

Abstract
The G309 allele of SNPs in the mouse double minute (MDM2) promoter locus is associated with a higher risk of cancer and proliferative vitreoretinopathy (PVR), but whether SNP G309 contributes to the pathogenesis of PVR is to date unknown. The clustered regularly interspaced short palindromic repeats (CRISPR)-associated endonuclease (Cas) 9 from Streptococcus pyogenes (SpCas9) can be harnessed to manipulate a single or multiple nucleotides in mammalian cells. Here we delivered SpCas9 and guide RNAs using dual adeno-associated virus-derived vectors to target the MDM2 genomic locus together with a homologous repair template for creating the mutation of MDM2 T309G in human primary retinal pigment epithelial (hPRPE) cells whose genotype is MDM2 T309T. The next-generation sequencing results indicated that there was 42.51% MDM2 G309 in the edited hPRPE cells using adeno-associated viral CRISPR/Cas9. Our data showed that vitreous induced an increase in MDM2 and subsequent attenuation of p53 expression in MDM2 T309G hPRPE cells. Furthermore, our experimental results demonstrated that MDM2 T309G in hPRPE cells enhanced vitreous-induced cell proliferation and survival, suggesting that this SNP contributes to the pathogenesis of PVR.
Keywords: apoptosis; cell growth; epithelial cell; ERK; genetic polymorphism; molecular biology; CRISPR/Cas9, MDM2T309G
Introduction
Proliferative vitreoretinopathy (PVR)3 is a vision-threatening disease resulting from surgical correction of rhegmatogenous retinal detachment and open ocular injury (1), and it is characterized by the formation of preretinal or epiretinal membranes (2). The epiretinal membranes consist of extracellular matrix proteins and cells, including retinal pigment epithelial (RPE) cells, retinal glial cells, fibroblasts, and macrophages. PVR occurs in 8–10% of patients who have undergone a surgical repair of rhegmatogenous retinal detachment and accounts for ∼75% of all primary failures following the surgery (2–8).
The oncogene protein murine double minute 2 (MDM2) is an E3 ubiquitin protein ligase whose human homologue (also called Hdm2) is an important negative regulator of the p53 tumor suppressor (9–11). The phenotype of murine embryonic lethality of MDM2 null can be prevented by knocking out the p53 gene (12, 13). Vitreous from experimental rabbits preferentially activates platelet-derived growth factor receptor (PDGFR). This activation in turn triggers the downstream signaling pathway of PI3K/Akt, which phosphorylates MDM2, thereby enhancing p53 degradation (14). Blocking MDM2 binding to p53 with a small molecule, Nutlin-3, protects rabbits against retinal detachment in a PVR rabbit model (3).
Intriguingly, the G allele of SNPs (rs2279744) in the MDM2 promoter locus has subsequently been found to be associated with a higher risk of PVR for rhegmatogenous retinal detachment patients (2, 15). This SNP is also associated with an increased risk of carcinogenesis (15–21). The SNP T309G (a T-to-G change at the 309th nucleotide) at the MDM2 first intron promoter locus enhances the affinity of the transcriptional activator specificity protein 1 (Sp1), leading to a heightened expression of MDM2 and the subsequent attenuation of p53 expression in cancer cells (15). However, whether or not this SNP contributes to the pathogenesis of PVR has not been explored.
The system of clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated nucleases (Cas) in bacteria and archaea provides adaptive immunity against viruses and plasmids when their CRISPR RNAs (crRNAs) are used to guide the Cas cleavage of foreign nucleic acids (22–24). In Streptococcus pyogenes (Sp), the Cas9 (SpCas9) contains two nuclease domains, RuvC and HNH, each of which can cleave one strand of the double-stranded target DNA when directed by the crRNA and transactivating crRNA (24, 25). This SpCas9 can be reprogrammed to target specific genomic loci in mammalian cells using the processed single guide (sg) RNAs that consist of crRNA and transactivating crRNA (24). The double-stranded DNA breaks at the specific genomic loci produced by CRISPR/Cas9 can be repaired by endogenous repair machinery for either non-homologous end joining (NHEJ) or homology-directed repair (HDR), which depends on the cell state and presence of a repair template (26, 27). NHEJ and HDR are two distinct competent repair pathways in cells. NHEJ can introduce unpredictable insertions and deletions, and it may repair the lesion by simply rejoining the two double-stranded DNA break ends (26). HDR can use an exogenous single- or double-stranded DNA template with the desired changes to make mutations in the genomic loci (26). However, HDR is less frequently used than NHEJ because it occurs only during S and G2 phases, whereas NHEJ can be found throughout the cell cycle (26, 28). The CRISPR/Cas9 technology has recently been used in a variety of genome-editing applications in eukaryotic cells and mice (26, 29–32), and it provides a unique opportunity to demonstrate whether MDM2 T309G contributes to the pathogenesis of PVR.
Here we generated the mutation of T309G in the MDM2 genomic locus in human primary retina pigment epithelial (hPRPE) cells using CRISPR/Cas9 technology. We demonstrated that the vitreous from experimental rabbits (RV) increased expression of MDM2 and subsequent attenuation of p53 expression in hPRPE cells with MDM2 T309G. Furthermore, we found that MDM2 T309G in hPRPE cells promoted RV-induced cell proliferation and survival, which are intrinsic to the development of PVR.
Results
Creation of MDM2 T309G in the Genomic Locus Using CRISPR/Cas9
The SNP MDM2 T309G is associated with a higher risk of PVR (2); however, it is not known whether this SNP contributes to PVR. Because RPE cells are believed to be the major cell type in PVR membranes that causes retinal detachment in the development of PVR (2, 5–7), we attempted to create this SNP in hPRPE cells using CRISPR/Cas9 technology. Because the ultimate goal of this research is to explore a novel therapeutic approach to PVR, and AAVs do not cause any disease (33), we chose AAV-derived viral vectors to deliver CRISPR/Cas9 into our target cells. However, because of the packaging size limitation of the AAV-derived vectors, we had to adapt a dual vector system that packages SpCas9 and sgRNA expression cassettes (SpGuide) in two separate viral vectors, pAAV-SpCas9 and pAAV-SpGuide, respectively (30). To separate hPRPE cells transduced by pAAV-SpGuide, we replaced the GFP promoter hSyn (30) with the promoter of CMV (Fig. 1A). There are four types of cells in the PVR membrane, including RPE cells. Therefore, we substituted the promoter pMecp2 (30) for the promoter of the RSV to drive expression of SpCas9 (34) (Fig. 1B).
FIGURE 1.
