

Abstract
The potential for recombining intact polyketide synthase (PKS) modules has been extensively explored. Both enzyme-substrate and protein-protein interactions influence chimeric PKS activity, but their relative contributions are unclear. We now address this issue by studying a library of 11 bimodular and 8 trimodular chimeric PKSs harboring modules from the erythromycin, rifamycin, and rapamycin synthases. Although many chimeras yielded detectable products, nearly all had specific activities below 10% of the reference natural PKSs. Analysis of selected bimodular chimeras, each with the same upstream module, revealed that turnover correlated with the efficiency of intermodular chain translocation. Mutation of the acyl carrier protein (ACP) domain of the upstream module in one chimera at a residue predicted to influence ketosynthase-ACP recognition led to improved turnover. In contrast, replacement of the ketoreductase domain of the upstream module by a paralog that produced the enantiomeric ACP-bound diketide caused no changes in processing rates for each of six heterologous downstream modules compared with those of the native diketide. Taken together, these results demonstrate that protein-protein interactions play a larger role than enzyme-substrate recognition in the evolution or design of catalytically efficient chimeric PKSs.
Keywords: enzyme turnover, polyketide, protein chimera, protein engineering, protein-protein interaction
Introduction
The assembly line architecture of multimodular polyketide synthases (PKSs)5 represents a promising catalytic framework for combinatorial biosynthesis (1). A particularly well studied example of this family of multienzyme systems is the 6-deoxyerythronolide B synthase (DEBS), which produces 6-deoxyerythronolide B (Fig. 1), the parent aglycone of the macrolide antibiotic erythromycin (2). DEBS is comprised of three distinct homodimeric proteins: DEBS1, DEBS2, and DEBS3, each containing two PKS modules, with each module being responsible for a distinct round of polyketide elongation and modification. DEBS uses propionyl-CoA to prime the loading didomain (LDD) of module 1 and six methylmalonyl-CoA-derived extender units in catalysis of the six cycles of polyketide chain elongation, followed by terminal release of the mature polyketide chain by thioesterase (TE)-catalyzed macrolactone formation.
FIGURE 1.

Architecture of DEBS and structure of the resulting product 6-deoxyerythronolide B (6-dEB, 1). The modular architecture of the three constituent proteins (DEBS1, DEBS2, and DEBS3) is shown in cartoon form, together with the product of each catalytic module attached to its acyl carrier protein (ACP) domain. AT, acyltransferase; KS, ketosynthase; KR, ketoreductase (KR0, inactive KR); DH, dehydratase; ER, enoylreductase; TE, thioesterase. Black tabs represent docking domains, short C- and N-terminal polypeptides that enable selective interactions between specific pairs of individual polypeptides.
Since the original genetic characterization of DEBS over two decades ago (3, 4), extensive in vivo and in vitro analysis has revealed that specific protein-protein interactions play a critical role in the proper vectorial channeling of biosynthetic intermediates from one PKS module to the next (5). A particularly effective mini-assembly line for mechanistic analysis has been a simple bimodular derivative of DEBS (Fig. 2) harboring modules 1 and 2 with a fused TE domain. This bimodular PKS has served as a prototype for many analogous constructs.
FIGURE 2.

Catalytic cycle of a bimodular DEBS derivative. This mini assembly line is comprised of three proteins: the LDD (shown in red), DEBS module 1 (M1, shown in yellow), and DEBS module 2 fused to the TE domain (M2+TE, shown in blue/gray). The acyltransferase (AT) of the LDD specifically transfers the propionyl moiety of propionyl-CoA (step 1) to the terminal thiol of the phosphopantetheinylated ACP (step 2) of the LDD. This primer unit is then translocated by acylation of the active site Cys-SH of the KS domain of DEBS M1 (step 3). Meanwhile, the ACP of DEBS M1 is loaded with a methylmalonyl extender unit by the action of the acyltransferase domain of DEBS M1. KS-catalyzed chain elongation by decarboxylative Claisen condensation yields an ACP-bound β-ketoacyl-diketide intermediate (step 4). In DEBS M1, the KR domain then catalyzes an epimerization of the C-2 methyl group followed by diastereospecific reduction (step 5) to give the mature (2S,3R)-diketide, which is then translocated to DEBS M2 via a thioester to thiol transacylation. There it undergoes another round of chain elongation and KR-catalyzed reduction (without epimerization), followed by (TE)-catalyzed release and lactonization (steps 6 and 7).
The potential for engineering chimeric PKSs by recombining modules from paralogous polyketide biosynthetic pathways has been explored for nearly two decades. Early studies revealed the importance of ACP-KS interactions (6, 7), as well as the utility of intermodular linker interactions (8), in the design of productive chimeric assembly lines. At the same time, the relatively broad, but not unrestricted, substrate tolerance of typical KS domains was also recognized (9). In one particularly thorough study, Menzella et al. (10) carried out a large scale assessment of the feasibility of combinatorial biosynthesis by pairwise combinations of 14 modules derived from 8 different PKS clusters, thereby allowing the in vivo characterization of 154 chimeric bimodular PKSs, with the bimodular DEBS derivative (Fig. 2) serving as the benchmark system. Approximately 50% of the resulting chimeras yielded measurable quantities of the expected polyketide product when expressed in Escherichia coli (>0.1 mg/liter culture; ∼1% of the productivity of the benchmark DEBS system). These researchers then extended their investigation to the generation and analysis of trimodular PKSs, using the corresponding trimodular DEBS construct (DEBS modules 1, 2, and 3+TE) as a reference standard (11). Although this extensive study succeeded in revealing many functional PKS combinations, the low yields of the resulting engineered tetraketides suggested that inactive or low producing chimeras most likely resulted from some undefined combination of poor recognition of the diketide by the KS domain of heterologous downstream module or defective interaction of the donor ACP and acceptor KS domains at the junction between heterologous modules.
