

Abstract
The key requirement for a portable store of natural gas is to maximize the amount of gas within the smallest possible space. The packing of methane (CH4) in a given storage medium at the highest possible density is, therefore, a highly desirable but challenging target. We report a microporous hydroxyl-decorated material, MFM-300(In) (MFM = Manchester Framework Material, replacing the NOTT designation), which displays a high volumetric uptake of 202 v/v at 298 K and 35 bar for CH4 and 488 v/v at 77 K and 20 bar for H2. Direct observation and quantification of the location, binding, and rotational modes of adsorbed CH4 and H2 molecules within this host have been achieved, using neutron diffraction and inelastic neutron scattering experiments, coupled with density functional theory (DFT) modeling. These complementary techniques reveal a very efficient packing of H2 and CH4 molecules within MFM-300(In), reminiscent of the condensed gas in pure component crystalline solids. We also report here, for the first time, the experimental observation of a direct binding interaction between adsorbed CH4 molecules and the hydroxyl groups within the pore of a material. This is different from the arrangement found in CH4/water clathrates, the CH4 store of nature.
Introduction
In recent years, natural gas, comprised primarily of methane, CH4 (87–96%),1 has become an important economic and technological target as a bridging fuel to a low carbon future. Although not renewable, large reserves (approximately two-thirds of available fossil fuel reserves) and advances in recovery make natural gas an incredibly abundant energy source. This high availability, coupled with the lowest CO2 emissions of any hydrocarbon fuel2 and a low sulfur content,3 ideally place natural gas as a replacement for coal and oil. Currently the widespread use of natural gas is restricted because under ambient conditions, gaseous CH4 has a low energy density and therefore requires constant access to a pipeline. The energy density of bulk CH4 can be increased by liquefaction or, to a lesser extent, compression, using either complex cryogenic tanks or heavy-walled high-pressure vessels with expensive compressors, precluding their use in mass-market applications.4
An alternative approach to increasing the storage density of CH4 is by adsorption on the surface of a porous material, reducing the intermolecular distance and therefore increasing the packing density. Metal–organic framework (MOF) materials have been widely investigated for their ability to store significantly higher volumes of gas than a conventional tank under the same conditions.4−6 This has pronounced effects in a number of materials, which have been reported to adsorb gas species (upon saturation uptakes) at densities comparable to their respective liquid phase. To date, this effect has been observed primarily in MOFs showing high CO2 or H2 adsorption under extreme conditions, typically low temperatures, and/or high pressures.7−11 High volumetric CH4 uptakes [v(STP)/v] have been reported for a number of state-of-the-art MOFs at ambient temperature and 35 bar. MOF-74-Ni [230],4 HKUST-1 [225],4 PCN-14 [202],4 MOF-519 [200],12 and NOTT-220 [189]13 all have significantly higher capacity than bulk CH4 [33] under the same conditions. This approach offers promise for more efficient CH4 storage. It is worth noting that, throughout this Article, the volumetric uptake is derived from the bulk material density on the basis of single crystals, and when the efficiency of powder packing is considered, the uptakes will be reduced accordingly. This is subject to the different compressibility and mechanical stability in different materials.
Variation of metal ions/clusters and organic ligands enables a multitude of design possibilities for MOF materials allowing incorporation of functional groups14,15 and/or desirable metal binding sites16−18 for specific guest binding interactions. To design future materials with improved gas adsorption properties, it is vital to understand the intermolecular contacts, location, and binding interactions of adsorbed guest molecules within the cavity of the host materials. Determination of the binding of molecular H2 and CH4 within porous materials is a very challenging proposition due the short scattering length of hydrogen in X-ray experiments. In addition, adsorbed H2 and CH4 molecules have low enthalpies of adsorption, resulting in them being highly disordered within the pores of MOFs. Indeed, H2 and CH4 have been shown to behave as a “free-rotor” in the solid state when condensed at cryogenic temperatures. To date, determination of the location of adsorbed H2 and CH4 molecules has only been achieved in exceptional cases,19 typically within MOF systems incorporating strong binding sites such as open metal sites20,21 or geometrically confined pores in which the movement (translational motion) of adsorbed gas molecules is highly constrained.22 The nature of the potential interaction(s) of pendant functional groups with adsorbed CH4 within pores at a molecular level remains unclear. Herein, we report the high volumetric uptake of CH4 in an In(III)-tetracarboxylate material, MFM-300(In) [202 v/v at 298 K and 35 bar; 328 v/v at 110 K and 0.6 bar]. The material also shows high volumetric uptake for H2 (488 v/v at 77 K and 20 bar). Direct observation and quantification of adsorbed H2 and CH4 molecules within MFM-300(In) have been achieved using a combination of neutron diffraction and scattering techniques coupled with computational investigations. These complementary results suggest that the adsorbed CH4 molecules form specific interactions with metal-bound hydroxyl groups within the pore, supplemented by intermolecular dipole interactions between adsorbed CH4 molecules and the phenyl-ring lined walls of the pore. These cooperative supramolecular interactions allow packing of CH4 to very high density upon saturation (0.37 g cm–3) reminiscent of liquid CH4 (0.42 g cm–3 at 111 K)23 and 71% of that of the solid (0.52 g cm–3 at 20.4 K).24,25 Interestingly, to our best knowledge, the H4Cguest···H–Ohost interaction reported here represents the first example of such molecular contact in MOFs, and contrasts with the structures observed in CH4/water clathrate materials in which the CH4 molecule is completely orientationally disordered and not observed to interact with the H···OH cage-forming linkage.26 Similar binding has also been observed in H2-loaded MFM-300(In).
Materials and Methods
Synthesis of MFM-300(In)
H4L (biphenyl-3,3′,5,5′-tetracarboxylic acid 330 mg, 1.00 mmol) and In(NO3)3·5H2O (585 mg, 1.50 mmol) were mixed and dispersed in a DMF/MeCN mixture (30 mL, 2:1 v/v) in a 250 mL glass pressure reactor. The white slurry was acidified with conc. nitric acid (65% 1.0 mL), and the vessel sealed and heated at 80 °C for 48 h. The resultant flaky white precipitate was washed with DMF and dried in air. Yield: 347 mg (42% yield based upon solvent content from microanalysis). Anal. Calcd for [In2(OH)2(C16H6O8)]·1.28H2O·2.87(C3H7NO): C, 35.92; H, 3.75; N, 4.88. Found: C, 35.92; H, 3.53; N, 4.88.
The acetone-exchanged material was prepared by suspending the as-synthesized sample in an excess of acetone for 5 days with frequent exchange of solvent. Anal. Calcd for [In2(OH)2(C16H6O8)]·1.10H2O·1.76(C3H6O): C, 35.91; H, 2.94; N, 0.00. Found: C, 35.91; H, 2.94; N, 0.00. Selected IR(ATR): ν/cm–1 = 3501 (br) O–H, 1705 (s), 1610 (s), 1550 (br), 1418 (br), 1359 (s), 1220 (s), 1089 (w), 782 (s), 748 (s).
