

Abstract
Solar water-splitting represents an important strategy toward production of the storable and renewable fuel hydrogen. The water oxidation half-reaction typically proceeds with poor efficiency and produces the unprofitable and often damaging product, O2. Herein, we demonstrate an alternative approach and couple solar H2 generation with value-added organic substrate oxidation. Solar irradiation of a cyanamide surface-functionalized melon-type carbon nitride (NCNCNx) and a molecular nickel(II) bis(diphosphine) H2-evolution catalyst (NiP) enabled the production of H2 with concomitant selective oxidation of benzylic alcohols to aldehydes in high yield under purely aqueous conditions, at room temperature and ambient pressure. This one-pot system maintained its activity over 24 h, generating products in 1:1 stoichiometry, separated in the gas and solution phases. The NCNCNx–NiP system showed an activity of 763 μmol (g CNx)−1 h–1 toward H2 and aldehyde production, a Ni-based turnover frequency of 76 h–1, and an external quantum efficiency of 15% (λ = 360 ± 10 nm). This precious metal-free and nontoxic photocatalytic system displays better performance than an analogous system containing platinum instead of NiP. Transient absorption spectroscopy revealed that the photoactivity of NCNCNx is due to efficient substrate oxidation of the material, which outweighs possible charge recombination compared to the nonfunctionalized melon-type carbon nitride. Photoexcited NCNCNx in the presence of an organic substrate can accumulate ultralong-lived “trapped electrons”, which allow for fuel generation in the dark. The artificial photosynthetic system thereby catalyzes a closed redox cycle showing 100% atom economy and generates two value-added products, a solar chemical, and solar fuel.
Introduction
Splitting water into the fuel H2 and byproduct O2 using sunlight represents a sustainable strategy to produce renewable and storable energy. The design of solar water splitting systems remains an enormous challenge, and no commercial application of such technology exists despite extensive efforts over several decades. However, proof-of-principle water splitting has already been demonstrated by coupling a photovoltaic unit to a water electrolyzer, in photoelectrochemical (PEC) cells, and colloidal systems.1−4 The intricate nature of the water oxidation half-reaction, involving multiple electron and proton transfers,5,6 is believed to be the main obstacle in achieving this goal. In addition, O2 has only limited commercial application and its production in complete water-splitting is potentially unwanted as most of the proton reduction catalysts are O2 sensitive,7,8 and system-damaging reactive oxygen species can form through incomplete water oxidation or from uncontrolled O2 reduction.9
Replacing the water oxidation half-reaction with valuable substrate oxidation reactions would bypass O2 production and enable the synthesis of a high value organic chemical, a so-called solar chemical, in addition to the solar fuel H2 in a closed redox cycle.10 A useful process is the selective oxidation of benzyl alcohols to carbonyl compounds, which is a fundamental reaction both in the laboratory and on an industrial scale. For example, carbonyl derivatives such as aldehydes and ketones are widely used as precursors in the pharmaceutical and fragrance industries and for complex syntheses.11−13 Since many classical oxidation reactions are currently carried out in organic solvents at high pressure and temperature and/or employ hazardous stoichiometric oxidants, such as MnO4– or CrO3, there is clear scope for greener synthetic routes.14
Simultaneous photoreduction of aqueous protons and organic substrate oxidation in a single compartment requires coupling between light harvester and catalysts, catalyzing two redox reactions, and the accumulation of products without their interference in the opposite half-reaction. To simplify this demanding task, the two halves of the system are typically studied separately with a sacrificial reagent closing the catalytic cycle.
Solar-light driven H2 production from water has been reported in the presence of sacrificial electron donors by employing solution based homogeneous and semiheterogeneous colloidal systems comprising a light harvester combined with synthetic, enzymatic, and metallic catalysts.15−21 Photocatalytic oxidations of organic substrates containing alkene,22,23 amine,24 alcohol,25,26 and sulfide26,27 groups have also been reported in the presence of electron scavengers in homogeneous and suspension systems.
Molecular catalyst-driven simultaneous substrate oxidation and H2 production has been previously reported under fully homogeneous conditions as well as in PEC cells.10,28,29 However, these reported nonsacrificial systems were carried out in organic solvents, employed multiple expensive and fragile noble metal based dyes and catalysts, leading to complicated schemes with low efficiencies, and thereby limited practical applications.
