

Summary
Cardiac myocytes from the mdx mouse, the mouse model of Duchenne muscular dystrophy, exhibit t-tubule disarray and increased calcium sparks, but a unifying molecular mechanism has not been elucidated. Recently, improper trafficking of junctophilin (JPH)-2 on an altered microtubule network caused t-tubule derangements and calcium mishandling in a pressure-overload heart failure model. Mdx cardiac myocytes have microtubule abnormalities, but how this may affect JPH-2, t-tubules, and calcium handling has not been established. Here, we investigated the hypothesis that an inverse relationship between microtubules and JPH-2 underlies t-tubule disruptions and calcium mishandling in mdx cardiac myocytes. Confocal microscopy revealed t-tubule disorganization in mdx cardiac myocytes. Quantitative Western blot analysis demonstrated JPH-2 was decreased by 75% and showed an inverse hyperbolic relationship with α- and β-tubulin, the individual components of microtubules, in mdx hearts. Colchicine-induced microtubule depolymerization normalized JPH-2 protein levels and localization, corrected t-tubule architecture, and reduced calcium sparks. In summary, these results suggest microtubule-mediated misregulation of JPH-2 causes t-tubule derangements and altered calcium handling in mdx cardiac myocytes.
Key Words: calcium handling, Duchenne cardiomyopathy, junctophilin-2, microtubules
Abbreviations and Acronyms: DMD, Duchenne muscular dystrophy; JPH-2, junctophilin-2; PBS, phosphate-buffered saline; SR, Sarcoplasmic reticulum; TT, transverse tubules; VGCC, voltage-gated calcium channel; WT, wild-type
Visual Abstract
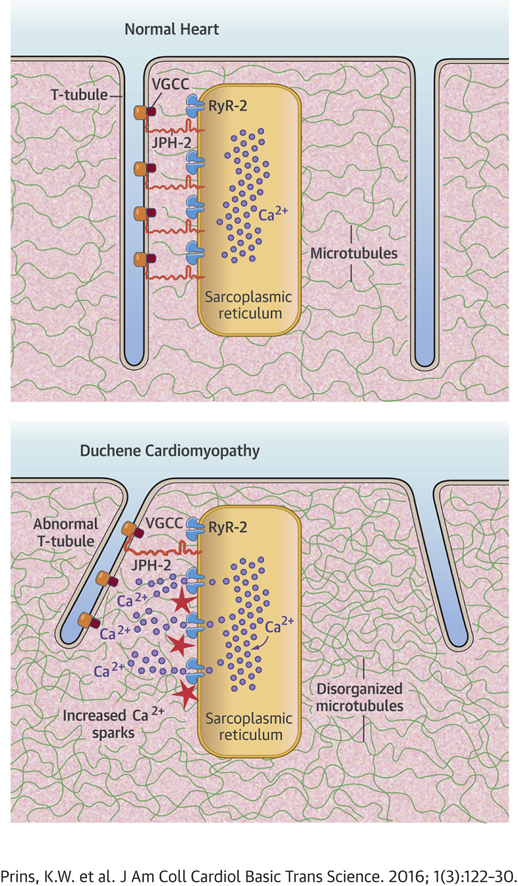
Highlights
-
•
Decreased junctophilin-2 levels are associated with cardiac t-tubule derangements in mdx mice, the mouse model of Duchenne muscular dystrophy (DMD).
-
•
Reduced junctophilin-2 protein levels correlate with increases in total microtubule content in mdx hearts.
-
•
Colchicine-mediated microtubule depolymerization increases junctophilin-2 protein levels and improves localization patterns which, in turn, are associated with t-tubule reorganization and reduced calcium sparks.
-
•
This study identifies microtubule-mediated misregulation of junctophilin-2 as a novel molecular mechanism in Duchenne cardiomyopathy.
Loss of dystrophin causes Duchenne muscular dystrophy (DMD), an X-linked disease characterized by striated muscle dysfunction resulting in a life expectancy of only 20 years to 30 years (1). Heart failure is the cause of death for 20% to 25% of DMD patients (2); therefore research aimed at understanding the molecular and cellular phenotypes underlying the cardiomyopathy of DMD has been conducted. Altered calcium homeostasis marked by increased calcium sparks 3, 4, 5 and t-tubule disarray were documented in cardiac myocytes from the dystrophin-deficient mdx mouse 6, 7, but a unifying molecular mechanism has not been identified.
Junctophilin (JPH)-2, the protein that links the plasma membrane of t-tubules to the ryanodine receptor, is essential for proper t-tubule structure and function (8). Recently, it was reported that JPH-2 mislocalization due to abnormalities in microtubule cytoskeleton caused pathological t-tubule remodeling and abnormal calcium handling in the pressure overload-induced heart failure model (9). However, the relationship between microtubules and JPH-2 in other models of heart failure or cardiomyopathy has not been analyzed. Because previous studies documented microtubule derangements in mdx cardiac myocytes 5, 10, we tested the hypothesis that microtubule alterations cause JPH-2 misregulation and result in t-tubule disruptions and calcium mishandling in mdx mice.
