

Abstract
We report that the (4S)-4,5-dihydroxy-2,3-pentanedione (DPD) can undergo a previously undocumented non-enzymatic glycation reaction. Incubation of DPD with viral DNA or the antibiotic gramicidin S resulted in significant biochemical alterations. A protein labeling method was consequently developed that facilitated the identification of unrecognized glycation targets of DPD in a prokaryotic system. Our results open new avenues toward tracking and understanding the fate and function of the elusive quorum-sensing signaling molecule.
Keywords: quorum sensing, DPD, glycation, protein modification, proteomics
Graphical Abstract
The quorum-sensing signaling molecule DPD assumes a yet unrecognized biochemical role capable of glycating DNA and proteins in a rapid and stereospecific manner.
Over the past decade our laboratory and others have investigated the bacterial biochemical properties of (4S)-4,5-dihydroxy-2,3-pentanedione (DPD).[1] Our interest in this molecule originally stemmed from its distinct 5-carbon densely oxygenated makeup and reports touting its ability to coordinate gene expression in both gram-negative and gram-positive bacteria in a population-dependent manner, termed as autoinducer-2 quorum sensing (AI-2 QS). Moreover, we have shown that DPD can exist in a complex, multispecies equilibrium whereby its unique architecture allows it to undergo cyclization and ring opening reactions similar to saccharides (Scheme 1A).[2]
Scheme 1.

A) DPD and its equilibrium species in aqueous solution. B) Fructose undergoing the Heyns rearrangement with lysine.
Enormous resources have been invested in AI-2 QS trying to connect QS pathogenic events, however, these research efforts have only yielded sporadic reports of AI-2 QS regulated behaviors.[3] Fundamental to AI-2 QS on one hand is DPD’s biosynthetic manufacture and receptors found in over 70 bacteria, yet, a consistent link among DPD signal, receptor, mechanism and target output within this plethora of bacterial species has been lacking.[4] Additional questions on DPD’s cross-species signaling role have also been damped by the view that the luxS gene, which was thought to be dedicated to AI-2 production, also assumes an integral function in the activated methyl cycle.[5] Taken in sum, while DPD has been portrayed as being central to bacterial interspecies quorum sensing, its importance in bacterial pathophysiological control remains elusive. Herein, an additional piece to this molecule’s “yin and yang” chemical biology is presented. We describe an intrinsic reactivity of DPD that enables DNA- and protein-glycation, a previously undocumented biochemical role for this signaling molecule in the bacterial cell.
It has been established that the carbonyl moiety of a reducing sugar is capable of reacting non-enzymatically with the side chains of lysine/arginine residues embedded within a protein (Maillard reaction), which is initiated by the formation of a transient Schiff base. This Schiff base intermediate can then undergo an Amadori (in aldoses) or a Heyns rearrangement (in ketoses) (Scheme 1B), which can ultimately lead to the formation of advanced glycation end products (AGEs),[6] and subsequent alteration of the protein’s normal function. As such, AGEs have been implicated in a number of pathologies associated with aging, diabetes, and arthritis.[7] Research findings demonstrating that nucleotide bases could also participate in advanced glycosylation reactions have offered additional insights into the pathophysiology of AGE-related diseases. Prior studies have shown that DNA-modification by glucose 6-phosphate (G-6-P) leads to a significant decrease in transfection efficiency and an increase in plasmid mutations in vivo.[8] Incorporation of the stable adduct N2-1-(carboxyethyl)-2′-deoxyguanosine (which can form on prolonged incubation of DNA with glucose or methylglyoxal) within template DNA blocked polymerase activity and induced mutagenesis.[9] The accumulation of DNA glycation products over time may thus promote genetic instability and facilitate disease progression.