Recent biochemical investigations in our own laboratories have revealed that protein-protein recognition between ACP and KS domains during intermodular polyketide chain translocation involves a set of protein-protein interactions that are orthogonal to those implicated in intramodular polyketide chain elongation (12, 13). This critical distinction has provided a critical mechanistic framework for factoring the contribution of imperfect intermodular ACP-KS interactions to the turnover efficiency of chimeric assembly line PKSs. We have also established that the KR domain of DEBS M2, which generates the enantiomeric (2R, 3S)-2-methyl-3-hydroxy diketide diastereomer of the natural (2S, 3R) product of DEBS M1, has comparable specificity for the ACP domain of DEBS M1 compared with its natural ACP2 substrate (14). This finding has now allowed us to investigate the influence of the recognition of substrate stereochemistry by an acceptor KS on the overall turnover by otherwise identical chimeric PKSs. We have therefore undertaken a carefully controlled, quantitative analysis of the relative contribution of these protein-protein and protein-substrate recognition to the turnover efficiency of chimeric PKS assembly lines generated by recombining intact modules obtained from heterologous PKSs.
Results
Engineering of Chimeric Bimodular and Trimodular PKSs
Eleven chimeric derivatives of the bimodular PKS shown in Fig. 2 were constructed in which DEBS M2 was replaced by DEBS M3, M5, or M6, or alternatively by each of six modules from the rifamycin synthase (RIFS (15)) or two modules from the rapamycin synthase (RAPS (16); Fig. 3). The predicted structures of each of the corresponding triketide products (structures 2-8 (17–19)) are shown in Fig. 3. To facilitate intermodular interactions between heterologous modules, each acceptor module was fused to the N-terminal docking domain of DEBS2 (Fig. 1). (The numbers in parentheses define the identity of the docking domains. For example, the benchmark bimodular derivative of DEBS consists of LDD(4), (5)M1(2), and (3)M2+TE, where parenthetical numbers refer to the parent DEBS modules that are normally adjacent to the corresponding N- or C-terminal docking domains). These helical docking domains mediate weak but specific non-covalent interactions between donor and acceptor modules (KD = ∼100 μm) (20).
FIGURE 3.

Chimeric bimodular PKSs. Each PKS included LDD(4) and DEBS (5)M1(2) in combination with (3)Module+TE as the variable acceptor. Acceptor modules were derived from DEBS, rifamycin synthase (RIFS), or rapamycin synthase (RAPS). Compatible docking domains from DEBS (depicted as black tabs or numbers in parentheses) were fused to the corresponding C- and N-terminal ends of the respective donor and acceptor modules to enhance the specificity and efficiency of intermodular chain translocation. The predicted triketide products 2–8 generated in the presence of propionyl-CoA, methylmalonyl-CoA (and malonyl-CoA, in the case of RIFS M2+TE), and NADPH, are shown for each chimeric module pair. All acceptor modules are specific for methylmalonyl-CoA, except for RIFS M2, which prefers malonyl-CoA but can also accept methylmalonyl-CoA.
An additional eight chimeric trimodular PKSs were also engineered in an analogous manner using LDD(4), DEBS (5)M1(2), an intervening heterologous (3)Module(2), and DEBS (3)M3+TE as the terminal acceptor module. As before, appropriate docking domains were fused to the C- and N-terminal ends of each protein so as to enable effective protein-protein interactions. In addition to the benchmark trimodular PKS with DEBS (3)M2(2) as the intervening module, six modules from RIFS and two modules from RAPS were also tested (Fig. 4). The structures of each of the predicted tetraketide products (structures 9-15) are shown above each trimodular series (Fig. 4). The yield and purity of individual proteins used in this study are documented in Fig. 5.
FIGURE 4.

Chimeric trimodular PKSs. Each PKS includes LDD(4), DEBS (5)M1(2), a variable (3)Module(2), and DEBS (3)M3+TE as the terminal module. The variable intervening modules were derived from DEBS, RIFS, or RAPS. Compatible docking domains from DEBS (depicted as black tabs or numbers in parentheses) were fused to the C and N termini of each module. The predicted triketide products 9–15, generated in the presence of propionyl-CoA, methylmalonyl-CoA (and malonyl-CoA, in the case of RIFS M2+TE), and NADPH, are shown above each trimodular combination. All acceptor modules were specific for methylmalonyl-CoA except for RIFS M2, which prefers malonyl-CoA but can also accept methylmalonyl-CoA.
FIGURE 5.

Yields and purity of proteins used. A and B, SDS-PAGE analysis of individual PKS modules used to reconstitute chimeric bimodular (A) and trimodular (B) assembly lines are shown. C, SDS-PAGE analysis of mutant and hybrid modules used in this study is shown. The yield of each protein from a recombinant E. coli culture is summarized in the panel to the right.
Activity of Chimeric Bimodular and Trimodular PKSs
To test the in vitro activity of each chimeric PKS, a previously developed UV340 spectrophotometric assay was performed using propionyl-CoA, methylmalonyl-CoA (and malonyl-CoA in case of RIFS M2), and NADPH as substrates (21). The stoichiometric coupling between polyketide formation and NADPH consumption at steady state, in which the stoichiometric coefficient corresponds to the number of catalytically active ketoreductase domains in the assembly line, allows convenient and sensitive monitoring of the steady-state turnover rate of the multimodular PKS assembly line (Fig. 6) (21).
FIGURE 6.