Finally, to investigate the translational aspects of our hypothesis, we examined the cardiomyopathy of mdx mice via echocardiography and isoproterenol stress tests as previous studies showed mildly reduced systolic function 11, 12, 13, 14 and excessive mortality with isoproterenol administration 10, 15, 16 in mdx mice. Because Zhang et al. (9) and Guo et al. (17) showed improvement in systolic function with normalization of JPH-2, we hypothesized colchicine-induced JPH-2 normalization would reduce the severity of mdx cardiomyopathy.
Methods
Mice
Control C57BL/10 and mdx mice were purchased from Jackson Laboratories. All animals were housed and treated following the guidelines set forth by the University of Minnesota Institutional Animal Care and Use Committee.
Antibodies
Polyclonal antibodies for voltage-gated calcium channel (VGCC) (Sigma, Waltham, Massachusetts) and JPH-2 (ThermoScientific) and monoclonal antibodies for α-tubulin (Sigma), β-tubulin (Sigma), and dystrophin (Leica, Buffalo Grove, Illinois) were purchased from the identified vendors. Alexa-Fluor-488 or Alexa-Fluor-568−conjugated anti-rabbit antibodies were purchased from Molecular Probes (Eugene, Oregon). Infrared dye-conjugated anti-mouse and anti-rabbit antibodies were purchased from LICOR Biosciences (Lincoln, Nebraska).
Isolation of cardiac myocytes
Isolation of ventricular cardiac myocytes was performed as described previously (18).
T-tubule assessment
Freshly isolated cardiac myocytes were fixed in 4% paraformaldehyde for 10 min at 37°C, washed with phosphate-buffered saline (PBS) 2 times for 5 min, incubated with AlexaFluor 488 conjugated Wheat Germ Agglutinin (Sigma) for 10 min at room temperature, and then washed in PBS 2 times for 5 min. Cells were mounted in Antifade (Molecular Probes) and imaged on Bio-Rad MRC 1000 scan head mounted on an upright Nikon Optishot microscope (Tokyo, Japan) at the University Imaging Centers at the University of Minnesota. Z-stacks were collected and converted into a z-projection using ImageJ (Bethesda, Maryland). T-tubule quantification was performed using the TTPower plugin on ImageJ as described (7).
Immunofluorescence analysis
Primary cardiac myocytes were fixed in 4% paraformaldehyde for 10 min at 37°C, permeabilized with 1% Triton X-100 (Sigma) in PBS, blocked in 5% BSA for 10 min 3 times, and incubated with primary antibodies overnight at 4°C. Sections were then washed and blocked with 5% bovine serum albumin for 10 min 3 times and then incubated with Alexa-Fluor-488- or Alexa-Fluor-568−conjugated secondary antibodies for 30 min at 37°C. Then, cardiac myocytes were washed with PBS and mounted in Anti-Fade Reagent (Molecular Probes).
Calcium transient measurements
Freshly isolated cardiac myocytes were loaded with Fura-2AM (a ratiometric Ca2+ indicator; 2 μM [Molecular Probes]) for 10 min at room temperature after a de-esterification period of 20 min in M199 medium (Sigma). Cells were incubated with 10 μm colchicine or vehicle (double-distilled water [ddH2O]) for 2 h and then subjected to calcium analysis. Fura-2 fluorescence was measured using a spectrophotometer (Stepper Switch, IonOptix). Initially, Fura-2 was excited at 360 nm (the isosbestic point independent of Ca2+) and then continuously at 380 nm (Ca2+-dependent fluorescence). Emission was collected at >510 nm by a photomultiplier tube. Ratiometric data were collected and analyzed online using commercial software (IonOptix).
Calcium spark analysis
Freshly isolated adult cardiac myocytes were plated on glass coverslips coated with 10 μg/ml laminin and in M199 medium and allowed to attach for 2 h. Then, colchicine (10 μM) or vehicle was added for 2 h. Cardiac myocytes were loaded with Ca2+ indicator Fluo 8-AM (5 μM, AAT Bioquest, Sunnyvale, California) for 10 min at room temperature followed by washout. For confocal imaging, we used an inverted confocal microscope (Leica TCS SP8, Wetzlar, Germany) with a 40×, 1.3 NA oil-immersion objective. Fluo 8-AM was excited at 488 nm and the emission was collected at >505 nm. For Ca2+spark imaging, line scan was performed at a speed of 1.43 ms/line for 1,000 lines.