Gas Adsorption Isotherms
Gravimetric isotherms (0–20 bar) were recorded at 77 and 87 K under liquid nitrogen and argon, respectively, for H2, and at 110, 215, and 245 K (liquid nitrogen cryo-furnace), 195 K (dry ice/acetone), and 273, 283, 293, 303, and 308 K (temperature controlled water-bath) for CH4. Data were collected using an IGA-003 system (Hiden Isochema, Warrington, UK) equipped with a turbomolecular pumping system. Acetone exchanged samples were loaded into the system and degassed at 120 °C and 1 × 10–6 mbar for 20 h to give a dry, desolvated material of typical mass ca. 50 mg. 99.995+% grade H2 and CH4 were purchased from Air Liquide and used as received. High pressure (0–50 bar) gravimetric gas adsorption and desorption data were collected using a Xemis system (Hiden Isochema, Warrington, UK) equipped with a turbomolecular pumping system backed by a diaphragm pump. CH4 isotherms (0–50 bar) were recorded at 273, 283, 293, 298, and 303 K by a temperature controlled water bath.
Neutron Powder Diffraction
Neutron powder diffraction experiments were undertaken at the WISH diffractometer at the ISIS facility. MFM-300(In) was loaded into a 6 mm diameter vanadium sample can and outgassed at 1 × 10–7 mbar and 100 °C for 1 day. The sample was loaded into a liquid helium cryostat and cooled to 7 K for data collection. D2 and CD4 gas were introduced by warming the samples to 50 and 150 K, respectively, and the gas was dosed volumetrically from a calibrated volume. The gas-loaded sample was then cooled to 7 K over a period of 2 h to ensure good mobility of adsorbed D2 and CD4 within the crystalline structure of MFM-300(In). The sample was kept at 7 K for an additional half an hour before date collection to ensure the thermal equilibrium.
Inelastic Neutron Scattering
Inelastic neutron scattering (INS) experiments were undertaken using the TOSCA spectrometer at the ISIS facility. MFM-300(In) was loaded into an 11 mm diameter vanadium sample can and outgassed at 1 × 10–7 mbar and 100 °C for 1 day. The sample was loaded into a helium closed cycle refrigerator (CCR) cryostat and cooled to 11 K for data collection. H2 and CH4 gas were introduced by warming the sample to 50 and 150 K, respectively, and the gas was dosed volumetrically from a calibrated volume. The gas-loaded sample was then cooled to 7 K over a period of 2 h to ensure good mobility of adsorbed D2 and CD4 within the crystalline structure of MFM-300(In). The sample was kept at 7 K for an additional half an hour before date collection to ensure the thermal equilibrium.
Density Functional Theory (DFT) Calculations
Density functional theory (DFT) calculations were performed using CASTEP.27 Three types of calculations were performed: geometry optimization (GO), lattice dynamics (LD), and molecular dynamics (MD). GO is used to determine the ground-state structure, binding energies of adsorbed CH4 molecules, and rotational potentials for CH4 molecules when combined with single point energy (SPE) calculations. LD was used to determine the molecular vibrations and thus the hydrogen vibrational density of states for comparison with the INS data. Physisorbed CH4 is particularly problematic for LD methods due to the weak intermolecular interactions. MD simulations were, therefore, performed to determine the vibrational density of states from the velocity autocorrelation function. This analysis of the MD trajectories was performed with the nMoldyn code.28
All calculations used the GGA-PBE functional,29 and some tests were performed with semiempirical dispersive corrections (DFT-D) using the Grimme parametrization.30 The calculations were performed on a single unit cell of size ∼15 × ∼15 × ∼12 Å3 and therefore used a k-point set including only the Gamma point. Ultrasoft pseudopotentials were used to separate the core and (pseudo) valence electrons (1 for H, 4 for C, 6 for O, and 13 for In were treated explicitly in the calculations), and the plane wave cutoff was set to 340 eV for all calculations except MD, for which a lower cutoff of 260 eV was used to reduce computational time. MD simulations were performed in the NVT ensemble using a time step of 1 fs and ran for 10 000 steps, therefore covering a time frame of 10 ps. For GO calculations, energy convergence was set to 10–5 eV/atom, and the maximum residual force per atom was typically 0.02 eV/atom. LD calculations were limited to the Gamma point, Brillouin zone center modes.
Results and Discussion
Synthesis and Crystal Structure Analysis
The solvated framework complex [In2(OH)2(C16H6O8)]·1.28H2O·2.87(DMF) [MFM-300(In)-solvate] was prepared via solvothermal reaction of H4L (H4L = biphenyl-3,3′,5,5′-tetracarboxylic acid) and In(NO3)3·5H2O in a 2:1 ratio mixture of dimethylformamide (DMF) and acetonitrile and was isolated as a microcrystalline powder. Structure solution by synchrotron powder X-ray diffraction confirmed that this material crystallizes in the tetragonal space group I4122, and is isostructural to the related MFM-300(Al).31,32 We note that this MOF has been previously reported in a different preparation, which yields large single crystals.33 In our hands, these two forms of MFM-300(In) are equivalent despite the difference in morphology. MFM-300(In) exhibits an open structure comprising chains of [InO4(OH)2] moieties bridged by tetracarboxylate ligands L4– to afford a porous structure with channels formed by corner-sharing [InO4(OH)2] octahedra linked via two mutually cis-μ2-OH groups. The [InO4(OH)2]∞ chain in MFM-300(In) is similar to that found in MIL-53(In),34 although the latter features trans-μ2-OH groups, resulting in pronounced framework flexibility. The cis-μ2-OH groups in MFM-300(In) tie the framework into a rigid “wine-rack” arrangement. Desolvated MFM-300(In) displays a surface area of 1071 m2 g–1 and a total pore volume of 0.419 cc g–1 as determined from the N2 isotherm at 77 K. These values are consistent with the porosity derived from the single-crystal structure, and confirm the complete activation and phase purity of the material.
H2 and CH4 Sorption Analysis
H2 and CH4 sorption isotherms confirm that MFM-300(In) displays completely reversible uptake over a wide range of temperatures (77–308 K) and pressures (Figure 1). The H2 adsorption properties of this material are commensurate with the measured pore volume and BET surface area, reaching 275 v/v at 77 K and 1 bar, rising to 488 v/v at 20 bar, comparable with other MOF materials with similar BET surface areas.35,36 The heat of adsorption at zero loading is estimated to be 6.8 kJ mol–1, within the range (4–8 kJ mol–1) typically observed for MOF materials with comparable uptakes.35,36
Figure 1.
