dashed box). Close-ups of these sites are depicted to the right. In YiiP, red sticks denote site B or site C residues from one monomer; gray stick denotes residue His-261, which contributes to site C from the adjacent monomer, and yellow spheres denote zinc ions. In SLC30A10, red sticks denote residues that correspond to site B or site C of YiiP; although Phe-68 is not a part of site B, it is depicted here due to the close proximity of its aromatic ring to that of Tyr-74. In all residues depicted as sticks, nitrogen atoms are shown in blue.
We performed sequence alignments and closely analyzed the predicted structure of SLC30A10. We discovered that, in SLC30A10, residues corresponding to site B of YiiP were Gly-67, Ala-70, and Tyr-74 (the aromatic ring of Tyr-74 was in proximity to that of Phe-68) (Fig. 8). Because of the lack of conservation of these residues with site B of YiiP, we did not analyze them further. In contrast, two of the residues of site C of YiiP were conserved in SLC30A10; His-232 and His-248 of YiiP were represented by His-333 and His-350 in SLC30A10, respectively (Fig. 8; note that His-261, His-283, and Asp-285 of YiiP were not conserved in SLC30A10 and not studied further). Analyses of the predicted structure of SLC30A10 revealed that side chains of His-333 and His-350 were surface-exposed and in proximity to each other (Fig. 8). We felt that these residues may potentially be involved in metal coordination in a manner similar to that seen in site C of YiiP and thereby regulate transport. Unfortunately, despite several attempts, we were unable to detect manganese binding using an epitope-tagged version of the C-terminal domain of SLC30A10. It may be that metal binding studies will require purification of the full-length protein. Nevertheless, we decided to test the requirement of residues His-333 and His-350 in manganese transport. However, before making point mutations, we felt it was prudent to quickly test whether the entire C-terminal domain of SLC30A10 was required for manganese efflux. For this, we generated a chimeric construct in which the transmembrane domain of SLC30A10 was fused to the C-terminal domain of ZnT1 (SLC30A10N-termZnT1C-term). This chimeric construct trafficked to the cell surface (Fig. 9, A and B), indicating that, for purposes of intracellular trafficking, the C-terminal domain of ZnT1 could compensate for that of SLC30A10. We then performed the GPP130 degradation assay to assess the ability of SLC30A10N-termZnT1C-term to mediate manganese efflux. Expression of SLC30A10N-termZnT1C-term failed to protect against the manganese-induced loss of GPP130 (Fig. 9, C and D). Residual GPP130 levels in manganese-treated cells expressing SLC30A10N-termZnT1C-term were significantly lower than those expressing SLC30A10WT and comparable with transfection control (Fig. 9, C and D). This result suggested that the C-terminal domain of SLC30A10 was required for manganese efflux and justified closer analyses of specific residues within this domain.
FIGURE 9.

Analyses of the manganese efflux activity of C-terminal domain mutants of SLC30A10. A and B, HeLa cells were transfected with FLAG-tagged SLC30A10N-termZnT1C-term (A) or cotransfected with SLC30A10N-termZnT1C-term and HA-tagged DPP4 (B). Two days later, cells were processed for immunofluorescence, exactly as described in Fig. 2. P denotes the Pearson's coefficient for colocalization between the SLC30A10 construct and calnexin or DPP4 (mean ± S.E.; n = 12–15 cells for each colocalization analysis). Scale bars, 10 μm. C, HeLa cells were transfected with SLC30A10WT or SLC30A10N-termZnT1C-term. Two days after transfection, cultures were processed for the GPP130 degradation assay, exactly as described in Fig. 3. Scale bar, 10 μm. D, quantification of the amount of GPP130 remaining in cells after manganese treatment from C. Untransfected cells did not express any SLC30A10 construct. For each transfection condition, percent GPP130 remaining after manganese was calculated as described in Fig. 3 (mean ± S.E.; n = 15 cells per manganese treatment and transfection condition; *, p < 0.05 for the difference between WT and other transfection conditions by one-way ANOVA and Dunnett's post hoc test). One-way ANOVA and Dunnett's post hoc test also revealed that there was a significant difference between untransfected and SLC30A10WT groups (p < 0.05), but there was no difference between untransfected and SLC30A10N-termZnT1C-term groups (p > 0.05). E and F, HeLa cells were transfected with FLAG-tagged SLC30A10H333A,H350A (E) or cotransfected with SLC30A10H333A,H350A and HA-tagged DPP4 (F). Further processing was as described in A and B. P denotes the Pearson's coefficient for colocalization between the SLC30A10 construct and calnexin or DPP4 (mean ± S.E.; n > 10 cells for each colocalization analysis). Scale bars, 10 μm. G, HeLa cells were transfected with SLC30A10WT or SLC30A10H333A,H350A and processed for the GPP130 degradation assay, as described in C. Scale bar, 10 μm. H, quantification of the amount of GPP130 remaining in cells after manganese treatment from G. Untransfected cells did not express any SLC30A10 construct. For each transfection condition, percent GPP130 remaining after manganese was calculated as described in Fig. 3 (mean ± S.E.; n = 12–15 cells per manganese treatment and transfection condition; *, p < 0.05 for the difference between WT and other transfection conditions by one-way ANOVA and Dunnett's post hoc test). One-way ANOVA and Dunnett's post hoc test also revealed that there was a significant difference between untransfected and the two other groups (p < 0.05; the difference between untransfected and H333A,H350A groups is shown by an asterisk). I, intracellular manganese measurements to validate results of the GPP130 degradation assay presented in G above. Identical numbers of HeLa cells were plated in 35-mm dishes and transfected with a control construct (denoted as Control), SLC30A10WT, or SLC30A10H333A,H350A. After this, cultures were used to assay for intracellular manganese using inductively coupled plasma mass spectrometry exactly as described in Fig. 5. Absolute amount of intracellular manganese in control-transfected cells was normalized to 100 (mean ± S.E.; n ≥ 3 for each construct; *, p < 0.05 for the difference between SLC30A10WT and other transfection conditions by one-way ANOVA and Dunnett's post hoc test). One-way ANOVA and Dunnett's post hoc test also showed that there was a significant difference between control and the two other groups (p < 0.05; the difference between control and H333A,H350A groups is depicted with an asterisk).
