

Abstract
Hepatitis B virus (HBV) is a stealth virus, minimally inducing the interferon system required for efficient induction of both innate and adaptive immune responses. However, 90% of acutely infected adults can clear the virus, suggesting the presence of other, interferon-independent pathways leading to viral clearance. Given the known ability of helicases to bind viral nucleic acids, we performed a functional screening assay to identify helicases that regulate HBV replication. We identified the superkiller viralicidic activity 2-like (SKIV2L) RNA helicase (a homolog of the Saccharomyces cerevisiae Ski2 protein) on the basis of its direct and preferential interaction with HBV X-mRNA. This interaction was essential for HBV X-mRNA degradation at the RNA exosome. The degradation of HBV X-mRNA at the RNA exosome was also mediated by HBS1L (HBS1-like translational GTPase) protein, a known component of the host RNA quality control system. We found that the redundant HBV-precore translation initiation site present at the 3′-end of HBV X-mRNA (3′ precore) is translationally active. The initiation of translation from this site without a proper stop codon was identified by the non-stop-mediated RNA decay mechanism leading to its degradation. Although 3′ precore is present in the five main HBV-RNA transcripts, only X-mRNA lacks the presence of an upstream start codons for large, middle, and small (L, M, and S) HBV surface proteins. These upstream codons are in-frame with 3′ precore translation initiation site, blocking its translation from the other HBV-mRNA transcripts. To our knowledge, this is the first demonstration of the anti-viral function of the non-stop-mediated RNA decay mechanism.
Keywords: exosome complex; hepatitis B virus (HBV, Hep B); RNA-binding protein; RNA degradation; RNA helicase; HBS1L; Non-Stop mediated RNA Decay (NSD); RNA Exosome; SKIV2L; virus restriction factor
Introduction
Viral infections are initially detected and counteracted by the host through the innate immune response (1). The activation of the innate response is required for the production of interferon (IFN) and other inflammatory cytokines that act both directly (by suppressing the viral infection) and indirectly (by bridging the shift from innate to adaptive immune response, permitting the efficient elimination of the infection) (2). Some of the innate antiviral immune responses are continuously expressed, not amplified, permitting the restriction of incoming viruses immediately after infection. These processes are constitutive and are not induced by the virus or external ligands (3). Such processes depend on host factors that detect “non-self” signature in the incoming virus particles or components thereof.
HBV3 is a DNA virus that infects 3 billion people worldwide, of whom ∼350–400 million harbor chronic infections (4, 5). HBV infection leads to a variety of liver diseases ranging from acute or fulminant hepatitis to liver cirrhosis and hepatocellular carcinoma (6). The HBV genome is partially double-stranded relaxed circular DNA of 3200 base pairs with overlapping open reading frames (ORFs). Inside the nucleus the relaxed circular DNA is converted by the host DNA repair pathway to form the covalently closed circular DNA. Although HBV covalently closed circular DNA is transcribed into five main RNA species with different lengths, all of these transcripts share the same polyadenylation signal and have a common 3′-terminus (7). The smallest of these transcripts (0.7 kb) encodes the HBV X protein, which is a transcription transactivator that is important for HBV life cycle. Several studies have showed that HBV-X induces HBV-replication through the activation of transcription of its mRNAs (8–11).
Although HBV is known to act like a stealth virus, evading the innate immune response and the induction of IFN-stimulated genes (ISGs) (12), 90% of acutely HBV-infected adults clear the virus spontaneously. This observation suggests the possible presence of other, IFN-independent host mechanisms that facilitate HBV clearance.
Through targeting of viral-RNA for clearance, the host RNA-decay machineries have been implicated in the host defense against viral infection. In plants and insects, the major anti-viral mechanism is the RNA interference (RNAi). The RNAi processes viral RNAs into short interfering RNAs (siRNAs) leading to endonucleolytic cleavage and degradation of complementary target mRNAs (13). In mammalian cells, however, RNAi does not appear to play a significant antiviral role (14). Zinc-finger antiviral protein (ZAP) was found to degrade human immunodeficiency virus (HIV), filoviruses, Sindbis virus, HBV, and murine gamma herpesvirus 68 (MHV68) viral RNAs containing ZAP-responsive element (ZRE). ZAP binding of ZREs recruits cellular RNA-decay machinery, including that of the RNA exosome (13, 15).
RNA quality control mechanisms are thought to have evolved to screen for, and degrade, abnormal RNA transcripts, thereby maintaining the integrity of the flow of genetic information (from DNA to RNA to functional protein) (13). Viral RNAs, which may exhibit abnormal RNA structures, may be identified by these mechanisms, leading to degradation of these transcripts. One of these RNA quality control mechanisms, nonsense-mediated RNA decay (NMD), has been shown to participate in the host anti-viral defense by identifying foreign viral RNAs and targeting such transcripts for degradation in both plants and mammals (16, 17). In yeast, aberrant mRNAs lacking in-frame termination codons are recognized and degraded by the non-stop decay (NSD) pathway. The NSD mechanism requires the interaction of RNA exosome with the Ski complex, a multiprotein structure that includes the Ski2p helicase and (notably) Ski7p; this combination activates the degradation of aberrant mRNAs (18, 19). The eukaryotic RNA exosome is a ubiquitous endo- and 3′ → 5′ exoribonuclease that cooperates with multiple co-factors, including the SKIV2L helicase, to direct the processing, quality control, and degradation of virtually all classes of RNAs. It includes highly conserved nine-subunit core proteins that share structural similarity with phosphorolytic exoribonucleases such as bacterial polynucleotide phosphorylase (20). Because mammalian cells lack an apparent Ski7p homolog, the occurrence of NSD in mammals has been a matter of debate. Recently, NSD was reported to occur in mammals, where the process was shown to depend on Hbs1, Dom34 (a binding partner of Hbs1), and components of the exosome-Ski complex (including, SKIV2L helicase, the human homolog of the yeast Ski2) (21).
Results
Screening for Helicases Suppressing HBV Replication
Screening for human helicases that suppress HBV replication was performed as mentioned under “Experimental Procedures” (Fig. 1A); in short, we used Hep38.7-Tet cells, a subclone derived from HepAD38 in which HBV replication is controlled by a tetracycline-regulated promoter and a pre-arrayed library of 909 different lentiviruses carrying shRNA sequences targeting the genes encoding each of 133 human helicases. HBV replication was initiated by withdrawal of tetracycline treatment 24 h after shRNA transfection. Medium was changed every 2 days after a wash with PBS. Spent medium was collected on days 6 and 7. Cell viability on day 7 was measured using the XTT assay. Helicase gene-suppressing shRNA clones with increased HBV-DNA levels in the culture medium were subjected to confirmatory screening by measuring the intracellular level of HBV-DNA.
FIGURE 1.

SKIV2L suppresses HBV replication. A, Diagram representing the screening approach. B, helicases that suppress HBV replication. Two or more different shRNAs targeting the genes encoding DDX3X, DDX3Y, DDX41, DHX35, DDX60, SKIV2L, or HelB helicases (or non-targeting control shRNAs) were transfected into Hep38.7-Tet (+) cells 2 days before tetracycline withdrawal, and stable transfectants were selected using puromycin. After 1 week of selection, HBV-DNA titers were quantified (separately) in the cells and medium using real-time PCR. Titers were plotted relative to those obtained in the control cells (transfected with non-targeting shRNAs). Three independent experiments were performed in triplicate, and data are presented as the mean ± S.D. C, HepG2 cells were co-transfected with HBV-D60 together with pEF4-SKIV2L-MycHis or empty vector (pEF4-MycHis). Four days after transfection, HBV core-associated DNA was quantified by real-time PCR. Data are plotted as -fold difference compared with mock-treated cells. Three independent experiments were performed in triplicate, and data are presented as the mean ± S.D. D, HepG2 cells were co-transfected with HBV-D60 plasmid or empty vector (pUC19; mock) together with pEF4-SKIV2L-MycHis or pEF4-MycHis. Three days later intracellular HBV-RNA levels were assessed by Northern blot. Northern blot analysis of HBV-RNA is shown in the upper panel. The lower panel shows the cellular ribosomal RNA stained with ethidium bromide. E, firefly luciferase reporter plasmids harboring HBV promoters (or no insert), Renilla luciferase reporter plasmids, and either SKIV2L or empty vector constructs were co-transfected into HepG2 cells. Dual luciferase assays were performed at 48 h after transfection. Relative luciferase units are plotted as -fold difference relative to mock-treated cells. Three independent experiments were performed in triplicate, and data are presented as the mean ± S.D. Three independent experiments were performed in triplicate, and data are presented as the mean ± S.D. Statistical significance was measured using two-tailed Student's t test. ** = p < 0.01.
Helicases whose HBV suppression activity was confirmed in the secondary screen (DDX3X, DDX3Y, DDX41, DHX35, DDX60, SKIV2L, and HELB) showed more than 2-fold induction of HBV-DNA titers in both the cells and medium when the corresponding genes were independently silenced with two or three different shRNAs (Fig. 1B). Notably, DDX3 previously was reported to bind HBV-polymerase protein and block its reverse transcriptase activity (22). Additionally, DDX41 and DDX60 were reported as possible viral DNA sensors that induce anti-viral processes (23, 24). The anti-viral function of DHX35, SKIV2L, and HELB do not appear to have been previously reported.
SKIV2L RNA Helicase Shows anti-HBV Function
SKIV2L is the human homologue of the yeast SKI2 helicase. Because SKI2P previously was reported to suppress viral replication in yeast (25), we assessed whether SKIV2L possesses a similar activity in HepG2 cells, a line known to support HBV replication. Silencing of SKIV2L expression with two different shRNAs resulted in increased HBV-DNA titers in both cells and medium (Fig. 1B). SKIV2L overexpression in pUC-HBV-D-IND60 (HBV-D60)-transfected cells yielded decreased levels of both HBV core-associated DNA (p < 0.01) and HBV-RNA, as shown by real-time PCR (Fig. 1C) and Northern blot analysis (Fig. 1D), respectively. SKIV2L overexpression in this background also slightly induced HBV core promoter activity while exhibiting no effect on pre-S, S, and X promoters (Fig. 1E). These data suggest that SKIV2L suppresses HBV at a post-transcriptional level.