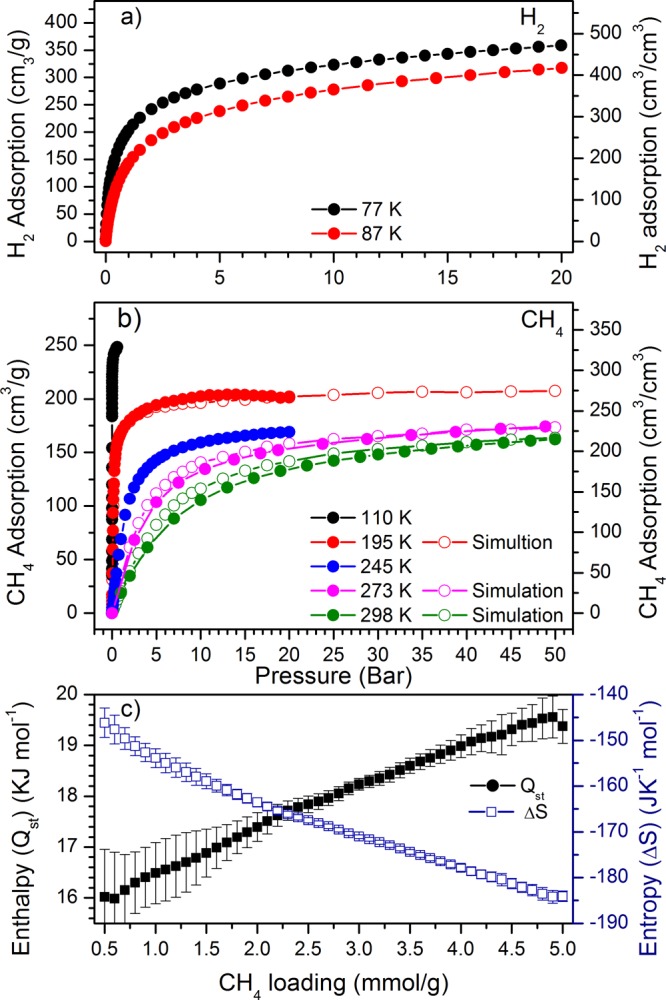
(a,b) Adsorption isotherms of H2 and CH4 in MFM-300(In), respectively. (c) Variation of the isosteric heat (Qst) and entropy (ΔS) of adsorption of CH4 in MFM-300(In).
CH4 adsorption in MFM-300(In) shows a maximum uptake of 328 v/v at 110 K and 0.6 bar, dropping gradually with increasing temperature, with uptakes at 20 bar of 266, 224, and 198 v/v at 195, 245, and 273 K, respectively. High pressure CH4 adsorption isotherms revealed uptake capacities of 220 and 202 v/v at 273 and 298 K, respectively, at 35 bar, rising to 228 and 213 v/v, respectively, at 50 bar. Grand Canonical Monte Carlo (GCMC) simulations show excellent agreement with these experimental data (Figure 1b). The CH4 uptake of MFM-300(In) at 35 bar and 298 K is one of the highest values reported for a MOF to date, surpassed only by MOF-74(Ni, Co)4 and HKUST-14 (Table 1). The isosteric heat of adsorption of CH4 was estimated using the van’t Hoff isochore to be 16 kJ mol–1, which gradually rises to 20 kJ mol–1 with increasing CH4 loading (Figure 1), comparable to that of other MOFs with similar uptake capacities (12–21 kJ mol–1).4 The steady increase in the heat of adsorption reflects the presence of intermolecular interactions between adsorbed CH4 molecules within the pores.
Table 1. Summary of Gas Uptake and Density for Adsorbed CH4 in the Best Performing MOFs.
| m2/g |
35 bar
ambient |
highest reported |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| MOF | BET | Langmuir | pore vol, cm3/g | density, g/cm3 | uptake, v/v | adsorbed density, g/cm3 | uptake, v/v | adsorbed density, g/cm3 | ref |
| MFM-300(In) | 1071 | 0.42 | 1.35 | 202 | 0.231 | 328a | 0.375 | this work | |
| Ni-MOF-74 | 1438 | 0.51 | 1.195 | 230 | 0.245 | 267b | 0.284 | (4) | |
| HKUST-1 | 1977 | 0.69 | 0.881 | 225 | 0.240 | 272b | 0.290 | (4) | |
| MFM-220 | 1014 | 0.39 | 1.46 | 189 | 0.215 | 287c | 0.327 | (13) | |
| MOF-519 | 2400 | 0.938 | 0.953 | 200 | 0.145 | 355d | 0.258 | (12) | |
| MFM-122 | 3286 | 1.41 | 0.589 | 180 | 0.141 | 400e | 0.312 | (17, 39) | |
| MOF-520 | 3290 | 1.277 | 0.586 | 162 | 0.140 | 302d | 0.262 | (12) | |
| MOF-5 | 3320 | 1.38 | 0.605 | 126 | 0.098 | 328d | 0.255 | (40) | |
| MOF-177 | 4500 | 1.89 | 0.427 | 122 | 0.098 | 350d | 0.281 | (40) | |
| MOF-205 | 4460 | 2.16 | 0.38 | 120 | 0.095 | 345d | 0.273 | (40) | |
| MOF-210 | 6240 | 3.6 | 0.25 | 82 | 0.059 | 377d | 0.272 | (40) | |
| PCN-14 | 2360 | 0.83 | 0.819 | 200 | 0.191 | (4) | |||
| AX-21 | 4880 | 1.64 | 0.487 | 153 | 0.124 | (4) | |||
0.6 bar, 111 K.
80 bar, 298 K.
20 bar, 195 K.
Langmuir model, 250 bar, 298 K.
1 bar, 125 K.
The density of adsorbed CH4 in MFM-300(In) can be estimated by using the total pore volume (0.419 cc g–1) derived from the N2 isotherm at 77 K (assuming no distortion of the pore structure). At 110 K and 0.6 bar, the density of adsorbed CH4 in MFM-300(In) was calculated to be 0.37 g cm–3, comparable to that of liquid CH4 (0.42 g cm–3 at 111 K), suggesting an ultraclose packing of the adsorbed CH4 molecules in the pore, which could be a result of strong geometric confinement and/or specific host binding. Significantly, to our best knowledge, this density (0.37 g cm–3) found in MFM-300(In) represents the highest observed value in a porous MOF (Table 1). The density of adsorbed CH4 in MFM-300(In) at 298 K and 35 bar is estimated as 0.231 g cc–1 (55% of the liquid density). Similarly, the density of adsorbed H2 approaches that of the liquid, 0.069 g cc–1 at 77 K and 20 bar (liquid hydrogen density = 0.0708 g cc–1 at 20 K). The high density packing of the adsorbed gas molecules indicates that adsorbed CH4 and H2 molecules in MFM-300(In) pack very efficiently, resulting in a very high storage density in a given pore volume. This led us to study the packing of CH4 and H2 molecules within MFM-300(In) at crystallographic resolution and investigate vibrational and rotational motions within the pore to determine the intermolecular interactions, which account for this observation.
Determination of the Binding Sites for Adsorbed Hydrogen and Methane Molecules
The locations of adsorbed H2 and CH4 molecules within MFM-300(In) were determined by in situ neutron powder diffraction (NPD) as a function of gas loading (Figure 2). NPD patterns were recorded at 7 K for the desolvated material and at loadings of 1.0, 1.5, 3.0, 4.5, and 6.0 D2/In and 1.0, 2.0, and 3.0 CD4/In. Fourier difference map analysis of the NPD data of the desolvated MOF indicates no residual nuclear density peaks within the pore, thus confirming the complete activation and structural stability of the desolvated sample. The bare material was then dosed volumetrically to the desired loading of D2 at 50 K and CD4 at 150 K, accompanied by significant changes in the relative peak intensities of the diffraction patterns. Sequential Fourier difference map analysis of the NPD patterns revealed the position of the center of gravity of the adsorbed D2 and CD4 molecules, which were further developed by Rietveld refinement of these data.