As the next step, we substituted His-333 and His-350 with alanine to generate SLC30A10H333A,H350A. The double mutant was strongly expressed, and colocalization assays showed that it trafficked to the cell surface (Fig. 9, E and F). After treatment with manganese, residual GPP130 levels in cells expressing SLC30A10H333A,H350A (32 ± 3%) were significantly lesser than those expressing SLC30A10WT (81 ± 8%) (Fig. 9, G and H), suggesting that the efflux activity of SLC30A10H333A,H350A was less than SLC30A10WT. Importantly, however, statistical analyses revealed that the residual GPP130 remaining in H333A,H350A-expressing cells (32 ± 3%) was significantly greater than that in untransfected controls (15 ± 1.5%) (Fig. 9H), suggesting that substitution of His-333 and His-350 with alanine did not completely abolish the manganese efflux activity of SLC30A10. Consistent with the results of the GPP130 degradation assay, normalized levels of intracellular manganese in cells expressing SLC30A10H333A,H350A (48 ± 4) were significantly greater than those expressing SLC30A10WT (29 ± 7) but significantly lesser than control-transfected cultures (100 ± 0) (Fig. 9I). We noted that, between SLC30A10H333A,H350A and SLC30A10WT groups, the percent difference in values for residual GPP130 remaining was 87% but that for intracellular manganese was 49% (Fig. 9, H and I). The smaller difference between groups in the intracellular manganese measurement assay may be explained by the fact that GPP130 is highly sensitive to changes in intracellular and intra-Golgi manganese levels (18, 40, 43). Indeed, a prior study showed that GPP130 may be degraded when cells are treated with exceedingly low amounts of manganese that do not lead to a measurable increase in intracellular manganese (43). The modest increase in intracellular manganese that occurs in cells expressing SLC30A10H333A,H350A (Fig. 9I) was likely sufficient to increase intra-Golgi manganese to a level that induced degradation of a substantial amount of GPP130 (Fig. 9H). Despite the difference in the magnitude of the effect, results of the GPP130 degradation and manganese measurement assays were congruous and implied that SLC30A10H333A,H350A retained some manganese efflux activity, but the activity was less than that of SLC30A10WT. Thus, residues His-333 and His-350 are required for optimal or complete manganese efflux activity of SLC30A10.
C-terminal Domain of SLC30A10 Cannot Transfer Manganese Transport Capability to ZnT1
As an additional test of the role of the C-terminal domain of SLC30A10 in mediating manganese transport, we wanted to determine whether this domain could transfer manganese transport capability to ZnT1. We expressed ZnT1WT or a chimeric construct that had the transmembrane domain of ZnT1 followed by the C-terminal domain of SLC30A10 in cells (ZnT1N-termSLC30A10C-term). Colocalization analyses revealed that ZnT1WT trafficked to the cell surface (Fig. 10, A and B), consistent with prior studies (32, 41). The ZnT1N-termSLC30A10C-term chimera also localized to the cell surface (Fig. 10, A and B). Importantly, unlike SLC30A10WT, manganese treatment induced robust GPP130 degradation in cells that expressed either ZnT1WT or ZnT1N-termSLC30A10C-term (Fig. 10, C and D). Residual GPP130 levels in cells expressing ZnT1WT or the chimera were significantly lesser than SLC30A10WT but comparable with untransfected controls (Fig. 10, C and D). Furthermore, intracellular manganese levels in cells expressing ZnT1WT or ZnT1N-termSLC30A10C-term were comparable with transfection control and significantly greater than SLC30A10WT (Fig. 10E). Moreover, unlike SLC30A10WT, expression of ZnT1N-termSLC30A10C-term failed to protect cells against manganese-induced cell death (Fig. 10F). These results implied that both ZnT1WT and ZnT1N-termSLC30A10C-term failed to mediate manganese efflux. Thus, the C-terminal domain of SLC30A10 is not sufficient to confer manganese efflux activity to ZnT1.
As part of these studies, we also performed a viability assay after treatment of cells with zinc and observed that although ZnT1WT protected cells against zinc-induced death, ZnT1N-termSLC30A10C-term failed to protect (Fig. 10G), implying that ZnT1N-termSLC30A10C-term lacked zinc transport activity. This result provides additional evidence supporting the requirement of the C-terminal domain of ZnT1 in mediating zinc transport, which has been previously reported (32). Thus, results in Figs. 9 and 10 suggest that the C-terminal domains of SLC30A10 and ZnT1 are required for optimal transport activities of the respective proteins.
In sum, sequences in the transmembrane and C-terminal domains together confer full manganese efflux capability to SLC30A10.