It is worth noting that SKIV2L-induced suppression of HBV replication was IFN-independent, as shown in Fig. 2. Although interferon-α (IFN-α) induced ISG15, no change in SKIV2L expression was observed (Fig. 2A). This result suggests that SKIV2L is not an IFN-induced gene. We also investigated whether SKIV2L can induce IFN. Although IFN-β induction was observed when HepG2 cells were transfected with TANK-binding kinase 1 (TBK1), no induction was observed with SKIV2L overexpression (Fig. 2B). IFN-I and IFN-III have been shown to induce Janus kinase (JAK) signal transducer and activator of transcription (STAT) signaling, leading to the activation of interferon-stimulated response element (ISRE) promoter activity (26). IFN-α treatment significantly induced ISRE-promoter activity (p < 0.01) (Fig. 2C). However, neither HBV nor SKIV2L activated ISRE-promoter activity (p > 0.05) (Fig. 2D). We extended this analysis by using SNC-IR cells, a subclone (derived from HCV subgenomic replicon cells SNC#2; Ref. 27) that is resistant to IFN due to the lack of IFNAR2 expression (Fig. 2E). SNC-IR cells were cured of HCV by treatment with cyclosporine A (SNC-IRC cells) and used for analysis of the SKIV2L effect on HBV replication. Transfection with SKIV2L significantly suppressed HBV replication in SNC-IRC cells (p < 0.01) (Fig. 2F).
FIGURE 2.

SKIV2L suppression of HBV replication is independent from the IFN system. A, HepG2 cells were treated with the indicated dose of IFN-α or subjected to mock treatment. After 24 h, the expression of SKIV2L and ISG15 was quantified by real-time RT-PCR; values in IFN-treated cells are presented relative to those in mock-treated cells. Three independent experiments were performed in triplicate, and data are presented as the mean ± S.D. B, HepG2 cells were transfected with pEF4-SKIV2L-MycHis, pEF4-MycHis, pcDNA3-TBK1, or pcDNA3 together with both the IFN-β luciferase reporter plasmid and the Renilla luciferase reporter plasmid (pTK-Rluc). Dual luciferase assays were performed at the indicated time points. Firefly luciferase activities are plotted as -fold difference relative to level at 0 h. Three independent experiments were performed in triplicate, and data are presented as the mean ± S.D. C, HepG2 cells were transfected with empty vector (pEF4-MycHis; Mock), ISRE-driven luciferase plasmid (pISRE-TA-Luc), and Renilla luciferase (pTK-Rluc) reporter plasmid in a 3:1:0.2 ratio (respectively). 24 h later, cells were treated with either PBS or IFN-α 100 IU/ml. 24 h later, dual luciferase assays were performed. Relative luciferase units were plotted as -fold difference relative to PBS-treated cells at the respective time point. Three independent experiments were performed in triplicate, and data are presented as the mean ± S.D. D, HepG2 cells were transfected with ISRE-driven luciferase plasmid (pISRE-TA-Luc) and Renilla luciferase (pTK-Rluc) reporter plasmid together with one of the following combinations: pEF4-MycHis + pUC19 (Mock), pEF4-SKIV2L-MycHis + pUC19 (SKIV2L), or pUCHBV-D60 + pEF4-MycHis (HBV). Dual luciferase assays were performed at the indicated time points. Relative luciferase units were plotted as -fold difference relative to 0- mock-treated cells. Three independent experiments were performed in triplicate, and data are presented as the mean ± S.D. E, total-RNA preparations from SNC#2, SNC-IR, and SNC-IRC replicon cells were extracted and subjected to RT-PCR to determine the expression of IFNAR1, IFNAR2, HCV replicon RNA, and GAPDH. F, SNC-IRC cells were co-transfected with HBV-D60 together with pEF4-SKIV2L-MycHis or pEF4-MycHis. 4 days after transfection, HBV core-associated DNA was quantified by real-time PCR. Data are plotted as -fold difference relative to pEF4-MycHis-treated cells. Three independent experiments were performed in triplicate, and data are presented as the mean ± S.D. Statistical significance was measured was measured using two-tailed Student's t test. ** = p < 0.01.
SKIV2L Preferentially Binds HBV X-mRNA
SKIV2L encodes a RNA helicase (28). We hypothesized that SKIV2L-induced HBV suppression might be mediated through SKIV2L interaction with one or more HBV RNAs. Notably, the HBV genome encodes at least five primary mRNA transcripts. To investigate a possible interaction between SKIV2L and any of the HBV RNAs, we co-transfected HepG2 hepatoma cells with a SKIV2L-Myc construct together with HBV-D60 and performed RNA immunoprecipitation (RIP) experiments (Fig. 3A) (the SKIV2L-Myc construct encodes a Myc-tagged SKIV2L protein, permitting use of anti-Myc antibody for immunoprecipitation of the helicase and bound transcripts). To exclude nonspecific interactions of the Myc antibody used for RIP, or HBV-RNAs with the purified protein, we performed another RIP experiment with anti-Myc antibody after transfecting HepG2cells with a plasmid expressing another Myc-tagged protein (TSSK2-Myc) together with HBV-D60 (Fig. 3B). We used two sets of primers to quantify HBV-mRNA transcripts by real-time RT-PCR analysis. One set was designed to amplify a sequence (nt 64–357) shared by HBV precore, pre-genomic, pre-S1, and pre-S2 mRNA transcripts; the other set of primers was designed to amplify another sequence (nt 1386–1625) shared by HBV X-mRNA and the above-mentioned 4 transcripts (Fig. 3A) (nucleotide numbering is based on the HBV-D_IND60 sequence, GenBankTM accession number AB246347.1 (29). We compared the abundance of RNA transcripts containing the nt 1386–1625 or nt 64–357 sequences in the Myc antibody-precipitated samples to the original levels of the same transcripts in the input samples by real-time RT-PCR using the ΔΔCT method in which the host GAPDH, mRNAs titers in both the input and the Myc antibody-precipitated samples, were used as endogenous controls. RNA transcripts containing the nt 1386–1625 or nt 64–357 sequences were found to be enriched >10-fold or 2-fold, respectively, in the Myc antibody-precipitated samples from SKIV2L-Myc (Fig. 3C, lower panel) but not TSSK2-Myc transfected cells (Fig. 3C, upper panel). The output samples pulled down by the control isotype antibody (mouse IgG) did not show a significant change in HBV RNA titers relative to those of GAPDH mRNAs (Fig. 3C). These data indicate that (in general) HBV RNA transcripts are bound at higher levels by SKIV2L than are other cellular mRNAs like those encoding GAPDH (Fig. 3, B and C). Notably, the HBV-X transcript containing nt 1386–1625 (but not that containing nt 64–357) showed higher (1-log) preferential interaction with SKIV2L helicase (Fig. 3C, lower panel).
FIGURE 3.

SKIV2L preferentially binds HBV X-mRNA. A, HepG2 cells were transfected with either pEF4-TSSK2-MycHis or pEF4-SKIV2L-MycHis and with HBV-D60 plasmids as indicated. Lysates were extracted at 48 h after transfection. Aliquots (1/10 volumes) were used for the detection of TSSK2, SKIV2L and β-actin (loading control) by immunoblotting (left upper and lower panels); separate aliquots (1/10 volumes) were used for RNA extraction, and the detection of HBV RNA titers by real-time RT-PCR is shown in B. The remaining lysate volumes were subjected to RIP assay using either isotype control antibody (mouse IgG) or mouse anti-Myc IgG to pull down TSSK2-Myc or SKIV2L-Myc proteins. Immunoblotting analysis was performed for the detection of TSSK2 or SKIV2L-Myc in the output samples (right panel). B, HBV mRNA titers in the indicated samples were quantified relative to GAPDH mRNA levels. Three independent experiments were performed in triplicate, and data are presented as the mean ± S.D. C, a diagram representing the sites of the primers used to quantify HBV mRNAs associated with SKIV2L after SKIV2L pulldown by RIP assay. Primers HBV-D/MS-F (64-) and HBV-D/S-R (-357) were used to quantify (collectively) the 4 largest HBV transcripts, including HBV-pg, precore, Pre-S1, and Pre-S2 transcripts; primers HBV-D/X-F (1386-) and HBV-D/X-R (-1625) were used to quantify (collectively) all 5 transcripts, consisting of the above 4 as well as the HBV X mRNA (nucleotide numbering is based on the HBV-ADR sequence). Three independent experiments were performed data are presented as the mean ± S.D.
HBV X-mRNA Binding to RNA Exosome Is SKIV2L-dependent
SKIV2L is thought to deliver RNA substrates to the catalytic core of the RNA exosome (30). We, therefore, investigated the association of HBV X-mRNA with the RNA exosome complex. Because the RIP assay previously has been reported to identify RNAs associated with the RNA exosome (31), we pulled down either EXOSC4 (Fig. 4A) or EXOSC5 (Fig. 4B) proteins (known components of the RNA exosome) and studied their association with HBV X-mRNA. For efficient production of detectable quantities of the HBV-X transcript, we cloned a sequence corresponding to the HBV X-mRNA sequence (nt 1237–1933); nucleotide numbering is based on the HBV-D_IND60 sequence (GenBankTM accession number AB246347.1) lacking the X promoter region along with a 12-nt tail after the poly(A) signal (TATAAA)) into the pcDNA3.1 plasmid; this construct (pcDNA3.1-X) placed the inserted sequence under control of the CMV promoter. pcDNA3.1-X was used to study the HBV X-mRNA interaction with two different RNA exosome proteins; we performed the assay with the two different proteins to avoid possible protein-specific interactions. Although siRNA silencing of SKIV2L yielded increased HBV X-mRNA titers (Fig. 4, C (p < 0.05) and D (p < 0.05)), the RIP assay showed that silencing of SKIV2L suppressed HBV X-mRNA interaction with RNA exosome components EXOSC4 (Fig. 4E) and EXOSC5 (Fig. 4F). These results show that SKIV2L is required for the HBV X-mRNA/RNA exosome interaction. We further performed an intracellular localization of the interaction between SKIV2L and the RNA exosome complex. Cytoplasmic interaction between SKIV2L and EXOSC4 or between SKIV2L and EXOSC5 was demonstrated both by co-localization (Fig. 5, A and B) and by proximity ligation assay (Fig. 5C) using immunofluorescence analysis. These data suggest that the cytoplasm is the site for the interaction of these components and the subsequent HBV X-mRNA degradation.
FIGURE 4.