There remains a need to find inexpensive and robust photosensitizers, which can couple proton reduction and substrate oxidation, while eliminating the need to consume noninnocent and unsustainable sacrificial reagents.30,31 A promising photocatalytic material is a polyheptazine-based carbon nitride, as it is nontoxic, metal-free, and easily synthesized from inexpensive precursors and shows thermal and chemical stability.32,33 The well positioned band edges also make it feasible to drive many photocatalytic reactions,33 including full water splitting.34−36 Polymeric carbon nitride has been applied for photocatalytic H2 evolution in the presence of metallic and molecular catalysts.20,37−39 Mesoporous carbon nitride has also been reported to act as a photocatalyst for mild radical oxidation of benzyl alcohols, without the need for an oxidation catalyst, under an O2 (via O2•–) saturated environment as a sacrificial electron acceptor, in organic solvents.40,41
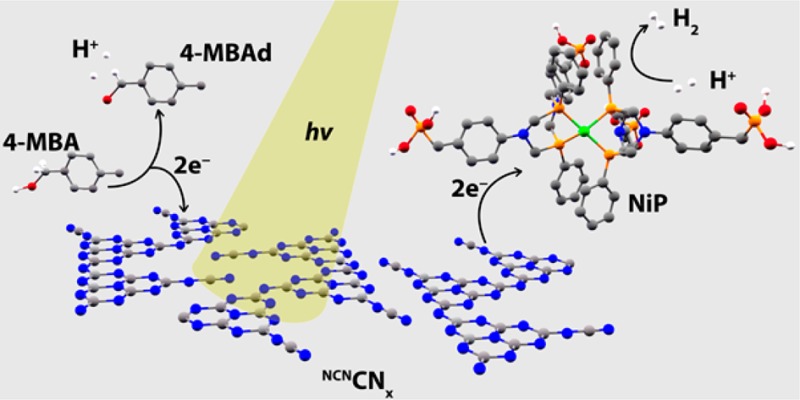
Herein, we report a complete redox cycle that photocatalyzes the production of H2 coupled to the selective oxidation of benzyl alcohols to aldehydes. As a light harvester, we employed a carbon nitride with a cyanamide surface functionalization notated as NCNCNx (Figure 1), which has been recently reported to be significantly more active for H2 production than the unfunctionalized carbon nitride, H2NCNx, under sacrificial conditions.42 To enable platinum-free conditions, we employed a molecular nickel(II) bis(diphosphine) based H2-evolution catalyst, NiP (Figure 1), and the NCNCNx–NiP system allowed for visible-light driven benzyl alcohol oxidation and simultaneous H2 production in purely aqueous solution at room temperature and ambient pressure, in the absence of organic cosolvents and sacrificial reagents (Figure 1).
Figure 1.

Schematic representation of a closed redox system for solar-driven simultaneous proton reduction and alcohol oxidation in aqueous solution. Irradiation of surface functionalized carbon nitride, NCNCNx, results in the formation of a photoexcited state in which the holes are quenched by alcohol for aldehyde formation and the photoexcited electrons are effectively transferred from NCNCNx to the molecular catalyst NiP (bromide counterions omitted for clarity) resulting in H2 formation.
Results and Discussion
Preparation and Characterization of the Photosystem
We have previously reported a platinum-free sacrificial H2 evolution system with H2NCNx and the molecular Ni catalyst, NiP.20 Amorphous polymeric H2NCNx, known as melon, is a metal-free photocatalyst, which can be easily synthesized from nitrogen rich precursors, and it displays long-term activity and stability.37NiP belongs to a family of hydrogenase-inspired molecular catalysts with a Ni-bis(diphosphine) catalytic core,43,44 and it has been shown to operate effectively in heterogeneous hybrid systems,20 as well as under homogeneous conditions.15,21 Here, we have used the more active NCNCNx (for characterization see Figures S1–S3),42 to investigate full redox cycle systems for solar light driven simultaneous alcohol oxidation and proton reduction (Figure 1).
Photocatalytic systems were prepared by dispersing NCNCNx (5 mg), NiP, and 4-methylbenzyl alcohol (4-MBA) in an aqueous phosphate (KPi) solution (3 mL), in a photoreactor with a total volume of 7.74 mL. The photoreactor was sealed with a rubber septum, purged with N2 (containing 2% CH4), and then irradiated at 25 °C using a solar light simulator equipped with an air mass 1.5 global (AM 1.5G) filter at 1 sun intensity (100 mW cm–2), unless specified otherwise. The headspace H2 gas was quantified in regular intervals by gas chromatography (using CH4 as internal standard), while 4-MBA and other possible oxidation products were analyzed by 1H NMR spectroscopy (see Experimental Section for details). The conversion yield was determined based on the percentage conversion of benzyl alcohol to oxidation products (Figure S4).