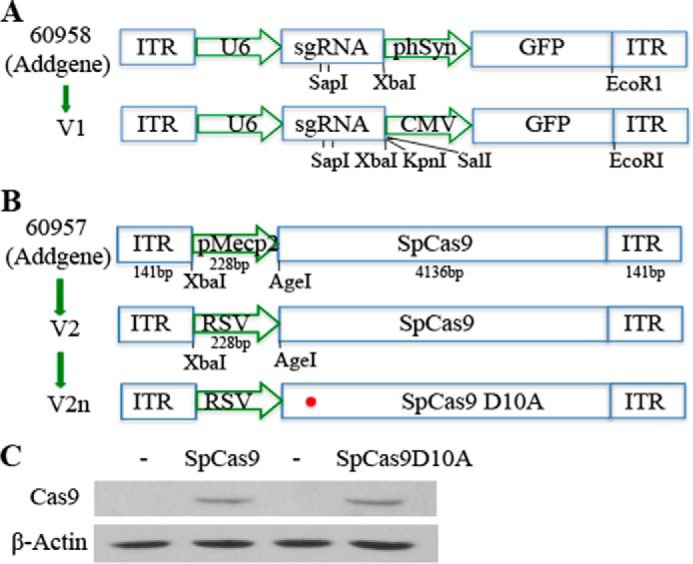
Schematic of the AAV-SpGuide and AAV-SpCas9 vectors. A, schematic of the AAV-SpGuide vector. ITR, inverted terminal repeat; U6, polymerase III promoter; phSyn, human synapsin 1 gene promoter for GFP expression in the vector from Addgene (catalog no. 60958); SapI, a restriction endonuclease for cloning the 20-nt targeted sequence into the AAV-derived vector backbone. CMV-GFP replacement of phSyn-GFP was achieved by XbaI/EcoRI. B, schematic of the AAV-SpCas9 vector. pMecp2, neuron-specific Mecp2 promoter. RSV promoter replacement of pMecp2 was accomplished by XbaI/AgeI. The 60957 vector was purchased from Addgene. C, 293T cells were transfected with plasmids of pAAV-SpCas9 or pAAV-SpCas9D10A. After 48-h of transfection, the transfected cells were lysed, and the lysates were subjected to Western blotting analysis with the indicated antibodies. Non-transfected 293T cell lysates were used as negative controls. This is representative of three independent experiments.
The SpCas9 contains two conserved nuclease domains, HNH and RuvC, which cleave the target DNA strand complementary and non-complementary to the guide RNA, respectively. A mutation of aspartate to alanine (D10A) in the RuvC catalytic domain can convert SpCas9 into the DNA nickase (SpCas9D10A). Two SpCas9 D10A-nicking enzymes directed by a pair of sgRNAs targeting opposite strands of a target locus can mediate double DNA strand breaks while minimizing off-target activity because single-strand nicks are preferentially repaired by the high-fidelity base excision repair pathway (35). Thus, the SpCas9 in the AAV vector was mutated to SpCas9 D10A with an in situ mutagenesis kit (Fig. 1B). To test the effectiveness of the DNA constructs, we transfected these two vectors separately into 293T cells. Western blotting analysis showed that the SpCas9 and SpCas9 D10A were successfully expressed in the transfected 293T cells (Fig. 1C).
We next sought to test the efficiency of SpCas9-mediated editing of the genomic MDM2 locus around the SNP in RPE cells. To identify RPE cells that were suitable for our experimental purpose, we isolated the genomic DNA from hPRPE cells, PCR-amplified a region around the MDM2 SNP for Sanger DNA sequencing, and found that there was T309T in the MDM2 SNP in hPRPE cells, as shown in Fig. 2A. These cells were then used to introduce MDM2 T309G in the genomic locus. In synthesizing sgRNAs, the two 20-nt targeted sequences (30, 36–38) (Fig. 2B) were cloned into the pAAV-SpGuide backbone (Fig. 1A), and the clones were verified by DNA sequencing.
FIGURE 2.

Identification of MDM2-sgRNAs. A, the genomic DNA was isolated from hPRPE cells and subjected to PCR amplification of a region around the MDM2 SNP 309. The PCR products were purified from a 2% agarose gel for DNA sequencing. B, graphic representation of the MDM2-targeted loci. 20-nt target 1 (612–631) and target 2 (583–675) in the human genomic MDM2 loci (NC_000012.12) are indicated by a green arrow for nicking at sites (red arrows) by SpCas9D10A. The protospacer-adjacent motifs (PAM) are marked in blue. C, genomic DNA was extracted from transfected cells using the QuickExtract DNA extraction solution system following the protocol of the manufacturer for amplification of a region around MDM2 SNP 309. The gel-purified PCR products were subjected to DNA sequencing analysis. MDM2-sgRNA1 or 2 or LacZ-sgRNA, partial sequencing from the DNA extracted from the transfected cells with pAAV-SpCas9 and pAAV-MDM2-sgRNA1 or 2 (or LacZ-sgRNA)-CMV-GFP. Arrows indicate the expected SpCas9 cleavage sites.
To edit the genomic MDM2 locus, we transfected hPRPE cells with the dual vectors of pAAV-SpCas9 plus pAAV-MDM2-sgRNA1 or 2 using electroporation. pAAV-LacZ-sgRNA was used as a control. The transduction efficiency was about 60%, as estimated by immunofluorescence. To determine whether any insertions and deletions were mediated by the CRISPR/Cas9 system, we isolated genomic DNA from the transfected hPRPE cells and amplified the region around the MDM2 SNP using a high-fidelity Herculase II fusion polymerase. The amplified DNA fragments were then subjected to Sanger DNA sequencing (39). As shown in Fig. 2C, there were mutations in front of protospacer-adjacent motifs (MDM2-sgNRA1, CGG; MDM2-sgRNA2, AGG) from the PCR products derived from the transduced hPRPE cells with SpCas9 plus MDM2-sgRNA1 or 2 but not from those with LacZ-sgRNA. These results demonstrate that the two MDM2 sgRNAs efficiently guided the SpCas9 to induce insertions and deletions in hPRPE cells.