Discussion
YiiP serves as a model for structure-function studies of cation diffusion facilitators. In YiiP, side chains of residues Asp-45, Asp-49, His-153, and Asp-157, which constitute site A, directly coordinate zinc and mediate transport (23, 24). Mutations in residues that correspond to site A of YiiP in other zinc transporters, including the mammalian transporters SLC30A1/ZnT1 and SLC30A5, block activity (15, 31, 32). In SLC30A10, residues that correspond to site A of YiiP are Asn-43, Asp-47, His-244, and Asp-248. The substitution of Asp-45 in YiiP to Asn-43 in SLC30A10 led us to hypothesize that this single amino acid change may confer manganese transport activity to SLC30A10. This idea was supported by the fact that asparagine has a higher propensity to coordinate with manganese than with zinc (36). Furthermore, the zinc transporters SLC30A5 and SLC30A8 gain cadmium transport capacity when a histidine residue in their putative site A is changed to aspartate (22), indicating that a single amino acid change in the transmembrane ion coordinating site can affect substrate specificity. Surprisingly, we discovered that in SLC30A10, of the four residues that correspond to site A of YiiP, the side chain of only one residue, Asp-248, is required for activity. This result implies that the mechanism of ion coordination within the transmembrane domain of SLC30A10 is likely different from that of YiiP and other zinc-transporting cation diffusion facilitators. Zinc prefers tetrahedral coordination, whereas manganese prefers octahedral coordination (36). Therefore, it is perhaps not surprising that the means by which ion coordination is achieved in the transmembrane domain may differ between SLC30A10 and zinc transporters.
In addition to Asp-248, our studies identified Glu-25 and Asn-127 in the transmembrane domain to be required for manganese efflux via SLC30A10. Interestingly, these required residues are conserved in other SLC30 family proteins that transport zinc. Moreover, studies of prokaryotic zinc transporters show that residues Glu-31 of CzcD and Asn-135 of ZitB, which correspond to Glu-25 and Asn-127 of SLC30A10, respectively, are required for activity (33, 42). Thus, our identification of required residues in the transmembrane domain does not provide a clear explanation for the substrate specificity of SLC30A10. Currently, in the absence of a crystal structure, we can only speculate about the underlying mechanisms. Prior studies on the zinc transporter ZRC1, which is the yeast homolog of SLC30A10, suggest that even single amino acid changes near the transmembrane ion coordination site may affect substrate specificity (44). Primary sequence analyses show that a number of residues of SLC30A10 that are near the required Asp-248, Glu-25, and Asn-127 amino acids differ from those of other SLC30 proteins. Thus, one possibility is that, due to these differences, residues in the transmembrane domain of SLC30A10 are oriented in a manner that favors manganese and simultaneously disfavors zinc coordination. As transporters function under severe substrate-limiting conditions in cells, a small change in the dissociation constant for a metal may have a profound effect on transport capability. Clearly, structural studies are now essential to further understand the mechanism of ion coordination in, and transport by, SLC30A10. In the above discussion, the following additional points must be noted. Our studies do not imply that any of the above required residues directly mediate manganese coordination in SLC30A10; it is possible that the introduced mutations induce changes in the orientation of other amino acids that actually mediate metal coordination. The alanine substitutions performed in this study cannot rule out a role for the backbone oxygen atoms of Asn-43, Asp-47, or His-244 in SLC30A10 in coordinating or transporting manganese. Our assays were not designed to elucidate changes in transport kinetics, and at this point, we do not know whether the Michaelis constant of transport is altered when Asn-43, Asp-47, or His-244 is mutated. Finally, our work does not imply that, in other manganese-transporting cation diffusion facilitators, residues that correspond to Asp-47, Asn-43, or His-244 of SLC30A10 will be dispensable for activity. Indeed, as one example, in the bacterial manganese transporter MntE, the residue that corresponds to Asn-43 of SLC30A10 is also an asparagine residue, but unlike SLC30A10, the asparagine residue of MntE is required for activity (34).
In addition to residues in the transmembrane domain, we discovered that residues His-333 and His-350 in the cytoplasmic domain of SLC30A10 are required for complete manganese efflux activity. This result fits with prior studies showing that residues in the cytoplasmic domain of SLC30A1/ZnT1, YiiP, CzcD, and ZitB are required for optimal activity (23, 32, 42). The precise mechanism by which the cytoplasmic domain of SLC30A10 influences manganese efflux remains to be elucidated; it cannot be the sole manganese specificity determinant as it cannot transfer manganese transport capacity to SLC30A1/ZnT1. It may be that this domain assumes a structure that enhances the ability of the transmembrane domain to bind and transport manganese. Alternatively, the cytoplasmic domain may play a role in mediating interactions with chaperone proteins that transfer manganese to the transmembrane domain. Further analyses, such as metal binding assays, structural studies, and proteomic screens, are now essential to elucidate the role of the C-terminal domain in mediating manganese transport. It is important to note that, in cells, the C-terminal domain of SLC30A10 is essential for trafficking to the plasma membrane (13), and the C-terminal domains of other cation diffusion facilitators may similarly be involved in targeting the proteins to the correct intracellular location.