Photocatalytic Performance
The reaction conditions were optimized systematically for H2 production activity per catalyst, expressed as NiP-based turnover frequency (TOFNiP; mol H2 (mol NiP)−1 h–1 determined after 1 h of irradiation) and turnover number (TONNiP; mol H2 (mol NiP)−1 determined after 24 h of irradiation, unless specified otherwise), as well as the conversion yield and selectivity of alcohol conversion to oxidation products after 24 h. The parameters of optimization were the amount of NiP, the amount of 4-MBA, and the pH of the solution (Table S1, Figures S5–S8).
The optimized conditions for the NCNCNx–NiP system are 5 mg of NCNCNx suspended in 3 mL of KPi (0.02 M, pH 4.5) with 50 nmol of NiP and 30 μmol of 4-MBA under simulated solar light irradiation (Figure 2). There was a linear increase in both H2 and aldehyde production for the first 6 h of irradiation, whereupon the rate started to decrease over time (Table S2). A TOFNiP of 31.1 ± 3.1 h–1 and TONNiP of 425.4 ± 42.5 was achieved with full solar spectrum irradiation, while oxidation of 4-MBA afforded 4-methylbenzaldehyde (4-MBAd) selectively in 66.0 ± 6.6% yield without further oxidation to form the carboxylic acid. Thus, H2 and 4-MBAd were photogenerated cleanly and in a 1:1 stoichiometry over time and reached 21.3 ± 2.1 μmol and 19.8 ± 2.0 μmol after 24 h, respectively (Figure 2a).
Figure 2.
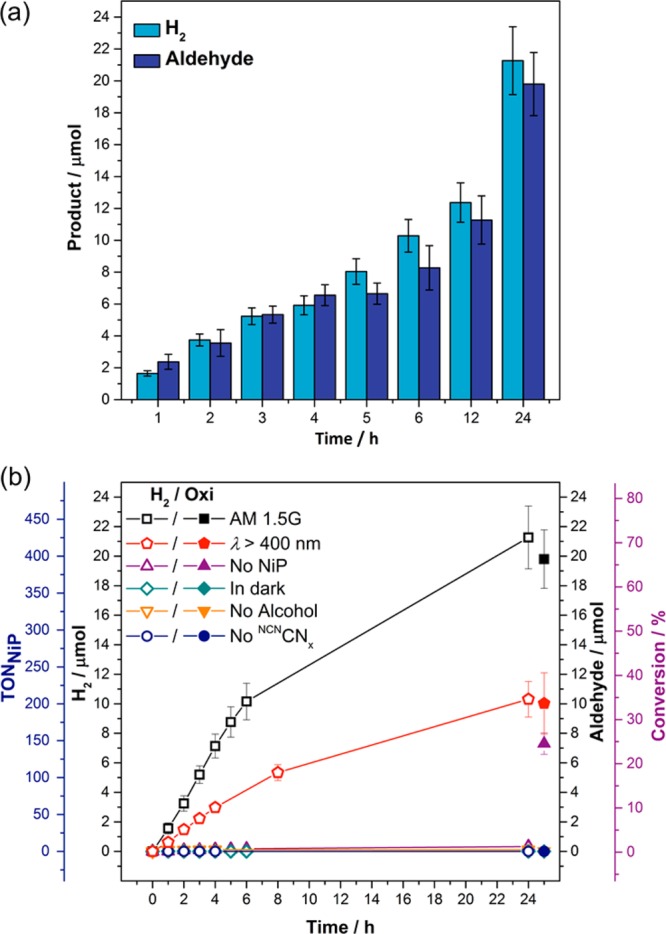
(a) Time-dependent photocatalytic aldehyde and H2 production with NCNCNx (5 mg), NiP (50 nmol), 4-MBA (30 μmol) in KPi (0.02 M, pH 4.5, 3 mL) under 1 sun irradiation (AM 1.5G) at 25 °C. For each time-point, three vials were irradiated for a specified number of hours and then worked-up. (b) Simultaneous H2 and aldehyde production under optimized reaction conditions in the absence (AM 1.5G) and in the presence of a UV-filter (λ > 400 nm). The pair of hollow and filled symbols of the same shape and color corresponds to H2 and aldehyde production, respectively, under the specified reaction conditions. Control experiments in the absence of NiP, 4-MBA, NCNCNx and in the dark are also shown.