Sarcoplasmic reticulum load analysis
Freshly isolated cardiac myocytes plated on laminin-coated coverslips were treated with 10 μM colchicine or vehicle for 2 h and were then loaded with 2 μM Fura-2AM for 10 min and de-esterified for 10 min at room temperature. After de-esterification, coverslips were mounted onto a perfusion chamber and perfused with modified Tyrode’s solution (in mM: 140 NaCl, 0.5 MgCl2, 5 HEPES, 5.5 glucose, 1.8 CaCl2, 5 KCl; pH 7.4) at 30°C. Myocytes were paced at 0.5 Hz for at least 20 s after which pacing was stopped and the perfusate was switched to Tyrode’s solution with 20 mM caffeine to measure the sarcoplasmic reticulum calcium load. Finally, myocytes were again perfused with Tyrode’s solution and paced at 0.5 Hz to ensure they were still viable. No more than 2 myocytes were measured per coverslip. Transient data were collected by measuring the 360:380 ratio using the Ionoptix Calcium and Contractility System and data were analyzed using IonWizard software (Westwood, Massachusetts).
Image processing
Confocal images were collected and equivalently processed using Adobe Photoshop Version CS6 (San Jose, California).
Western blot analysis and quantification
Whole protein extracts from isolated ventricles from mice were performed as described (19). Protein concentration was determined using a BCA protein assay kit (Pierce, Waltham, Massachusetts). Cardiac extracts (25 μg) were subjected to sodium dodecyl sulfate−polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to nitrocellulose membrane. Membranes were washed/blocked in 5% milk in PBS for 1 h, and incubated with primary antibodies for 1 h at room temperature. Membranes were then washed/blocked twice for 10 min in 5% milk in PBS and then incubated with infrared secondary antibodies for 30 min at room temperature. Membranes were washed in Tris-buffered saline containing 0.1% Tween (Sigma) twice for 5 min. Imaging and quantification of Western blots was performed on an Odyssey Infrared Imaging system. SDS-PAGE was performed in parallel and stained with Coomassie brilliant blue stain and was imaging at the 700 nm wavelength on the Odyssey Imaging system (Lincoln, Nebraska) to serve as the loading control as described (20).
Chronic colchicine treatment
Mice were treated with intraperitoneal injection of either filter-sterilized PBS or 0.5 mg/kg of filter-sterilized colchicine (Sigma) dissolved in PBS every other day for 2 weeks.
Echocardiography
Echocardiographic analysis was conducted using a VisualSonics 2100 ultrasound machine (Toronto, Ontario, Canada) with mice anesthetized with 2% to 3% inhaled isoflurane. Heart rate had to be >400 beats/min during echocardiographic analysis. Systolic and diastolic dimensions and fractional shortening were determined from M-mode images in the parasternal short-axis view at the level of the mitral valve.
Isoproterenol stress tests
Eight-month to 10-month-old wild-type (WT) and mdx mice (n = 8 mice to 10 mice per treatment group) were treated with either PBS or colchicine for 2 weeks and then administered isoproterenol at a dose 10 mg/kg at 9am, 1pm, and 5pm via intraperitoneal injection for 3 consecutive days. Survival was assessed at 8-h intervals for the first 5 days and then daily thereafter.
Statistical analysis
All data are represented as mean ± standard error of the mean. Student t tests were used to compare 2 groups with assumed equivalent variance if sample size was <5 with an F-test to assess variance if sample size was >5 and 1-way analysis of variance with Tukey post-hoc test was used to compare means between 3 groups. Kaplan-Meier survival curve was used to determine survival differences after initial treatment with isoproterenol with log-rank test to assess differences in survival. Statistical significance was defined as p ≤ 0.05.
Results
T-tubule disruptions in mdx cardiac myocytes are associated with reduced JPH-2 protein levels and altered localization
We first analyzed the t-tubule network in isolated cardiac myocytes from young (2- to 3-months old) and older (8- to 10-months old) WT and mdx mice. The 8- to 10-month time was used because that is the age when mdx mice begin to show signs of cardiomyopathy 12, 14. When compared to 8-month to 10-months of age, WT and young mdx cardiac myocytes had no discernable difference in t-tubule architecture, but older mdx cardiac myocytes showed disrupted t-tubules (Figure 1A). When quantified using TTPower analysis, older mdx t-tubule structural integrity (39.2 ± 2.2 AU) was significantly different than WT (57.3 ± 1.3 AU) and young mdx mice (56.8 ± 2.3 AU) (Figure 1B).
Figure 1.