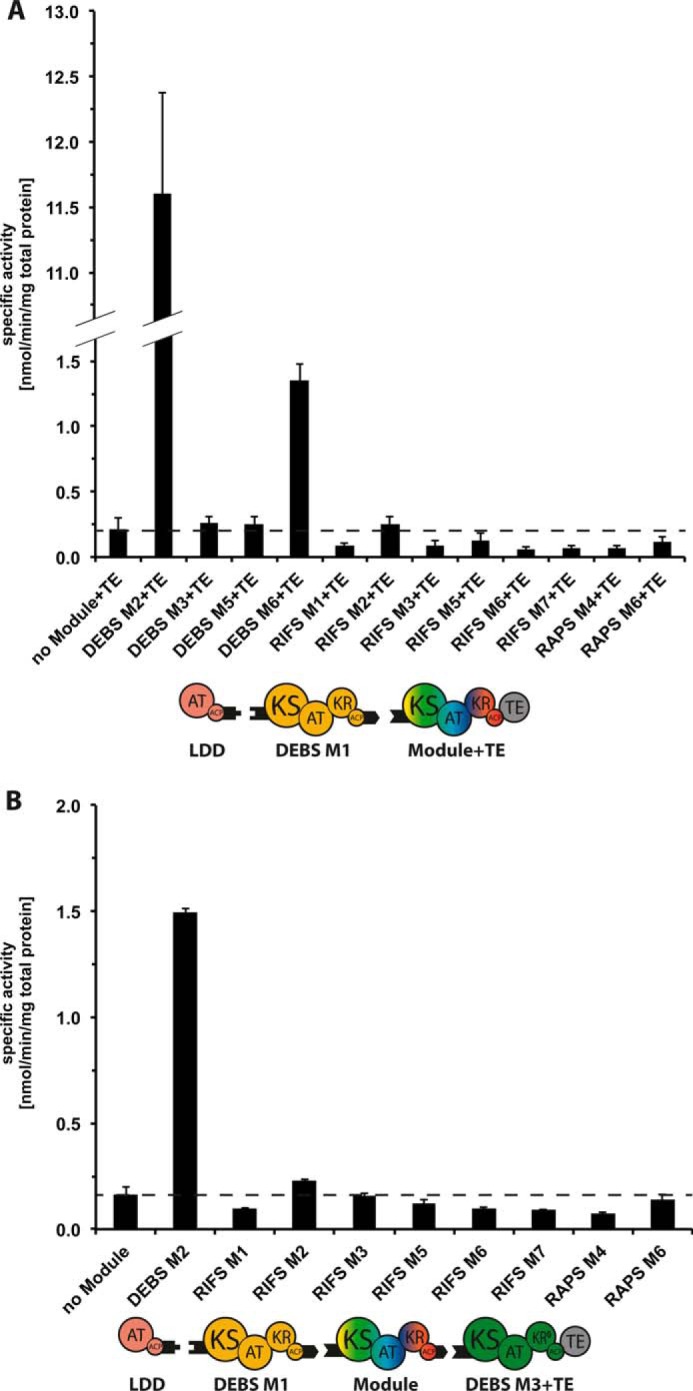
Turnover rates of chimeric bimodular (A) and trimodular (B) PKSs. All initial rate data were obtained at individual PKS protein concentrations of 4 μm and non-limiting concentrations of propionyl-CoA, methylmalonyl-CoA, and NADPH. In assays containing RIFS M2+TE, malonyl-CoA was also included because this module prefers malonyl extender units, although exclusion of malonyl-CoA did not affect the turnover rate of this system. Dashed lines indicate the threshold rate of NADPH consumption in the absence of the chimeric module. Error bars indicate averages of two measurements (each performed in triplicate) obtained with independent protein preparations. Numerical values for all bars are provided in supplemental Tables S6 and S7.
For the bimodular PKSs, the reference DEBS M1+M2+TE construct showed a steady-state rate of triketide formation rate of 11.6 nmol/min/mg total protein, in good agreement with that previously reported for this system (21). All the other chimeric bimodular assembly lines exhibited significantly reduced rates of turnover, the highest being that using DEBS M6 as the surrogate acceptor module (1.4 nmol/min/mg total protein; Fig. 6A). Background NADPH consumption was measured in a control reaction containing only LDD(4) and (5)DEBSM1(2) in the absence of a downstream acceptor module (Fig. 6A, dashed line). Only the chimeras derived from DEBS M3, DEBS M5, DEBS M6, and RIFS M2 showed detectable activity above this threshold background.
A similar decrease in polyketide synthase activity was observed for the chimeric trimodular PKSs. The reference DEBS M1+M2+M3+TE construct showed a steady-state rate of tetraketide formation rate of 1.5 nmol/min/mg total protein, whereas the remaining nine chimeric trimodular PKSs were 6–20-fold less active (Fig. 6B). The baseline rate of NADPH consumption was established by a control reaction lacking DEBS M2 and containing only LDD(4), (5)M1(2), and DEBS (3)M3+TE. Importantly, this control also established the extent to which aberrant intermodular chain translocation between DEBS M1 and M3 could be facilitated solely by compatible docking domains. Relative to this threshold (Fig. 6B, dashed line), only the trimodular construct harboring RIFS M2 showed detectable activity, albeit only 15% of the reference DEBS M1+M2+M3+TE system. The close correspondence between the turnover rates of the corresponding bimodular and trimodular PKSs argues against any potential influence of the appended TE domain on the measured relative activities of the chimeric bimodular PKSs.
The polyketide product profiles of each of the above reaction mixtures were analyzed by LC-MS after overnight incubation. As shown in Fig. 7, 6 of the 11 chimeric bimodular PKSs synthesized detectable quantities of their expected product. Although the chimeras harboring RIFS M5 and RIFS M7 showed only close to background NADPH consumption, they still produced traces of their expected triketide products in an overnight incubation. Likewise, as shown in Fig. 8, 4 of 8 chimeric trimodular PKSs yielded detectable amounts of the expected tetraketide.
FIGURE 7.

LC-MS analysis of triketide products. A, lactones 2 and 4 (C9H16O3, calculated molecular weight 172.110) were detected in reaction mixtures containing the reference module (DEBS (3)M2+TE), as well as DEBS (3)M5+TE and (3)M6+TE, and RIFS (3)M5+TE and (3)M7+TE. B, ketolactone 3 (C9H14O3, calculated molecular weight 170.090) was detected in reaction mixtures containing DEBS (3)M3+TE and RIFS (3)M2+TE. Ketolactone 8 (C8H11O3, calculated molecular weight 156.080) was detected in reaction mixtures containing RIFS (3)M2+TE with malonyl-CoA. For all systems the extracted ion chromatograms, obtained by extraction of the [M + Na]+ species, are shown, and the peak of interest is marked by an arrow based on its characteristic mass spectrum, shown explicitly in the case of DEBS (3)M2+TE. (Labeled peaks from left to right correspond to [M + H-H2O]+, [M + H]+, and [M + Na]+ ions.)