Given the structural similarity between DPD and monosaccharides, it seemed plausible to consider that DPD could undergo similar chemically centric processes. As a first step toward probing this unexplored reactivity of DPD, we incubated DPD (10 mM) with a model nucleotide base 9-methylguanine (9-MG, 1 mM, Scheme 2A) in PBS pH 7.4 at reflux temperature for 16 h, and monitored the reaction by LC/MS. Mass spectral analysis of the crude reaction showed a mixture of 9-MG-DPD adducts (see Figure S1 for details), although no attempt was made at their isolation and identification. Having established that DPD modifies the model nucleotide in vitro, we next investigated the biochemical effect of glycation by evaluation of DNA transfection into E. coli. Earlier reports have documented the loss of transfection potential of glycated single stranded DNA (ssDNA) resulting from its reaction with G-6-P.[8a] Taking a similar approach, we isolated ssDNA from bacteriophage M13 and incubated it with DPD (0.5 and 5 mM) at 37°C. At different incubation times, reaction mixtures (100 ng ssDNA + DPD) were transfected into TG1 competent cells, followed by quantitation of plaque-forming units after an overnight growth. As evident in Figure S2A, exposure of the single stranded viral genome with 5 mM DPD led to a dramatic decrease in plaque formation (up to three orders of magnitude in a span of 4 days), that is first order with respect to incubation time (Figure S2B). Control incubations of ssDNA or DPD alone were performed to account for potential degradation effects, but were not found to significantly alter transfection during the course of the reaction (Figure S2A). We note that the initial experiment with 9-MG was undertaken using “accelerated” conditions for a facile assessment of the purine amino group reactivity.[8b] Incubations with ssDNA were however performed under “long-term” conditions at physiological pH/temperature, to emulate the long-lived DNA molecules in vivo, which could undergo gradual and time-dependent glycation reactions.[8a]
Scheme 2.

Structures of A) 9-methylguanine and B) Gramicidin S. C) Formation of a DPD-lysine covalent-adduct that contains an α,β–diketone functionality.
To further explicate on the glycating ability of DPD, we utilized the antibiotic gramicidin S (GS, Scheme 2B) as a model system of protein glycosylation, as previously described.[10] GS is a cyclic decapeptide containing two proximal ornithine residues that could promote glycation. As expected, multiple adducts were observed by LC/MS upon incubation of DPD (5 mM) with GS (50 μM) at 37°C (see Figure S3 for details). This DPD-induced modification invariably resulted in a substantial decrease in GS potency (up to eight-fold increase in MIC against a selected panel of bacteria, see Table S1), and suggests a previously unrecognized mechanism of DPD-mediated antibiotic resistance.
The observation that the signaling molecule can remarkably alter the biochemical properties of the model DNA and peptide in vitro provides a whole new exciting perspective on DPD fate and function. Yet, our quest to decipher the physiological implications of DPD glycation was hampered by the dearth of methods that can distinctly identify AI-2 molecular targets/receptors. In hindsight, we reckoned on the assumption that a nucleophilic attack on DPD’s C2-α-hydroxyketone by a primary amine would afford a Schiff-base intermediate, which could then undergo a series of tautomerizations/rearrangements yielding multiple isomers including the Heyns product 2 (Scheme 2C & Figure S4). As a means to test this hypothesis, we incubated Nα-acetyl-L-lysine methyl ester (1 mM) with DPD (10 mM) at 70 °C (pH 8.0) for 48 h and monitored the reaction via LC/MS analysis. As expected, a DPD-lysine covalent monoadduct (m/z 317.1696) was observed that fits the structure of product 2, albeit in very low yield (Scheme 2C and Figure S5). This diminutive yield is most likely due to the reversibility of the transient DPD-Lysine-Schiff base formation, and reduced nucleophilicity of the simple lysine analogue (pKa = 10.4) at pH 8.[11] Indeed, Maillard-type reactions involving lysines are typically thought to be significantly suppressed under physiological conditions.[12] In contrast, lysine residues found within a protein’s microenvironment can possess depressed pKa values (~6.0), which can translate to the increased reactivity via general acid/base proton transfer.[11b]
Using this chemical rationale, we envisaged that a DPD-Heyns product could be evoked in a rapid manner within the correct protein microenvironment. An additional sobering thought that would also need our attention was that Heyns rearrangement products are subject to forming amino aldoses, which are known to be unstable, thus making this reaction extremely difficult to follow.[13] Despite these undeniable challenges, we envisioned that a successful Heyns rearrangement would at some point present an α,β-diketone handle that could be trapped with a phenylenediamine tag via irreversible quinoxaline ring formation (Figure 1A).[2b, 14] To this end, we synthesized two chemical tags for further studies: phenylenediamine-TAMRA 3 and phenylenediamine-biotin 4 (Figures 1B and S6). The value of both probes being that trapped protein-DPD-TAMRA or protein-DPD-biotin conjugate could be analyzed by in-gel fluorescence or subjected to affinity purification, respectively, thus providing invaluable information regarding which proteins rapidly undergo DPD glycation.