SKIV2L is required for the interaction between HBV-RNA and RNA exosome. A, 293T cells were transfected with either nonspecific siRNA or SKIV2L-specific siRNAs. 24 h later, cells were transfected with pEF-BOS and pcDNA3.1 (Mock) or with pEF-BOS-EXOSC4-FLAG and pcDNA3.1-X plasmids as indicated. 48 h later cell lysates were extracted. Aliquots (1/10 volumes) of the inputs were used for the detection of EXOSC4 (left three lanes), endogenous β-actin, and endogenous SKIV2L (left lower two panels) by immunoblotting; separate aliquots ( volumes) were used for RNA extraction, and the detection of HBV X-mRNA titers by real-time RT-PCR is shown in B. For the remaining lysate volumes from pEF-BOS-EXOSC4-FLAG- and pcDNA3.1-X-transfected cells, RIP assays were performed using either mouse anti-FLAG antibody or isotype control (mouse IgG). EXOSC4-FLAG was detected in the output samples by immunoblotting with anti-FLAG tag antibody (right four lanes). B, 293T cells were transfected with either nonspecific siRNA or SKIV2L-specific siRNAs. 24 h later, cells were transfected with pEF-BOS and pcDNA3.1 (Mock) or with pEF-BOS-EXOSC5-FLAG and pcDNA3.1-X plasmids as indicated. 48 h later cell lysates were extracted. Aliquots (1/10 volumes) of the inputs were used for the detection of EXOSC5 (left three lanes), endogenous β-actin, and endogenous SKIV2L (left lower two panels) by immunoblotting; separate aliquots (1/10 volumes) were used for RNA extraction and the detection of HBV X-mRNA titers by real-time RT-PCR as shown in E. For the remaining lysate volumes from pEF-BOS-EXOSC5-FLAG- and pcDNA3.1-X-transfected cells, RIP assays were performed using either mouse anti-FLAG antibody or isotype control (Mouse IgG). EXOSC5-FLAG was detected in the output samples by immunoblotting with anti-FLAG tag antibody (right four lanes). C, HBV X-mRNA titers in the indicated input samples from A were quantified using GAPDH mRNA titers as the internal control. Data are plotted as -fold difference compared with titers in the cells co-transfected with the combination of EXOSC4-FLAG, pcDNA3.1-X, and nonspecific siRNA. Data from three different experiments are presented as the mean ± S.D. D, HBV X-mRNA titers in the indicated input samples from D were quantified using GAPDH mRNA titers as the internal control and plotted as -fold difference compared with titers in the cells co-transfected with the combination of EXOSC5-FLAG, pcDNA3.1-X, and nonspecific siRNA. Data from three experiments are presented as the mean ± S.D. E, HBV X-mRNA titers in the indicated output samples from A were quantified relative to GAPDH mRNA titers in the same sample. Data are plotted as -fold difference compared with levels in samples pulled down with isotype control antibody (Mouse IgG). Data from three experiments are presented as the mean ± S.D. F, HBV X-mRNA titers in the indicated output samples from D were quantified relative to GAPDH mRNA titers in the same sample. Data are plotted as -fold difference compared with levels in samples pulled down with isotype control antibody (Mouse IgG). Data from three experiments are presented as the mean ± S.D. Statistical significance was measured was measured using two-tailed Student's t test (paired). * = p < 0.05. ** = p < 0.01.
FIGURE 5.
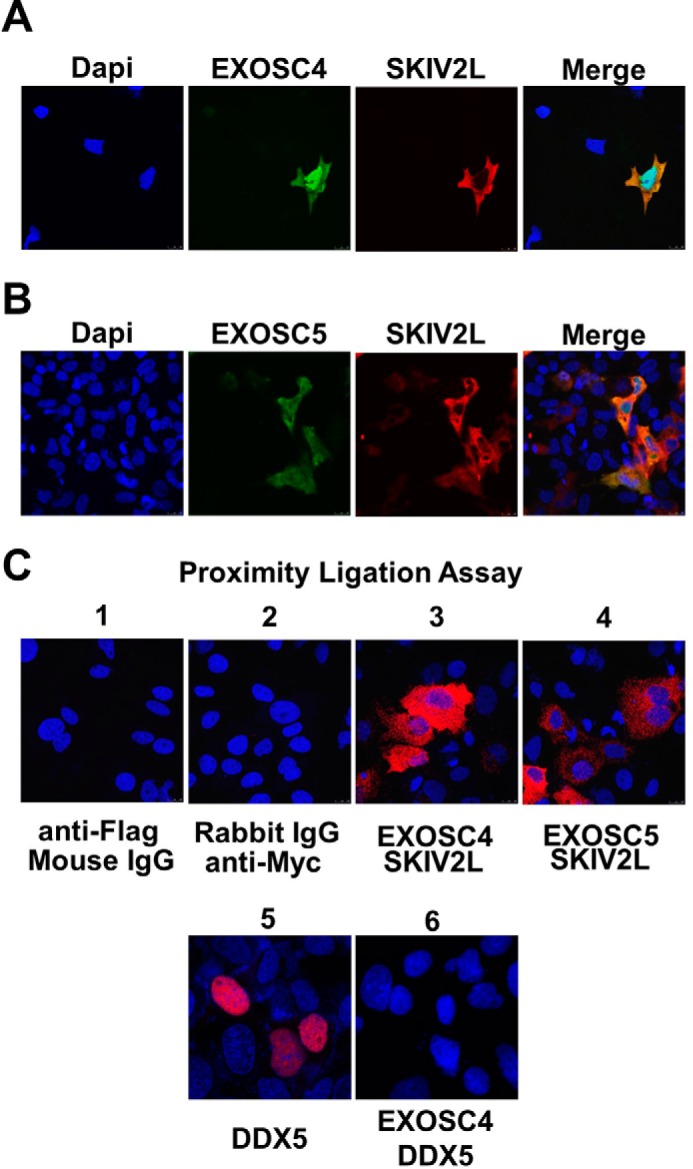
Intracellular localization of SKIV2L/RNA exosome interaction. HuH-7 cells were transfected with pEF4-SKIV2L-MycHis in combination with either pEF-BOS-EXOSC4-FLAG (A) or pEF-BOS-EXOSC5-FLAG plasmids (B). Intracellular localization of SKIV2L-Myc and EXOSC4-FLAG or EXOSC5-FLAG proteins was visualized by immunofluorescence analysis using rabbit anti-SKIV2L and mouse anti-FLAG antibodies. The left panels show the nuclear staining with DAPI (blue). EXOSC4-FLAG and EXOSC5-FLAG intracellular expression was detected by Alexa 488-conjugated secondary antibody (green). SKIV2L intracellular expression was detected using Alexa 555-conjugated secondary antibody (red). The right panels show a merged images of EXOSC4 or 5 (green) with SKIV2L (red) staining C, proximity ligation assay showing the interaction between SKIV2L and EXOSC4 (panel C3) and between SKIV2L and EXOSC5 (panel C4) proteins as red dots. Panels C1 and C2 show the staining observed with combinations of isotype control antibodies; the lack of red staining in these panels confirms that the assay does not detect nonspecific binding. As a control for a nonspecific interaction between over-expressed proteins at the PLA assay, panel C5 shows the nuclear expression of the overexpressed Halo tagged DDX5 helicase using mouse anti-Halo antibody, whereas panel C6 shows no interaction between DDX5 and EXOSC4 protein in PLA assay as evident by the lack of the red dots.
HBS1L-dependent Decay of HBV X-mRNA at the RNA Exosome
RNA quality control mechanisms are known to utilize the SKIV2L-RNA exosome system for the degradation of aberrant RNA and can be classified into UPF1-dependent (NMD) or HBS1L-dependent (NSD and No-Go RNA decay) mechanisms (13). Therefore, we analyzed the effect of either UPF1 or HBS1L on SKIV2L-mediated suppression of HBV X-mRNA. SKIV2L overexpression yielded a significant decrease in HBV X-mRNA titers (p < 0.05) (Fig. 6A). Silencing of UPF1 did not alter this suppression; however, silencing of HBS1L restored HBV X-mRNA titers to levels statistically indistinguishable (p > 0.05) from those seen in the absence of SKIV2L overexpression (Fig. 6A). HBS1L and UPF1 siRNA knockdown efficiency is shown (Fig. 6B). Similarly, EXOSC4 and -5 interaction with HBV X-mRNA was significantly reduced (p < 0.05 and p < 0.01, respectively) when HBS1L expression was silenced by the inclusion of a HBS1L-specific siRNA (Fig. 6, C and D). These data suggest that the mechanism of RNA exosome-mediated interaction with (and suppression of) HBV X-mRNA is HBS1L-dependent. Because the interaction of HBV X-mRNA with the RNA exosome may eventually lead to its degradation, we investigated the effect of SKIV2L, HBS1L, and RNA exosome on the stability of HBV X-mRNA. The central channel of the RNA exosome is formed by six evolutionary conserved subunits, including EXOSC4 and EXOSC5. Deletion of any of these proteins has been shown to significantly affect RNA exosome structure and assembly (32). De novo RNA transcription was stopped by actinomycin D treatment, and HBV X-mRNA decay overtime was detected by Northern blot analysis. In our hands silencing of either EXOSC4 or EXOSC5 expression stabilized HBV X-mRNA at 8 h after actinomycin D treatment, confirming the role of the RNA exosome in HBV X-mRNA degradation. This effect also was seen with silencing of SKIV2L and HBS1L expression, showing involvement of the products of these genes in HBV X-mRNA decay (Fig. 7, A and B). EXOSC4 and -5 siRNA knockdown efficiency is shown (Fig. 7C). Because mRNA decay by the SKIV2L-activated RNA exosome occurs via a nonsense (3′-end)-mediated decay process, we thus conclude that HBV X-mRNA decay is a 3′-end process that is dependent on HBS1L, SKIV2L, and the RNA exosome.
FIGURE 6.