FIGURE 8.

LC-MS analysis of tetraketide products. A, lactone 10 (C12H20O4, calculated molecular weight 228.140) was detected in reaction mixtures containing the reference module (DEBS (3)M2(2)), as well as RIFS (3)M5(2). B, pyrone 9 (C12H18O4, calculated molecular weight 226.120) was detected in reaction mixtures containing RIFS (3)M2(2) and (3)M3(2), as well as RAPS (3)M6(2). For all systems the extracted ion chromatograms, obtained by extraction of the [M + Na]+ species, are shown, and the peak of interest is marked by an arrow based on its characteristic mass spectrum, shown explicitly in the case of DEBS (3)M2(2). (Labeled peaks from left to right correspond to [M + H-H2O]+, [M + H]+, and [M + Na]+ ions.)
Role of ACP-KS Interactions at the Fusion Site in Chimeric PKSs
Because the majority of the chimeric PKS constructs shown in Fig. 6 had very low levels of activity, we sought to identify the specific step in assembly line biosynthesis at which the growing polyketide chain might have become stalled. For this purpose, radio-SDS-PAGE analysis was performed using [14C]propionyl-CoA to prime the LDD. By quantifying the extent of labeling of each protein module in the presence of methylmalonyl-CoA and NADPH, the site of polyketide accumulation could be deduced. To enhance the sensitivity of the experiment and thereby allow the detection of rare intermodular chain translocation events in these low activity systems, the TE domains of each acceptor module were inactivated by active site Ser → Ala mutations.
As seen in Table 1, the acceptor module of the reference system (DEBS LDD+M1+M2+TE0) was rapidly labeled, reaching saturation within 3 min. In contrast, the single turnover occupancy level of the acceptor modules of representative chimeric bimodular PKSs correlated quite well with their catalytic turnover (compare Figs. 6A and Table 1). Only DEBS M6+TE0 showed significant labeling, reaching 42% of the reference system after 3 min. Other acceptor modules (DEBS M3+TE0, RIFS M2+TE0, RAPS M4+TE0, and RAPS M6+TE0) showed negligible levels of diketide occupancy on this time scale.
TABLE 1.
Quantification of the occupancy of acceptor modules in chimeric bimodular PKSs
All bimodular PKSs included DEBS LDD and M1 plus the designated acceptor module. Only labeling data for the acceptor module are shown, because the upstream proteins are the same in all cases. Labeling of each acceptor module (2 μm) was quantified at 1 and 3 min in the presence of both LDD (2 μm) and M1 (2 μm). Each measurement was performed in duplicate. The entire experiment was repeated with independently prepared proteins to verify the data trends. The TE domain of each acceptor module was inactivated by site-directed mutagenesis to abolish TE-catalyzed triketide chain release and consequent multiple turnover from this module.
| Acceptor module | Normalized area counts |
|
|---|---|---|
| After 1 min | After 3 min | |
| DEBS M2+TE0 | 0.83 ± 0.11 | 1.00 ± 0.08 |
| DEBS M3+TE0 | <0.02 | <0.02 |
| DEBS M6+TE0 | 0.21 ± 0.07 | 0.42 ± 0.17 |
| RIFS M2+TE0 | 0.04 ± 0.01 | 0.08 ± 0.00 |
| RAPS M4+TE0 | <0.02 | <0.02 |
| RAPS M6+TE0 | <0.02 | 0.05 ± 0.00 |
To ascertain the cause-effect relationship between ineffective chain translocation and turnover, we sought to enhance the rate of chain translocation in a representative chimeric PKS via site-directed mutagenesis of the ACP domain of DEBS M1. We recently carried out a detailed analysis of the structural elements that control ACP-KS recognition at the DEBS M2-M3 interface (13). Our findings revealed that the N terminus of helix I of ACP2 is the primary specificity determinant during intermodular chain translocation. By contrast, chain elongation within DEBS M3 was controlled by a loop connecting helices I and II of ACP3.
Based on this earlier observation, we predicted that chain translocation from DEBS M1 to M3 in a chimeric bimodular PKS would be enhanced by introducing the E23K mutation (corresponding to E1424K, based on whole module numbering) into helix I of ACP1, because a cationic residue is also found at the corresponding position of ACP2. Indeed, turnover of the E23K mutant of DEBS M1+M3+TE was increased more than 2-fold over the corresponding bimodular PKS derived exclusively from wild-type modules (0.6 ± 0.05 nmol/min/mg total protein, compared with 0.3 ± 0.05 nmol/min/mg total protein) under experimental conditions identical to those of Fig. 6). Taken together, these results underscore the pivotal role of ACP-KS interactions during translocation of the growing polyketide chain between heterologous PKS modules.
Role of Substrate-KS Recognition at Fusion Junctions in Chimeric PKSs
Having examined the influence of ACP-KS interactions on chain translocation at chimeric PKS junctions, we sought to interrogate the relative contribution of substrate-KS recognition on the turnover efficiency of these systems. To this end we rationalized that presentation of the enantiomer of the native (2S,3R)-2-methyl-3-hydroxy-diketide-ACP product of DEBS M1 (Fig. 2) to the downstream modules of selected chimeric bimodular PKSs from Fig. 3 would enable a controlled comparison of the relative importance of specific protein-protein and protein-substrate recognition. Because we have recently shown that the KR domain of DEBS M2 has comparable specificity for ACP1 as for its cognate ACP2 domain (14), we engineered DEBS M1 by replacing its endogenous KR domain with the paralogous KR2 from DEBS M2. The resulting hybrid module, DEBS (5)M1-KR2(2), was expressed as a soluble protein (Fig. 5, 2 mg/liter in E. coli) with purification behavior on an ion exchange chromatography column that was indistinguishable from most wild-type PKS modules.