T-Tubule Derangements in mdx Mice Are Associated With JPH-2 Misregulation
(A) Representative confocal images of cardiac myocytes stained with wheat germ agglutinin to delineate t-tubules. Older (8 months to 10 months) but not young (2 months to 3 months) mdx mice showed loss of regularity of the t-tubule network. Scale bar equals 5 μm. (B) Quantification of t-tubule organization in older (8 months to 10 months of age) WT, young, and older mdx mice. There was a significant reduction in t-tubule regularity in aged mdx mice. An asterisk (*) indicates p ≤ 0.05 when compared to WT; A pound symbol (#) indicates p ≤ 0.05 when compared to young mdx as determined by 1-way ANOVA with Tukey post-hoc analysis. N = 17 cells to 20 cells per genotype from 2 animals to 3 animals per genotype. Representative Western blot analyses and subsequent quantification from cardiac extracts of young (ages 2 months to 3 months) (C and D) and older (8 months to 10 months) (E and F) WT and mdx mice along with CBB stained SDS-PAGE, which served as a loading control. There was no difference in VGCC but there was a significant 75% reduction in JPH-2 levels in older mdx mice (p = 0.0003). N = 3 animals to 4 animals per genotype per age. (G) Representative confocal micrographs of cardiac myocytes from aged-matched (8 months to 10 months) control and mdx cardiac myocytes stained for JPH-2. Scale bar equals 5 μm. (H) Quantification of JPH-2 localization using TTPower. Older mdx mice showed altered localization of JPH-2 in cardiac myocytes (p < 0.0001). N =17 cells to 19 cells from 2 animals to 3 animals per treatment. An asterisk (*) indicates p ≤ 0.05 as determined using Student t test. Values are presented as mean ± SEM. ANOVA = analysis of variance; CBB = Coomassie brilliant blue; JPH-2 = junctophilin-2; SDS-PAGE = sodium dodecyl sulfate−polyacrylamide gel electrophoresis; VGCC = voltage-gated calcium channel; WT = wild-type.
To provide insight into the mechanism of t-tubule disruptions in older mdx mice, we investigated 2 key t-tubule proteins: the VGCC and JPH-2. Protein levels of VGCC were not altered in either young or older mdx heart extracts (Figures 1C to 1F). However, JPH-2 protein levels were markedly reduced by 75% in older mdx hearts (WT: 1 ± 0.9 AU and mdx: 0.024 ± 0.4 AU) but not significantly altered in young mice (WT: 1.0 ± 0.1 AU and mdx: 1.3 ± 0.2 AU) (Figures 1C to 1F), a pattern that mirrored the t-tubule phenotype. Immunofluorescence analysis of isolated cardiac myocytes from older WT (74.2 ± 1.9 AU) and mdx mice (57.8 ± 1.7 AU) revealed altered localization of JPH-2 in mdx mice (Figures 1G and 1H). In summary, JPH-2 misregulation was associated with t-tubule disruptions in mdx hearts.
Inverse hyperbolic relationship between JPH-2 and α- and β-tubulin
Because a previous report demonstrated that an altered microtubule cytoskeleton caused mislocalization of JPH-2 (9), we analyzed the relationship between α- and β-tubulin, the individual components of microtubules, and JPH-2 in mdx hearts. At a young age when JPH-2 levels were normal, α-tubulin (WT: 1.0 ± 0.1 AU and mdx: 1.2 ± 0.1 AU) and β-tubulin (WT: 1.0 ± 0.2 AU and mdx: 1.3 ± 0.1 AU) protein expression was not significantly different in mdx cardiac extracts (Figures 2A and 2B). However, in older mdx hearts when JPH-2 protein was reduced, α-tubulin (WT: 1.0 ± 0.1 AU and mdx: 2.7 ± 0.3 AU) and β-tubulin (WT: 1.0 ± 0.9 AU and mdx: 2.5 ± 0.2 AU) content was increased approximately 2.6-fold (Figures 2C and 2D). When the relationships between α- and β-tubulin and JPH-2 in older mice was analyzed, we observed a significant inverse hyperbolic relationships (r2 = 0.95 for both curve fitting models) (Figures 2E and 2F). These data suggested increased microtubule content was associated with reduced JPH-2 protein levels.
Figure 2.

Negative Hyperbolic Relationship Between JPH-2 and Tubulin in Aged mdx Cardiac Extracts
Quantitative Western blot analysis of α- and β-tubulin levels in young (A and B) and aged (C and D)mdx mice. There was no significant difference in tubulin levels in young (2 months to 3 months of age) mice but older (8 months to 10 months of age) mdx mice showed a significant 2.6-fold increase in α- and β-tubulin protein expression. (For α-tubulin comparison, p = 0.0008. For β-tubulin comparison, p = 0.0007.) N = 3 animals to 4 animals per genotype per age point. (E and F) A strong negative hyperbolic relation between JPH-2 and α- and β-tubulin in older mdx cardiac extracts exists (r2 = 0.95 for α-tub and JPH-2 curve-fitting, and r2 = 0.95 for β-tubulin and JPH-2 curve fitting). ∗p ≤ 0.05 as determined using Student t test. Values are presented as mean ± SEM. Abbreviations as in Figure 1.