Control experiments in the dark or in the absence of NCNCNx did not yield H2 or aldehyde (Table S3). In the absence of NiP, only negligible amounts of H2 were detected during irradiation, but selective 4-MBA oxidation to 4-MBAd was observed with a 24.3 ± 2.4% conversion, suggesting that 4-MBA oxidation may be the first step in the catalytic redox cycle (see below for more details). In the absence of 4-MBA only a minimal amount of H2 was detected.
The NCNCNx–NiP photosystem was also studied under visible only irradiation (λ > 400 nm). A TOFNiP of 12.3 ± 1.2 h–1 and a TONNiP of 206.3 ± 24.1 were observed, with 33.7 ± 6.5% 4-MBA conversion, corresponding to approximately 50% of the activity observed under full spectrum irradiation (Figure 2b). The visible light response of the NCNCNx–NiP system showed improvement when compared to the previously reported sacrificial H2NCNx–NiP photosystem, where only 16% of the activity observed under full spectrum irradiation was preserved under visible-light irradiation.20
The activity of NiP in this closed redox cycle compares well with previously reported sacrificial H2 production hybrid systems using colloidal light absorbers. A photocatalytic system comprising H2NCNx and NiP in sacrificial electron donor ethylenediaminetetraacetic acid (EDTA) solution at pH 4.5 resulted in a TOFNiP of 109.3 ± 10.9 h–1 and TONNiP of 155. Sacrificial photo-H2 generation was also demonstrated with NiP and ruthenium tris(bipyridine) (RuP) dye sensitized TiO2, which showed a TOFNiP of 72 ± 5 h–1 and TONNiP of 278 ± 19.15 More recently, carbon quantum dots were used as a photosensitizer in a sacrificial system with NiP, and a TOFNiP of 41 h–1 and TONNiP of 64 were reported.21
The external quantum efficiency (EQE) of the system was determined using a solar light simulator equipped with a monochromator (λ = 360 ± 10 nm, I = 4.43 mW cm–2). The highest EQE of 15.2 ± 1.5 % was obtained with NCNCNx (5 mg), 4-MBA (30 μmol) in 3 mL of KPi (0.1 M) buffer at pH 4.5 after 1 h of irradiation. The EQE measured was significantly higher than that of the previously reported H2NCNx–NiP hybrid system with an EQE of 0.37 ± 0.02 %,20 as well as the homogeneous RuP–NiP system with an EQE of 9.7 ± 1.2 %.15
The activity of NCNCNx for benzyl alcohol oxidation also compares well with systems reported for substrate oxidation with a sacrificial electron acceptor. Photocatalytic 4-MBA oxidation in the presence of a carbon nitride, under a high pressure of O2, resulted in 77% 4-MBAd formation with 99% selectivity, but the oxidation was carried out in fluorinated organic solvent to promote O2 solubility and at 100 °C.40 Another system with carbon nitride and O2 as a sacrificial acceptor reported 59% 4-MBA oxidation with 66% selectivity for 4-MBAd in water,41 but required 100 °C and strongly acidic conditions (pH 0).41 The NCNCNx provides a platform for selective benzyl alcohol oxidation, whereas substrate oxidation reactions on metal oxides such as TiO2 are limited by low selectivity due to formation of highly oxidizing holes and reactive radicals in water.45−48 The NCNCNx–NiP colloidal suspension system reported in this work combines selective substrate oxidation and H2 production in a single compartment, thereby eliminating the need for a sacrificial reagent, remaining functional under fully aqueous solution and at room temperature as well as ambient pressure for up to 24 h.
Analysis of Individual Components
A series of benzyl alcohols with different para-substituents were studied to probe the effect of substrate structure on photocatalytic performance (Table S3). Conversion yields observed did not show a direct correlation with respect to the electron-donating or -withdrawing nature of the substituents. Only with the strongest electron-withdrawing substituent tested, −CF3, was a significant reduction in conversion yield to 6.1 ± 2.0% observed. All of the benzyl alcohol derivatives underwent selective conversion to the aldehyde, except for with the strongest electron-donating group, −C(CH3)3, in which 64% selectivity for aldehyde formation was observed and further oxidation to 4-tert-butylbenzoic acid also occurred. The results indicate that alcohol oxidation is not the overall activity-limiting step in the redox cycle (see below). When 4-MBA (30 μmol) was replaced with methanol (30 μmol) under otherwise identical conditions (NCNCNx and NiP in KPi), no H2 was observed, in contrast to systems using an excess amount of aliphatic alcohols as a sacrificial reagent for photocatalytic proton reduction.