In addition to evaluating the turnover efficiency of this hybrid module in the presence of DEBS LDD(4) and (3)M2+TE, we also measured turnover in the presence of six other acceptor PKS modules (Fig. 9). The products (Fig. 9A, structures 16–18) of each of these chimeric PKSs were predicted to be the corresponding (2R, 3S)-diastereomers of the previously characterized triketide lactones 2, 3, and 8. Consistent with the previously established 7-fold preference of DEBS M2 for its natural (2S,3R)-diketide substrate over the enantiomeric (2R,3S)-diketide analog (22), the bimodular construct harboring the hybrid donor module showed a comparable 7-fold reduction in turnover rate compared with its wild-type bimodular counterpart (Fig. 9B). Remarkably, none of the other bimodular PKSs containing heterologous downstream modules was significantly affected in response to the change in the configuration of the diketide intermediate (Fig. 9B), indicating that enzyme-substrate recognition is not as important as specific ACP-KS protein-protein recognition during intermodular polyketide chain translocation. To verify the predicted properties of the bimodular PKSs in Fig. 9, the corresponding triketide products were each analyzed by LC-MS (Fig. 10).
FIGURE 9.

Influence of substrate-KS recognition on chimeric PKSs. A, chimeric bimodular PKSs harboring the DEBS M1-KR2 mutant. Each PKS included LDD(4) and DEBS (5)M1-KR2(2) in combination with (3)Module+TE as the variable acceptor. The predicted triketide products 16–18 generated in the presence of propionyl-CoA, methylmalonyl-CoA (and malonyl-CoA, in the case of RIFS M2+TE), and NADPH, are shown for each chimeric module pair. B, turnover rates of bimodular constructs consisting of DEBS LDD, DEBS M1 plus the designated acceptor module (black bars) and turnover rates of the same systems with DEBS M1-KR2 mutant (gray bars) in place of DEBS M1. All initial rate data were obtained at individual PKS protein concentrations of 4 μm and non-limiting concentrations of propionyl-CoA, methylmalonyl-CoA (and malonyl-CoA, in the case of RIFS M2+TE), and NADPH. The dashed line indicates the threshold rate of NADPH consumption in the absence of the chimeric module. Error bars indicate averages of three measurements. Numerical data from this bar graph are summarized in supplemental Table S8.
FIGURE 10.

LC-MS analysis of diastereomeric triketide lactones produced by bimodular chimeric PKS harboring either DEBS M1 (left) or DEBS M1-KR2 (right). Diastereomeric triketide lactones 2 and 17 (C9H16O3, calculated molecular weight 172.110) were detected in reaction mixtures containing DEBS (3)M2+TE, as well as DEBS (3)M5+TE and (3)M6+TE. A reference sample of lactone 17 was produced by self-priming of DEBS3 with methylmalonyl-CoA (28); the two diastereomers are distinguishable by LC-MS analysis (supplemental Fig. S1). Diastereomeric triketide lactones 3 and 16 (C9H14O3, calculated molecular weight 170.090) were detected in reaction mixtures containing DEBS (3)M3+TE and RAPS (3)M6+TE. In all cases the extracted ion chromatograms corresponding to the [M + Na]+ species are shown, and the peak of interest is marked by an arrow.
Discussion
Combinatorial assembly of modules from different assembly line PKSs is, in principle, a powerful strategy for complex molecule biosynthesis. The relative importance of protein-protein interactions and enzyme-substrate recognition in such engineered systems remains unclear, however. We have now carried an extensive in vitro analysis that addresses these issues for the first time in a systematic, carefully controlled manner.
Purification and analysis of 11 chimeric bimodular PKSs and 8 chimeric trimodular PKSs harboring modules from the erythromycin, the rifamycin, and the rapamycin synthases vividly demonstrated that most chimeric PKSs suffer severe impairment of catalytic activity (Fig. 6), notwithstanding the grafting of demonstrated compatible C- and N-terminal docking sites to their intermodular interfaces (8, 23). The 10–100-fold reduction in the catalytic efficiency and productivity between wild-type and chimeric systems was particularly striking, given that the choice of bimodular and trimodular PKS constructs was based on published reports that demonstrated measurable in vivo product forming capacity for several of these chimeras (10, 11). We therefore sought to exploit the power of biochemical reconstitution to dissect the mechanistic basis for the observed loss in catalytic effectiveness.
Analysis of the occupancy of the acceptor modules in chimeric bimodular PKSs showed that in the majority of cases, polyketide chain translocation represented a serious bottleneck (Table 1). Earlier studies have highlighted a role for ACP-KS interactions in the channeling of polyketide chains between modules (13, 18); this conclusion is reinforced by our observation that a Glu → Lys charge reversal mutation in the ACP domain of the donor module DEBS M1 improved turnover in the presence of DEBS M3+TE. The structural logic of these protein-protein interactions is starting to become apparent (24). We note that, because interactions between the donor ACP and acceptor KS domains have not been explicitly interrogated in the experiments summarized in Table 1, the possibility of additional unproductive and therefore catalytically silent binding between chimeric PKS modules cannot be ruled out. Indeed, an important feature of these studies is that they focus only on catalytically relevant protein-protein interactions.
To assess the relative importance of substrate-KS recognition in the same chimeric bimodular PKSs, we engineered a variant DEBS M1 donor module harboring the KR2 domain derived from DEBS M2. In place of the natural (2S, 3R)-2-methyl-3-hdyroxydiketide product of DEBS M1, this hybrid module generates the enantiomeric (2R,3S)-diketide-ACP intermediate. Replacement of KR1 by KR2 was not expected to significantly alter the rate of reduction of the ACP-bound β-ketoacylthioester intermediate (Fig. 2) (14), thereby enabling a comparison with the corresponding bimodular PKSs harboring wild-type DEBS M1 (Fig. 9). The observed 7-fold decrease in the turnover rate of the reference system is in excellent agreement with the differences in kcat previously reported when DEBS M2+TE was presented with chemo-enzymatically synthesized (2S, 3R)-diketide-ACP versus (2R, 3S)-diketide-ACP (22). In sharp contrast, none of the intrinsically slower chimeric bimodular PKSs harboring DEBS M3, DEBS M5, DEBS M6, RIFS M2, RIFS M5, or RAPS M6 as acceptor modules were significantly further affected by the change in configuration of the diketide intermediate. Thus, recognition of the incoming substrate by the KS domain of the acceptor module does not appear to influence intermodular chain translocation to the same extent as protein-protein recognition of the donor ACP domain by the acceptor KS. Future efforts to design catalytically efficient chimeric assembly lines must therefore focus on optimizing protein-protein interactions at the junctions between heterologous PKS modules.