To create the SNP MDM2 T309G in the genome of hPRPE cells, we chose to use SpCas9D10A nickase activity, as there was no off-target DNA sequence found for the pair of sgRNA1 and 2 based on the double nickase design tool. Thus, U6-sgRNA2 was PCR-amplified and cloned into the pAAV-U6-sgRNA1 vector. pAAV-SpCas9D10A, pAAV-MDM2-sgRNA1 and 2, and ssHRT (Fig. 3A) together were transfected into the hPRPE cells by electroporation. The HDR donor template consisted of a single-strand, 96-bp genomic sequence homologous to a region encompassing the SNP with a G309 replacement of T309. However, the mutagenesis efficiency using this HDR strategy is very low (only 0.5–20%) (26, 28, 40, 41). To increase the efficiency of HDR-mediated genome editing, we immediately treated the post-transfected cells with 0.5 μm Scr7, which can inhibit NHEJ, a competent HDR pathway (26). On the third day after transfection, the GFP-expressing hPRPE cells were sorted by FACS, and the genomic DNA fragments around the SNP from some of the sorted cells were amplified by PCR using high-fidelity Herculase II DNA polymerase. The Surveyor nuclease assay of the PCR products indicated that there was a DNA fragment (about 100 bp) released (Fig. 3B) in MDM2-sgRNA-transfected cells, but there was none in control LacZ-sgRNA-transfected cells, suggesting that there were insertions and deletions in the middle of the PCR fragment (about 200 bp). Sanger DNA sequencing (Fig. 3C) confirmed that there were heterozygous MDM2 T309 and G309 in the selected hPRPE cells transduced with the SpCas9 and MDM2-sgRNA. Next-generation sequencing analysis indicated that there were 42.51% MDM2 G309 and 57.19% MDM2 T309 in GFP-positive hPRPE cells transfected with SpCas9D10A, MDM2-sgRNA1 and 2, and the HDR template (Fig. 3D). Some of the transduced cells were sorted into PCR tubes (one cell per tube), and the genomic DNA from single cells was subjected to PCR amplification of the fragment around the SNP for Sanger DNA sequencing. The sequencing results indicate that, in the 309 position, there were 20% (2 of 10) cells containing T/T, 70% (7 of 10) cells with T/G, and 10% (1 of 10) cells with G/G (Fig. 3E). These results demonstrate that the genomic MDM2 T309G was successfully created in hPRPE cells using CRISPR/Cas9 technology.
FIGURE 3.
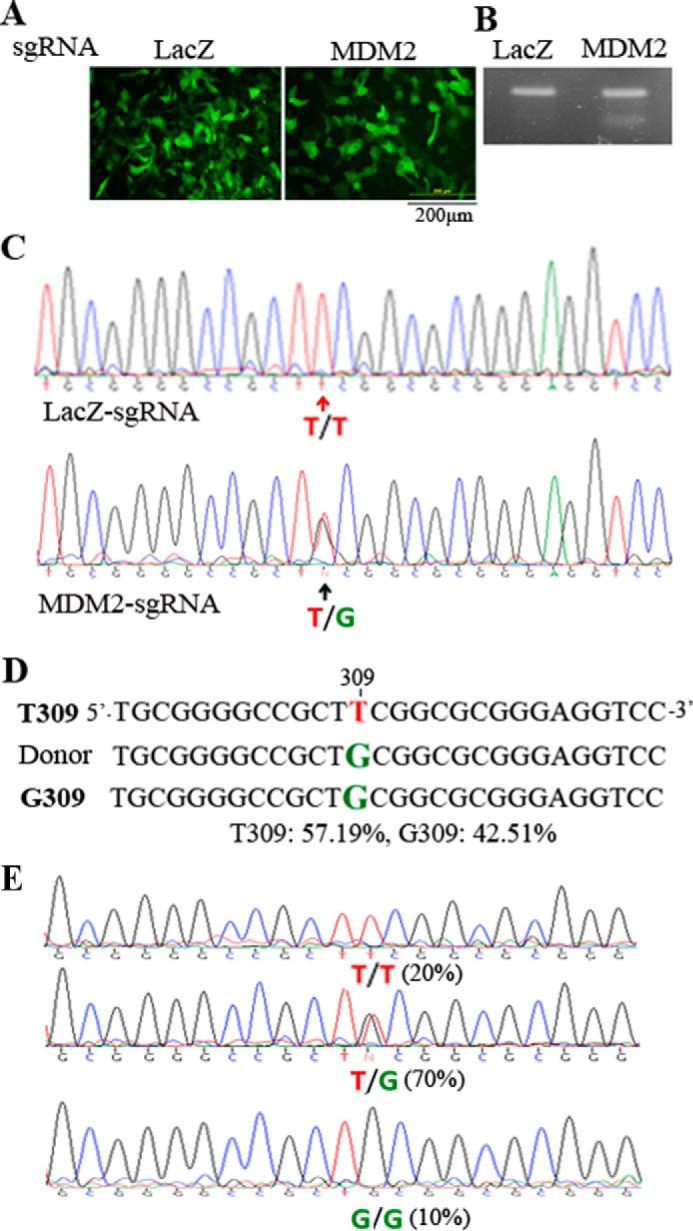
Creation of the genomic MDM2 T309G mutation. A, transfection of hPRPEs with dual AAV vectors and an ssHRT. The dual vectors of pAAV-SpCas9D10A plus pAAV-MDM2-sgRNA1 and 2 or pAAV-LacZ-sgRNA and the ssHRT were transfected into hPRPE cells by electroporation as described under “Experimental Procedures.” Subsequently, the cells were treated with Scr7 (5 μm). After transfection for 16 h, the cells were photographed under a fluorescence microscope. Scale bar = 200 μm. This is representative of three independent experiments. B, genomic DNA extracted from the transfected cells was used to amplify the region containing MDM2 SNP 309. The gel-purified PCR products were subjected to a Surveyor nuclease assay. This is representative of two independent experiments. C, the purified PCR products in B were also subjected to Sanger DNA sequencing. The top and bottom panels were from hPRPE cells transfected with LacZ-sgRNA and MDM2-sgRNA1 and 2, respectively. D, the purified PCR products from B were also analyzed by next-generation sequencing. The results showed that there was 57.19% MDM2 T309 and 42.51% MDM2 G309 in transduced cells with SpCas9D10A plus MDM2-sgRNA1 and 2 and ssHRT. E, single cells from transduced cells (B) were sorted by FACS into PCR tubes containing 5 μl of QuickExtract DNA extraction buffer, and the genomic DNA was isolated for PCR amplification of DNA fragments around the SNP. The purified DNA fragments from the 10 single cells were subjected to Sanger DNA sequencing. There were two 309T/T cells (20%), seven 309T/G cells (70%), and one 309G/G cell (10%) in the 10 sorted single cells.
MDM2 T309G Promotion of Vitreous Stimulated an Increase in MDM2 and a Decrease in p53
The MDM2 intron promoter region SNP T309G (rs2279744) has been shown to increase the affinity of the transcriptional activator Sp1, resulting in elevated expression of MDM2 in some cancer cell lines (15). In addition, this SNP is associated with a higher risk of PVR (2). Therefore, we tested whether the CRISPR/Cas9-created MDM2 T309G would elevate the expression of MDM2 in hPRPE cells and whether RV would influence this change in MDM2 and p53 between hPRPE cells with the MDM2 T309G SNP and WT T309T (WT). Unexpectedly, the SNP failed to enhance MDM2 expression in MDM2 T309G hRPE cells in comparison with that in WT cells (Fig. 4A). However, the vitreous enhanced the expression of MDM2 and effected a decrease in p53 in hPRPE cells with the SNP compared with the WT (Fig. 4A). These results suggested that RV induced Sp1 association with the MDM2 promoter motif with the SNP and thus enhanced expression of MDM2 and subsequent suppression of p53. If this prediction was correct, blocking the association between MDM2 and p53 should recover the p53 loss. Thus, we treated hPRPE cells for 16 h with both RV and Nutlin-3. Western blotting analysis of the treated cell lysates (Fig. 4A) indicated that Nutlin-3 prevented RV-induced reduction in p53 in the MDM2 T309G hPRPE cells. This result indicates that the vitreous-induced decrease in p53 was due to vitreous-stimulated interaction between MDM2 and p53.