HBS1L mediated interaction and suppression of HBV X-mRNA. A, HepG2 cells were transfected with either nonspecific siRNAs (NS) or with UPF1-specific (si-UPF1) or HBS1L-specific (si-HBS1L) siRNAs. 24 h later, the cells were co-transfected with pcDNA3.1-X together with pEF4-SKIV2L-MycHis or empty vector (pEF4-MycHis; mock). After 48 h, HBV-RNA levels were quantified by real-time RT-PCR. Data are plotted as -fold difference relative to mock-treated cells. Three independent experiments were performed in triplicate, and data are presented as the mean ± S.D. B, HepG2 cells were transfected with either nonspecific siRNAs or with UPF1-specific (si-UPF1) (upper panel) or HBS1L-specific (si-HBS1L) siRNAs (lower panel). 72 h later the cells were lysed, the cellular proteins were extracted, and the effect of the siRNAs silencing on the protein levels of its target gene were analyzed by immunostaining using the specific antibodies. C, 293T cells were transfected with either nonspecific or HBS1L (si-HBS1L) siRNAs. 24 h later, cells were transfected with pcDNA3.1-X plasmids as indicated. After 48 h, cell lysates were extracted. Aliquots (1/10 volumes) of the inputs were used for the detection of endogenous EXOSC4 (upper left panel), endogenous HBS1L and β-actin (loading control) (middle left panel), or endogenous EXOSC5 (lower left panel) by immunoblotting; separate aliquots (1/10 volumes) were used for RNA extraction and determination of HBV X-mRNA titers by real-time RT-PCR as shown in B. For the remaining lysate volumes, RIP assays were performed using either anti-EXOSC4, anti-EXOSC5 IgG, or isotype control antibody (rabbit IgG). EXOSC4 or EXOSC5 were detected in the output samples by immunoblotting (right upper and lower panels, respectively). D, HBV X-mRNA titers in EXOSC4, and -5 pulled down samples (upper and lower panels, respectively) were quantified relative to GAPDH mRNA titers in the same sample. HBV X-mRNA levels are plotted as -fold difference compared with levels in samples pulled down with isotype control antibody (mouse IgG). Data from three experiments are presented as the mean ± S.D. Statistical significance was measured was measured using two-tailed Student's t test. * = p < 0.05, ** = p < 0.01.
FIGURE 7.

Silencing of genes encoding SKIV2L, HBS1L, or exosome complex proteins increases the stability of HBV X mRNA. A, two different experiments (Exp. 1 and Exp. 2) in which HepG2 cells were transfected with nonspecific siRNA (si-NS), si-SKIV2L, si-HBS1L, si-EXOSC4, and si-EXOSC5 as indicated, and 24 h later, cells were transfected with pcDNA3.1-X. After 48 h the cells were treated with actinomycin D (8 μg/ml). Total cellular RNA was collected at the indicated time points, and HBV X mRNA was detected by Northern blot analysis (upper panels of Exp. 1 and 2). As a loading control, the 28S and 18S ribosomal rRNAs in the input samples were visualized (after electrophoresis) by staining with ethidium bromide and imaging under UV illumination (lower panels of Exp. 1 and 2). B, three independent experiments reproducing the Northern blot analysis shown in A were performed, and HBV X-mRNA densities were calculated by Image J software and plotted as -fold difference compared with the density at 0 h. Data are presented as the mean ± S.D. C, HepG2 cells were transfected with nonspecific siRNA, si-EXOSC4 (left panel), or si-EXOSC5 (right panel) siRNAs. After 48 h, the cells were lysed, and total proteins were isolated. The expression of each gene was quantified by immunostaining. Statistical significance was measured was measured using two-tailed Student's t test. * = p < 0.05; ** = p < 0.01.
The Secondary Structure of HBV-epsilon at the 3′-End of HBV X-mRNA Does Not Affect Its Degradation
The complex RNA secondary structure (the HBV-epsilon structure) present at the 5′-end of HBV-pre-genomic RNA (pgRNA) recently was reported to be sensed by RIG-I, leading to the induction of interferon λ (33). This result suggests that HBV-epsilon may be identified by host anti-viral factors as a pathogen-associated molecular pattern (PAMP). Because the epsilon structure also is present at the 3′-end of the HBV X-mRNA, we analyzed the role of HBV-epsilon on HBV X-mRNA decay. We disrupted the secondary structure of HBV-epsilon by using constructs in which we replaced the 5′-GUCC-3′ sequence (present on the more-5′ strand of the lower stem loop (nt 1854–1857)) with the complementary 5′-CAGG-3′ sequence or by mutating the complementary sequence (on the other strand (nt 1897–1900)) to 5′-CCUG-3′ (Fig. 8A). Nucleotide numbering is based on the HBV-D_IND60 sequence (GenBankTM accession number AB246347.1). In cells harboring the wild type or mutated HBV-epsilon, transcription was stopped by exposure to actinomycin D, and the decay of HBV X-mRNA from the wild-type HBV X, 5′-CAGG-3′, and 5′-CCTG-3′ mutants was followed over time by Northern blot analysis (Fig. 8B). The time series of HBV X-mRNA levels was used to calculate the half-life of each transcript, and these values were plotted as -fold difference compared with the half-life of wild-type HBV X-mRNA (Fig. 8C). There was no statistically significant difference between the half-lives of the three different HBV X-mRNA transcripts, suggesting the absence of a significant effect of HBV-epsilon structure on the rate of HBV X-mRNA decay.
FIGURE 8.

A, schematic showing the mutations introduced to disrupt HBV-epsilon's secondary structure. Either the sequence from nt 1854 to 1857 on the 5′ strand was mutated to 5′-CAGG-3′ or the sequence from nt 1897 to1900 on the 3′ strand was mutated to 5′-CCUG-3′ (nucleotide numbering is based on the HBV-D_IND60 sequence. GenBankTM accession number AB246347.1). B, HepG2 cells were transfected with pcDNA3.1-X, pcDNA3.1-X (5′-CAGG-3′), or pcDNA3.1-X (5′-CCTG-3′). 48 h later the cells were treated with actinomycin D (8 μg/ml). Total cellular RNA was collected at the indicated time points, and HBV X-mRNA levels were determined by Northern blot analysis (upper panels). As a loading control, the 28S and 18S ribosomal RNAs in the input samples were stained with ethidium bromide (lower panels). C, the density of the different HBV X-mRNA bands detected by the Northern blot analysis shown in A were quantified and used to calculate the half-lives of HBV X-mRNA. The half-lives of HBV X-mRNA encoded by the different constructs were plotted as the -fold difference compared with that of wild-type HBV X-mRNA. Data from three experiments are presented as the mean ± S.D. NS, not significant.
NSD-mediated Decay of HBV X-mRNA
Analysis of the HBV X-mRNA structure shows the presence of redundant precore translation initiation sites at its 3′-end (3′ precore), upstream of the polyadenylation signal and without any in-frame stop codon; hence, any translation from these sites will result in a stalled ribosome. Such transcripts are known to be identified by the HBS1L-dependent NSD RNA quality control mechanism, leading to the degradation of the identified RNA in SKIV2L/RNA exosome-dependent manner. To demonstrate that translation occurs from a 3′ precore initiation site, we re-engineered plasmid pcDNA3.1-X to replace the small redundant sequence of HBV-core corresponding to the 3′-end of the HBV X-mRNA coding sequence with a Gaussia luciferase (GLuc)-encoding sequence lacking a start codon. In the resulting construct, the GLuc coding sequence was in-frame with only one of the known start codons, the ATG of the 3′ precore, making GLuc a reporter for translation from this ATG (Fig. 9A). As a negative control, we mutated the 3′ precore ATG to TTG. We transfected the resulting constructs into HepG2 cells and monitored luciferase activity. The presence of an active start codon (ATG) in the 3′ precore translation initiation site yielded 10-fold higher luciferase activity than that obtained with the TTG negative control (Fig. 9B). These data confirm that translation can be initiated from this redundant precore translation initiation site at the 3′-end of HBV X-mRNA. Although the 3′ precore translation site is present in all HBV-mRNAs, it is expected to be translationally active only in the HBV X-mRNA, leading to the initiation of NSD. The 3′ precore and HBV-surface proteins (S) start codons (L/M/S) are in-frame; the presence of an in-frame translation initiation site (S proteins) upstream of the 3′ precore will prevent the scanning ribosome from translating the 3′ precore (Fig. 9C).
FIGURE 9.
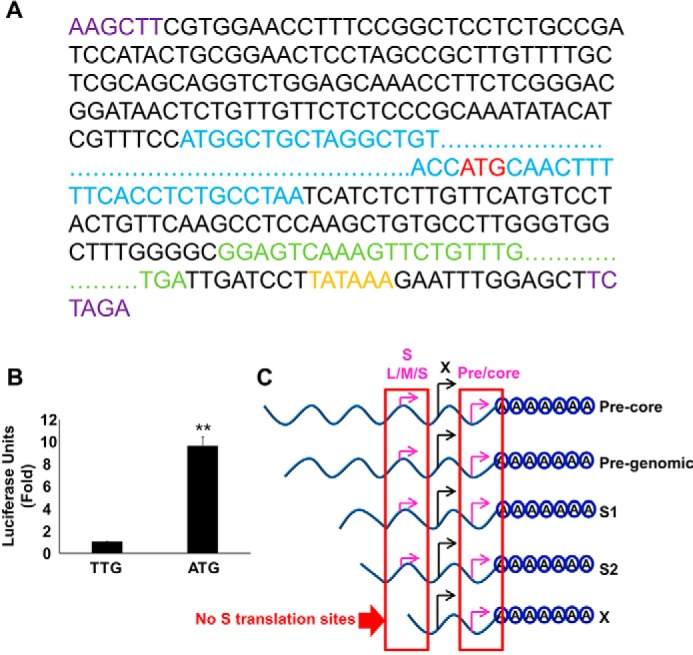
Translation from the 3′ precore regulates the stability of HBV X-mRNA. A, schematic of the HBV X mRNA-Gaussia luciferase (GLuc) coding sequence translation fusion. The GLuc sequence (green) was inserted in place of the redundant core sequence corresponding to the 3′-end of the X mRNA; a HBV-poly(A) signal (orange) also was inserted after the GLuc coding sequence. The ATG start codon (red) of the 3′ precore was left intact or was mutated to TTG (to prevent possible translation from this site). This entire sequence then was subcloned (via flanking HINDIII and XBAI sites (purple)) into the pcDNA3.1 vector, placing the construct under the control of the CMV promoter. Because the GLuc coding sequence is only in-frame with the 3′ precore ATG start codon, GLuc expression should result only if the ribosome skips the first ATG (X ORF) and starts translation at the 3′ precore ATG start codon (red). The X protein ORF is shown in blue. For clarity, extended sequences are not shown and are instead indicated by dots. B, the 3′ precore translation initiation site is active in HBV X-mRNA. HepG2 cells were transfected with either pcDNA3.1-X (ATG-GLuc) or pcDNA3.1-X (TTG-GLuc) plasmids containing the GLuc sequence in frame with an active (ATG) or mutated (TTG) 3′ precore translation start codon, respectively. 48 h later GLuc activity was measured in the medium and plotted as -fold difference relative to the activity from pcDNA3.1-X (TTG-GLuc)-transfected cells. Three independent experiments were performed in triplicate, and the data are was presented as the mean ± S.D. C, a diagram showing the in-frame sequence of HBV-3′ precore and HBV-S (L/M/S) start codons; as a result of this translation is only expected to be initiated from the HBV-3′ precore in the X transcript because it lacks the S (L/M/S) start codons. Statistical significance was measured was measured using two-tailed Student's t test. ** = p < 0.01.