Experimental Procedures
Reagents
Phusion Hot Start II polymerase was from Thermo Scientific. T4 DNA Ligase was from Invitrogen. Restriction enzymes were from New England Biolabs. All primers were synthesized by Elim Biopharm. For DNA purification, the GeneJET plasmid miniprep kit and the GeneJET gel extraction kit from Thermo Scientific were used. XL10-Gold ultracompetent cells were from Agilent; MAX Efficiency® DH5α competent cells and One Shot® BL21 (DE3) cells were from Invitrogen. All chemicals for buffer preparations were from Sigma-Aldrich. Isopropyl-β-d-1-thiogalactopyranoside, kanamycin sulfate, and carbenicillin were from Gold Biotechnology. LB-Miller broth for cell cultures was from Fisher Scientific. Nickel-nitrilotriacetic acid affinity resin was from MC Lab. For anion exchange chromatography, the HiTrapQ column was from GE Healthcare. For all SDS-PAGE analyses, SDS-PAGE Mini Protean TGX precast gels were from Bio-Rad (4–20% and 7.5%). For protein concentration, Amicon Ultra centrifugal filters were from Millipore. CoA, reduced NADPH, sodium propionate, propionic acid, methylmalonic acid, malonic acid, and magenesium chloride hexahydrate were from Sigma-Aldrich. ATP was from Teknova. Reducing agent tris(2-carboxyethyl)phosphine (TCEP) was from Thermo Scientific. UVette cuvettes (2 × 10-mm path) were from Eppendorf. Sodium [1-14C]-propionate was from Moravek Biochemicals.
Strains
Glycerol stocks were prepared by mixing LB medium containing 30% glycerol with the appropriate overnight culture in a 1:2 v/v ratio and were stored at −80 °C. Seed cultures were inoculated with cells from the cell stock and grown overnight.
Plasmids
Plasmids harboring genes encoding individual PKS modules from different organisms were amplified from primary cosmid DNA of the corresponding PKS gene clusters (DEBS, RIFS, RAPS). Supplemental Table S1 specifies the primer sequences used for this purpose. All plasmids were constructed using the two-fragment Gibson assembly method or infusion cloning (Clontech), and their sequences were verified by DNA sequencing. Supplemental Tables S2 and S3 summarize the plasmid construction details for bimodular and trimodular PKS systems, respectively. Templates and primers used for the construction of DEBS M1-KR2 are shown in supplemental Table S5.
Site-directed Mutagenesis
To alter individual amino acid residues in expressed modules, either QuikChange site-directed mutagenesis (Agilent) or a site-directed mutagenesis strategy coupled to restriction-ligation was used. Mutagenesis primers are defined in supplemental Table S4. All mutations were verified by DNA sequencing.
Bacterial Cell Culture and Protein Purification
All proteins were expressed and purified using similar protocols. For holo-proteins (where the ACP domain is post-translationally modified with a phosphopantetheine arm), E. coli BAP1 cells (25) were used as the host. All proteins contained a C-terminal His6 tag for purification. Cultures were grown on a 1-liter scale at 37 °C to an A600 of 0.6. Protein production was induced with 0.25 mm isopropyl-β-d-1-thiogalactopyranoside, whereupon the temperature was adjusted to 18 °C, and the cells were grown for another 16 h. The cells were harvested by centrifugation at 4420 × g for 20 min and lysed by sonication in a buffer consisting of 50 mm sodium phosphate, 10 mm imidazole, 450 mm NaCl, and 10% glycerol, pH 7.6. The cell debris was removed by centrifugation at 25,000 × g for 1 h. The supernatant was added to nickel-nitrilotriacetic acid-agarose resin (2 ml of resin/liter of culture), and the resulting slurry was incubated at 4 °C for 1 h. Thereafter, the mixture was applied to a Kimble-Kontes Flex column and washed with the above lysis buffer (25 column volumes). Additional washing was performed with 12.5 column volumes of wash buffer (50 mm phosphate, 25 mm imidazole, 300 mm NaCl, 10% glycerol, pH 7.6). Proteins were eluted in two steps (each with 6 column volumes). The first elution step utilized 75 mm phosphate, 150 mm imidazole, 40 mm NaCl, 10% glycerol, pH 7.6, whereas the second elution step was performed with 75 mm phosphate, 500 mm imidazole, 40 mm NaCl, 10% glycerol, pH 7.6.
Further purification was performed by anion exchange chromatography on an ÄKTA FPLC system using a HiTrapQ column. Buffer A contained 50 mm phosphate, 10% glycerol, pH 7.6, whereas buffer B contained 50 mm phosphate, 500 mm NaCl, 10% glycerol, pH 7.6. Protein concentrations were measured with the BCA protein assay kit (Thermo Scientific). Samples were stored as aliquots at −80 °C until further use.