Replacing the phosphate with acetate buffer (pH 4.5, 0.1 M) did not alter the photocatalytic rate of H2 and aldehyde generation during the first few hours, indicating that the activity of the NCNCNx–NiP system is not significantly buffer dependent, although the photoactivity decayed faster in the acetate buffered system (Table S4). Increasing the phosphate concentration from 20 mM to 0.1 and 0.5 M led to an enhancement in initial activity from TOFNiP = 31.1 ± 3.1 to 76.3 ± 7.6 and 111.4 ± 11.1 h–1, respectively. However, after 24 h, the system performances equalized with a TONNiP of approximately 400 and a 4-MBA-to-4-MBAd conversion yield between 60% to 83% observed for the three systems (Tables S1 and S4). Further experiments in 20 mM KPi solution with 80 mM KCl or 80 mM K2SO4 showed a slight increase in the photocatalytic rate of H2 production with a TOFNiP of 53.9 ± 5.9 and TOFNiP of 49.2 ± 4.9 h−1, respectively (Figure S9). These results suggest that the enhanced photocatalytic activity is partially due to the ionic strength of the buffer used but also the significant increase in phosphate ion concentration that can potentially act as a proton relay, improving the efficiency of proton transfer to the molecular catalyst.49,50
The efficiencies of the individual half-reactions of the photocatalytic system were also examined separately in sacrificial schemes (Figure 3). Solar-light-driven H2 production was tested with NCNCNx (5 mg) and NiP (50 nmol) in aqueous EDTA electron donor solution at pH 4.5, in the absence of 4-MBA. A TOFNiP of 53.2 ± 5.3 h–1 and a TONNiP of 424.18 ± 42.4 were recorded in EDTA solution, similar to the 4-MBA system, suggesting that the NCNCNx–NiP hybrid system is not limited by the electron donation of 4-MBA in the longer time regime (Table S4).
Figure 3.
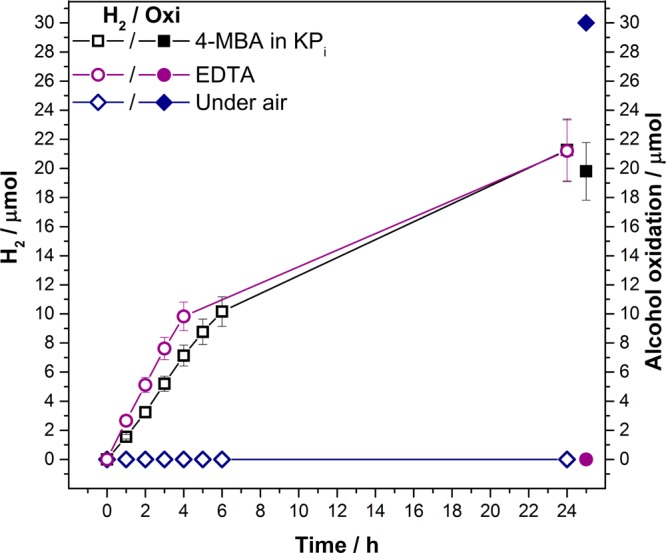
Photocatalytic H2 production and 4-MBA oxidation in the presence of NCNCNx (5 mg), 4-MBA (30 μmol), and NiP (50 nmol) in an aqueous KPi solution (0.02 M, pH 4.5, 3 mL). Sacrificial H2 production with NCNCNx (5 mg) and NiP (50 nmol) in EDTA (0.1 M, pH 4.5, 3 mL) in the absence of 4-MBA. Sacrificial alcohol oxidation with NCNCNx (5 mg) and 4-MBA (30 μmol) in an aqueous KPi solution (0.02 M, pH 4.5, 3 mL) under air, as a sacrificial electron acceptor, in the absence of NiP. All samples were irradiated with 1 sun irradiation (100 mW cm–2, AM 1.5G, 25 °C). The pair of hollow and filled symbols of the same shape and color corresponds to H2 and aldehyde production, respectively, under the specified reaction conditions.