Precore Premature Stop Codon Increases HBV X-mRNA Stability
We next investigated the effect of translation termination after initiation from the precore ATG on HBV X-mRNA stability. Specifically, HBV nt 1896 was mutated from G to A, converting a TGG triplet to a TAG stop codon in-frame with the 3′ precore ATG. This mutation, which leads to a precore premature stop, is commonly observed in HBV e antigen-negative patients (34), who typically exhibit higher serum levels of HBV-DNA (35). We analyzed the effect of this mutation on HBV X-mRNA degradation over time by Northern blot analysis after stopping transcription by exposure to actinomycin D. In clones harboring the premature precore stop codon, HBV X-mRNA exhibited increased stability compared with HBV X-mRNA produced from wild-type clones (Fig. 10, A and B). In contrast to the effect of silencing HBS1L or EXOSC4 on the increased stability of wild type HBV X-mRNA (Fig. 7, A and B), it did not affect the stability of HBV X-mRNA from the clones harboring the premature precore stop codon (Fig. 10, C and D). These data suggest that wild type HBV X-mRNA decay is regulated by NSD RNA quality control and that the introduction of the premature precore stop codon suppresses this mechanism and induces HBV X-mRNA stability.
FIGURE 10.

Precore premature stop codon increases HBV X-mRNA stability. A, HepG2 cells were transfected with pcDNA3.1-X or pcDNA3.1-X (G1896A), a construct harboring a stop codon in-frame with the 3′ precore ATG. 48 h later the cells were treated with actinomycin D (8 μg/ml). Total cellular RNA was collected at the indicated time points, and detection of HBV X-mRNA was performed by Northern blot analysis (upper panels). As a loading control, the 28S and 18S ribosomal RNAs in the input samples were stained with ethidium bromide (lower panels). Northern blot data from two independent experiments (1 and 2) were shown. B, HBV X-mRNA density was calculated by Image J software, plotted in the lower panel as -fold difference compared with the density at 0 h. Data from three independent experiments are presented as the mean ± S.D. C, HepG2 cells were transfected with si-NS, si-HBS1L, or si-EXOSC4 as indicated. 24 h later, cells were transfected with pcDNA3.1-X (G1896A). After 48 h, the cells were treated with actinomycin D (8 μg/ml). Total cellular RNA was collected at the indicated time points, and HBV X mRNA was detected by Northern blot analysis (upper panels). As a loading control, the 28S and 18S ribosomal rRNAs in the input samples were visualized (after electrophoresis) by staining with ethidium bromide and imaging under UV illumination (lower panels). D, HBV X-mRNA bands density shown in C were calculated by Image J software, plotted as -fold difference compared with the density at 0 h. Data from three independent experiments are presented as the mean ± S.D. NS, not significant. E, a diagram explaining the possible effect of G1896A mutation leading to the escape of HBV X mRNA from NSD RNA quality control. Statistical significance was measured was measured using two-tailed Student's t test. ** = p < 0.01
Discussion
RNA quality control mechanisms can identify aberrant structures in the mRNA transcripts inducing a stall of the translation machinery (13). Given their compact genomes, many viruses produce RNAs with atypical features. For example, some viruses encode multiple proteins within one polycistronic mRNA molecule (13). Translation of open reading frames at the 5′-end of such transcripts terminates well upstream from the poly(A) tail. The large distance between the ribosome present at the stop codon and the poly(A) tail precludes interaction of the ribosome with the poly(A)-binding protein (PABP), a situation known to induce UPF1-dependent NMD RNA quality control mechanism (36) leading to viral RNA degradation at the SKIV2L/RNA exosome system. NMD RNA quality control recently has been shown to identify and degrade positive-strand viral RNA viruses like Sindbis virus and Semliki Forest virus (16). A similar scenario can be applied on the translation of the HBV-core protein from the HBV-pgRNA or precore transcripts, in which a long distance (around 2600 nucleotides) is present between the stop codon of the core's coding ORF and the poly(A) tail. NMD is dependent on the function of UPF1 helicase (37). Notably, knocking down of UPF1 with any of five different shRNAs in the course of our helicase screening experiment did not affect HBV replication, and silencing of UPF1 with gene-specific siRNAs did not rescue HBV X-mRNA from SKIV2L-induced suppression. These results suggest that NMD does not play a role in the degradation of HBV mRNAs.
Overlapping ORFs and mRNA transcripts are the result of the compact structure of HBV genome; such a structure may lead to some abnormalities in translation that can be identified in a translation-dependent pattern and targeted by the host. All HBV mRNA transcripts include (at the 3′-end just before the poly(A) tail) redundant translation initiation sites for HBV-precore (3′ precore) and core (3′ core) ORFs. Because these redundant sites do not have an in-frame stop codon, translation from these sites will end in a stalled ribosome, a situation known to induce HBS1L-dependent NSD RNA quality control, thereby leading to the degradation of the affected RNA (21). Given that the 3′ precore and 3′ core translation initiation codons are in-frame, a translation from the first start codon (precore) will mask and prevent translation from the subsequent start codon (core). In a similar pattern, the translation initiation codon of the 3′ precore is in-frame with the upstream translation initiation sites of large, middle, and small ORFs; hence, it is expected not to be translated from any HBV mRNA transcript containing these sites. The HBV X-mRNA transcript is the only HBV transcript that lacks the start codons for small ORFs; additionally, it is notable that the start codon for the X ORF is out-of-frame with respect to the 3′ precore ORF start codon. Thus, a ribosome scanning the HBV X-mRNA in the same frame as the 3′ precore ATG will skip the first ATG (for the X ORF) and proceed to translate the 3′ precore ORF. Because the 3′ precore ORF of the HBV X-mRNA lacks a stop codon, the resulting ribosome is expected to read through the end of the transcript and become stalled, presumably invoking NSD RNA quality control. In accordance with this hypothesis, we found that HBV X-mRNA transcript showed the highest interaction with SKIV2L, and its degradation was mediated by HBS1L-dependent mechanism at the RNA exosome. Using luciferase assay, we also showed that translation from 3′ precore is active in the HBV X-mRNAs transcripts. Furthermore, introduction of a stop codon (via the G1896A mutation) to the 3′ precore ORF increased the stability of this transcript. All in all, these data suggest that HBV X-mRNA is identified and degraded by the NSD RNA quality control pathway.
Interestingly, the G1896A mutation is frequently observed in HBV e antigen-negative patients, a subgroup characterized by high levels of HBV replication and more severe liver disease (35). We hypothesize that this mutation enables the virus to escape the degradation of HBV X-mRNA; the increased accumulation of the X transcript (and of the X protein) could be responsible for the increased severity of HBV symptoms in these patients (Fig. 10E).
The ZAP protein was previously shown to target nt 1820–1918 of HBV-pgRNA, leading to nuclear degradation of this transcript by the RNA exosome (15). In contrast, we demonstrated that degradation of HBV X-mRNA occurs via a cytoplasmically localized translation-dependent NSD RNA quality control mechanism. Both of these differences (the higher affinity of the SKIV2L-HBV X-mRNA interaction and the cytoplasmic location) suggest the existence of a novel, ZAP-independent mechanism that suppresses HBV replication by targeting HBV X-mRNA for degradation via the RNA exosome.
HBV-X protein is a transcription transactivator and is important for HBV life cycle. Several studies have showed that HBV-X induces HBV replication through the activation of transcription of its mRNAs (8–11). Human hepatocytes chimeric mice injected with HBx-deficient HBV virus developed measurable viremia only in HBx-expressing livers (38). Moreover, using primary human hepatocyte and differentiated HepaRG cells demonstrated that HBx is essential to initiate and constantly required to maintain HBV infection (39). Although HBV X-mRNA is not the replicating HBV-RNA transcript (HBV-pgRNA), through the suppression of HBV-X by the degradation of its mRNA the host will effectively attenuate HBV life cycle.
In conclusion, we identified the novel anti-HBV function of the NSD-dependent RNA quality control mechanism that preferentially targets HBV X-mRNA for degradation at the RNA exosome complex. Fig. 11 summarizes this mechanism.
FIGURE 11.

The degradation of HBV-RNA by the SKIV2L/RNA exosome pathway. A ribosome may skip the first HBV X ORF translation site and instead initiate translation from the 3′ precore start codon. Due to the lack of an in-frame stop codon, such a ribosome will stall in the poly(A) tail of the transcript. The stalled ribosome will be recognized by HBS1L, which in turn will induce NSD RNA quality control, leading to the degradation of the HBV X-mRNA by the SKIV2L-RNA exosome system.
Experimental Procedures
Cell Culture, Reagents, siRNAs, and Transfection
HepG2 and Hep38.7-Tet (40), HuH7 cells (41), and LucNeo#2 cells (42) were cultured as previously described. Hep38.7-Tet (+) cells were treated with tetracycline (Sigma, 400 ng/ml). Hep38.7-Tet (−) were withdrawn from tetracycline treatment for 5 days (to induce HBV replication) except where a different withdrawal time is indicated. SNC-IR cells were derived from HCV subgenomic replicon cells SNC#2 (27) as a subclone that is resistant to IFN-α. The subclone was cured of HCV by exposure to cyclosporine A (Sigma; 1 μg/ml) for 1 week. IFN-α was obtained from Schering-Plough. si-SKIV2L, si-UPF1, si-HBS1L, si-EXOSC4, and si-EXOSC5 siRNAs were obtained as Silencer Select reagents from Life Technologies. Silencer Select were transfected using Lipofectamine RNAiMAX (Life Technologies); plasmid DNA transfection of HepG2 and 293T cells was performed using TransIT-LTI (Mirius) or Lipofectamine 2000 (Life Technologies), respectively. Reverse transfection was used to transfect siRNA into HepG2 and 293T cells.