PKS Enzymatic Assays
The stoichiometric relationship between polyketide production and NADPH consumption in a reconstituted enzyme system has been previously verified, thereby enabling the use of a UV assay procedure to monitor PKS turnover (21). Reactions were performed on a 70-μl scale and contained 400 mm sodium phosphate (pH 7.2), 5 mm TCEP, 10 mm MgCl2, 1 mm CoA, and 8 mm ATP. Methylmalonate/malonate (1 mm) was converted to racemic methylmalonyl-CoA using the enzymes MatB (2 μm) and methylmalonyl-CoA epimerase (4 μm) (26). Propionyl-CoA was synthesized from propionate using PrpE (1 μm) (27). The concentration of these enzymes and cofactors was selected to assure that the acyl-CoA supply was not limiting the rate of product formation. Reactions were initiated by the addition of PKS proteins (4 μm each) along with a mixture of propionate (0.5 mm), methylmalonate/malonate (1 mm), and NADPH (0.5 mm). The rate was monitored at 340 nm over 20 min. For product analysis by LC-MS, reactions were quenched with ethyl acetate, extracted twice with 450 μl, dried in vacuo, and stored at −20 °C.
Liquid Chromatography-Mass Spectrometry Analysis of Polyketides
Dried samples were reconstituted in 100 μl of methanol, separated on a Gemini-NX C18 column (Phenomenex, 5 μm, 2 × 100 mm) connected to an Agilent 1260 HPLC over a 28-min linear gradient of acetonitrile from 3% to 95% in water, and subsequently injected into a 6520 Accurate-Mass QTOF mass spectrometer. Reduced and unreduced triketides and tetraketide products were located by searching for the theoretical m/z for the [M + Na]+ and [M + H]+ ion. Unreduced triketides: [M + Na]+ = 193.080 and [M + H]+ = 171.098, reduced triketides [M + Na]+ = 195.100 and [M + H]+ = 173.118, acyclic triketides: [M + Na]+ = 177.090 and [M + H]+ =155.108, unreduced triketides using malonyl-CoA: [M + Na]+ = 179.070 and [M + H]+ = 157.088. Unreduced tetraketides: [M + Na]+ = 249.110 and [M + H]+ = 227.128, reduced tetraketides: [M + Na]+ = 251.130 and [M + H]+ = 229.148, acyclic tetraketides: [M + Na]+ = 252.130 and [M + H]+ = 230.148, unreduced tetraketides using malonyl-CoA: [M + Na]+ = 235.090 and [M + H]+ = 213.108.
14C Radioisotopic SDS-PAGE Labeling Assay
[1-14C]Propionate was used to interrogate intermodular chain translocation from LDD(4) to DEBS (5)M1(2) and subsequently to an acceptor (3)Module+TE. Substrates were generated by mixing 0.4 mm [1-14C]propionate, 2 mm methylmalonate, 2.4 mm CoASH, 3.5 μm PrpE, 2 μm MatB, and 4 μm SCME in a 30-min reaction containing 400 mm sodium phosphate, pH 7.2, 5 mm TCEP, 10 mm MgCl2, and 8 mm ATP. To quantify the efficiency of channeling of the labeled propionyl group successively from the LDD to each downstream module, the substrate mixture was diluted 10-fold into individual reaction mixtures containing 2 μm LDD(4), 2 μm DEBS (5)M1(2), and 2 μm of each acceptor module in the presence of 400 mm sodium phosphate, pH 7.2, 5 mm TCEP, and 0.75 mm NADPH. By minimizing the amount of labeled propionyl-CoA in the reaction mixture, nonspecific transfer of the radiolabel directly from the LDD to an acceptor module was minimized. At specified time points, reactions were quenched by addition of Laemmli buffer, and samples were separated via 7.5% SDS-PAGE at 200 V for 44 min. The gel was washed with water for 5 min and stained with SimplyBlueTM SafeStain (Invitrogen) for 10 min. Following destaining with water for 5 min, the gel was mounted on a filter paper and dried in vacuo for 2 h using a Bio-Rad 543 gel dryer. The dried gel was imaged for 1 h lane by lane to quantify 14C on individual protein bands using a Rita Star TLC Analyzer (Raytest). Peaks were integrated to quantify the radiolabel bound to each protein.
Author Contributions
The manuscript was written through contributions of all authors. M. K., M. P. O., J. A., T. R., and B. L. designed the study. M. K. purified the proteins, constructed TE-knockout mutants, and carried out the NADPH consumption, LC-MS analysis, and 14C radiolabeling experiments. J. A. constructed the plasmid library. M. P. O. constructed the DEBS M1-KR2 mutant, purified it, and performed experiments for Fig. 9. All authors have given approval to the final version of the manuscript.
Supplementary Material
This work was supported by National Institutes of Health Grants GM 087934 (to C. K.) and GM 022172 (to D. E. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Tables S1–S8 and Fig. S1.
- PKS
- polyketide synthase
- ACP
- acyl carrier protein
- DEBS
- 6-deoxyerythronolide B synthase
- KR
- ketoreductase
- KS
- ketosynthase
- RAPS
- rapamycin synthase
- RIFS
- rifamycin synthase
- TE
- thioesterase
- LDD
- loading didomain
- TCEP
- tris(2-carboxyethyl)phosphine.