The ability of the system to carry out alcohol oxidation was investigated by replacing the molecular catalyst NiP with air (O2) as an electron acceptor. Solar-light driven alcohol oxidation with NCNCNx (5 mg) and 4-MBA (30 μmol) resulted in quantitative alcohol oxidation with 70% selectivity toward 4-methylbenzoic acid formation after 24 h, with respect to the products detected by 1H NMR (Table S4). This implies that the degree of oxidation of alcohols with this material can be tuned by varying the nature of the electron acceptor and demonstrates an advantage of using NiP over the sacrificial acceptor O2. The ability of O2 to act as an electron acceptor in this system also highlights a key advantage of substrate oxidation over full water splitting in one-pot systems, where the O2 formed would compete with and inhibit proton reduction.
The photocatalytic activity of NCNCNx–NiP was compared with conventional unfunctionalized melon-type carbon nitride, H2NCNx (Figures 4 and S10). The H2 and aldehyde production activity of NCNCNx–NiP was one order of magnitude greater than H2NCNx–NiP under the same experimental conditions (TOFNiP= 11.7 ± 1.2 h–1 and TONNiP= 49.79 ± 14.2; Table S5), indicating that surface functionalization of the carbon nitride is vital. The enhanced photocatalytic activity of NCNCNx compared to H2NCNx has been previously attributed to the improved interaction and charge transfer via the cyanamide moieties of the NCNCNx and a Pt catalyst.42 We demonstrate here that this enhanced activity with NCNCNx is also observed when the Pt was replaced with the molecular NiP catalyst.42
Figure 4.
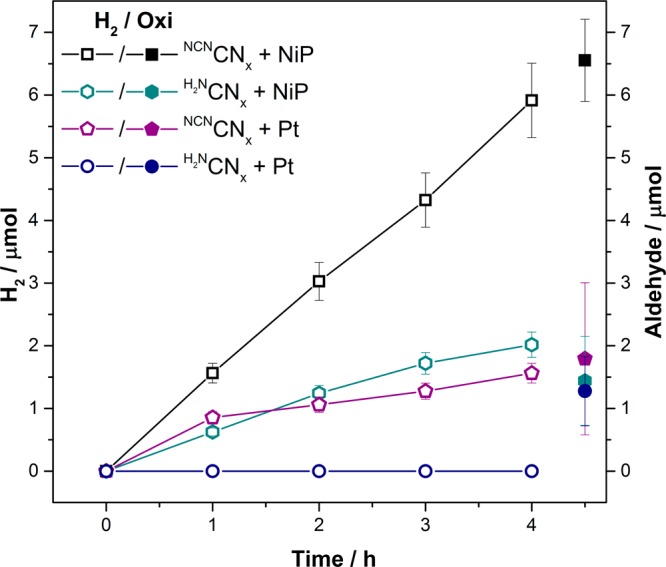
Photocatalytic H2 and aldehyde production with NCNCNx or H2NCNx (5 mg), in the presence of 4-MBA (30 μmol) and NiP (50 nmol) in KPi (0.02 M, pH 4.5, 3 mL) were conducted under 1 sun irradiation (100 mW cm–2, AM 1.5G, 25 °C). Photocatalytic experiments in the presence of NCNCNx or H2NCNx (5 mg), 4-MBA (30 μmol), and H2PtCl6 (10 μL, 8 wt %) in KPi (0.02 M, pH 4.5, 3 mL) were also carried out. The pair of hollow and filled symbols of the same shape and color corresponds to H2 and aldehyde production, respectively, under the specified reaction conditions.
The in situ photodeposition of H2PtCl6 to platinize NCNCNx or H2NCNx was carried out following an optimized Pt loading procedure,42 resulting in the formation of NCNCNx–Pt and H2NCNx–Pt systems for photo-H2 generation.37 These systems were then studied for simultaneous alcohol oxidation and proton reduction (Figure 4, Table S5) in the absence of NiP under otherwise identical conditions. NCNCNx–Pt showed one-third of the photocatalytic activity observed using NCNCNx–NiP toward H2 production during the initial 4 h of irradiation, whereas H2NCNx–Pt was not active for H2 production at all. Both NCNCNx–Pt and H2NCNx–Pt showed only approximately 5% 4-MBA oxidation to aldehyde after 4 h of irradiation. The initially low photocatalytic activity of Pt could be attributed to an induction period associated with the photodeposition of the active catalyst.37 Even after 24 h of irradiation, the overall photocatalytic activity of NCNCNx–NiP (H2:4-MBAd = 21:20 μmol) was still better than that with NCNCNx–Pt (14:12 μmol), representing the clear advantage of the molecular Ni catalysts over Pt in this system. A similar trend was previously reported with a sacrificial system containing carbon quantum dots and NiP.21
A control experiment was carried out to confirm that Pt does not interfere with the 4-MBAd formed by the oxidation of 4-MBA. In sacrificial EDTA solution, NCNCNx–Pt and 4-MBAd (30 μmol) were combined in the absence of NiP (Table S5). After 24 h of irradiation, 18% of the 4-MBAd was oxidized to carboxylic acid, whereas 4-MBA was not detected by 1H NMR spectroscopy, indicating that Pt is not able to reduce 4-MBAd to 4-MBA, but may also act as unselective catalyst for 4-MBAd oxidation.