Screening for Human Helicases That Suppress HBV Replication
The screen for suppressors of HBV replication was performed using Hep38.7-Tet cells, which were previously reported to efficiently support HBV replication and covalently closed circular DNA formation (40). To maximize the effect of shRNA silencing on HBV replication, these cells were transfected with shRNA during growth in the presence of tetracycline (Tet (+)); tetracycline subsequently was withdrawn (Tet (−)) to initiate HBV replication. We used a pre-arrayed library (Sigma) of 909 different lentiviruses carrying shRNA sequences targeting the genes encoding each of 133 human helicases; each lentivirus also harbored a gene endowing transfectants with puromycin resistance. 24 h post-transfection, medium containing tetracycline was replaced with medium containing 2 μg/ml puromycin (Life Technologies). Subsequently, medium was changed every 2 days after a wash with PBS. Spent medium was collected on days 6 and 7. Cell viability on day 7 was measured using the XTT assay (Roche Applied Science). Helicase gene-suppressing shRNA clones with increased HBV-DNA levels in the culture medium were subjected to confirmatory screening by measuring the intracellular level of HBV-DNA. Targets that yielded reproducible 2-fold or greater increases in HBV-DNA levels in both medium and cells with ≥2 shRNA sequences and with cellular viability ≥80% of control shRNA transfected cells were selected for further investigation.
Plasmid Construction
SKIV2L expressing vector pFN21AE4596 was purchased from flexi ORF clones (Promega). SKIV2L-cDNA was then excised and inserted into pFC14K-HaloTag Flexi Vector (Promega) using carboxyl Flexi system (Promega) forming the pFC14K-SKIV2L-Halo. To construct pEF4-SKIV2L-MycHis, SKIV2L cDNA was cloned from pFC14K-SKIV2L-Halo using the primers SKIV2L-EcoRI-S (5′-AGGTAGAATTCACCATGATGGAGACAGAGCGAC-3′) and SKIV2L-XbaI-AS (5′-CTTCTTCTAGATCACTGGGTGTAGAGGCTGGC-3′). The obtained fragment is cut by the corresponding restriction enzymes and ligated into EcoRI/XbaI sites of pEF4-MycHis plasmid. HBV-D-IND60 sequences corresponding to the following nucleotide sequence of HBV subtype ADW2, accession number AM282986, S1 promoter (2710–2828), S2 promoter (2927–3115), and X promoter (951–1310) were PCR cloned. The primers used were: for S1 promoter: S1-SacI-S 5′-AAGCAGAGCTCAACATCTAGTTAATCATTAC-3′, and S1-ECOR5-AS 5′-CTTCTGATATCAGAATATGGTGACCCACAAAATG-3′; for S2 promoter: S2-SacI-S 5′-AAGCAGAGCTCCAGCAAATCCAGATTGGGAC-3′, and S2-ECOR5-AS 5′-CTTCTGATATCTCTGGCGATTGGTGGAGGCAG-3′; for X promoter X-SACI-S 5′-AAGCAGAGCTCAAAACTTCCTATTAACAGGCC-3′, and X-ECOR5-AS 5′-CTTCTGATATCGACCTGCTGCGAGCAAAAC-3′. The obtained fragments were cut by the corresponding restriction enzymes and ligated into the SACI/ECOR5 restriction sites of pGL4.28 vector (Promega). For efficient production of detectable amount of HBV X-mRNA transcript, the HBX X-mRNA sequence (nt 1237- 1933 of HBV-D-IND60) lacking the X promotor region and including 12 nucleotides after the poly(A) signal (TATAAA) was obtained by PCR amplification using the primers XTr-HindIII-S (5′-TCCAGAAGCTTCGTGGAACCTTTGTGGCTCC-3) and XTr-XbaI-AS (5′-GTCCTTCTAGAAGCTCCAAATTCTTTATAAGGATCAA-3′) and inserted into pcDNA3.1 plasmid under control of CMV promoter after digestion with HindIII/XbaI to construct the pcDNA3.1-X. For the disruption of HBV-epsilon secondary structure at the lower stem loop, HBV X-mRNA (nt 1237- 1933 of HBV-D-IND60) was amplified from pcDNA3.1-X using the primers XTr-HindIII-S (5′-TCCAGAAGCTTCGTGGAACCTTTGTGGCTCC-3′) and one of the following antisense primers, XTr-CCTG-AS (5′-CTTGGTCTAGAAGCTCCAAATTCTTTATAAGGGTCAATGTCCATCAGGCAAAGCCACCCAAGGCACAGCTTGGAGGCTTGAACAGTGGGACATGTACA-3′) or XTr-CAGG-AS (5′-CTTGGTCTAGAAGCTCCAAATTCTTTATAAGGGTCAATGTCCATGCCCCAAAGCCACCCAAGGCACAGCTTGGAGGCTTGAACAGTGCCTGATGTACA-3′). The obtained fragments were inserted into the HindIII/XbaI restriction sites of pcDNA3.1 plasmid to construct pcDNA3.1-X (5′-CCTG-3′) and pcDNA3.1-X (5′-CAGG-3′), respectively. To construct the precore premature stop codon (G1896A mutations), two different DNA fragments were amplified from pcDNA3.1-X using the primers XTr-HindIII-S (5′-TCCAGAAGCTTCGTGGAACCTTTGTGGCTCC-3′) together with G1896AS (5′-CAATGTCCATGCCCTAAAGCC-3′) and G1896A-S (5′-GGCTTTAGGGCATGGACATTG-3′) together with XTr-XbaI-AS (5′-GTCCTTCTAGAAGCTCCAAATTCTTTATAAGGATCAA-3′). These fragments were joined together using the XTr-HindIII-S and XTr-XbaI-AS primers and inserted into pcDNA3.1 plasmid after digestion with HindIII and XbaI to construct the pcDNA3.1-X (G1896A). To insert GLuc under translation control of the HBV X-mRNA 3′ precore translation initiation site, the GLuc sequence lacking ATG was cloned from pCMV-GLuc using the primers XTr-GLuc-S (5′-GGTGGCTTTGGGGCGGAGTCAAAGTTCTG-3′) and GLuc-XbaI-AS (5′-GGCCCTCTAGAAGCTCCAAATTCTTTATATTAGTCACCACCGGCCCCC-3′). Another fragment was cloned from pcDNA3.1-X using XTr-HindIII-S and XTr-GLuc-AS (5′-CAGAACTTTGACTCCGCCCCAAAGCCACC-3′). The obtained two fragments were joined together using XTr-HindIII-S and GLuc-XbaI-AS primers. The resulting fragment was inserted into the pcDNA3.1 plasmid after digestion with HindIII and XbaI to construct the pcDNA3.1-X (ATG-GLuc) plasmid. To construct a similar plasmid with a silenced 3′ precore translation initiation site, two different DNA fragments were amplified from pcDNA3.1-X (ATG-GLuc) using the following primers: XTr-HindIII-S together with TTG-AS (5′-GTTGCAAGGTGCTGGTGCGC-3′) and TTG-S (5′-GCGCACCAGCACCTTGCAAC-3′) together with XTr-XbaI-AS. These fragments were joined together using the XTr-HindIII-S and GLuc-XbaI-AS primers and inserted into pcDNA3.1 plasmid after digestion with HindIII and XbaI to construct the pcDNA3.1-X (TTG-GLuc) (Fig. 9A). pUCHBV-D_IND60 (HBV-D60) (29), core promoter reporter plasmid (43), and IFN-β reporter plasmid (44) were described previously. TSSK2 cDNA was obtained from HepG2 total cellular RNA using superscript III first-strand synthesis kit (Invitrogen); TSSK2 coding sequence was then amplified using TSSK2-BAMHI-S (5′-AAGCAGGATCCGCCACCATGGACGATGCCACAGTCC-3′) and TSSK2-XBAI-AS (5′-GGTCCTCTAGAGGTGCTTGCTTTCCCCACCTC-3′). The obtained fragments were inserted into the BAMHI/XbaI restriction sites of pEF4-MycHis plasmid to form pEF4-TSSK2-MycHis. pFN21AB4122 plasmid expressing N-terminal Halo-tagged DDX5 was purchased from Promega. Using the carboxyl Flexi system transfer (Promega), DDX5 ORF was switched into pFC14A plasmid to express the Halo tag at the C-terminal end.
RNA Extraction and Real-time RT-PCR
Primers used are listed in Table 1. For Real time PCR, we used Beta Actin primer-probe set (TaqMan Gene Expression Assay, Applied Biosystems). We also used predesigned KiCqStart primers from (H_SKIV2L_2 for SKIV2L).
TABLE 1.
The primers utilized in this study
| Gene | Description | Sequence |
|---|---|---|
| Real-time RT-PCR | ||
| IFN-β | Sense | 5′-GGCCATGACCAACAAGTGTCTCCTCC-3′ |
| Anti-sense | 5′-ACAGGTTACCTCCGAAACTGAGCGC-3′ | |
| GAPDH | Sense | 5′-CCACTCCTCCACCTTTGAC-3′ |
| Anti-sense | 5′-ACCCTGTTGCTGTAGCCA-3′ | |
| ISG15 | Sense | 5′-CATGGGCTGGGACCTGACG-3′ |
| Anti-sense | 5′-CGCCAATCTTCTGGGTGATCTG-3′ | |
| HBV-RNA | HBV-D/MS-F (64-) | 5′-TGGCTCCAGTTCAGGAACAG-3′ |
| Pre-S2 | HBV-D/S-R (-357) | 5′-GTTGGAGGACAGGAGGTTGG-3′ |
| HBV-RNA | HBV-D/X-F (1386-) | 5′-CTGTGCTGCCAACTGGATCC-3′ |
| X | HBV-D/X-R (-1625) | 5′-GTTCACGGTGGTCTCCATGC-3′ |
| RT-PCR | ||
| IFNAR1 | Sense | 5′-AGTGTTATGTGGGCTTTGGATGGTTTAAGC-3′ |
| Anti-sense | 5′-TCTGGCTTTCACACAATATACAGTCAGTGG-3′ | |
| IFNAR2 | Sense | 5′-ATTTCCATCTATTGTTGAGG-3′ |
| Anti-sense | 5′-CACTTTCTTCTTTCTGTTGA-3′ | |
| SNC#2-NS5A | Sense | 5′-GGACTTGCTGGAAGACACTG-3′ |
| Anti-sense | 5′-GGCATTCACCAGGAACTCGA-3′ | |
| GAPDH | Sense | 5′-CATGTTCGTCATGGGTGTGAACCA-3′ |
| Anti-sense | 5′-AGTGATGGCATGGACTGTGGTCAT-3′ | |
| Real time PCR | ||
| HBV-DNA | Sense (2983-) | 5′-AAGGTAGGAGCTGGAGCATTCG-3′ |
| Anti-sense (-3093) | 5′-AGGCGGATTTGCTGGCAAAG-3′ | |
| Probe | 5′FAM-AGCCCTCAGGCTCAGGGCATAC-3′TAMRA | |
Total cellular RNA was extracted using TRIzol reagent (Life Technologies). RNA was treated with RQ1 RNase-free DNase (Promega) to exclude residual DNA contamination. RNA then was re-purified using the RNeasy Mini kit (Qiagen). For real-time RT-PCR, reverse transcription for RIP output samples was performed using the Superscript Vilo cDNA Synthesis kit (Thermo Fisher); reverse transcription for other samples was performed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems). For HBV, real-time PCR was performed with the TaqMan Gene Expression Master Mix (Applied Biosystems). For SKIV2L, ISG15, UPF1, HBS1L, EXOSC4, and EXOSC5, the Fast SYBR® Green Master Mix (Life Technologies) was used. Expression of HBV-RNA was normalized to that of the human β-actin-encoding gene (Applied Biosystems); expression of other loci was normalized to that of the endogenous GAPDH-encoding gene. Relative real-time quantification was performed using the Step One Plus Real-Time PCR systems (Life Technologies).