Figure 1.

A) Labeled protein via quinoxaline ring formation. B) Phenylenediamine-TAMRA/biotin tags. C) Labeling of proteome from wild-type S. typhimuirum. FL, fluorescence. CBB, Coomassie Brilliant Blue
Previously, glycation involving common saccharides was only considered to be relevant for eukaryotic organisms as it is chemically challenging (taking weeks to occur), however, Ivanov and coworkers have provided evidence that AGE formation also occurs in prokaryotes despite its short-life span.[15] As a first demonstration of the utility of the probes to examine DPD-linked protein glycation, we set out to label the whole lysate derived from the wild-type Salmonella enterica serovar Typhimurium (S. typhimurium) strain 14028, as DPD’s role in the AI-2 QS of S. typhimurium has been well documented.[1a, 1c, 1d] This strain was grown to stationary phase (OD600 = 3.6) and the whole cell lysate was incubated with DPD (4 mM) at pH 7.0 at 25 °C for 1 h. Following protein precipitation, the protein pellets were re-solubilized and then incubated with phenylenediamine-TAMRA 3 (100 μM) at 25 °C for 1 h. As shown in Figure 1C, a comparison of the fluorescence image with the Coomassie Brilliant Blue reveals that the proteins in this milieu are rapidly glycated. Importantly, when the lysate was incubated without DPD, no labeling was observed, indicating that the phenylenediamine tag is selective for DPD-glycated proteins (See Figure S7).
Encouraged by these results, we sought to identify which proteins are glycated by DPD. As such, we used a combination strategy utilizing phenylenediamine-biotin 4 and mass spectrometry-based proteomic-profiling techniques.[16] Here, whole cell lysate (S. typhimurium strain 14028) was used with or without denaturing prior to DPD labeling (“denatured” or “native” experiment, respectively). We anticipated that a comparison of these two samples would enable us to determine which proteins are modified by DPD, and thus possess the specific structural interactions necessary to form the Heyns-protein adduct. Labeled proteins were enriched using streptavidin beads, trypsinized, and analyzed by tandem ESI-MS/MS (See SI for detailed protocols). To exclude statistically irrelevant hits, positive hits were defined based on the criteria as follows: (1) a minimum of two unique peptides per protein in all three replicates, (2) p value <0.05, and (3) >2-fold more spectral intensity detected in the native sample than in the denatured sample.[17]
In the native sample, 63 proteins met the criteria for positive hits (Table S2 and Figure S8). Interestingly, several are known to regulate AI-2 QS; the lsr repressor protein LsrR is one of such examples. According to previous biochemical studies[1d, 18] and crystallographic data of LsrR derived from Escherichia coli,[19] it is the phosphorylated form of DPD (phospho-DPD) that binds to this protein, provoking destabilization and derepression of the lsr operon. However, it has been proposed that intact linear DPD can also bind to LsrR, resulting in altered biofilm architecture through a distinct signaling pathway in E. coli.[20] In addition, it is noteworthy that the periplasmic protein LsrB, which recognizes and internalizes the AI-2 signal,[1c] was not detected as a positive hit. However, it is the cyclic form of DPD that LsrB recognizes, which accounts for the lack of reactivity in this labeling.
Other positive hits in the above-mentioned proteome labeling included the phosphoenolpyruvate phosphotransferase PT1[21] and phosphoenolpyruvate synthase regulatory protein PsrP. These comprise the phosphoenolpyruvate phosphotransferase system (PTS), which is required for initial internalization of AI-2 signals into the cytosol, followed by lsr operon activation.[22] In addition, the NAD(P)H-dependent glycerol-3-phosphate dehydrogenase GpsA was detected. This protein reduces dihydroxyacetone phosphate, which inhibits the regulatory activity of LsrR.[23]
To gain a greater appreciation of DPD-protein labeling efficiency, LsrR from S. typhimurium LT2 was cloned and overexpressed to afford whole cell lysate; to this, BSA was added as a control protein, as its use as a glycation model is well documented.[24] The obtained protein solution was labeled and visualized with DPD and 3 as described vide supra (Figures 2 & S7). As expected, labeling of LsrR was dependent on concentration, temperature, and incubation time (Figures 2A–B). Labeling efficiency was reduced when LsrR was denatured prior to incubation with DPD (Lanes 2 and 3, Figure 2C). Quite remarkably, the unnatural (4R)-DPD (ent-DPD) exhibited significantly reduced labeling efficiency (Lanes 2 and 4, Figure 2C). In contrast, labeling of BSA was marginal compared to that of LsrR, suggesting that the former does not structurally recognize DPD, thus underlining the selectivity of our labeling probe. To rule out additional spurious artifacts, the specificity of the phenylenediamine probe was examined using fructose and glucose instead of DPD, which did not give fluorescently labeled LsrR (See Figure S9). Collectively, our labeling method allows for selective detection of proteins that could recognize and react with DPD. These results also illustrate that the protein must possess a unique tertiary structure microenvironment for Heyns-DPD-protein marking to transpire. In particular, the observed stereoselectivity is in contrast with protein glycation by natural sugars, where reactivity is not dictated by stereochemistry.[25]
Figure 2.