FIGURE 4.

Vitreous induction of an increase in MDM2 in MDM2 T309G hPRPE cells. A, hPRPE cells (with MDM2 T309 only: T/T or MDM2 T309 plus G309: T/G) that had been serum-starved overnight were treated with RV and Nutlin-3 (10 μm) for 16 h. The cell lysates were then Western-blotted using the indicated antibodies. Fold was calculated by first normalizing to the level of β-Actin and then calculating the ratio of the stimulated over the basal (i.e. unstimulated cells). This is representative of three independent experiments. B, hPRPE cells (described above), serum-starved overnight, were subsequently treated with RV, mithramycin A (200 nm), or PD98059 (50 μm) for 16 h. The cell lysates were then subjected to Western blotting analysis using the antibodies indicated. Fold was calculated by first normalizing to the level of either β-Actin or Erk or Sp1 and then calculating the ratio of the stimulated over the basal (i.e. unstimulated). This is representative of three independent experiments.
To examine whether the vitreous-induced increase in MDM2 was due to the vitreous-stimulated association of Sp1 with the MDM2 GC-rich promoter, we treated hPRPE cells with a Sp1 inhibitor, mithramycin A, an aurelic antibiotic that has been shown to selectively inhibit Sp1 transcription factor-mediated transcriptional activation by blocking Sp1 binding to the GC-rich promoter motif. As shown in Fig. 4B, treatment with mithramycin A blunted an RV-stimulated increase in MDM2 and a decrease in p53 in hPRPE cells with MDM2 T309G, suggesting that RV enhanced Sp1 association with the MDM2 GC-rich promoter.
Next, we asked how the vitreous could induce the Sp1-dependent increase in MDM2. Because Erk can phosphorylate Sp1 at Thr-936 (42), thereby enhancing its transcriptional activity, we hypothesized that vitreous could induce Erk phosphorylation of Sp1, which would increase the Sp1 association with the GC-rich motif and promote its target expression. As predicted, treatment of hPRPE cells with an Erk inhibitor, PD98059, for 16 h blocked the vitreous-stimulated phosphorylation of Erk and Sp1 and prevented the vitreous-induced increase in MDM2 in PRPE cells with the SNP (Fig. 4B). These results demonstrate that the MDM2 T309G in PRPE cells leads to an enhanced RV-induced, Erk/Sp1-dependent increase in MDM2 and a decrease in p53. These biochemical reactions could promote vitreous-induced cellular responses intrinsic to PVR.
MDM2 T309G Enhancement of RV-induced Cell Proliferation and Survival
Increased MDM2 leads to a decrease in p53 and thus promotes cell proliferation and survival against apoptosis (15, 43). RV stimulated an increase in MDM2 in hPRPE cells with MDM2 T309G but not with the WT (Fig. 5). Therefore, we suspected that, after surgical attachment of the retina, RPE cells enter the vitreous and that the RPE cells with T309G or G309G will survive better than those with WT cells in the MDM2 intron promoter locus. To test this, we treated hPRPE cells with RV for 3 days and analyzed them for apoptosis. As shown in Fig. 5, with RV treatment, hPRPE cells with MDM2 T309G underwent less apoptosis than those with the WT. In addition, hPRPE cells with the SNP proliferated more in response to vitreous than those with WT cells (Fig. 6).
FIGURE 5.
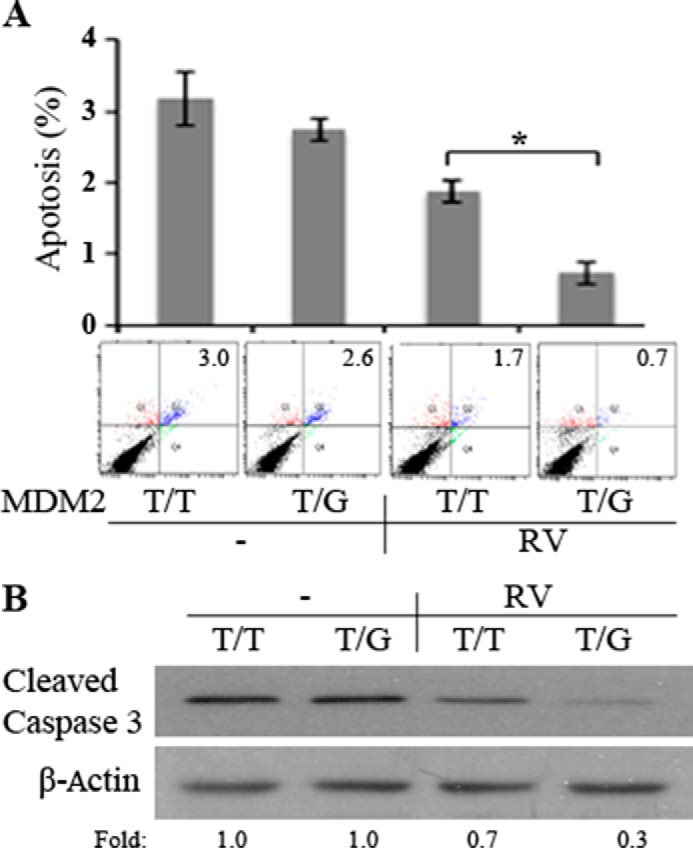
MDM2 T309G enhancement of RV-induced cell survival against apoptosis. A, hPRPE cells, as described in Fig. 3, were seeded into 60-mm dishes at a density of 100,000 cells/dish in DMEM/F12 plus 10% FBS. After the cells attached to the dishes (about 8 h), the medium was changed to either DMEM/F12 or RV (diluted to 1:2 in DMEM/F12). The media were replaced every day. On day 3, the cells were stained with FITC-conjugated annexin V and propidium iodide in an apoptosis assay kit following the instructions of the manufacturer. Cells that were stained with annexin V and/or propidium iodide were detected and quantified by flow cytometry in a Coulter Beckman XL instrument. The mean ± S.D. of three independent experiments is shown. One example of the experimental raw data is shown below the bar graphs. B, the lysates of the cells treated as in A were subjected to Western blotting with the antibodies indicated. Fold was calculated by first normalizing to the level of β-Actin and then calculating the ratio of the stimulated over the basal (i.e. unstimulated) cells. This is representative of three independent experiments. *, p < 0.05, unpaired t test.