Colchicine-induced microtubule depolymerization normalized JPH-2 levels and localization which corrected T-tubule morphology in mdx mice
To further probe the relationship between microtubules and JPH-2 in mdx hearts, we treated older mdx mice for 2 weeks with colchicine to induce microtubule depolymerization and examined the effects on JPH-2 and t-tubules. Colchicine treatment effectively depolymerized microtubules in cardiac myocytes in vivo (Figure 3A) and was associated with a 7.6-fold increase in JPH-2 protein levels (mdx-PBS: 1.0 ± 0.08 AU and mdx-colchicine: 7.6 ± 1.0 AU) (Figures 3B and 3C). Moreover, JPH-2 localization patterns in mdx cardiac myocytes were significantly improved with colchicine treatment (mdx-PBS: 62.6 ± 1.6 AU and mdx-colchicine: 68.1 ± 1.9 AU) (Figure 3D and 3E). Finally, colchicine-mediated restoration of JPH-2 improved t-tubule organization in older mdx hearts (mdx-PBS: 44.5 ± 2.0 AU and mdx-colchicine: 52.5 ± 2.2 AU) (Figures 3F and 3G).
Figure 3.

Microtubule Depolymerization Normalized JPH-2 Levels and Localization and t-Tubule Morphology in mdx Mice
(A) Confocal micrographs of cardiac myocytes stained with an α-tubulin antibody to show the microtubule cytoskeleton. Colchicine depolymerized microtubule in vivo. (B) Representative Western blots and CBB-stained SDS-PAGE of extracts from mdx mice treated with either PBS or colchicine. (C) JPH-2 was upregulated 7.6-fold in mice treated with colchicine (p = 0.0007). (D) Representative confocal micrographs showing JPH-2 levels and localization. (E) Quantification of JPH-2 localization using TTPower. JPH-2 localization improved with colchicine treatment (p = 0.04). N = 20 cells to 22 cells from 2 animals to 3 animals per treatment. (F) Representative images of t-tubules stained with wheat germ agglutinin from cardiac myocytes isolated from mdx treated with either PBS or colchicine. (G) Quantification of t-tubule organization using TTPower. Colchicine resulted in a significant improvement in t-tubule organization (p = 0.02). N = 19 cells to 20 cells per treatment from 2 animals to 3 animals. An asterisk (*) indicates p ≤ 0.05 as determined using Student t test. Scale bar is 5 μm in all images. Values are presented as mean ± SEM. PBS = phosphate-buffered saline; other abbreviations as in Figure 1.
Acute colchicine treatment improved calcium handling in mdx cardiac myocytes
Because JPH-2 was shown to be an important regulator of the ryanodine receptor 21, 22, 23, we investigated how colchicine-mediated JPH-2 restoration affected calcium handling in isolated mdx cardiac myocytes treated acutely with colchicine. Acute colchicine treatment did not affect diastolic calcium, onset kinetics, or amplitude of calcium transients, but there was accelerated calcium decay at 25% to baseline (mdx: 59.4 ± 2.5 ms and mdx-colchicine: 51.1 ± 3.0 ms) but not at 75% to baseline (mdx: 219.9 ± 20.4 ms and mdx-colchicine: 202.2 ± 25.0 ms) (Figures 4A to 4F), which suggested improved ryanodine receptor gating. To further investigate ryanodine receptor activity, we measured calcium spark frequency and found acute colchicine treatment reduced calcium sparks by more than 50% (mdx: 0.84 ± 0.2 sparks/s/100 μm and mdx-colchicine: 0.31 ± 0.08 sparks/s/100 μm) (Figures 4G and 4H). Acute colchicine treatment caused a nonsignificant alteration in sarcoplasmic reticulum calcium load (Supplemental Figure 1), which would not explain the reduction in calcium sparks. All together, these data suggest that acute colchicine treatment improved ryanodine receptor gating in mdx cardiac myocytes.
Figure 4.

Colchicine Treatment Improved Calcium Handling in mdx Cardiac Myocytes
(A) Confocal micrographs of mdx cardiac myocytes stained with an α-tubulin antibody to show effective microtubule depolymerization with colchicine treatment. (B) Representative curves of calcium transients. (C–E) Colchicine treatment did not affect baseline calcium, peak calcium, or kinetics of onset of calcium transients, but increased calcium transient decay to 25% of baseline calcium levels (p = 0.03). (F). N = 20 cells to 22 cells from 2 animals to 3 animals per treatment. (G) Representative calcium spark images from isolated mdx cardiac myocytes. Arrows indicate calcium sparks. (H) Quantification of calcium sparks frequency. Colchicine treatment significantly reduced calcium sparks in mdx cardiac myocytes (p = 0.04). N = 17 cells to 20 cells from 3 animals per treatment. ∗p ≤ 0.05 as determined by Student t test. Values are presented as mean ± SEM.