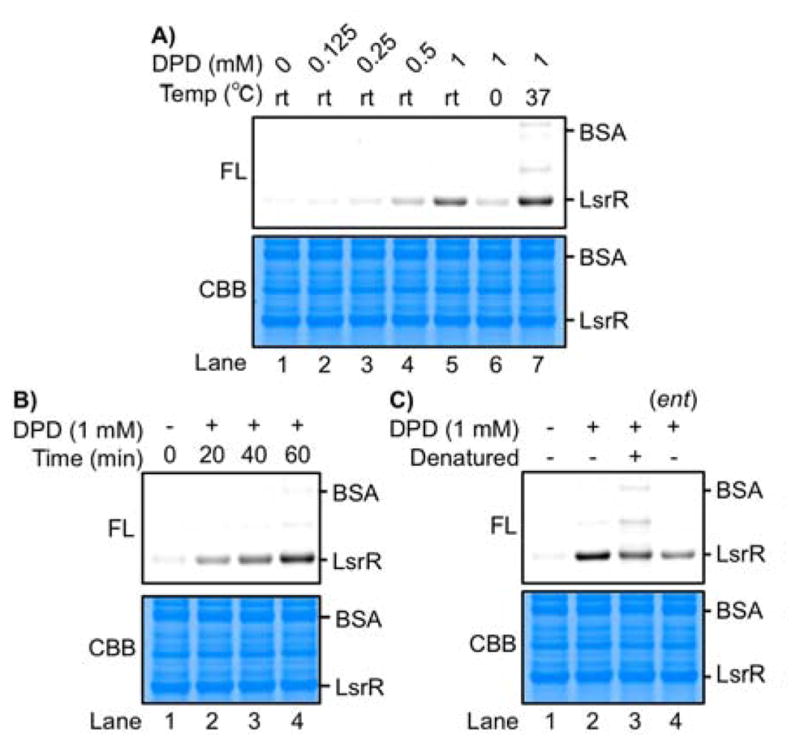
DPD labelling of LsrR under various conditions: A) concentration of DPD and incubation temperature; B) incubation time; C) denatured LsrR and ent-DPD. FL, fluorescence. CBB, Coomassie Brilliant Blue
The significance of these proteomics findings to bacterial physiology must be viewed with caution. In our labeling experiments, we have used millimolar concentrations of DPD that may raise questions for its physiological relevance. Quantitative detection of QS molecules in real time is a rather challenging task as they are produced at different concentrations throughout the bacterial life cycle. AI-2 has been reported to accumulate at a maximal intracellular level of 2 mM in E. coli,[26] however, we speculate that it could exist at a higher concentration given the transient and, as our current data imply, highly reactive nature of DPD. Although our results offer the tantalizing possibility that DPD acts as a post-translational modifier for regulation of AI-2 QS, the fortuity that this biochemical processing is non-QS-2-related could not be discounted and remains to be demonstrated. What is clear is that DPD has the hallmarks of a glycating agent and we have disclosed evidences characterizing this previously unrecognized reactivity. Using ssDNA and gramicidin S as model systems, we have shown that DPD can react with both to produce conspicuous biochemical alterations. We subsequently developed a labeling method that enabled the explicit identification of proteins that can recognize and react with DPD, and as a first application, successfully labeled a prokaryotic proteome. A fascinating aspect is the rapid and stereospecific nature of DPD-induced glycation, requiring bacterial proteins to possess a requisite tertiary structure microenvironment for the DPD-Heyns reaction to ensue. Future studies from our laboratory will focus upon unraveling the chemical makeup of these Heyns products as well as the structural requirements and downstream effects of DPD glycation. More importantly, we are endeavoring to expand the utility of our labeling probes in eukaryotic systems, as this will provide further insights into the physiological implications of the quorum-sensing signaling molecule beyond quorum sensing.