FIGURE 6.
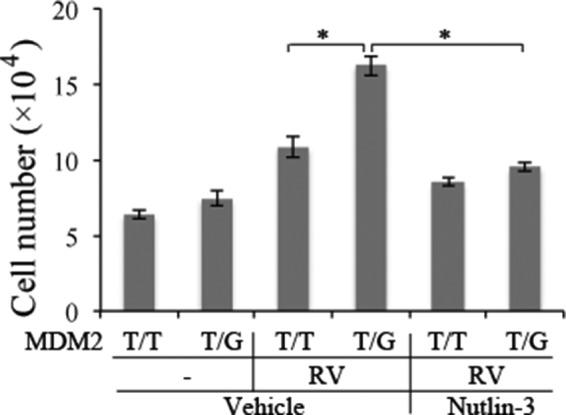
MDM2 T309G promotion of RV-induced cell proliferation. hPRPE cells were seeded into a 24-well plate at a density of 3 × 10e4 cells/well in DMEM/F12 plus 10% FBS. After the cells had attached to the plates, the medium was changed to either DMEM/F12 or RV (diluted 1:2 in DMEM/F12) with or without Nutlin-3 (10 μm). The media were replaced every day. Cells were counted with a hemocytometer under a light microscope on day 3. Mean ± S.D. of three independent experiments is shown. *, p < 0.05, unpaired t test.
Discussion
In this article, we demonstrate that MDM2 T309G in hPRPE cells enhances an RV-stimulated increase in MDM2 concomitant with a decrease in p53 as well as a cellular responses intrinsic to PVR compared with the WT. These results are consistent with a two-hit hypothesis (44). Although the polymorphisms of T or G alleles (rs2279744) in the MDM2 309 position did not influence MDM2 expression, they did lead to a differential response to the vitreous stimulation. Notably, MDM2 G309 enhances Sp1-dependent expression of MDM2 in cancer cells in comparison with MDM2 T309 (15). Taken together, our results and clinical findings of MDM2 SNP association with PVR (2) suggest that this SNP contributes to the development of PVR.
We speculate that MDM2 G309 RPE cells respond to vitreous with increased survival, proliferation, and elevated expression of fibrotic proteins (e.g. fibronectin and collagen). We found that the inhibitors of Sp1 and Erk did not completely block the vitreous-induced p53 decrease, suggesting that vitreous may activate another pathway to suppress p53 expression. The vitreous could activate Akt (Fig. 7A) (14), which could phosphorylate MDM2, enhancing its association with p53 and promoting its degradation (Fig. 7) (45, 46).
FIGURE 7.
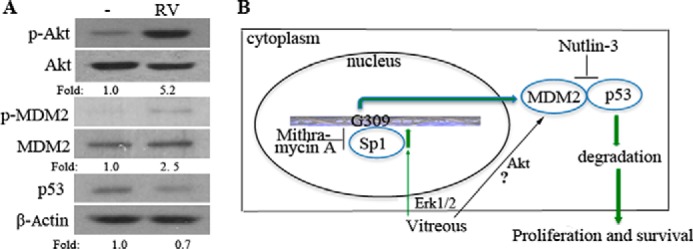
Potential RV-induced signaling pathways that trigger cellular responses. A, serum-starved hPRPE cells were treated with RV for 16 h, and then the cell lysates were subjected to Western blotting analysis using the antibodies indicated. Fold was calculated by first normalizing to the level of either Akt, MDM2, or β-Actin and then calculating the ratio of stimulated over basal (i.e. unstimulated) cells. This is representative of three independent experiments. B, pathway 1 indicates that vitreous stimulation leads to the activation of Erk, which phosphorylates Sp1, enhancing its association with the GC-rich motif in the MDM2 promoter. Pathway 2 indicates that vitreous stimulation leads to the activation of Akt and phosphorylation of MDM2, leading, in turn, to an increased association between MDM2 and p53. Both pathways resulted in degradation of p53, thereby enhancing cell proliferation and survival.
In this study, our efforts in raising the percentage of the hPRPE cells with the mutant MDM2 T309G in the whole population were made in two ways. One was the treatment of AAV-CRISPR/Cas9-transduced cells with the drug Scr7, which targets the DNA binding domain of DNA ligase IV for inhibiting the ligase IV-dependent NHEJ and enhancing the frequency of HDR (26). The CRISPR/Cas9 system we used is a reprogrammed one from microbial type II CRISPR systems and has been harnessed to facilitate facile genetic manipulations in a variety of cell types and organisms (29). The reprogrammed Cas9 can be guided to generate targeted double-stranded DNA breaks that stimulate genome editing via one of the two DNA damage repair pathways: NHEJ, resulting in insertions and deletions, or HDR, resulting in precise sequence substitution in the presence of a repair template (26). SpCas9 D10A, the mutant form of SpCas9, has been shown previously to facilitate HDR at on-target sites (47), but its efficiency is substantially lower than that of WT Cas9. In our research, we have utilized the double-nicking strategy, which maintains a high on-target efficiency while reducing off-target modifications to background levels. Even so, the HDR efficiency might be still low. Thus, the drug Scr7 was subjected to our research for increasing the HDR rate. The other strategy was to sort out the transduced hPRPE cells by FACS because, in the pAAV-SpGuide vector, there was a CMV-GFP expression cassette. Importantly, hPRPE cells could continuously grow and proliferate after cell sorting. In summary, we have shown that the CRISPR/Cas9-created MDM2 T309G in hPRPE cells enhanced vitreous-induced expression of MDM2 and cell proliferation and survival.
Experimental Procedures
Major Reagents
The antibodies against Cas9, Erk, phospho (p)-Erk, Sp1, p-Sp1 (Thr-739), Akt, p-Akt (Ser-473), cleaved Caspase 3, p-MDM2 (Ser-166), and p53 were purchased from Cell Signaling Technology (Danvers, MA). The antibody against MDM2 was from Abgent (San Diego, CA). The primary antibodies against β-Actin and the secondary antibodies of the HRP-conjugated goat anti-rabbit IgG and anti-mouse IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Enhanced chemiluminescent substrate for the detection of HRP was from Pierce Protein Research Products. Carbenicillin was purchased from Sigma, and Scr7 was from Xcessbio Biosciences, Inc. (San Diego, CA). Mithramycin A and Nutlin-3 were from Cayman (Ann Arbor, Michigan), and PD98059 was from Cell Signaling Technology.