Chronic colchicine treatment did not alter organ-level assessments of cardiomyopathy in mdx mice
Next, we determined if 2 weeks of colchicine treatment led to whole-organ level improvements in older mdx mice. M-mode echocardiography did not reveal any significant changes in cardiac geometry or systolic function after 2 weeks of colchicine treatment in mdx mice (Supplemental Figure 2), but both mdx groups showed signs of mild cardiomyopathy when compared to age-matched WT mice. Finally, colchicine treatment did not confer a significant survival benefit when older mdx mice were subjected to isoproterenol stress test (WT survival: 75%, mdx-colchicine survival: 40%, mdx-PBS survival: 10%) (Supplemental Figure 3).
Discussion
Our main new findings are the following: 1) t-tubule disarray in mdx cardiac myocytes is associated with altered localization and decreased protein levels of JPH-2; 2) there is a significant inverse hyperbolic relationship between JPH-2 and α- and β-tubulin, the individual subunits of microtubules, in mdx hearts; 3) disruption of the JPH-2/microtubule relationship, using colchicine to depolymerize microtubules, not only normalizes JPH-2 localization, but we show for the first time that it restores JPH-2 protein levels which corrects t-tubule morphology; and 4) colchicine treatment can effectively reduce calcium spark frequency in isolated mdx cardiac myocytes, which we propose is mediated through stabilization of the ryanodine receptor. Collectively, these results provide evidence that microtubule-dependent misregulation of JPH-2 underlies t-tubule derangements and abnormal calcium handling in Duchenne cardiomyopathy, and implicates JPH-2 as a new molecular mediator in the pathophysiology of Duchenne cardiomyopathy.
Importantly, our results provide additional mechanistic insight into 2 secondary pathological cardiac phenotypes associated with dystrophin-deficiency: t-tubule disruptions and calcium mishandling. First of all, we show that normalization of JPH-2 protein levels and localization improves t-tubule architecture, a cellular phenotype that has not had a proposed molecular mechanism. Secondly, colchicine treatment reduces calcium spark frequency which was in agreement with results of Kerr et al. (24), which we propose is due to normalization of JPH-2 after microtubule depolymerization leading to improved ryanodine receptor gating. These results provide an additional molecular mechanism by which the ryanodine receptor gating is altered in mdx cardiac myocytes, in addition to the loss of a calstabin-ryanodine receptor interaction as previously defined (4). In summary, our results implicate JPH-2 as the molecular mediator of t-tubule abnormalities and abnormal gating of the ryanodine receptor in Duchenne cardiomyopathy.
Next, we provide further support for the hypothesis that JPH-2 regulates ryanodine receptor activity. Several studies have documented increased calcium spark frequency when JPH-2 is decreased or altered in localization 9, 21, 22, 23, suggesting the ryanodine receptor is improperly gated when JPH-2 is disrupted. In particular, Zhang et al. (9) showed that microtubule depolymerization-induced normalization of JPH-2 localization patterns reduced calcium spark frequency in cultured mouse cardiac myocytes, a result very similar to our findings. However, unlike Zhang et al. (9), we did not observe changes in calcium transient amplitude with colchicine treatment. The discrepancy between our results and those of Zhang et al. (9) may be due in-part to SR load as JPH-2 knockdown causes reduced SR calcium content, but calcium transients normalized to SR load are actually increased (22). Mdx mice have normal SR calcium content (25) and are not significantly altered by colchicine treatment (Supplemental Figure 1), which might explain why colchicine treatment did not increase calcium transient amplitude in mdx cardiac myocytes. Another explanation for the differences between our results and those of Zhang et al. (9) could relate to the timing of experiments as we analyzed calcium transients in freshly isolated cardiac myocytes whereas Zhang et al. (9) showed normalization of calcium transients in cells cultured for >48 h. Nonetheless, our results support a crucial role for JPH-2 in regulating ryanodine receptor activity.
Although we were able to document restoration of t-tubule organization with colchicine treatment, we did not observe organ-level cardiac improvements in mdx mice. One explanation is that t-tubule corrections do not always lead to improved whole organ function; a finding that was documented in previous publications. Firstly, t-tubule derangements precede left ventricular dysfunction in the thoracic aortic banded rat model (26). Moreover, while transgenic overexpression of JPH-2 could prevent pathological t-tubule remodeling and blunted the effects of aortic banding; it did not completely prevent the onset of heart failure (17). Another reason for the lack of improvement of cardiomyopathy in mdx mice treated with colchicine could be the relatively mild cardiomyopathy of mdx mice. Whereas Guo et al. (17) showed improvements in ejection fraction with JPH-2 overexpression, there was not a complete normalization of ejection fraction, which could explain why echocardiography did not reveal improvements in mdx mice treated with colchicine as the mdx mice had only mild reduced fractional shortening (Supplemental Figure 2). Finally, the lack of a significant improvement in isoproterenol stress test may be explained by the underlying pathophysiology that was corrected with colchicine treatment. Previously, membrane permeability was associated with increased death in mdx mice when treated with isoproterenol 10, 15, 16, and thus the nonsignificant improvement in survival with colchicine may have resulted from an inability to correct cardiac myocytes permeability in mdx hearts. In summary, there may be several reasons why colchicine treatment did not improve mdx cardiomyopathy.