Figure 2.

View of the crystal structure of H2/CH4 loaded MFM-300(In) showing (a,b) view of the corner-sharing extended [InO4(OH)2] chain highlighting interactions between the framework structure with D2 and CD4 molecules, respectively; and (c,d) view of the c-crystallographic axis showing the positions of the adsorbed D2 and CD4 molecules, respectively. D2 molecules in (a) and (c) are colored according to the scheme: Site I, green; Site II, orange; Site III, yellow; Site IV, purple; Site V, blue; Site VI, gray; Site VII, light blue. CD4 molecules in (b) and (d) are colored according to the following scheme: Site I, green; Site II, orange; Site III, yellow; Site IV, purple; Site V, blue; Site VI, gray; Site VII, light blue.
At the first loading of D2, two independent binding sites (D2I and D2II) were located within the one-dimensional pore of MFM-300(In)·(D2)1.80. The initial binding site, D2I, is located near the bridging hydroxyl group of the [InO4(OH)2] backbone [D2I···H–O = 2.54(1) Å] with an occupancy of 0.73. D2II is located interstitially between two phenyl rings of neighboring ligand molecules [D2II···phenyl ring = 3.78(1) Å]. D2II is 3.54(2) Å from the edge of the pore channel, with an occupancy of 0.17, with an intermolecular distance between D2I and D2II of 5.20(2) Å. Upon the second loading, MFM-300(In)·(D2)3.20, the occupancy of D2I increases to 0.91, accompanied by the occupancy of D2II tripling from 0.17 to 0.50 and moving closer to the edge of the pore channel [D2II···phenyl ring = 2.78(1) Å]. Additional adsorbed D2 molecules now fill a third site (D2III) at an occupancy of 0.19, sandwiched between two adjacent D2II sites [D2II···D2III = 2.50(1) Å].
On additional D2 dosing to MFM-300(In)·(D2)5.98, the occupancy of D2I remains at 0.92, and the occupancy of D2II increases to 0.76 and shifts closer to the edge of the pore channel [D2II···phenyl ring = 2.59(2) Å]. Site III (D2III) remains in the same position, with an increase of occupancy from 0.19 to 0.55. An additional D2 binding site D2IV is observed interstitial between a D2I and D2III, with an occupancy of 0.80 between sites D2I and D2III [D2I···D2IV··· D2III = 3.21(1) and 4.06(2) Å, respectively]. Upon the fourth gas dosing (4.8 D2/In), the positions of the previously determined D2 binding sites are unchanged, with the addition of two further adsorption sites. The first new site, D2V, is located toward the center of the pore, 2.05(1) Å from D2IV and 3.46(1) Å from the central carbon atom of the organic ligand with an occupancy of 0.75. The second new site, D2VI, is located between sites D2IV and D2V with a distance of 2.72(1) Å for D2VI···D2V, with an occupancy of 0.36. At the final loading of D2, MFM-300(In)·(D2)12.10, the occupancies of D2II–VI increase further toward saturation (Table 2). The position of the newly observed D2IV site is shifted considerably toward the center of the pore channel at this loading, increasing the distances to D2V to 3.13(4) Å, accompanied by an increase in occupancy to 1.40 (2). At this saturation loading, a further adsorption site, D2VII, interstitial between D2II and D2III is observed [D2II···D2VII = 2.95(3) Å] at an occupancy of 0.12. At this final loading, MFM-300(In) is saturated with D2, with a gas loading corresponding to an uptake of 689 cc cc–1. This high uptake capacity indicates very close packing of molecular D2 at 7 K. Quantum effects may also account for the high saturation uptake for deuterium in this MOF material.11
Table 2. Summary of NPD Occupancies of the Adsorbed D2 Molecules in MFM-300(In) at Different Sites as a Function of Loadinga.
| site occupancy |
||||||||
|---|---|---|---|---|---|---|---|---|
| loading | site I/D2I | site II/D2II | site III/D2III | site IV/D2IV | site V/D2V | site VI/D2VI | site VII/D2VII | uptake v(STP)/v |
| 0.90 D2/In | 0.73 (2) | 0.17 (1) | 102 | |||||
| 1.60 D2/In | 0.91 (3) | 0.50 (1) | 0.19 (7) | 181 | ||||
| 2.99 D2/In | 0.92 (2) | 0.72 (1) | 0.55 (1) | 0.80 (2) | 339 | |||
| 4.83 D2/In | 1.23 (4) | 0.50 (2) | 0.76 (2) | 1.23 (4) | 0.75 (2) | 0.36 (2) | 548 | |
| 6.08 D2/In | 1.27 (2) | 0.62 (1) | 1.00 (3) | 1.40 (2) | 1.09 (4) | 0.45 (2) | 0.25 (2) | 689 |
D2 is refined as a single atom with occupancy 2.0 because the orientation could not be determined from the NPD data. After taking into account the site multiplicity, the occupancies are shown as D2 molecule per indium ion.
Upon loading the regenerated material with methane, two independent CD4 binding sites (CD4I and CD4II) were located in MFM-300(In)·(CD4)2.18 (Table 3). The positions are similar to that observed in MFM-300(In)·(D2)1.80. CD4I is located near the bridging hydroxyl group [CI···H–O = 3.881(12) Å] with an occupancy of 0.796(4); CD4II is located interstitially between the phenyl rings from two ligand molecules [CII···phenyl ring = 3.424(8) Å] and is 3.060(8) Å from the edge of the 1D channel with a lower occupancy of 0.292(5). The intermolecular distance between CD4I and CD4II is 5.284(3) Å. On further loading to MFM-300(In)·(CD4)4.18, the occupancy for CD4I increases slightly to 0.872(4), and its distance to the bridging hydroxyl group is also increased [CI···H–O = 4.040(8) Å]. The location of CD4II remains the same [CD4I···CD4II = 5.248(2) Å], and the occupancy doubles from 0.292(5) to 0.596(6). Additional adsorbed CD4 molecules now fill a third site (CD4III), which is sandwiched by sites CD4I and CD4II [CI···CIII and CIII···CII distances of 3.327(8) and 5.089(8) Å, respectively] and 4.123(8) Å from the edge of the 1D channel with an occupancy of 0.284(4). In the final loading, MFM-300(In)·(CD4)6.22, the occupancy of CD4I increases to saturation with a refined occupancy of 1.008(12) [CI···H–O = 3.989(8) Å], while CD4II is unchanged. The occupancy of CD4III increases to 0.478(6), and its location moves slightly toward the pore center [4.629(10) Å from the edge of the 1D channel; CIII···CII distance of 5.649(11) Å]. Interestingly, a fourth site (CD4IV) appears to accommodate the remaining CD4 molecules. CD4IV is sandwiched between two CD4III sites with an intermolecular separation of 3.984(11) Å for CIV···CIII and an occupancy of 0.564(14). Thus, the adsorbed molecules CD4I–IV form a 1D alternating molecular chain in the 1D channels of MFM-300(In) via intermolecular interactions (Figure 2b), consistent with the steady increase in the heat of adsorption on loading of CH4.