Supplementary Material
Acknowledgments
We gratefully acknowledge Prof. Bonnie Bassler (Princeton University) for providing us with S. typhimurium strains and Sanford-Burnham Proteomics Facility (Sanford-Burnham Medical Research Institute) for proteomic analysis. We thank Prof. Amanda L. Garner (University of Michigan), Dr. Bin Zhou, Dr. Sacha Javor, Dr. Karen C. Collins, and Dr. Chisato M. Yamazaki for technical support and insightful discussions. We also acknowledge financial support from the NIH (Grant AI077644 to K.D.J.) and the Skaggs Institute for Chemical Biology. This is manuscript # 29280 from The Scripps Research Institute.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Lowery CA, Park J, Kaufmann GF, Janda KD. J Am Chem Soc. 2008;130:9200. doi: 10.1021/ja802353j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lowery CA, Matamouros S, Niessen S, Zhu J, Scolnick J, Lively JM, Cravatt BF, Miller SI, Kaufmann GF, Janda KD. Chem Biol. 2013;20:903. doi: 10.1016/j.chembiol.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Miller ST, Xavier KB, Campagna SR, Taga ME, Semmelhack MF, Bassler BL, Hughson FM. Mol Cell. 2004;15:677. doi: 10.1016/j.molcel.2004.07.020. [DOI] [PubMed] [Google Scholar]; d) Roy V, Smith JAI, Wang J, Stewart JE, Bentley WE, Sintim HO. J Am Chem Soc. 2010;132:11141. doi: 10.1021/ja102587w. [DOI] [PubMed] [Google Scholar]
- 2.a) Bassler BL, Greenberg EP, Stevens AM. J Bacteriol. 1997;179:4043. doi: 10.1128/jb.179.12.4043-4045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Globisch D, Lowery CA, McCague KC, Janda KD. Angew Chem Int Ed. 2012;51:4204. doi: 10.1002/anie.201109149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo M, Gamby S, Zheng Y, Sintim HO. Int J Mol Sci. 2013;14:17694. doi: 10.3390/ijms140917694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schauder S, Bassler BL. Genes Dev. 2001;15:1468. doi: 10.1101/gad.899601. [DOI] [PubMed] [Google Scholar]
- 5.Vendeville A, Winzer K, Heurlier K, Tang CM, Hardie KR. Nat Rev Microbiol. 2005;3:383. doi: 10.1038/nrmicro1146. [DOI] [PubMed] [Google Scholar]
- 6.Hodge JE. J Agric Food Chem. 1953;1:928. [Google Scholar]
- 7.a) Iwashige K, Kouda K, Kouda M, Horiuchi K, Takahashi M, Nagano A, Tanaka T, Takeuchi H. J Physiol Anthropol Appl Human Sci. 2004;23:19. doi: 10.2114/jpa.23.19. [DOI] [PubMed] [Google Scholar]; b) Nass N, Bartling B, Navarrete Santos A, Scheubel RJ, Börgermann J, Silber RE, Simm A. Z Gerontol Geriatr. 2007;40:349. doi: 10.1007/s00391-007-0484-9. [DOI] [PubMed] [Google Scholar]
- 8.a) Bucala R, Model P, Cerami A. Proc Natl Acad Sci U S A. 1984;81:105. doi: 10.1073/pnas.81.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Papoulis A, al-Abed Y, Bucala R. Biochemistry. 1995;34:648. doi: 10.1021/bi00002a032. [DOI] [PubMed] [Google Scholar]
- 9.a) Cao H, Jiang Y, Wang Y. J Am Chem Soc. 2007;129:12123. doi: 10.1021/ja072130e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wuenschell GE, Tamae D, Cercillieux A, Yamanaka R, Yu C, Termini J. Biochemistry. 2010;49:1814. doi: 10.1021/bi901924b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shakkottai VG, Sudha R, Balaram P. J Pept Res. 2002;60:112. doi: 10.1034/j.1399-3011.2002.02901.x. [DOI] [PubMed] [Google Scholar]