Finally, our results add to several other publications that document a crucial role for JPH-2 in t-tubule integrity. The first link between JPH-2 and t-tubules was shown when knockout of JPH-2 resulted in embryonic lethality due to lack and cardiac contractility and improper t-tubule formation (27). Several other studies document that alterations of JPH-2 via miR-24 28, 29, RNA interference 21, 22, 30, or calpain-mediated protein cleavage (23) cause t-tubule derangements and rescue of JPH-2 protein levels or localization via transgenic overexpression (17), inhibition of miR-24 (28), calpain-inhibition (23), or microtubule depolymerization (9) prevent pathological t-tubule remodeling. Taken together with our new findings, JPH-2 is a critical mediator of t-tubule structure in cardiac myocytes.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: Cardiomyopathy accounts for approximately one-quarter of deaths in Duchenne muscular dystrophy. Currently, our knowledge of the mechanistic pathophysiology of the Duchenne cardiomyopathy is incomplete, but these data provide a molecular explanation for the disruption of t-tubules and calcium mishandling in Duchenne cardiomyopathy.
TRANSLATIONAL OUTLOOK: Future studies that examine the effects of normalization of JPH-2 through multiple mechanisms including colchicine treatment, gene transfer, or micro-RNA inhibition in large animal models of Duchenne cardiomyopathy with a more severe phenotype could be conducted to gain further information about how JPH-2 affects outcomes in Duchenne cardiomyopathy. If results are positive, a clinical trial examining the effects of colchicine in Duchenne muscular dystrophy could be considered.
Acknowledgements
The authors would like to thank Drs. Amit Gaggar and Thenappan Thenappan for assistance in editing the manuscript.
Footnotes
This work was supported by the University of Minnesota Lillehei Heart Institute.
Dr. Metzger has received grants from the National Institutes of Health (NIH) and MDA. Dr. Prins has received NIH T32 (HL069764) and F32 (HL129554) grants. Dr. Asp has received the NIH F32 (HL115876) grant. Dr. Wang has received the NIH RO1 (HL114760) grant. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Appendix
References
- 1.Moser H. Duchenne muscular dystrophy: pathogenetic aspects and genetic prevention. Hum Genet. 1984;66:17–40. doi: 10.1007/BF00275183. [DOI] [PubMed] [Google Scholar]
- 2.Fayssoil A., Nardi O., Orlikowski D., Annane D. Cardiomyopathy in duchenne muscular dystrophy: pathogenesis and therapeutics. Heart Fail Rev. 2010;15:103–107. doi: 10.1007/s10741-009-9156-8. [DOI] [PubMed] [Google Scholar]
- 3.Wang X., Weisleder N., Collet C. Uncontrolled calcium sparks act as a dystrophic signal for mammalian skeletal muscle. Nat Cell Biol. 2005;7:525–530. doi: 10.1038/ncb1254. [DOI] [PubMed] [Google Scholar]
- 4.Fauconnier J., Thireau J., Reiken S. Leaky RyR2 trigger ventricular arrhythmias in duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2010;107:1559–1564. doi: 10.1073/pnas.0908540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prosser B.L., Ward C.W., Lederer W.J. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–1445. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 6.Lorin C., Gueffier M., Bois P., Faivre J.F., Cognard C., Sebille S. Ultrastructural and functional alterations of EC coupling elements in mdx cardiomyocytes: an analysis from membrane surface to depth. Cell Biochem Biophys. 2013;66:723–736. doi: 10.1007/s12013-013-9517-8. [DOI] [PubMed] [Google Scholar]
- 7.Pasqualin C., Gannier F., Malecot C.O., Bredeloux P., Maupoil V. Automatic quantitative analysis of t-tubule organization in cardiac myocytes using ImageJ. Am J Physiol Cell Physiol. 2015;308:C237–C245. doi: 10.1152/ajpcell.00259.2014. [DOI] [PubMed] [Google Scholar]
- 8.Landstrom A.P., Beavers D.L., Wehrens X.H. The junctophilin family of proteins: from bench to bedside. Trends Mol Med. 2014;20:353–362. doi: 10.1016/j.molmed.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang C., Chen B., Guo A. Microtubule-mediated defects in junctophilin-2 trafficking contribute to myocyte transverse-tubule remodeling and Ca2+ handling dysfunction in heart failure. Circulation. 2014;129:1742–1750. doi: 10.1161/CIRCULATIONAHA.113.008452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strakova J., Dean J.D., Sharpe K.M., Meyers T.A., Odom G.L., Townsend D. Dystrobrevin increases dystrophin's binding to the dystrophin-glycoprotein complex and provides protection during cardiac stress. J Mol Cell Cardiol. 