DNA Constructs
To generate single guide RNAs (sgRNAs) for SpCas9 targets, we used the CRISPR design tool to select the two 20-nt target sequences preceding a 5′-NGG of a protospacer-adjacent motif sequence around the MDM2 SNP309 in the MDM2 genomic locus (Rs2279744, NC_000012.12) (30). The target sequences used were target 1 (5′-CGGGAGGTCCGGATGATCGC-3′) and target 2 (5′-CGAAGCGGCCCCGCAGCCCC-3′). The control sgRNA sequence was designed to target the LacZ gene from Escherichia coli (target sequence, 5′-TGCGAATACGCCCACGCGATGGG-3′) (30). The annealed oligos were cloned into the pAAV-U6-sgRNA-CMV-GFP vector (V1) by SapI. All clones were confirmed by DNA sequencing using a primer (5′-GGACTATCATATGCTTACCG-3′) from the sequence of the U6 promoter, which drives the expression of sgRNAs.
hSyn-GFP in an AAV vector (Addgene, catalog no. 60958, Cambridge, MA) (30) was replaced with the PCR-amplified CMV-GFP from the pEGFP-C1 vector (Clontech, Mountain View, CA, catalog no. 6084-1,) using Xbal/EcoRI. The PCR primers for this amplification were as follows: forward, 5′-CGTCTAGAgGGTACCgGGGCCCgGTCGACTAGTTATTAATAGTAATCAATTACGG-3′; reverse 5′-CAGAATTCGCTGCAGGTTATCGAGATCTGAGTCCGGACTTGTA-3′. pAAV-RSV-SpCas9 (V2) was derived from an AAV vector (Addgene, catalog no. 60957) by replacing the promoter pMecp2 with the PCR-amplified Rous sarcoma virus (RSV) from a pLKO.1-shMDM2 vector (GE Dharmacon, Lafayette, CO) using XbaI/AgeI. The PCR primers for this amplification were as follows: forward, 5′-CGGTCTAGAAATGTAGTC TTATGCAATAC-3′; reverse, 5′-CGGA CCGGTTTTATGTATCGAGCTAGGCAC-3′. The SpCas9 mutation (D10A) was generated in the vector V2 using the following set of mutagenic primers: 5′-CAGCATCGGCCTGGCCATCGGCACCAACT-3′ and its complimentary oligonucleotide according to instructions provided with the QuikChange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). To express SpGuides in hPRPE cells, the top oligos: 5′-ACCG-20 nt (target MDM2 DNA sequences 1, 2 or LacZ sgRNA sequence) and bottom oligos: 5′-AAC-20 nt-C (20 nt: complimentary target MDM2 DNA sequences or LacZ sgRNA sequence) were annealed and cloned into the V1 vector, respectively, and sequencing of PCR products, clones, and mutations was done by the Massachusetts General Hospital DNA Core Facility (Cambridge, MA).
Cell Culture and Transfection
hPRPE cells were purchased from Lonza (Walkersville, MD) and cultured in DMEM: nutrient mixture F-12 medium (F12) (Invitrogen) supplemented with 10% FBS and 1% penicillin/streptomycin. HEK293T cells (293 cells containing SV40 T-antigen) from Dana-Farber Cancer Institute/Harvard Medical School (Boston, MA) were cultured in DMEM (high-glucose, 4.5g/ml) supplemented with 10% FBS. All cells were cultured at 37 °C in a humidified incubator with 5% CO2 (4).
The constructs of pAAV-RSV-SpCas9 and pAAV-RSV-SpCas9D10A were transfected into 293T cells using Lipofectamine. Briefly, during transfection, Lipofectamine 2000 (Invitrogen) (3 μl) was mixed with the plasmid (1 μg) and incubated at room temperature for 30 min. The transfection mixture was then transferred to 293T cells that were ∼70% confluent. After 48 h, the resulting lysates were subjected to Western blotting analysis using antibodies against Cas9 and β-Actin (4).
Western Blotting Analysis
Cells at 90% confluence in either a 24- or 48-well plate were deprived of serum for 24 h under continuous incubation and were then treated with or without RV (diluted 1:2 in DMEM/F12) for 16 h. After the cells were washed twice with ice-cold PBS, they were lysed in 1× sample buffer, which was diluted with extraction buffer (10 mm Tris-HCl (pH 7.4), 5 mm EDTA, 50 mm NaCl, 50 mm NaF, 1% Triton X-100, 20 μg/ml aprotinin, 2 mm Na3VO4, and 1 mm phenylmethylsulfonyl fluoride) from 5× protein sample buffer (25 mm EDTA (pH 7.0), 10% SDS, 500 mm dithiothreitol, 50% sucrose, 500 mm Tris HCl (pH 6.8), and 0.5% bromphenol blue). The samples were boiled for 5 min and then centrifuged for 5 min at 13,000 × g. Proteins in the centrifuged and heated samples were separated by 10% SDS-PAGE, transferred to PVDF membranes, and subjected to Western blotting analysis using the appropriate antibodies. Signal intensity was determined by densitometry using National Institutes of Health ImageJ software (4).
Transfection of hPRPE Cells
hPRPE cells were transfected with pAAV-SpCas9D10A and pAAV-SpGuide (MDM2-sgRNAs or LacZ-sgRNA) supplemented with a single-strand homology repair template (ssHRT) (5′-GGAGTTCAGGGTAA AGGTCACG GG GGCCGGGGGCTGCGGGGCCGCTGCG GCGCGGGAGGTCCGGATGATCGCAGGTGCCTGTCGGGTCACTAG-3′, Integrated DNA Technologies, Coralville, IA) using electroporation (Amaxa Biosystems). Briefly, 5 × 105 cells were centrifuged at 100 × g for 10 min at room temperature, and the cell pellet was carefully resuspended in 100 μl of room temperature Nucleofector solution (Lonza). The cell solution was mixed with 2 μg of pAAV-SpCas9D10A, 2 μg of pAAV-SpGuide vector, and 0.2 μg of ssHRT and then transferred into a certified cuvette placed in the nucleofector cuvette holder. The transfection was then started in a predetermined program. Subsequently, 500 μl of the pre-equilibrated culture medium was added into the cuvette, and then transfected cells were transferred to one well of a 6-well plate for continuous culture, supplemented with Scr7 (5 μm). At 72 h post-transfection, GFP-positive hPRPE cells were sorted by FACS to enrich the population of transduced cells or into PCR tubes (one cell per tube with 5 μl of QuickExtract DNA extraction solution (Epicenter Biotechnologies, Madison, WI) (33).