Table 3. Summary of NPD Occupancies of the Adsorbed CD4 Molecules in MFM-300(In) at Different Sites as a Function of Loadinga.
| site occupancy |
|||||
|---|---|---|---|---|---|
| loading | site I/CD4I | site II/CD4II | site III/CD4III | site IV/CD4IV | uptake v(STP)/v |
| 1.09 CD4/In | 0.796(4) | 0.292(6) | 121 | ||
| 2.09 CD4/In | 0.872(8) | 0.596(6) | 0.568(8) | 214 | |
| 3.11 CD4/In | 1.008(12) | 0.582(9) | 0.956(12) | 0.564(14) | 340 |
After taking into account the site multiplicity, the occupancies are shown as CD4 molecule per In ion.
In MFM-300(In)·(CD4)6.22, the material is saturated with a total CD4 occupancy corresponding to an uptake of 340 v/v and with an adsorbed CD4 density of 0.63 g cm–3, 92% of that of solid deuterated CH4 (0.68 g cm–3).31 This result is in excellent agreement with the measured CH4 isotherm uptake (328 v/v) at 110 K. The slightly higher value observed for CD4 adsorption is probably due to a quantum effect as observed in H2/D2 adsorption studies.11 Comparison of the densities of adsorbed and solid CD4 indicates a very efficient intermolecular packing of CH4 molecules in MFM-300(In), considering that the density of adsorbed CD4 may be underestimated by the use of the pore volume derived from N2 isotherm at 77 K, as the total void space of MFM-300(In) as measured and determined by the N2 BET experiment may not be entirely accessible to the bulkier CH4 molecules. The high packing efficiency is also evidenced by the observation of the comparable intermolecular distance between the adsorbed CD4 molecules at different sites when compared to that in solid CH4 (3.95–4.28 Å).37,38
This NPD study confirms that the free bridging hydroxyl group within the pore of MFM-300(In) is the primary binding site for adsorbed H2 and CH4 molecules, leading to high occupancy at low gas loadings. This suggests that adsorbed H2 and CH4 molecules populate at site D2I/CD4I at low surface coverage as a direct result of a CxDyguest···H–Ohost binding interaction. D2I forms a direct binding interaction with the bridging hydroxyl group in the pore, and the binding distance is slightly longer than that observed in MOFs with open metal sites,9 consistent with the nature of the different binding mechanisms. Upon higher loadings of D2, multiple sites appear at the center of the pore, forming a 3D intermolecular binding network similar to that observed in solid H2. At higher loadings of CD4, the occupancy of this initial site CD4I increases in occupancy from 0.796(4) to 0.872(4), reaching a maximum of 1.008(12) from the first to the third loading. This suggests that adsorbed CH4 molecules populate site CD4I at low surface coverage as a direct result of D4Cguest···H–Ohost binding. With increased CH4 loading, site CD4I reaches saturation rapidly with residual CH4 molecules filling other sites stabilized, in principle, by intermolecular interaction between adsorbed CH4 molecules. The D4Cguest···H–Ohost distance for the first adsorption site does not vary significantly with loading [3.881(12)–4.040(8) Å], indicating that the strength of the D4Cguest···H–Ohost interaction is constant regardless of the presence of additional CD4···CD4 interactions. This distance is very similar to that observed in CH4/water clathrates (Cguest···Ohost = 3.77–3.92 Å).26 Closer examination reveals an interesting structural difference between the D4Cguest···H–Ohost binding in MFM-300(In) and that in CH4/water clathrates. In the latter case, water molecules form two different types of cages via intermolecular hydrogen bonds: (i) six large cages having 12 pentagonal and 2 hexagonal faces (denoted by 51262) formed by 24 water molecules and (ii) two small cages having 12 pentagonal faces (denoted by 512) formed by 20 water molecules. The large and small cages have an average cavity size of 4.33 and 3.95 Å, respectively, and host one CH4 molecule per cage, giving a stoichiometry of (CH4)0.174·H2O (Figure 3). The distance between two trapped CH4 molecules in the adjacent cages is ca. 5 Å,26 and this is very similar to the intermolecular distance observed for adsorbed CD4 molecules in MFM-300(In), confirming the high packing efficiency in the MOF. The stoichiometries for adsorbed CD4 molecules in MFM-300(In) with respect to the HO-M group are (CD4)1.09·HO-M, (CD4)2.09·HO-M, and (CD4)3.11·HO-M for the first, second, and third loadings, respectively. Although these numbers are much higher than that observed for clathrates, it is worth noting that the nature of the host–guest complexes (clathrate and MOF) is not strictly comparable. Indeed, this result suggests that, in addition to the HO-M group, the geometrical confinement due to the phenyl rings in the pore also plays a significant role in trapping CH4 in the pore. On both cages of the clathrate, the protons on water molecules all point toward each other, assembling cage “edges” with a fully disordered CH4 molecule at the center. Thus, the dominating binding interaction in clathrates will be H3C–Hguest···Ohost–H. In contrast, adsorbed CD4 molecules bind to the hydroxyl groups along the H–O bond in MFM-300(In), and thus the dominating binding interaction is H3C–Hguest···H–Ohost. Indeed, the position of the proton on the hydroxyl group was refined in the NPD data, and no distortion of the HO-M group was observed upon CD4 inclusion, as is also evidenced by further neutron spectroscopic studies and corresponding modeling (see below). Thus, this study may open new investigations of binding interactions in clathrate materials when strong space confinement is considered.
Figure 3.

Views of (a) the cage structure and H3C–Hguest···Ohost interaction of CH4 in a clathrate; and (b) the channel and H4Cguest···H–Ohost interaction of CH4 in MFM-300(In). Distances do not take into account van der Waals radii; the equivalent distances taking this into account are 4.12 and 7.5 Å for the CH4–water clathrate and MFM-300(In), respectively.
Studies of the Rotational Motion of Adsorbed H2 and CH4 Molecules by INS/DFT/MD
The absolute orientation for all of the adsorbed D2 and CD4 molecules cannot be unambiguously determined from the NPD data due to the thermal motion of the host–guest system, despite data collection at 7 K. It is widely accepted that both of these adsorbates (H2 and CH4) act as a “free rotor” in bulk phase,39,40 but studies on the rotational motions and dynamics of adsorbed H2 are still in their infancy and the dynamics of CH4 molecules in MOFs remain unexplored. To gain insights into these properties, inelastic neutron scattering (INS) spectra were recorded for bare MFM-300(In) as a function of H2 and CH4 loading to investigate the rotational and vibrational properties of the host–guest system. INS is a very powerful neutron spectroscopic technique used to study the dynamics of H-containing systems, enabled by the very large scattering cross section of hydrogen (1–2 orders of magnitude) in comparison to most other elements. INS spectra of the bare MFM-300(In) and CH4 loaded material at loadings of 1.0, 2.0, 4.0, and 6.0 H2/In and 0.5 and 1.0 CH4/In were recorded at 11 K (Figures 4, 5, and S41–S45). The INS spectrum for the bare MFM-300(In) is very similar to that of the iso-structural MFM-300(Al)22 because metal ions (In and Al) have a negligible effect on the inelastic neutron scattering.