2014;76C:106–115. doi: 10.1016/j.yjmcc.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adamo C.M., Dai D.F., Percival J.M. Sildenafil reverses cardiac dysfunction in the mdx mouse model of duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2010;107:19079–19083. doi: 10.1073/pnas.1013077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quinlan J.G., Hahn H.S., Wong B.L., Lorenz J.N., Wenisch A.S., Levin L.S. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord. 2004;14:491–496. doi: 10.1016/j.nmd.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 13.Sarma S., Li N., van Oort R.J., Reynolds C., Skapura D.G., Wehrens X.H. Genetic inhibition of PKA phosphorylation of RyR2 prevents dystrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107:13165–13170. doi: 10.1073/pnas.1004509107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Erp C., Loch D., Laws N., Trebbin A., Hoey A.J. Timeline of cardiac dystrophy in 3-18-month-old MDX mice. Muscle Nerve. 2010;42:504–513. doi: 10.1002/mus.21716. [DOI] [PubMed] [Google Scholar]
- 15.Danialou G., Comtois A.S., Dudley R. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J. 2001;15:1655–1657. doi: 10.1096/fj.01-0030fje. [DOI] [PubMed] [Google Scholar]
- 16.Barnabei M.S., Sjaastad F.V., Townsend D., Bedada F.B., Metzger J.M. Severe dystrophic cardiomyopathy caused by the enteroviral protease 2A-mediated C-terminal dystrophin cleavage fragment. Sci Transl Med. 2015;7:294ra106. doi: 10.1126/scitranslmed.aaa4804. [DOI] [PubMed] [Google Scholar]
- 17.Guo A., Zhang X., Iyer V.R. Overexpression of junctophilin-2 does not enhance baseline function but attenuates heart failure development after cardiac stress. Proc Natl Acad Sci U S A. 2014;111:12240–12245. doi: 10.1073/pnas.1412729111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang W., Barnabei M.S., Asp M.L. Noncanonical EF-hand motif strategically delays Ca2+ buffering to enhance cardiac performance. Nat Med. 2013;19:305–312. doi: 10.1038/nm.3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanft L.M., Rybakova I.N., Patel J.R., Rafael-Fortney J.A., Ervasti J.M. Cytoplasmic gamma-actin contributes to a compensatory remodeling response in dystrophin-deficient muscle. Proc Natl Acad Sci U S A. 2006;103:5385–5390. doi: 10.1073/pnas.0600980103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eaton S.L., Roche S.L., Llavero Hurtado M. Total protein analysis as a reliable loading control for quantitative fluorescent western blotting. PLoS One. 2013;8:e72457. doi: 10.1371/journal.pone.0072457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen B., Guo A., Zhang C. Critical roles of junctophilin-2 in T-tubule and excitation-contraction coupling maturation during postnatal development. Cardiovasc Res. 2013;100:54–62. doi: 10.1093/cvr/cvt180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Oort R.J., Garbino A., Wang W. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–988. doi: 10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu C.Y., Chen B., Jiang Y.P. Calpain-dependent cleavage of junctophilin-2 and T-tubule remodeling in a mouse model of reversible heart failure. J Am Heart Assoc. 2014;3:e000527. doi: 10.1161/JAHA.113.000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerr J.P., Robison P., Shi G. Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat Commun. 2015;6:8526. doi: 10.1038/ncomms9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams I.A., Allen D.G. Intracellular calcium handling in ventricular myocytes from mdx mice. Am J Physiol Heart Circ Physiol. 2007;292:H846–H855. doi: 10.1152/ajpheart.00688.2006. [DOI] [PubMed] [Google Scholar]
- 26.Wei S., Guo A., Chen B. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takeshima H., Komazaki S., Nishi M., Iino M., Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 28.Li R.C., Tao J., Guo Y.B. In vivo suppression of microRNA-24 prevents the transition toward decompensated hypertrophy in aortic-constricted mice. Circ Res. 2013;112:601–605. doi: 10.1161/CIRCRESAHA.112.300806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu M., Wu H.D., Li R.C. Mir-24 regulates junctophilin-2 expression in cardiomyocytes. Circ Res. 2012;111:837–841. doi: 10.1161/CIRCRESAHA.112.277418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caldwell J.L., Smith C.E., Taylor R.F. Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1) Circ Res. 2014;115:986–996. doi: 10.1161/CIRCRESAHA.116.303448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.