- 11.a) Priego-Capote F, Scherl A, Müller M, Waridel P, Lisacek F, Sanchez JC. Mol Cell Proteomics. 2010;9:579. doi: 10.1074/mcp.M900439-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Isom DG, Castañeda CA, Cannon BR, García-Moreno BE. Proc Natl Acad Sci U S A. 2011;108:5260. doi: 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munanairi A, O’Banion SK, Gamble R, Breuer E, Harris AW, Sandwick RK. Carbohydr Res. 2007;342:2575. doi: 10.1016/j.carres.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wrodnigg T, Eder B. In: Glycoscience. Stütz A, editor. Vol. 215. Springer; Berlin Heidelberg: 2001. pp. 115–152. [Google Scholar]
- 14.a) Hara S, Yamaguchi M, Takemori Y, Yoshitake T, Nakamura M. Anal Chim Acta. 1988;215:267. [Google Scholar]; b) Campagna SR, Gooding JR, May AL. Anal Chem. 2009;81:6374. doi: 10.1021/ac900824j. [DOI] [PubMed] [Google Scholar]; c) Wang T, Douglass EF, Jr, Fitzgerald KJ, Spiegel DA. J Am Chem Soc. 2013;135:12429. doi: 10.1021/ja406077j. [DOI] [PubMed] [Google Scholar]; d) Glomb MA, Tschirnich R. J Agric Food Chem. 2001;49:5543. doi: 10.1021/jf010148h. [DOI] [PubMed] [Google Scholar]
- 15.a) Mironova R, Niwa T, Hayashi H, Dimitrova R, Ivanov I. Mol Microbiol. 2001;39:1061–1068. doi: 10.1046/j.1365-2958.2001.02304.x. [DOI] [PubMed] [Google Scholar]; b) Mironova R, Niwa T, Dimitrova R, Boyanova M, Ivanov I. J Biol Chem. 2003;278:51068. doi: 10.1074/jbc.M307470200. [DOI] [PubMed] [Google Scholar]
- 16.Cravatt BF, Simon GM, Yates JR. Nature. 2007;450:991. doi: 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- 17.Luber CA, Cox J, Lauterbach H, Fancke B, Selbach M, Tschopp J, Akira S, Wiegand M, Hochrein H, O’Keeffe M, Mann M. Immunity. 2010;32:279. doi: 10.1016/j.immuni.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 18.Xavier KB, Miller ST, Lu W, Kim JH, Rabinowitz J, Pelczer I, Semmelhack MF, Bassler BL. ACS Chem Biol. 2007;2:128. doi: 10.1021/cb600444h. [DOI] [PubMed] [Google Scholar]
- 19.Ha J-H, Eo Y, Grishaev A, Guo M, Smith JAI, Sintim HO, Kim E-H, Cheong H-K, Bentley WE, Ryu K-S. J Am Chem Soc. 2013;135:15526. doi: 10.1021/ja407068v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Attila C, Wang L, Wood TK, Valdes JJ, Bentley WE. J Bacteriol. 2007;189:6011. doi: 10.1128/JB.00014-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LiCalsi C, Crocenzi TS, Freire E, Roseman S. J Biol Chem. 1991;266:19519. [PubMed] [Google Scholar]
- 22.Pereira CS, Santos AJM, Bejerano-Sagie M, Correia PB, Marques JC, Xavier KB. Mol Microbiol. 2012;84:93. doi: 10.1111/j.1365-2958.2012.08010.x. [DOI] [PubMed] [Google Scholar]
- 23.Xavier KB, Bassler BL. J Bacteriol. 2005;187:238. doi: 10.1128/JB.187.1.238-248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Suarez G, Rajaram R, Oronsky AL, Gawinowicz MA. J Biol Chem. 1989;264:3674. [PubMed] [Google Scholar]; b) Lo TW, Westwood ME, McLellan AC, Selwood T, Thornalley PJ. J Biol Chem. 1994;269:32299. [PubMed] [Google Scholar]
- 25.a) Cagliero E, Roth T, Roy S, Lorenzi M. Diabetes. 1991;40:102. doi: 10.2337/diab.40.1.102. [DOI] [PubMed] [Google Scholar]; b) Yu Y, Li W, Wojciechowski B, Jenkins AJ, Lyons TJ. Am J Pharmacol Toxicol. 2007;2:148. [Google Scholar]
- 26.Zhu J, Pei D. ACS Chem Biol. 2008;3:110. doi: 10.1021/cb7002048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.