DNA Extraction, Core-associated HBV-DNA, and Real-time PCR
For the extraction of HBV-DNA from the medium, spent medium was combined with an equal volume of Sidestep Lysis and Stabilization Buffer (Agilent) and then assessed by real-time PCR. Cellular DNA was extracted using the DNeasy Blood and Tissue kit (Qiagen). The method for purification of cytoplasmic core-associated HBV-DNA was adapted from Pugh et al. (45). Purified DNA was subjected to real-time PCR using TaqMan Gene Expression Master Mix (Applied Biosystems) as previously described (46).
Northern Blot Analysis
Aliquots (20 μg) of total RNA were subjected to RNA electrophoresis in a 1% agarose gel (Seakem) containing formaldehyde. The 28S and 18S rRNAs were visualized in the gel by staining with ethidium bromide and illuminating with UV before transfer. The gel then was washed and transferred to Amersham Biosciences Hybond N+ membrane (GE Healthcare) using the gravity (downward transfer) method in 20× SSC buffer overnight. After transfer, the membrane was washed and cross-linked with a UV cross-linker. For use as a probe, the 3.2-kb HBV-D60 plasmid was linearized with EcoRI and then labeled with alkaline phosphatase enzyme using the AlkPhos Direct Labeling and Detection System (GE Healthcare). Hybridization was performed by overnight incubation at 55 °C and washed following the manufacturer's protocol. Chemiluminescent signals (indicative of hybridization to HBV-RNA) were generated using CDP-Star Detection Reagent (GE Healthcare) and detected using an ImageQuant LAS 4000 Mini (GE Healthcare). The probe utilized for the detection of HBV X-mRNA was constructed by excising the X-encoding fragment from pcDNA3.1-X plasmid using double-digestion with HINDIII + XBAI and then labeling the fragment using alkaline phosphatase as described above.
Immunoblot Analysis
Immunoblot analysis was performed as described previously (46). Primary antibodies were as follows: mouse anti-FLAG clone M2 (Sigma), mouse anti-cMyc clone 9E10 (Santa Cruz), mouse anti-β-actin clone AC-15 (Sigma), mouse anti-GAPDH clone GAPDH-71.1 (Sigma), rabbit polyclonal anti-SKIV2L (Proteintech), rabbit polyclonal anti-EXOSC4 (ab137250, Abcam), rabbit polyclonal anti-EXOSC5 (ab168804, Abcam), rabbit monoclonal anti-UPF1 (ab109363, Abcam), and rabbit anti-HBS1L (10359–1-AP, Proteintech). For secondary antibodies, we used Mouse TrueBlot ULTRA (anti-mouse Ig HRP) or rabbit TrueBlot (anti-rabbit IgG HRP) (Rockland).
Immunofluorescence and Proximity Ligation Assay
HuH-7 cells were used for immunofluorescence due to their larger size, which facilitated clear subcellular localization of the interaction between SKIV2L and the RNA exosome. SKIV2L-Myc was stained using mouse monoclonal anti-Myc (Santa Cruz) as the primary antibody followed by anti-mouse Alexa 555-conjugated secondary antibody (Molecular Probes). FLAG-tagged versions of the EXOSC4 and EXOSC5 proteins were stained using rabbit anti-FLAG antibody (Sigma) as the primary antibody followed by anti-rabbit Alexa 488-conjugated secondary antibody (Molecular Probes).
DDX5-Halo was stained using mouse monoclonal anti-Halo (Promega) as the primary antibody followed by anti-mouse Alexa 555-conjugated secondary antibody (Molecular Probes). Nuclei were visualized using DAPI. A proximity ligation assay showing the interaction between SKIV2L or DDX5 with EXOSC4 or EXOSC5 proteins was performed using the Duolink system (Sigma). For this assay the primary antibodies were the same as those used in immunofluorescence; the secondary antibodies were Duolink In-Situ PLA Probe Anti-Mouse PLUS Affinity-purified Donkey Anti-Mouse IgG (H+L) and Duolink In-Situ PLA Probe Anti-Rabbit MINUS Affinity-Purified Donkey Anti-Rabbit IgG (H+L).
RNA Co-immunoprecipitation
HepG2 and 293T cells were used for RIP analysis when indicated. The RIP assay was performed using the Magna RIP kit (Millipore; catalogue no. 17-700) according to the manufacturer's instructions, with minor modifications as follows. Live cells were cross-linked using formaldehyde (final concentration, 0.75%) for 10 min followed by glycine treatment (final concentration, 125 mm) before cell lysis. Lysates were subjected to reverse cross-linking for 2 h at 70 °C, then digested with Proteinase K before RNA purification. Aliquots (1/10 volume) of the input samples were for used immunoblotting of the desired proteins; separate aliquots (1/10 volume) were used for RNA extraction followed by assessment of RNA expression by RT-PCR. For pulldown Myc-tagged proteins (SKIV2L-MycHis and TSSK2-MycHis), FLAG-tagged proteins (EXOSC4-FLAG and EXOSC5-FLAG), and endogenous EXOSC4 and -5 proteins we used mouse anti-cMyc (clone 9E10; Santa Cruz), mouse anti-FLAG antibody (clone M2; Sigma), rabbit polyclonal anti-EXOSC4 (ab137250, Abcam), and rabbit polyclonal anti-EXOSC5 (ab168804, Abcam) antibodies, respectively. Normal mouse, or rabbit IgG (Santa Cruz) was used as the isotype control. The resulting output protein and RNA samples were utilized for immunoblotting and real time RT-PCR analysis, respectively.
Reporter Assays
HBV promoters reporter luciferase (Luc) plasmids, IFN-β-promoter Luc, or pISRE-TA-luc (Stratagene) express firefly luciferase driven by HBV core, pre-S, S, and X promoters, IFN-β-promoter, or ISRE respectively. Renilla luciferase in PRL-TK was driven by herpes simplex virus thymidine kinase promoter. Gaussia luciferase was translated by HBV X-mRNA redundant 3′ precore translation initiation site. After plasmid transfection or IFN-α treatment, cells were lysed, and luciferase activities were measured as described previously (46). To analyze translation from the HBV X-mRNA redundant 3′ precore translation initiation site, HepG2 cells were transfected with pcDNA3.1-X (ATG-GLuc) or pcDNA3.1-X (TTG-GLuc). 48 h later luciferase activity was measured in the medium. The effect of transfected plasmids on HCV replication was assessed in LucNeo#2 cells, which encode a HCV-1b replicon RNA carrying a luciferase-encoding gene; luciferase activity in these transfected cells was determined as described previously (42).
Statistical Analysis
Results from three independent experiments are expressed as the mean ± S.D. Statistical differences were calculated using two-tailed Student's t test (paired) using Microsoft Excel software. Levels of significance are indicated as follows: *, p < 0.05; **, p < 0.01.
Author Contributions
H. H. A., T. W., T. K., and K. C. designed the project. K. W., S.-i. H, and M. H. supplied key research materials and contributed to the preparation of the figures. H. H. A. and J. S. performed the experiments and analyzed the results. H. H. A. drafted the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We gratefully acknowledge all our collaborators: Dr. Masaya Sugiyama (National Center for Global Health and Medicine) for the pUCHBV-D_IND60 plasmid, Dr. Takayuki Hishiki (Tokyo Metropolitan Institute of Medical Science) for the SNC-IR cells, Dr. Hiroyuki Oshiumi (Kumamoto University) for the EXOSC4- and EXOSC5-encoding plasmids, Dr. K. Kuroki (Kanazawa University) for the pCSD3.5 plasmid, and all members of the Department of Virology II, NIID, for valuable discussions.
This study was supported by the Research Program on Hepatitis from the Japan Agency for Medical Research and development (AMED) Japan and the Miyakawa memorial research fund. The authors declare that they have no conflicts of interest with the contents of this article.
- HBV
- hepatitis B virus
- SKIV2L
- superkiller viralicidic activity 2-like (S. cerevisiae)
- NSD
- non-stop mediated RNA decay
- HBS1L
- HBS1-like translational GTPase
- EXOSC4
- exosome component 4
- EXOSC5
- exosome component 5
- UPF1
- regulator of nonsense transcripts homolog (yeast)
- ZAP
- Zinc-finger antiviral protein
- NMD
- non-sense-mediated RNA decay
- ISG
- IFN-stimulated gene
- ISRE
- interferon-stimulated response element
- RIP
- RNA immunoprecipitation
- nt
- nucleotide(s)
- pgRNA
- pre-genomic RNA.