Figure 4.
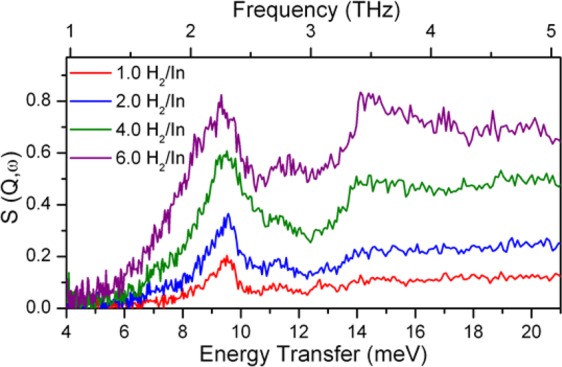
Comparison of the different INS spectra of MFM-300(In) at 1.0, 2.0. 4.0, and 6.0 H2 molecules per In. Additional INS spectra are shown in the Supporting Information.
Figure 5.
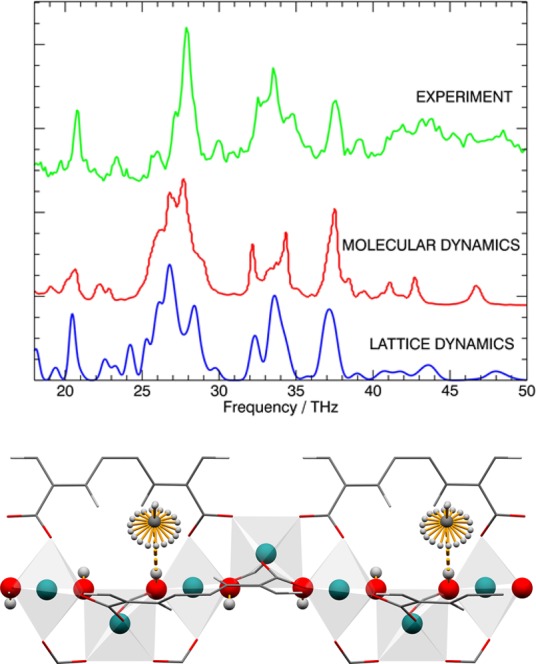
Comparison of the INS spectra for measurements of MFM-300(In) at TOSCA, calculations by MD, and calculations by LD; (bottom) view of the structural model showing the almost free rotation of CH4I molecules at the bridging hydroxyl group (1 THz = 4.14 meV = 33.4 cm–1). Additional INS spectra are shown in the Supporting Information.
Upon loading this material with para-H2, there is no distortion of the vibrational modes arising from the internal modes of the bare material, confirming the absence of structural change, consistent with the NPD analysis and the low enthalpy of adsorption. At a low H2 loading (1.0 H2/In) (Figure 4), a new peak was observed at 9.5 meV, lower than that of the solid H2 (14.7 meV), corresponding to the rotational motion of the adsorbed H2.41,42 In addition, a broad peak was observed at ca. 25 meV, corresponding to the liquid recoil motion of molecular H2.43 The appearance of this recoil motion indicates that there is little interaction between the adsorbed H2 and host material. The almost-free rotation of adsorbed H2 within this material is consistent with the crystal structure of pure solid H2, in which the molecular positions can be discerned, but the atoms are spherically disordered, even at temperatures as low as 2 K.40 At a higher H2 loading of 2.0 H2/In, there is an increase in the area of the peaks relating to the rotational and recoil motion of the adsorbed H2, consistent with the H2 binding at the low loading. Further increasing the H2 loading to 4.0 and 6.0 H2/In results in the consistent growth of the rotational and recoil peaks, accompanied by an additional peak at 14.7 meV, consistent with the formation of “solid-hydrogen-like” material within the pores of the MOF. This observation is in excellent agreement with the insight gained from the in situ crystallographic study, which reveals that at this loading, the intermolecular bond length between adsorbed H2 molecules (3–5 Å) is of an order similar to that observed in solid H2 (3.75 Å).44−46
INS spectra of CH4-loaded MFM-300(In) show a considerable increase in intensity with a broad peak at low energy transfer (below 40 meV) as a result of adsorbed CH4 molecules (Figures S44). Difference INS spectra exhibit an intense and broad peak at 10–30 meV as the dominating signal. No significant change to the vibrational peaks arising from the internal modes of the bare MOF was observed, indicating that there is no strong distortion of the local H modes from the porous material upon CH4 adsorption. This result suggests that adsorbed CH4 molecules in MFM-300(In) have a wide amplitude, weakly hindered rotational motion, even at a temperature that is well below its melting point (90.7 K). This is consistent with the structure of solid CH4-I that is shown to have well-defined molecular positions, but spherically disordered protons.33
Geometry optimization (GO) calculations were performed with both fixed and variable cell parameters, with an observed increase of the unit cell dimensions of 2–3%. However, we found this did not affect the calculated binding energies, rotational potentials, or vibrational spectra. Binding energies were calculated for the two most stable sites (CD4I and CD4II), which are populated with adsorbed CH4 molecules at low surface coverage. Density functional theory (DFT) calculations determine the binding energies to be 20.5 and 19.7 kJ mol–1 for CD4I and CD4II, respectively, in excellent agreement with that determined experimentally from the adsorption isotherms (16–20 kJ mol–1). The similarity of the calculated binding energies at two different sites also explains the absence of any marked steps in the adsorption profiles (even in logarithmic view as shown in Figure S12) that would be indicative of the presence of adsorption sites of significantly different stabilities.34 Rotating the CH4 molecule on the most stable site (CD4I) about a C–H bond aligned with the O–H group (Figure 5) and performing a series of single point energy (SPE) calculations enabled the rotational potential of adsorbed CH4 molecules to be determined. In this case, the rotational barrier is 5 meV (0.48 kJ mol–1), and therefore the rotation of adsorbed CH4 molecules is almost free, consistent with the observed strong recoil motion at low energy transfer in the INS spectra.
Lattice dynamics (LD) calculations of the bare MFM-300(In) enable the hydrogen density of states to be determined, which can be compared to the experimental spectrum. Above 18 THz or 74 meV, there are pronounced vibrational peaks, which show a good match between LD calculations of MFM-300(In) and the measured spectrum (Figure 5). The vibrational spectrum is a fingerprint of the solid-state structure, and reproducing the measured spectrum from the experimental structure using a high level of theory (DFT) validates the structural model.