While our manuscript was in revision, another interesting report on the manganese efflux activity of SLC30A10 was published (45). In this study, by Nishito et al. (45), the authors expressed human SLC30A10WT or mutants in a chicken cell line and assessed transport activity of SLC30A10 mutants by using a cell viability assay. There are two important differences between Nishito et al. (45) and our study: Nishito et al. show that the N43H mutation in SLC30A10 blocks manganese efflux activity (45), whereas a chimeric protein, which has the transmembrane domain of SLC30A10 followed by the cytoplasmic domain of ZnT1/SLC30A1 (likely similar to our SLC30A10N-termZnT1C-term construct), retains manganese efflux activity (45). How can the differences between the two studies be reconciled? We suggest the following explanations. First, Nishito et al. (45) does not include metal measurement data for the above SLC30A10 mutants; therefore, it is unclear whether these mutants protect against manganese-induced cell death by mediating manganese efflux or by an indirect mechanism (e.g. sequestering manganese). Second, the model systems used in the two studies are vastly different. Our study used HeLa cells. Although differences surely exist between hepatocytes and neuronal systems, which are the physiological site of action of SLC30A10, and HeLa cells, as HeLa cells are of human origin, they may better represent the conditions in which human SLC30A10 functions. Notably, the chicken cell line used by Nishito et al. (45) lacks SPCA1, the Golgi manganese pump required for the transport of manganese into the Golgi (46–48). Absence of SPCA1 alters glycosylation (49) and blocks the manganese-induced degradation of GPP130 (18); effects of these changes on SLC30A10 function are unknown. Furthermore, SLC30A10 localizes to the plasma membrane in relevant neuronal systems and in HeLa cells (13). However, Nishito et al. (45) note that a large pool of SLC30A10 is trapped in the Golgi in the chicken cell line and that it is not possible to determine whether the observed protective effect in viability assays is due to activity of SLC30A10 mutants on the cell surface or at the Golgi (45). Transport activities of SLC30A10 mutants may be dependent on intracellular localization, and a mutant, such as N43H, whose Michaelis constant may be different from WT, may be inactive when localized to the Golgi but gain activity at the plasma membrane. Overall, findings in this study and in Ref. 45 highlight the need, in the future, to assess the manganese efflux activity of WT and mutated forms of SLC30A10 using rigorous in vitro transport assays on the one hand and to validate results in challenging but physiologically relevant model systems, such as primary neurons, hepatocytes, and knock-out mice on the other hand.
To summarize, we show that residues in the transmembrane and C-terminal domains of SLC30A10 are required for complete manganese efflux activity. Furthermore, our work suggests that the mechanism of ion coordination in the transmembrane domain of SLC30A10 is likely different from that in related zinc transporters. These findings provide novel insights into an aspect of SLC30A10 biology that is directly relevant to its role in regulating manganese homeostasis and are likely to improve our understanding of the mechanisms of ion transport by SLC30 family proteins and other cation diffusion facilitators.
Experimental Procedures
Constructs
Plasmids encoding FLAG-tagged SLC30A10WT, HA-tagged DPP4 and GFP-tagged Rab5 (control construct) have been described by us (13). We previously demonstrated that the following parameters are comparable between cells expressing the control construct and those that are mock-transfected: absolute levels of intracellular manganese (measured using inductively coupled plasma mass spectrometry), manganese-induced degradation of GPP130, and cell viability after exposure to elevated manganese. These results imply that transfection of the control construct does not impact manganese homeostasis or transport (13). Mutations were introduced into FLAG-SLC30A10WT using QuikChange (Agilent Technologies, Santa Clara, CA). Plasmid encoding WT human SLC30A1/ZnT1 was obtained from the DNASU Plasmid Repository (Arizona State University, Tempe, AZ). The ZnT1 insert was cloned into the NotI and KpnI restriction sites of p3×-FLAG-CMV-10 vector (Sigma); the primers used were 5′ CTT GCG GCC GCG ATG GGG TGT TGG GGT CGG AAC 3′ (forward) and 5′ GTC GGT ACC CTA CAA AGA TGA TTC AGG TTG 3′ (reverse). Note that FLAG-SLC30A10WT is also in the p3×-FLAG-CMV-10 vector.
For generation of the ZnT1N-termSLC30A10C-term chimeric construct that had the transmembrane domain of ZnT1 followed by the C-terminal domain of SLC30A10, the DNA sequence CTCGAG, which constitutes a XhoI restriction site, was introduced into FLAG-ZnT1WT after residues coding for amino acid 344 of ZnT1WT (the C-terminal domain of ZnT1 is predicted to start at amino acid residue 345). The insertion was performed using the loop-in modification of the QuikChange protocol, as described by us previously (50); primers used were 5′ GCT CTT ATT CTT CTA CTC GAG CAA ACT GTT CCT AAA C 3′ (forward) and 5′ GTT TAG GAA CAG TTT GCT CGA GTA GAA G 3′ (reverse). The C-terminal domain of SLC30A10WT, beginning at amino acid 308 of SLC30A10, was PCR-amplified using the primers 5′ GAT CAG CTC GAG CAG ATG GTC CCA AAA GGA GTC 3′ (forward) and 5′ CTC ATC GGT ACC TTA AAA ATG CGT TCT GTT GAC 3′ (reverse). Both the PCR product and the FLAG-ZnT1WT vector were cleaved using XhoI and KpnI restriction enzymes. Finally, the cleaved PCR product was ligated into the vector to generate the chimera.