Surveyor Nuclease Assay and DNA Sequencing
During culture, some cells were pelleted for genomic DNA extraction using QuickExtract DNA extraction solution (Epicenter Biotechnologies) following the protocol of the manufacturer. In brief, the pelleted cells were resuspended in QuickExtract DNA extraction solution, vortexed for 15 s, at 65 °C for 6 min, vortexed for 15 s, and then at 98 °C for 5 min. The genomic region on either side of the MDM2 SNP309 site was PCR-amplified with high-fidelity Herculase II DNA polymerases (Agilent Technologies, Santa Clara, CA). The PCR primers (forward, 5′-GGGCGGGATTTCGGACGGC; reverse, 5′-CCACTGAACCGGCCCAATC) were synthesized by the Massachusetts General Hospital DNA core facility. The PCR products were separated in 2% agarose gel and purified with a GeneJET gel extraction kit (Thermo Scientific, Carlsbad, CA) for Sanger DNA sequencing and next-generation sequencing performed by the Massachusetts General Hospital DNA core facility and a Surveyor nuclease assay performed according to the instructions of the manufacturer (IDT). Briefly, PCR products (300 ng) from the agarose gel were incubated with the Surveyor nuclease and Surveyor Enhancer S with an additional 1/10 MgCl2 (0.15 m) for 30 min at 42 °C, following the protocol recommended by the manufacturer (IDT) (35).
Cell Proliferation Assay
hPRPE cells with MDM2 T309G or WT cells were seeded into 24-well plates at a density of 30,000 cells/well in DMEM/F12 (Invitrogen) supplemented with 10% FBS. After the cells had attached to the plates (∼8 h), the medium was aspirated. The cells were then rinsed twice with PBS and cultured in serum-free DMEM/F12 or RV (1:2 dilution in DMEM/F12) with or without Nutlin-3 (10 μm). Next, the cells were counted in a hemocytometer on day 3, and a minimum of three independent experiments were performed (4).
Apoptosis Assay
hPRPE cells with MDM2 T309G or WT cells were plated into 6-cm dishes at a density of 1 × 105 cells/dish in DMEM/F12 with 10% FBS. After the cells had attached, they were cultured on serum-free DMEM/F12 or RV (1:2 dilution in DMEM/F12) in the dishes. On day 3, the cells were harvested and stained with FITC-conjugated annexin V and propidium iodide according to the instructions provided with the apoptosis kit (BD Biosciences, Palo Alto, CA). The cells were analyzed by flow cytometry in a Coulter Beckman XL instrument (4).
Statistics
The data from the three independent experiments were analyzed using an unpaired t test in Prism 6 software. p Values of less than 0.05 were considered statistically significant.
Author Contributions
Y. D. and G. M. performed most of the experiments, analyzed the results, and made equal contributions to this paper. X. H. performed some of the experiments. P. A. D. revised the manuscript. F. Z. designed the experiments. H. L. conceived and conducted the experiments, analyzed the data, and wrote the manuscript.
Acknowledgments
We thank Randy Huang for assistance with flow cytometric analysis, Bianai Fan for histological sections, and Gale Unger for copyediting this article. We also thank Dr. Andrius Kazlauskas, Dr. Guoxiang Yuan, and Dr. Wenxing Zhao for valuable discussion on the topic of CRISPR/Cas9.
This work was supported in whole by the National Institutes of Health, NEI Grant R01 EY012509 (to H. L.) and in part by the National Institutes of Health, NEI Core Grant P30EY003790. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PVR
- proliferative vitreoretinopathy
- RPE
- retinal pigment epithelial
- CRISPR/Cas9
- clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9
- crRNA
- CRISPR RNA
- Sp
- Streptococcus pyogenes
- sgRNA
- single guide RNA
- NHEJ
- non-homologous end joining
- HDR
- homology-directed repair
- hPRPE
- human primary retinal pigment epithelial
- RV
- rabbit vitreous
- AAV
- adeno-associated virus
- RSV
- Rous sarcoma virus
- ssHRT
- single-strand homology repair template.
References
- 1. Morescalchi F., Duse S., Gambicorti E., Romano M. R., Costagliola C., and Semeraro F. (2013) Proliferative vitreoretinopathy after eye injuries: an overexpression of growth factors and cytokines leading to a retinal keloid. Mediators Inflamm. 2013, 269787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pastor-Idoate S., Rodríguez-Hernández I., Rojas J., Fernández I., García-Gutiérrez M. T., Ruiz-Moreno J. M., Rocha-Sousa A., Ramkissoon Y., Harsum S., MacLaren R. E., Charteris D., VanMeurs J. C., González-Sarmiento R., Pastor J. C., and Genetics on PVR Study Group (2013) The T309G MDM2 gene polymorphism is a novel risk factor for proliferative vitreoretinopathy. PLoS ONE 8, e82283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lei H., Rheaume M. A., Cui J., Mukai S., Maberley D., Samad A., Matsubara J., and Kazlauskas A. (2012) A novel function of p53: a gatekeeper of retinal detachment. Am. J. Pathol. 181, 866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lei H., Qian C. X., Lei J., Haddock L. J., Mukai S., and Kazlauskas A. (2015) RasGAP promotes autophagy and thereby suppresses platelet-derived growth factor receptor-mediated signaling events, cellular responses, and pathology. Mol. Cell Biol. 35, 1673–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Casaroli-Marano R. P., Pagan R., and Vilaró S. (1999) Epithelial-mesenchymal transition in proliferative vitreoretinopathy: intermediate filament protein expression in retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 40, 2062–2072 [PubMed] [Google Scholar]
- 6. Connor T. B. Jr., Roberts A. B., Sporn M. B., Danielpour D., Dart L. L., Michels R. G., de Bustros S., Enger C., Kato H., and Lansing M. (1989) Correlation of fibrosis and transforming growth factor-β type 2 levels in the eye. J. Clin. Invest. 83, 1661–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leaver P. K., and Billington B. M. (1989) Vitrectomy and fluid/silicone-oil exchange for giant retinal tears: 5 years follow-up. Graefes Arch. Clin. Exp. Ophthalmol. 227, 323–327 [DOI] [PubMed] [Google Scholar]
- 8. Cui J., Lei H., Samad A., Basavanthappa S., Maberley D., Matsubara J., and Kazlauskas A. (2009) PDGF receptors are activated in human epiretinal membranes. Exp. Eye Res. 88, 438–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haupt Y., Maya R., Kazaz A., and Oren M. (1997) Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 [DOI] [PubMed] [Google Scholar]
- 10. Oliner J. D., Kinzler K. W., Meltzer P. S., George D. L., and Vogelstein B. (1992) Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 358, 80–83 [DOI] [PubMed] [Google Scholar]
- 11. Vassilev L. T., Vu B. T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., Fotouhi N., and Liu E. A. (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 [DOI] [PubMed] [Google Scholar]
- 12. Jones S. N., Roe A. E., Donehower L. A., and Bradley A. (1995) Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378, 206–208 [DOI] [PubMed] [Google Scholar]
- 13. Montes de Oca Luna R., Wagner D. S., and Lozano G. (1995) Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378, 203–206 [DOI] [PubMed] [Google Scholar]