Molecular dynamics (MD) simulations were performed at temperatures ranging from 50 to 250 K. The simulation at 250 K gave the most appropriate sampling of the potential energy surface (PES). The hydrogen vibrational density of states from this run was in good agreement with the LD calculation and INS experiment (Figure 5). MD simulations of CH4-loaded MFM-300(In) allowed the vibrational density of states of the CH4 molecules to be determined, and this calculated spectrum for CH4 was compared to the experimental difference spectrum (Figure S43). In the low frequency part of the spectrum, where signals are related to whole molecule vibrations and low frequency liberational/rotational modes, the spectra are in good agreement. At higher frequency, the calculated spectrum shows the internal modes (bending and stretching) of CH4, but these are not observed in the experimental INS spectrum. The experimentally observed modes are attenuated by a Debye–Waller factor (exp(−Q2u2)), which is stronger for CH4 than for the MOF host, revealing a higher mean square displacement (u2) for the CH4 because it is relatively weakly bound and can undergo almost free rotations as suggested from SPE calculations.
Thus, this neutron spectroscopic study of MFM-300(In) has investigated the rotational modes and packing of adsorbed H2 and CH4 within this material. INS spectra of H2 loaded MFM-300(In) confirm the appearance of two types of adsorbed H2: one with reduced rotational energy and one with “almost-free” rotations; the latter is a reminiscent of solid H2. This study further confirms that the bridging hydroxyl is the strongest binding site for methane within MFM-300(In), forming H3C–Hguest···H–Ohost binding interactions, with the adsorbed CH4 molecules having wide amplitude, weakly hindered rotational motion in the confined cavity of the MOF, even at 11 K. To improve the CH4 storage capacity, further efforts are required to “immobilize” adsorbed CH4 molecules in the pore by increasing the binding energies and reducing their rotational motion via introduction of more specific host–guest interactions.
Conclusions
Several key insights into the binding of H2 and CH4 within the hydroxyl-decorated material MFM-300(In) have been revealed from the direct visualization and quantification of the host–guest interactions of these gases within the porous host at crystallographic resolution. This has implications for the design and discovery of future materials capable of storing H2 and CH4 at high capacity. The binding of H2 within this material is mediated by a combination of a number of weak intermolecular interactions at low loadings and a combination of adsorbate–adsorbent and adsorbent–adsorbent interactions at high loadings. The very high density of adsorbed CH4 molecules observed in MFM-300(In) was found to be dominated by a specific H4Cguest···H–Ohost interaction, which is supplemented further by CH4guest···CH4guest intermolecular dispersion interactions. The phenyl rings on the linker play a further role in providing space confinement for the adsorbed CH4 molecules, thereby inducing a change from the H3C–Hguest···Ohost–H binding mode observed in clathrates. These multiple relatively weak binding interactions are aligned in the pore with an optimal geometry, offering a highly cooperative binding environment for the adsorbed H2 and CH4 molecules. As a result, the adsorption and binding modes of H2 and CH4 within this material have been observed and rationalized by both experimental and theoretical investigations. We hope that the strategy developed here will help the development of improved materials with optimized pore environment for natural gas storage and separation.
Acknowledgments
We thank the Universities of Manchester and Nottingham for funding. M.S. acknowledges receipt of an EPSRC Program Grant and ERC Advanced Grant. E.B. acknowledges receipt of an ERC Starter Grant. We are especially grateful to STFC and the ISIS Facility for access to TOSCA and WISH Beamlines, to Diamond Light Source for access to Beamline I11, and to the European Synchrotron Radiation Facility for access to Beamline ID31. INS simulations were carried out using the high performance computing resources at the ILL.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b01323.
Author Present Address
▽ Physical Sciences and Engineering Division, King Abdullah University of Science & Technology, Thuwal, Makkah 23955, The Kingdom of Saudi Arabia.
The authors declare no competing financial interest.
Supplementary Material
References
- Danesh A.PVT and Phase Behaviour of Petroleum Reservoir Fluids; Elsevier: Oxford, 1998. [Google Scholar]
- Celzard A.; Fierro A. Energy Fuels 2005, 19, 573. 10.1021/ef040045b. [DOI] [Google Scholar]
- Lozano-Castelló J.; Alcañiz-Monge J.; de la Casa-Lillo M. A.; Cazorla-Amorós D.; Linares-Solano A. Fuel 2002, 81, 1777. 10.1016/S0016-2361(02)00124-2. [DOI] [Google Scholar]
- Mason J. A.; Veenstra M.; Long J. R. Chem. Sci. 2014, 5, 32. 10.1039/C3SC52633J. [DOI] [Google Scholar]
- Peng Y.; Krungleviciute V.; Eryazici I.; Hupp J. T.; Farha O. K.; Yildirim T. J. Am. Chem. Soc. 2013, 135, 11887. 10.1021/ja4045289. [DOI] [PubMed] [Google Scholar]
- Makal T. A.; Li J.-R.; Weigang L.; Zhou H.-C. Chem. Soc. Rev. 2012, 41, 7761. 10.1039/c2cs35251f. [DOI] [PubMed] [Google Scholar]
- Walton K. S.; Millward A. R.; Dubbeldam D.; Frost H.; Low J. J.; Yaghi O. M.; Snurr R. Q. J. Am. Chem. Soc. 2008, 130, 406. 10.1021/ja076595g. [DOI] [PubMed] [Google Scholar]
- Yang S.; Lin X.; Lewis W.; Suyetin M.; Bichoutskaia E.; Parker J. E.; Tang C. C.; Allan D. A.; Rizkallah P. J.; Hubberstey P.; Champness N. R.; Thomas K. M.; Blake A. J.; Schröder M. Nat. Mater. 2012, 11, 710. 10.1038/nmat3343. [DOI] [PubMed] [Google Scholar]