For generation of the SLC30A10N-termZnT1C-term chimera, an XhoI site was looped-in between residues coding for amino acids 307 and 308 of FLAG-SLC30A10WT, and the resulting construct was digested with XhoI and KpnI restriction enzymes. Similarly, the C-terminal domain of ZnT1 was cut using XhoI and KpnI restriction enzymes; the construct used was the one used above for generation of the ZnT1N-termSLC30A10C-term chimera. The C-terminal domain of ZnT1 was then ligated into the vector containing the N-terminal domain of SLC30A10 to generate SLC30A10N-termZnT1C-term.
Chemicals and Antibodies
Unless otherwise specified, chemicals were from ThermoFisher Scientific or Sigma. Rabbit polyclonal and mouse monoclonal antibodies against the FLAG tag (Sigma), mouse monoclonal antibody against the HA tag (Sigma), rabbit polyclonal antibody against calnexin (Abcam, Cambridge, UK), and custom mouse monoclonal antibody against GPP130 have been previously described by us (13).
Cell Culture, Transfections, and Manganese and Zinc Treatments
All experiments were performed in HeLa cells. As HeLa cells do not express endogenous SLC30A10, they provided an ideal system for overexpression studies where obtained results would not be impacted by activity of the endogenous protein. Cell culture, DNA transfections, and manganese and zinc treatments were performed exactly as described by us previously (13). To summarize here, cells were grown in minimum essential media (Corning Glass) that was supplemented with 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), 100 IU/ml penicillin-G, and 100 μg/ml streptomycin (both from Corning Glass). DNA transfections were performed using the JetPEI reagent (VWR International, Radnor, PA), following the manufacturer's recommendations. Manganese and zinc treatments were performed by the addition of freshly prepared MnCl2 or ZnCl2 to the media at concentrations indicated in the figure legends.
Structural Predictions
Predicted structure of SLC30A10 was generated using the PHYRE 2.0 server (37). All protein structure images were generated using the open source PyMOL software (The PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC).
Microscopy
For immunofluorescence, cells were fixed using 3% paraformaldehyde for 20 min at room temperature. Further processing was exactly as described by us previously (13). AlexaFluor-488- or -568-labeled secondary antibodies (ThermoFisher) were used. All images were captured using a Nikon swept-field confocal and a 100× oil immersion objective with a numerical aperture of 1.45 (both from Nikon Inc., Melville, NY). An iXon3 X3 DU897 EM-CCD camera (Andor Technology, Belfast, UK) was used for image capture. Images were captured as Z-stacks with 0.5-μm spacing between individual frames. Depicted images are maximum intensity projections from the stacks.
Image Analyses
Image quantification was performed using the NIS Elements software (Nikon), essentially as described by us previously (13). Within each experiment, all images were captured using identical settings.
For measurement of GPP130 fluorescence intensity, the ND Processing and ND Image Average (average all frames) functions were used to generate average intensity projections from individual Z-stacks. Background was separately subtracted for each image by drawing a region of interest in a part of the image that did not have any cellular component. An outline of the cell was then drawn using the Draw Bezier region of interest tool. Finally, total GPP130 fluorescence per cell was obtained using the region of interest statistics function.
For calculating Pearson's correlation coefficient, instead of average projections, for each image, one frame from the Z-stack was used. In each case, the selected frame had the largest expanse of the cell being analyzed. For quantification, an outline was drawn around the cell, and the Pearson's coefficient was calculated using the colocalization function of NIS elements.
GPP130 Degradation Assays
These were performed as described by us previously (13, 18, 39). To summarize here, cells were treated with or without 500 μm manganese for 4 h and then processed for immunofluorescence microscopy. GPP130 levels were quantified as described under “Image Analyses” above and in individual figure legends. Use of the above concentration and time of manganese treatment was based on our prior studies showing that, under these manganese treatment conditions, GPP130 was robustly degraded in transfection-control cells and in cells expressing transport-inactive, parkinsonism-causing mutants of SLC30A10; but expression of SLC30A10WT protected against the manganese-induced loss of GPP130 (13). Thus, the above treatment paradigm provided an effective although indirect means to rapidly evaluate the manganese efflux activity of SLC30A10 mutants generated in this study, relative to transfection control and SLC30A10WT. Note that, through our previous studies, we established that microscopy provides an accurate representation of manganese-induced changes in GPP130 levels and that results obtained by immunofluorescence assays are similar to those obtained by immunoblot (13, 18, 39).
Intracellular Metal Measurements Using Inductively Coupled Plasma Mass Spectrometry
Metal measurements were performed essentially as described by us previously (13). In each experiment, identical numbers of cells (∼200,000 cells per plate) were plated in 35-mm dishes and transfected 24 h after plating. Two days after transfection, cultures were treated with 500 μm manganese for 16 h. Further processing was as described previously (13). Briefly, cells were harvested by addition of trypsin and pelleted. We confirmed that, in each experiment, the pellet sizes for all transfection conditions were similar. Obtained pellets were washed two times with phosphate-buffered saline supplemented with 10 mm EDTA. After a final wash with EDTA-free phosphate-buffered saline, pellets were resuspended in 20 μl of phosphate-buffered saline (prior to resuspension in 20 μl of phosphate-buffered saline, we reconfirmed that, in each experiment, pellet sizes were similar between transfection conditions). These pellets were transferred to acid-washed vials and digested by the addition of 200 μl of 70% metal-free HNO3. After digestion, samples were diluted to 2% HNO3 using ultrapure water. Intracellular manganese was measured using an Agilent 7500ce quadrupole inductively coupled plasma mass spectrometer. In each experiment, the absolute amount of intracellular manganese (parts/billion) in control transfected cells was normalized to 100, and intracellular manganese levels in cells transfected with SLC30A10 constructs were expressed relative to control.