References
- 1. Walsh C. (2004) Polyketide and nonribosomal peptide antibiotics: modularity and versatility. Science 303, 1805–1810 [DOI] [PubMed] [Google Scholar]
- 2. Khosla C., Tang Y., Chen A. Y., Schnarr N. A., and Cane D. E. (2007) Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu. Rev. Biochem. 76, 195–221 [DOI] [PubMed] [Google Scholar]
- 3. Cortes J., Haydock S. F., Roberts G. A., Bevitt D. J., and Leadlay P. F. (1990) An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea. Nature 348, 176–178 [DOI] [PubMed] [Google Scholar]
- 4. Donadio S., Staver M. J., McAlpine J. B., Swanson S. J., and Katz L. (1991) Modular organization of genes required for complex polyketide biosynthesis. Science 252, 675–679 [DOI] [PubMed] [Google Scholar]
- 5. Khosla C., Herschlag D., Cane D. E., and Walsh C. T. (2014) Assembly line polyketide synthases: mechanistic insights and unsolved problems. Biochemistry 53, 2875–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McDaniel R., Kao C. M., Hwang S. J., and Khosla C. (1997) Engineered intermodular and intramodular polyketide synthase fusions. Chem. Biol. 4, 667–674 [DOI] [PubMed] [Google Scholar]
- 7. Ranganathan A., Timoney M., Bycroft M., Cortés J., Thomas I. P., Wilkinson B., Kellenberger L., Hanefeld U., Galloway I. S., Staunton J., and Leadlay P. F. (1999) Knowledge-based design of bimodular and trimodular polyketide synthases based on domain and module swaps: A route to simple statin analogues. Chem. Biol. 6, 731–741 [DOI] [PubMed] [Google Scholar]
- 8. Gokhale R. S., Tsuji S. Y., Cane D. E., and Khosla C. (1999) Dissecting and exploiting intermodular communication in polyketide synthases. Science 284, 482–485 [DOI] [PubMed] [Google Scholar]
- 9. Watanabe K., Wang C. C., Boddy C. N., Cane D. E., and Khosla C. (2003) Understanding substrate specificity of polyketide synthase modules by generating hybrid multimodular synthases. J. Biol. Chem. 278, 42020–42026 [DOI] [PubMed] [Google Scholar]
- 10. Menzella H. G., Reid R., Carney J. R., Chandran S. S., Reisinger S. J., Patel K. G., Hopwood D. A., and Santi D. V (2005) Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat. Biotechnol. 23, 1171–1176 [DOI] [PubMed] [Google Scholar]
- 11. Menzella H. G., Carney J. R., and Santi D. V (2007) Rational design and assembly of synthetic trimodular polyketide synthases. Chem. Biol. 14, 143–151 [DOI] [PubMed] [Google Scholar]
- 12. Kapur S., Chen A. Y., Cane D. E., and Khosla C. (2010) Molecular recognition between ketosynthase and acyl carrier protein domains of the 6-deoxyerythronolide B synthase. Proc. Natl. Acad. Sci. U.S.A. 107, 22066–22071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kapur S., Lowry B., Yuzawa S., Kenthirapalan S., Chen A. Y., Cane D. E., and Khosla C. (2012) Reprogramming a module of the 6-deoxyerythronolide B synthase for iterative chain elongation. Proc. Natl. Acad. Sci. U.S.A. 109, 4110–4115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ostrowski M. P., Cane D. E., and Khosla C. (2016) Recognition of acyl carrier proteins by ketoreductases in assembly line polyketide synthases. J. Antibiot. (Tokyo) 10.1038/ja.2016.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yu T. W., Shen Y., Doi-Katayama Y., Tang L., Park C., Moore B. S., Richard Hutchinson C., and Floss H. G. (1999) Direct evidence that the rifamycin polyketide synthase assembles polyketide chains processively. Proc. Natl. Acad. Sci. U.S.A. 96, 9051–9056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schwecke T., Aparicio J. F., Molnár I., König A., Khaw L. E., Haydock S. F., Oliynyk M., Caffrey P., Cortés J., Lester J. B., Böhm G. A., Staunton J., and Leadlay P. F. (1995) The biosynthetic gene cluster for the polyketide immunosuppressant rapamycin. Proc. Natl. Acad. Sci. U.S.A. 92, 7839–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park S. R., Yoo Y. J., Ban Y.-H., and Yoon Y. J. (2010) Biosynthesis of rapamycin and its regulation: past achievements and recent progress. J. Antibiot. 63, 434–441 [DOI] [PubMed] [Google Scholar]
- 18. You Y. O., Khosla C., and Cane D. E. (2013) Stereochemistry of reductions catalyzed by methyl-epimerizing ketoreductase domains of polyketide synthases. J. Am. Chem. Soc. 135, 7406–7409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hartung I. V., Rude M. A., Schnarr N. A., Hunziker D., and Khosla C. (2005) Stereochemical assignment of intermediates in the rifamycin biosynthetic pathway by precursor-directed biosynthesis. J. Am. Chem. Soc. 127, 11202–11203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Buchholz T. J., Geders T. W., Bartley F. E. 3rd, Reynolds K. A., Smith J. L., and Sherman D. H. (2009) Structural basis for binding specificitybetween subclasses of modular polyketide synthase docking domains. ACS Chem. Biol. 4, 41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lowry B., Robbins T., Weng C. H., O'Brien R. V., Cane D. E., and Khosla C. (2013) In vitro reconstitution and analysis of the 6-deoxyerythronolide B synthase. J. Am. Chem. Soc. 135, 16809–16812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu N., Tsuji S. Y., Cane D. E., and Khosla C. (2001) Assessing the balance between protein-protein interactions and enzyme-substrate interactions in the channeling of intermediates between polyketide synthase modules. J. Am. Chem. Soc. 123, 6465–6474 [DOI] [PubMed] [Google Scholar]
- 23. Tsuji S. Y., Cane D. E., and Khosla C. (2001) Selective protein-protein interactions direct channeling of intermediates between polyketide synthase modules. Biochemistry 40, 2326–2331 [DOI] [PubMed] [Google Scholar]
- 24. Robbins T., Liu Y.-C., Cane D. E., and Khosla C. (2016) Structure and mechanism of assembly line polyketide synthases. Curr. Opin. Struct. Biol. 41, 10–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pfeifer B. A., Admiraal S. J., Gramajo H., Cane D. E., and Khosla C. (2001) Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 291, 1790–1792 [DOI] [PubMed] [Google Scholar]
- 26. Hughes A. J., and Keatinge-Clay A. (2011) Enzymatic extender unit generation for in vitro polyketide synthase reactions: structural and functional showcasing of Streptomyces coelicolor MatB. Chem. Biol. 18, 165–176 [DOI] [PubMed] [Google Scholar]
- 27. Lowry B., Li X., Robbins T., Cane D. E., and Khosla C. (2016) A turnstile mechanism for the controlled growth of biosynthetic intermediates on assembly line polyketide synthases. ACS Cent. Sci. 2, 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jacobsen J. R., Cane D. E., and Khosla C. (1998) Spontaneous priming of a downstream module in 6-deoxyerythronolide B synthase leads to polyketide biosynthesis. Biochemistry 37, 4928–4234 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.