- Lin X.; Telepeni I.; Bake A. J.; Dailly A.; Brown C. M.; Simmons J. M.; Zoppi M.; Walker G. S.; Thomas K. M.; Mays T. J.; Hubberstey P.; Champness N. R.; Schröder M. J. Am. Chem. Soc. 2009, 131, 2159. 10.1021/ja806624j. [DOI] [PubMed] [Google Scholar]
- Yang W.; Lin X.; Jia J.; Blake A. J.; Wilson C.; Hubberstey P.; Champness N. R.; Schröder M. Chem. Commun. 2008, 44, 359. 10.1039/B712201B. [DOI] [PubMed] [Google Scholar]
- Yang S.; Lin X.; Dailly A.; Blake A. J.; Hubberstey P.; Champness N. R.; Schröder M. Chem. - Eur. J. 2009, 15, 4829. 10.1002/chem.200802292. [DOI] [PubMed] [Google Scholar]
- Gándara F.; Furukawa H.; Lee S.; Yaghi O. M. J. Am. Chem. Soc. 2014, 136, 5271. 10.1021/ja501606h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage M.; Yang S.; Suyetin M.; Bichoutskaia E.; Lewis W.; Blake A. J.; Barnett S. A.; Schröder M. Chem. - Eur. J. 2014, 20, 8024. 10.1002/chem.201304799. [DOI] [PubMed] [Google Scholar]
- Asmail N. H.; Suyetin M.; Cabot R.; Krap C. P.; Lü J.; Easun T. L.; Bichoutskaia E.; Lewis W.; Blake A. J.; Schröder M. Chem. - Eur. J. 2014, 20, 7317. 10.1002/chem.201304005. [DOI] [PubMed] [Google Scholar]
- An J.; Geib S. J.; Rosi N. L. J. Am. Chem. Soc. 2010, 132, 38. 10.1021/ja909169x. [DOI] [PubMed] [Google Scholar]
- Guo Z.; Wu H.; Srinivas G.; Zhou Y.; Xiang S.; Chem Z.; Yang Y.; Zhou W.; O’Keeffe M.; Chen B. Angew. Chem., Int. Ed. 2011, 50, 3178. 10.1002/anie.201007583. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Suyetin M.; Bichoutskaia E.; Blake A. J.; Allan D. R.; Barnett S. A.; Schröder M. Chem. Sci. 2013, 4, 1731. 10.1039/c3sc21769h. [DOI] [Google Scholar]
- Pham T.; Forrest K. A.; Nugent P.; Belmabkhout Y.; Luebke R.; Eddaoudi M.; Zaworotko M. J.; Space B. J. Phys. Chem. C 2013, 117, 9340. 10.1021/jp402304a. [DOI] [Google Scholar]
- Carrington E. J.; Vitórica-Yrezábel I. J.; Brammer L. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2014, B70, 404. 10.1107/S2052520614009834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Zhou W.; Yildirim T. J. Am. Chem. Soc. 2009, 131, 4995. 10.1021/ja900258t. [DOI] [PubMed] [Google Scholar]
- Wu H.; Simmons J. M.; Liu Y.; Brown C. M.; Wang X.-S.; Ma D.; Peterson V. K.; Southon P. D.; Kepert C. J.; Zhou H.-C.; Yildirim T.; Zhou W. Chem. - Eur. J. 2010, 16, 5205. 10.1002/chem.200902719. [DOI] [PubMed] [Google Scholar]
- Wu H.; Zhou W.; Yildirim T. J. Phys. Chem. C 2009, 113, 3029. 10.1021/jp8103276. [DOI] [Google Scholar]
- Setzmann U.; Wagner W. J. Phys. Chem. Ref. Data 1991, 20, 1061. 10.1063/1.555898. [DOI] [Google Scholar]
- Mooy H. H. Nature 1931, 127, 707. 10.1038/127707c0. [DOI] [Google Scholar]
- Greer S. C.; Meyer L. Z. Angew. Phys. 1969, 27, 198. [Google Scholar]
- Kirchner M. T.; Boese R.; Billups W. E.; Norman L. R. J. Am. Chem. Soc. 2004, 126, 9407. 10.1021/ja049247c. [DOI] [PubMed] [Google Scholar]
- Clark S. J.; Segall M. D.; Pickard C. J.; Hasnip P. J.; Probert M. I. J.; Refson K.; Payne M. C. Z. Kristallogr. - Cryst. Mater. 2005, 220, 567. 10.1524/zkri.220.5.567.65075. [DOI] [Google Scholar]
- Hinsen K.; Pellegrini E.; Stachura S.; Kneller G. R. J. Comput. Chem. 2012, 33, 2043. 10.1002/jcc.23035. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Emerzerhof M. Phys. Rev. Lett. 1996, 77, 3865. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Grimme S. J. Comput. Chem. 2006, 27, 1787. 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- Yang S.; Sun J.; Ramirez-Cuesta A. J.; Cellear S. K.; David W. I. F.; Anderson D. P.; Newby R.; Blake A. J.; Parker J. E.; Tang C. C.; Schröder M. Nat. Chem. 2012, 4, 887. 10.1038/nchem.1457. [DOI] [PubMed] [Google Scholar]
- Yang S.; Ramirez-Cuesta A. J.; Newby R.; Garcia-Sakai V.; Manuel P.; Callear S.; Campbell S. I.; Tang C. C.; Schröder M. Nat. Chem. 2014, 7, 121. 10.1038/nchem.2114. [DOI] [PubMed] [Google Scholar]
- Qian J.; Jiang F.; Yuan D.; Wu M.; Zhang S.; Zhanq L.; Hong M. Chem. Commun. 2012, 48, 9696. 10.1039/c2cc35068h. [DOI] [PubMed] [Google Scholar]
- Serra-Crespo P.; Stavitski E.; Kapteijn E.; Gascon J. RSC Adv. 2012, 2, 5051. 10.1039/c2ra20528a. [DOI] [Google Scholar]
- Suh M. P.; Park H. J.; Prasad T. K.; Lim D.-W. Chem. Rev. 2012, 112, 782. 10.1021/cr200274s. [DOI] [PubMed] [Google Scholar]
- Goldsmith J.; Wong-Foy A. G.; Cafarella M. J.; Siegel D. J. Chem. Mater. 2013, 25, 3373. 10.1021/cm401978e. [DOI] [Google Scholar]
- Baer D. R.; Frass B. A.; Riehl D. H.; Simmons R. O. J. Chem. Phys. 1978, 56, 1411. 10.1063/1.435960. [DOI] [Google Scholar]
- Newmann M. A.; Press W.; Nöldeke C.; Asmussen B.; Prager M.; Ibberson R. M. J. Chem. Phys. 2003, 119, 1586. 10.1063/1.1580809. [DOI] [Google Scholar]
- Press W. J. Chem. Phys. 1972, 56, 2597. 10.1063/1.1677586. [DOI] [Google Scholar]
- Kranendonk J. V.; Gush H. P. Phys. Lett. 1962, 1, 22. 10.1016/0031-9163(62)90265-2. [DOI] [Google Scholar]
- Sumida K.; Brown C. M.; Herm Z. R.; Chavan S.; Bordiga S.; Long J. R. Chem. Commun. 2011, 47, 1157. 10.1039/C0CC03453C. [DOI] [PubMed] [Google Scholar]
- Queen W. L.; Bloch E. D.; Brown C. M.; Hudson M. R.; Mason J. A.; Murray L. J.; Ramirez-Cuesta A. J.; Peterson V. K.; Long J. R. Dalton Trans. 2012, 41, 4180. 10.1039/c2dt12138g. [DOI] [PubMed] [Google Scholar]
- Yang S.; Ramirez-Cuesta A. J.; Schröder M. Chem. Phys. 2014, 428, 111. 10.1016/j.chemphys.2013.11.004. [DOI] [Google Scholar]
- Rabone J.; Yue Y.-F.; Chong S. Y.; Stylianou K. C.; Bacsa J.; Bradshaw D.; Darling G. R.; Berry N. G.; Khimyak Y. Z.; Ganin A. Y.; Wiper P.; Claridge J. B.; Rosseinsky M. J. Science 2010, 329, 1053. 10.1126/science.1190672. [DOI] [PubMed] [Google Scholar]
- Wilmer C. E.; Farha O. K.; Yildirim T.; Eryazici I.; Krungleviciute V.; Sarjeant A. A.; Snurr R. Q.; Hupp J. T. Energy Environ. Sci. 2014, 27, 1158. [Google Scholar]
- Furukawa H.; Ko N.; Go Y. B.; Artani N.; Choi S. B.; Choi E.; Yazaydin A. Ö.; Snurr R. Q.; O’Keeffe M.; Kim J.; Yaghi O. M. Science 2010, 329, 424. 10.1126/science.1192160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.