We used a longer duration of manganese treatment in the metal measurement studies (16 h) compared with the GPP130 degradation assay (4 h). This was essential because prior studies show that, in cell culture, when manganese is added to the extracellular medium, levels of intracellular manganese reach a peak within the first few hours and then decrease (in the continued presence of extracellular manganese) to achieve a steady state that is higher than the basal manganese level but lower than the peak (43). Steady-state levels are achieved between 8 and 24 h (43). For our studies, it was important to measure intracellular manganese once this steady state was achieved. We previously demonstrated that, in HeLa cells, basal manganese levels were low, but treatment with 500 μm manganese for 16 h increased intracellular manganese to a steady-state level that could be reliably detected (13). Furthermore, under this treatment condition, intracellular manganese levels in cells expressing SLC30A10WT were lower than transfection control cells and cells that expressed a transport-inactive mutant of SLC30A10 (13). Based on the above, in this study we retained the 16-h manganese treatment paradigm used for metal measurement assays in our previous study (13).
Immunoblot Assays
Immunoblot analyses, including cell lysis, were performed as described by us previously (13).
Viability Assays
Cell viability was assessed using the methylthiazolyldiphenyltetrazolium bromide assay, essentially as described by us previously (13). Briefly, cells were exposed to indicated amounts of manganese or zinc for 16 h in full media (i.e. minimum essential media supplemented with 10% fetal bovine serum, 100 IU/ml penicillin-G, and 100 μg/ml streptomycin). After this, cultures were washed with phosphate-buffered saline, incubated in Hanks' balanced salt solution (Sigma) containing 0.05% methylthiazolyldiphenyltetrazolium bromide (w/v; EMD Millipore) for 4 h at 37 °C, and lysed using 0.1 n hydrochloric acid in isopropyl alcohol with 1% Triton X-100. Absorption at 570 nm was subsequently measured using a plate reader.
Statistical Analyses
The Prism 6 software (GraphPad, La Jolla, CA) was used. All experiments were repeated at least three times, independently. Comparisons between groups were performed using one-way ANOVA and Dunnett's post hoc test. In general, p < 0.05 was considered to be significant. Asterisks in graphs, where present, denote statistically significant differences.
Author Contributions
S. M. conceived the project. C. E. Z. and S. M. designed experiments. C. E. Z. performed experiments and created data panels. C. E. Z. and S. M. analyzed data. S. M. wrote the manuscript with help from C. E. Z. and M. A.
Acknowledgments
We thank Dr. Nathaniel Miller (Jackson School of Geosciences, University of Texas, Austin) for performing all inductively coupled plasma mass spectrometry assays reported here; Dr. David Eide (University of Wisconsin, Madison), Dr. Adam Linstedt (Carnegie Mellon University), and Dr. Dennis Thiele (Duke University) for providing very useful advice and suggestions and for critical reading of the manuscript. We also thank Dr. Andrey Selyunin and Steven Hutchens (both from University of Texas, Austin) for technical assistance.
This work was supported by National Institutes of Health Grants R00-ES020844 and R01-ES024812 from NIEHS (to S. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ANOVA
- analysis of variance.
References
- 1. Aschner M., Erikson K. M., Herrero Hernández E., Hernández E. H., and Tjalkens R. (2009) Manganese and its role in Parkinson's disease: from transport to neuropathology. Neuromolecular Med. 11, 252–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Milatovic D., Zaja-Milatovic S., Gupta R. C., Yu Y., and Aschner M. (2009) Oxidative damage and neurodegeneration in manganese-induced neurotoxicity. Toxicol. Appl. Pharmacol. 240, 219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stanwood G. D., Leitch D. B., Savchenko V., Wu J., Fitsanakis V. A., Anderson D. J., Stankowski J. N., Aschner M., and McLaughlin B. (2009) Manganese exposure is cytotoxic and alters dopaminergic and GABAergic neurons within the basal ganglia. J. Neurochem. 110, 378–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Olanow C. W. (2004) Manganese-induced parkinsonism and Parkinson's disease. Ann. N.Y. Acad. Sci. 1012, 209–223 [DOI] [PubMed] [Google Scholar]
- 5. Perl D. P., and Olanow C. W. (2007) The neuropathology of manganese-induced Parkinsonism. J. Neuropathol. Exp. Neurol. 66, 675–682 [DOI] [PubMed] [Google Scholar]
- 6. Lucchini R. G., Guazzetti S., Zoni S., Donna F., Peter S., Zacco A., Salmistraro M., Bontempi E., Zimmerman N. J., and Smith D. R. (2012) Tremor, olfactory and motor changes in Italian adolescents exposed to historical ferro-manganese emission. Neurotoxicology 33, 687–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Butterworth R. F. (2013) Parkinsonism in cirrhosis: pathogenesis and current therapeutic options. Metab. Brain Dis. 28, 261–267 [DOI] [PubMed] [Google Scholar]
- 8. Gorell J. M., Johnson C. C., Rybicki B. A., Peterson E. L., Kortsha G. X., Brown G. G., and Richardson R. J. (1999) Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson's disease. Neurotoxicology 20, 239–247 [PubMed] [Google Scholar]