- 14. Lei H., Velez G., and Kazlauskas A. (2011) Pathological signaling via platelet-derived growth factor receptor α involves chronic activation of Akt and suppression of p53. Mol. Cell Biol. 31, 1788–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bond G. L., Hu W., Bond E. E., Robins H., Lutzker S. G., Arva N. C., Bargonetti J., Bartel F., Taubert H., Wuerl P., Onel K., Yip L., Hwang S. J., Strong L. C., Lozano G., and Levine A. J. (2004) A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 119, 591–602 [DOI] [PubMed] [Google Scholar]
- 16. Uhrinova S., Uhrin D., Powers H., Watt K., Zheleva D., Fischer P., McInnes C., and Barlow P. N. (2005) Structure of free MDM2 N-terminal domain reveals conformational adjustments that accompany p53-binding. J. Mol. Biol. 350, 587–598 [DOI] [PubMed] [Google Scholar]
- 17. Boersma B. J., Howe T. M., Goodman J. E., Yfantis H. G., Lee D. H., Chanock S. J., and Ambs S. (2006) Association of breast cancer outcome with status of p53 and MDM2 SNP309. J. Natl. Cancer Inst. 98, 911–919 [DOI] [PubMed] [Google Scholar]
- 18. Bougeard G., Baert-Desurmont S., Tournier I., Vasseur S., Martin C., Brugieres L., Chompret A., Bressac-de Paillerets B., Stoppa-Lyonnet D., Bonaiti-Pellie C., and Frebourg T. (2006) Impact of the MDM2 SNP309 and p53 Arg72Pro polymorphism on age of tumour onset in Li-Fraumeni syndrome. J. Med. Genet. 43, 531–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu H., Huang Y. J., Liu Z., Wang L. E., Li G., Sturgis E. M., Johnson D. G., and Wei Q. (2011) Effects of MDM2 promoter polymorphisms and p53 codon 72 polymorphism on risk and age at onset of squamous cell carcinoma of the head and neck. Mol. Carcinog. 50, 697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang L. H., Wang X., Xu W. T., and Hu Y. L. (2014) MDM2 rs2279744 polymorphism and endometrial cancer: a meta-analysis. Tumour Biol. 35, 3167–3170 [DOI] [PubMed] [Google Scholar]
- 21. Gao J., Kang A. J., Lin S., Dai Z. J., Zhang S. Q., Liu D., Zhao Y., Yang P. T., Wang M., and Wang X. J. (2014) Association between MDM2 rs 2279744 polymorphism and breast cancer susceptibility: a meta-analysis based on 9,788 cases and 11,195 controls. Ther. Clin. Risk Manag. 10, 269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jansen R., Embden J. D., Gaastra W., and Schouls L. M. (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 43, 1565–1575 [DOI] [PubMed] [Google Scholar]
- 23. Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D. A., and Horvath P. (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 [DOI] [PubMed] [Google Scholar]
- 24. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., and Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang T., Wei J. J., Sabatini D. M., and Lander E. S. (2014) Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maruyama T., Dougan S. K., Truttmann M. C., Bilate A. M., Ingram J. R., and Ploegh H. L. (2015) Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 33, 538–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sander J. D., and Joung J. K. (2014) CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 32, 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., and Church G. M. (2013) RNA-guided human genome engineering via Cas9. Science 339, 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hsu P. D., Lander E. S., and Zhang F. (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Swiech L., Heidenreich M., Banerjee A., Habib N., Li Y., Trombetta J., Sur M., and Zhang F. (2015) In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 33, 102–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xue W., Chen S., Yin H., Tammela T., Papagiannakopoulos T., Joshi N. S., Cai W., Yang G., Bronson R., Crowley D. G., Zhang F., Anderson D. G., Sharp P. A., and Jacks T. (2014) CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514, 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yin H., Xue W., Chen S., Bogorad R. L., Benedetti E., Grompe M., Koteliansky V., Sharp P. A., Jacks T., and Anderson D. G. (2014) Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 32, 551–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maguire A. M., Simonelli F., Pierce E. A., Pugh E. N. Jr., Mingozzi F., Bennicelli J., Banfi S., Marshall K. A., Testa F., Surace E. M., Rossi S., Lyubarsky A., Arruda V. R., Konkle B., Stone E., et al. (2008) Safety and efficacy of gene transfer for Leber's congenital amaurosis. N. Engl. J. Med. 358, 2240–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zarrin A. A., Malkin L., Fong I., Luk K. D., Ghose A., and Berinstein N. L. (1999) Comparison of CMV, RSV, SV40 viral and Vλ1 cellular promoters in B and T lymphoid and non-lymphoid cell lines. Biochim. Biophys. Acta 1446, 135–139 [DOI] [PubMed] [Google Scholar]
- 35. Ran F. A., Hsu P. D., Lin C. Y., Gootenberg J. S., Konermann S., Trevino A. E., Scott D. A., Inoue A., Matoba S., Zhang Y., and Zhang F. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Scherer S. E., Muzny D. M., Buhay C. J., Chen R., Cree A., Ding Y., Dugan-Rocha S., Gill R., Gunaratne P., Harris R. A., Hawes A. C., Hernandez J., Hodgson A. V., Hume J., Jackson A., et al. (2006) The finished DNA sequence of human chromosome 12. Nature 440, 346–351 [DOI] [PubMed] [Google Scholar]
- 37. Lander E. S., Linton L. M., Birren B., Nusbaum C., Zody M. C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Funke R., Gage D., Harris K., Heaford A., Howland J., et al. (2001) Initial sequencing and analysis of the human genome. Nature 409, 860–921 [DOI] [PubMed] [Google Scholar]
- 38. Lowe T. M., and Eddy S. R. (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang H., Yang H., Shivalila C. S., Dawlaty M. M., Cheng A. W., Zhang F., and Jaenisch R. (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang H., Wang H., Shivalila C. S., Cheng A. W., Shi L., and Jaenisch R. (2013) One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Milanini-Mongiat J., Pouysségur J., and Pagès G. (2002) Identification of two Sp1 phosphorylation sites for p42/p44 mitogen-activated protein kinases: their implication in vascular endothelial growth factor gene transcription. J. Biol. Chem. 277, 20631–20639 [DOI] [PubMed] [Google Scholar]
- 43. Guo A., Salomoni P., Luo J., Shih A., Zhong S., Gu W., and Pandolfi P. P. (2000) The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2, 730–736 [DOI] [PubMed] [Google Scholar]
- 44. Knudson A. G., Jr. (1971) Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U.S.A. 68, 820–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ofir-Rosenfeld Y., Boggs K., Michael D., Kastan M. B., and Oren M. (2008) Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol. Cell 32, 180–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ogawara Y., Kishishita S., Obata T., Isazawa Y., Suzuki T., Tanaka K., Masuyama N., and Gotoh Y. (2002) Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 277, 21843–21850 [DOI] [PubMed] [Google Scholar]
- 47. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., and Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]