References
- 1. Orzalli M. H., and Knipe D. M. (2014) Cellular sensing of viral DNA and viral evasion mechanisms. Annu. Rev. Microbiol. 68, 477–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schnare M., Barton G. M., Holt A. C., Takeda K., Akira S., and Medzhitov R. (2001) Toll-like receptors control activation of adaptive immune responses. Nat. Immunol. 2, 947–950 [DOI] [PubMed] [Google Scholar]
- 3. Yan N., and Chen Z. J. (2012) Intrinsic antiviral immunity. Nat. Immunol. 13, 214–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lavanchy D. (2004) Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral. Hepat. 11, 97–107 [DOI] [PubMed] [Google Scholar]
- 5. Yokosuka O., Kurosaki M., Imazeki F., Arase Y., Tanaka Y., Chayama K., Tanaka E., Kumada H., Izumi N., Mizokami M., and Kudo M. (2011) Management of hepatitis B: consensus of the Japan Society of Hepatology 2009. Hepatol. Res. 41, 1–21 [DOI] [PubMed] [Google Scholar]
- 6. Michitaka K., Nishiguchi S., Aoyagi Y., Hiasa Y., Tokumoto Y., and Onji M. (2010) Etiology of liver cirrhosis in Japan: a nationwide survey. J. Gastroenterol. 45, 86–94 [DOI] [PubMed] [Google Scholar]
- 7. Locarnini S., and Zoulim F. (2010) Molecular genetics of HBV infection. Antivir. Ther. 15, 3–14 [DOI] [PubMed] [Google Scholar]
- 8. Cha M. Y., Ryu D. K., Jung H. S., Chang H. E., and Ryu W. S. (2009) Stimulation of hepatitis B virus genome replication by HBx is linked to both nuclear and cytoplasmic HBx expression. J. Gen. Virol. 90, 978–986 [DOI] [PubMed] [Google Scholar]
- 9. Leupin O., Bontron S., Schaeffer C., and Strubin M. (2005) Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J. Virol. 79, 4238–4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tang H., Delgermaa L., Huang F., Oishi N., Liu L., He F., Zhao L., and Murakami S. (2005) The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J. Virol. 79, 5548–5556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Z., Protzer U., Hu Z., Jacob J., and Liang T. J. (2004) Inhibition of cellular proteasome activities enhances hepadnavirus replication in an HBX-dependent manner. J. Virol. 78, 4566–4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wieland S., Thimme R., Purcell R. H., and Chisari F. V. (2004) Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. U.S.A. 101, 6669–6674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rigby R. E., and Rehwinkel J. (2015) RNA degradation in antiviral immunity and autoimmunity. Trends Immunol. 36, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cullen B. R. (2014) Viruses and RNA interference: issues and controversies. J. Virol. 88, 12934–12936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mao R., Nie H., Cai D., Zhang J., Liu H., Yan R., Cuconati A., Block T. M., Guo J. T., and Guo H. (2013) Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathog. 9, e1003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Balistreri G., Horvath P., Schweingruber C., Zünd D., McInerney G., Merits A., Mühlemann O., Azzalin C., and Helenius A. (2014) The host nonsense-mediated mRNA decay pathway restricts mammalian RNA virus replication. Cell Host Microbe 16, 403–411 [DOI] [PubMed] [Google Scholar]
- 17. Garcia D., Garcia S., and Voinnet O. (2014) Nonsense-mediated decay serves as a general viral restriction mechanism in plants. Cell Host Microbe 16, 391–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lebreton A., Tomecki R., Dziembowski A., and Séraphin B. (2008) Endonucleolytic RNA cleavage by a eukaryotic exosome. Nature 456, 993–996 [DOI] [PubMed] [Google Scholar]
- 19. Brown J. T., Bai X., and Johnson A. W. (2000) The yeast antiviral proteins Ski2p, Ski3p, and Ski8p exist as a complex in vivo. RNA 6, 449–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu Q., Greimann J. C., and Lima C. D. (2006) Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell 127, 1223–1237 [DOI] [PubMed] [Google Scholar]
- 21. Saito S., Hosoda N., and Hoshino S. (2013) The Hbs1-Dom34 protein complex functions in non-stop mRNA decay in mammalian cells. J. Biol. Chem. 288, 17832–17843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang H., Kim S., and Ryu W. S. (2009) DDX3 DEAD-Box RNA helicase inhibits hepatitis B virus reverse transcription by incorporation into nucleocapsids. J. Virol. 83, 5815–5824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Z., Yuan B., Bao M., Lu N., Kim T., and Liu Y. J. (2011) The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 12, 959–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miyashita M., Oshiumi H., Matsumoto M., and Seya T. (2011) DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell. Biol. 31, 3802–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Widner W. R., and Wickner R. B. (1993) Evidence that the SKI antiviral system of Saccharomyces cerevisiae acts by blocking expression of viral mRNA. Mol. Cell. Biol. 13, 4331–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou Z., Hamming O. J., Ank N., Paludan S. R., Nielsen A. L., and Hartmann R. (2007) Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J. Virol. 81, 7749–7758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murata T., Ohshima T., Yamaji M., Hosaka M., Miyanari Y., Hijikata M., and Shimotohno K. (2005) Suppression of hepatitis C virus replicon by TGF-β. Virology 331, 407–417 [DOI] [PubMed] [Google Scholar]
- 28. Dangel A. W., Shen L., Mendoza A. R., Wu L. C., and Yu C. Y. (1995) Human helicase gene SKI2W in the HLA class III region exhibits striking structural similarities to the yeast antiviral gene SKI2 and to the human gene KIAA0052: emergence of a new gene family. Nucleic Acids Res. 23, 2120–2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sugiyama M., Tanaka Y., Kato T., Orito E., Ito K., Acharya S. K., Gish R. G., Kramvis A., Shimada T., Izumi N., Kaito M., Miyakawa Y., and Mizokami M. (2006) Influence of hepatitis B virus genotypes on the intra- and extracellular expression of viral DNA and antigens. Hepatology 44, 915–924 [DOI] [PubMed] [Google Scholar]
- 30. Schneider C., and Tollervey D. (2013) Threading the barrel of the RNA exosome. Trends Biochem. Sci. 38, 485–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shin J. H., Wang H. L., Lee J., Dinwiddie B. L., Belostotsky D. A., and Chekanova J. A. (2013) The role of the Arabidopsis Exosome in siRNA-independent silencing of heterochromatic loci. PLoS Genet. 9, e1003411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Estévez A. M., Lehner B., Sanderson C. M., Ruppert T., and Clayton C. (2003) The roles of intersubunit interactions in exosome stability. J. Biol. Chem. 278, 34943–34951 [DOI] [PubMed] [Google Scholar]
- 33. Sato S., Li K., Kameyama T., Hayashi T., Ishida Y., Murakami S., Watanabe T., Iijima S., Sakurai Y., Watashi K., Tsutsumi S., Sato Y., Akita H., Wakita T., Rice C. M., Harashima H., Kohara M., Tanaka Y., and Takaoka A. (2015) The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42, 123–132 [DOI] [PubMed] [Google Scholar]
- 34. Ulrich P. P., Bhat R. A., Kelly I., Brunetto M. R., Bonino F., and Vyas G. N. (1990) A precore-defective mutant of hepatitis B virus associated with e antigen-negative chronic liver disease. J. Med. Virol. 32, 109–118 [DOI] [PubMed] [Google Scholar]
- 35. Naoumov N. V., Schneider R., Grötzinger T., Jung M. C., Miska S., Pape G. R., and Will H. (1992) Precore mutant hepatitis B virus infection and liver disease. Gastroenterology 102, 538–543 [DOI] [PubMed] [Google Scholar]
- 36. Brogna S., and Wen J. (2009) Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Mol. Biol. 16, 107–113 [DOI] [PubMed] [Google Scholar]
- 37. Chamieh H., Ballut L., Bonneau F., and Le Hir H. (2008) NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat. Struct. Mol. Biol. 15, 85–93 [DOI] [PubMed] [Google Scholar]
- 38. Tsuge M., Hiraga N., Akiyama R., Tanaka S., Matsushita M., Mitsui F., Abe H., Kitamura S., Hatakeyama T., Kimura T., Miki D., Mori N., Imamura M., Takahashi S., Hayes C. N., and Chayama K. (2010) HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J. Gen. Virol. 91, 1854–1864 [DOI] [PubMed] [Google Scholar]
- 39. Lucifora J., Arzberger S., Durantel D., Belloni L., Strubin M., Levrero M., Zoulim F., Hantz O., and Protzer U. (2011) Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 55, 996–1003 [DOI] [PubMed] [Google Scholar]
- 40. Ogura N., Watashi K., Noguchi T., and Wakita T. (2014) Formation of covalently closed circular DNA in Hep38.7-Tet cells, a tetracycline inducible hepatitis B virus expression cell line. Biochem. Biophys. Res. Commun. 452, 315–321 [DOI] [PubMed] [Google Scholar]
- 41. Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z., Murthy K., Habermann A., Kräusslich H. G., Mizokami M., Bartenschlager R., and Liang T. J. (2005) Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Watashi K., Inoue D., Hijikata M., Goto K., Aly H. H., and Shimotohno K. (2007) Anti-hepatitis C virus activity of tamoxifen reveals the functional association of estrogen receptor with viral RNA polymerase NS5B. J. Biol. Chem. 282, 32765–32772 [DOI] [PubMed] [Google Scholar]
- 43. Tsukuda S., Watashi K., Iwamoto M., Suzuki R., Aizaki H., Okada M., Sugiyama M., Kojima S., Tanaka Y., Mizokami M., Li J., Tong S., and Wakita T. (2015) Dysregulation of retinoic acid receptor diminishes hepatocyte permissiveness to hepatitis B virus infection through modulation of sodium taurocholate cotransporting polypeptide (NTCP) Expression. J. Biol. Chem. 290, 5673–5684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aly H. H., Watashi K., Hijikata M., Kaneko H., Takada Y., Egawa H., Uemoto S., and Shimotohno K. (2007) Serum-derived hepatitis C virus infectivity in interferon regulatory factor-7-suppressed human primary hepatocytes. J. Hepatol. 46, 26–36 [DOI] [PubMed] [Google Scholar]
- 45. Pugh J. C., Yaginuma K., Koike K., and Summers J. (1988) Duck hepatitis B virus (DHBV) particles produced by transient expression of DHBV DNA in a human hepatoma cell line are infectious in vitro. J. Virol. 62, 3513–3516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Watashi K., Liang G., Iwamoto M., Marusawa H., Uchida N., Daito T., Kitamura K., Muramatsu M., Ohashi H., Kiyohara T., Suzuki R., Li J., Tong S., Tanaka Y., Murata K., Aizaki H., and Wakita T. (2013) Interleukin-1 and tumor necrosis factor-α trigger restriction of hepatitis B virus infection via a cytidine deaminase activation-induced cytidine deaminase (AID). J. Biol. Chem. 288, 31715–31727 [DOI] [PMC free article] [PubMed] [Google Scholar]