- 9. Lechpammer M., Clegg M. S., Muzar Z., Huebner P. A., Jin L. W., and Gospe S. M. Jr. (2014) Pathology of inherited manganese transporter deficiency. Ann. Neurol. 75, 608–612 [DOI] [PubMed] [Google Scholar]
- 10. Quadri M., Federico A., Zhao T., Breedveld G. J., Battisti C., Delnooz C., Severijnen L. A., Di Toro Mammarella L., Mignarri A., Monti L., Sanna A., Lu P., Punzo F., Cossu G., et al. (2012) Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am. J. Hum. Genet. 90, 467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tuschl K., Clayton P. T., Gospe S. M. Jr., Gulab S., Ibrahim S., Singhi P., Aulakh R., Ribeiro R. T., Barsottini O. G., Zaki M. S., Del Rosario M. L., Dyack S., Price V., Rideout A., Gordon K., et al. (2012) Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am. J. Hum. Genet. 90, 457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tuschl K., Mills P. B., Parsons H., Malone M., Fowler D., Bitner-Glindzicz M., and Clayton P. T. (2008) Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia–a new metabolic disorder. J. Inherit. Metab. Dis. 31, 151–163 [DOI] [PubMed] [Google Scholar]
- 13. Leyva-Illades D., Chen P., Zogzas C. E., Hutchens S., Mercado J. M., Swaim C. D., Morrisett R. A., Bowman A. B., Aschner M., and Mukhopadhyay S. (2014) SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J. Neurosci. 34, 14079–14095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang L., and Tepaamorndech S. (2013) The SLC30 family of zinc transporters–a review of current understanding of their biological and pathophysiological roles. Mol. Aspects Med. 34, 548–560 [DOI] [PubMed] [Google Scholar]
- 15. Kambe T., Tsuji T., Hashimoto A., and Itsumura N. (2015) The physiological, biochemical, and molecular roles of zinc transporters in zinc homeostasis and metabolism. Physiol. Rev. 95, 749–784 [DOI] [PubMed] [Google Scholar]
- 16. Kolaj-Robin O., Russell D., Hayes K. A., Pembroke J. T., and Soulimane T. (2015) Cation diffusion facilitator family: structure and function. FEBS Lett. 589, 1283–1295 [DOI] [PubMed] [Google Scholar]
- 17. Chen P., Bowman A. B., Mukhopadhyay S., and Aschner M. (2015) SLC30A10: a novel manganese transporter. Worm 4, e1042648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mukhopadhyay S., and Linstedt A. D. (2011) Identification of a gain-of-function mutation in a Golgi P-type ATPase that enhances Mn2+ efflux and protects against toxicity. Proc. Natl. Acad. Sci. U.S.A. 108, 858–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wahlberg K., Kippler M., Alhamdow A., Rahman S. M., Smith D. R., Vahter M., Lucchini R. G., and Broberg K. (2016) Common polymorphisms in the solute carrier SLC30A10 are associated with blood manganese and neurological function. Toxicol. Sci. 149, 473–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chao Y., and Fu D. (2004) Thermodynamic studies of the mechanism of metal binding to the Escherichia coli zinc transporter YiiP. J. Biol. Chem. 279, 17173–17180 [DOI] [PubMed] [Google Scholar]
- 21. Gupta S., Chai J., Cheng J., D'Mello R., Chance M. R., and Fu D. (2014) Visualizing the kinetic power stroke that drives proton-coupled zinc(II) transport. Nature 512, 101–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoch E., Lin W., Chai J., Hershfinkel M., Fu D., and Sekler I. (2012) Histidine pairing at the metal transport site of mammalian ZnT transporters controls Zn2+ over Cd2+ selectivity. Proc. Natl. Acad. Sci. U.S.A. 109, 7202–7207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu M., Chai J., and Fu D. (2009) Structural basis for autoregulation of the zinc transporter YiiP. Nat. Struct. Mol. Biol. 16, 1063–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu M., and Fu D. (2007) Structure of the zinc transporter YiiP. Science 317, 1746–1748 [DOI] [PubMed] [Google Scholar]
- 25. Wei Y., and Fu D. (2005) Selective metal binding to a membrane-embedded aspartate in the Escherichia coli metal transporter YiiP (FieF). J. Biol. Chem. 280, 33716–33724 [DOI] [PubMed] [Google Scholar]
- 26. Wei Y., and Fu D. (2006) Binding and transport of metal ions at the dimer interface of the Escherichia coli metal transporter YiiP. J. Biol. Chem. 281, 23492–23502 [DOI] [PubMed] [Google Scholar]
- 27. Wei Y., Li H., and Fu D. (2004) Oligomeric state of the Escherichia coli metal transporter YiiP. J. Biol. Chem. 279, 39251–39259 [DOI] [PubMed] [Google Scholar]
- 28. Coudray N., Valvo S., Hu M., Lasala R., Kim C., Vink M., Zhou M., Provasi D., Filizola M., Tao J., Fang J., Penczek P. A., Ubarretxena-Belandia I., and Stokes D. L. (2013) Inward-facing conformation of the zinc transporter YiiP revealed by cryoelectron microscopy. Proc. Natl. Acad. Sci. U.S.A. 110, 2140–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fujimoto S., Itsumura N., Tsuji T., Anan Y., Tsuji N., Ogra Y., Kimura T., Miyamae Y., Masuda S., Nagao M., and Kambe T. (2013) Cooperative functions of ZnT1, metallothionein and ZnT4 in the cytoplasm are required for full activation of TNAP in the early secretory pathway. PLoS One 8, e77445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fukunaka A., Kurokawa Y., Teranishi F., Sekler I., Oda K., Ackland M. L., Faundez V., Hiromura M., Masuda S., Nagao M., Enomoto S., and Kambe T. (2011) Tissue nonspecific alkaline phosphatase is activated via a two-step mechanism by zinc transport complexes in the early secretory pathway. J. Biol. Chem. 286, 16363–16373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohana E., Hoch E., Keasar C., Kambe T., Yifrach O., Hershfinkel M., and Sekler I. (2009) Identification of the Zn2+-binding site and mode of operation of a mammalian Zn2+ transporter. J. Biol. Chem. 284, 17677–17686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shusterman E., Beharier O., Shiri L., Zarivach R., Etzion Y., Campbell C. R., Lee I. H., Okabayashi K., Dinudom A., Cook D. I., Katz A., and Moran A. (2014) ZnT-1 extrudes zinc from mammalian cells functioning as a Zn2+/H+ exchanger. Metallomics 6, 1656–1663 [DOI] [PubMed] [Google Scholar]
- 33. Montanini B., Blaudez D., Jeandroz S., Sanders D., and Chalot M. (2007) Phylogenetic and functional analysis of the cation diffusion facilitator (CDF) family: improved signature and prediction of substrate specificity. BMC Genomics 8, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martin J. E., and Giedroc D. P. (2016) Functional determinants of metal ion transport and selectivity in paralogous cation diffusion facilitator transporters CzcD and MntE in Streptococcus pneumoniae. J. Bacteriol. 198, 1066–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Delhaize E., Gruber B. D., Pittman J. K., White R. G., Leung H., Miao Y., Jiang L., Ryan P. R., and Richardson A. E. (2007) A role for the AtMTP11 gene of Arabidopsis in manganese transport and tolerance. Plant J. 51, 198–210 [DOI] [PubMed] [Google Scholar]
- 36. Dokmanić I., Sikić M., and Tomić S. (2008) Metals in proteins: correlation between the metal-ion type, coordination number and the amino-acid residues involved in the coordination. Acta Crystallogr. D Biol. Crystallogr. 64, 257–263 [DOI] [PubMed] [Google Scholar]
- 37. Kelley L. A., and Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 38. Dunn K. W., Kamocka M. M., and McDonald J. H. (2011) A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 300, C723–C742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mukhopadhyay S., Bachert C., Smith D. R., and Linstedt A. D. (2010) Manganese-induced trafficking and turnover of the cis-Golgi glycoprotein GPP130. Mol. Biol. Cell 21, 1282–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mukhopadhyay S., and Linstedt A. D. (2012) Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science 335, 332–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Palmiter R. D., and Findley S. D. (1995) Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 14, 639–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Anton A., Weltrowski A., Haney C. J., Franke S., Grass G., Rensing C., and Nies D. H. (2004) Characteristics of zinc transport by two bacterial cation diffusion facilitators from Ralstonia metallidurans CH34 and Escherichia coli. J. Bacteriol. 186, 7499–7507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Masuda M., Braun-Sommargren M., Crooks D., and Smith D. R. (2013) Golgi phosphoprotein 4 (GPP130) is a sensitive and selective cellular target of manganese exposure. Synapse 67, 205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin H., Kumánovics A., Nelson J. M., Warner D. E., Ward D. M., and Kaplan J. (2008) A single amino acid change in the yeast vacuolar metal transporters ZRC1 and COT1 alters their substrate specificity. J. Biol. Chem. 283, 33865–33873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nishito Y., Tsuji N., Fujishiro H., Takeda T. A., Yamazaki T., Teranishi F., Okazaki F., Matsunaga A., Tuschl K., Rao R., Kono S., Miyajima H., Narita H., Himeno S., and Kambe T. (2016) Direct comparison of manganese detoxification/efflux proteins and molecular characterization of ZnT10 as a manganese transporter. J. Biol. Chem. 291, 14773–14787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lapinskas P. J., Cunningham K. W., Liu X. F., Fink G. R., and Culotta V. C. (1995) Mutations in PMR1 suppress oxidative damage in yeast cells lacking superoxide dismutase. Mol. Cell. Biol. 15, 1382–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Missiaen L., Raeymaekers L., Dode L., Vanoevelen J., Van Baelen K., Parys J. B., Callewaert G., De Smedt H., Segaert S., and Wuytack F. (2004) SPCA1 pumps and Hailey-Hailey disease. Biochem. Biophys. Res. Commun. 322, 1204–1213 [DOI] [PubMed] [Google Scholar]
- 48. Ton V. K., Mandal D., Vahadji C., and Rao R. (2002) Functional expression in yeast of the human secretory pathway Ca2+, Mn2+-ATPase defective in Hailey-Hailey disease. J. Biol. Chem. 277, 6422–6427 [DOI] [PubMed] [Google Scholar]
- 49. Ramos-Castañeda J., Park Y. N., Liu M., Hauser K., Rudolph H., Shull G. E., Jonkman M. F., Mori K., Ikeda S., Ogawa H., and Arvan P. (2005) Deficiency of ATP2C1, a Golgi ion pump, induces secretory pathway defects in endoplasmic reticulum (ER)-associated degradation and sensitivity to ER stress. J. Biol. Chem. 280, 9467–9473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Selyunin A. S., and Mukhopadhyay S. (2015) A conserved structural motif mediates retrograde trafficking of Shiga toxin types 1 and 2. Traffic 16, 1270–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]