Photoactivity and Stability Limiting Component
To gain better insights into the rate-limiting factors of the NCNCNx–NiP hybrid system, the amounts of NiP, 4-MBA, and light intensity were varied. Doubling the amount of NiP from 50 to 100 nmol per 5 mg of NCNCNx or the amount of 4-MBA from 30 to 60 μmol did not result in a significant change in photoactivity (Table S1). However, reducing the light intensity by 50% and 80% using neutral density filters resulted in an approximately 40% and 85% reduction in photocatalytic activity for H2 and 4-MBAd production after 1 h of irradiation (Table S6, Figure S11). The activity is therefore proportional and linearly dependent on the light intensity, which is also consistent with the reduced photoactivity of NCNCNx–NiP when filtering the UV light from the solar spectrum (see above).
The stability-limiting component of the system was identified by reactivation of a NCNCNx-NiP system after 25 h of irradiation (Table S7). Readdition of 4-MBA (30 μmol) did not reactivate the system, whereas addition of fresh NiP (50 nmol) restored solar-H2 production and enhanced the conversion yield for selective 4-MBA oxidation to 83.5 ± 9.7% (Figure S12). Therefore, the selectivity toward aldehyde formation is preserved despite the longer irradiation period (50 h), while the lifetime of the system is limited by the degradation of NiP.15,20,21
X-ray photoelectron spectroscopy (XPS) and Fourier Transform Infrared Spectroscopy (FTIR) characterization of the NCNCNx before and after irradiation for 24 h in the presence and absence of NiP showed negligible differences (Figures S13–S15). Thus, NCNCNx showed long-term activity and stability for simultaneous alcohol oxidation and H2 production. The XPS spectra also support that NiP is not physically adsorbed on the NCNCNx surface during the period of irradiation and acts as a homogeneous catalyst in the system.
Mechanistic Interpretation
The color of the colloidal suspension prepared with NCNCNx, NiP, and 4-MBA changed almost instantaneously from pale yellow to intense blue upon irradiation, and this color persisted during 24 h of continuous irradiation (Figure S16, inset). When NiP was replaced with a sacrificial electron acceptor, ammonium persulfate, or air (O2), the color of the suspension became yellow upon irradiation, whereas upon the substitution of 4-MBA with sacrificial electron donor EDTA, the color remained blue. The observed color change is therefore attributed to photogenerated electrons in the photosensitizer.
In order to characterize the color change upon irradiation of the NCNCNx, UV–vis absorption spectra were recorded for NCNCNx (5 mg) in KPi (3 mL, 0.02 M, pH 4.5) with and without 4-MBA (30 μmol) after simulated solar light irradiation (AM 1.5G) for 30 min. The suspension prepared with 4-MBA turned intense blue immediately upon irradiation, and consequently a broad absorption peak at λ = 650 nm was obtained (Figure S16), a direct spectroscopic observation of the trapped electrons in the NCNCNx. In the absence of 4-MBA no color change was observed, indicating that 4-MBA reductively quenches the photoexcited state of the NCNCNx.
NiP (50 nmol) was added to the blue pre-irradiated suspension using an airtight syringe under N2, and the peak at λ = 650 nm was monitored every 15 min for 1 h, while the suspension was stirred in the dark (Figure 5a). A significant decay in the absorption peak was observed in the first 15 min (τ1/2 = 10 min), indicating transfer of the trapped electrons from NCNCNx to NiP. After 30 min, the broad peak at λ = 650 nm disappeared. The photoexcited electrons were thus collected by NiP, and the color of the suspension became yellow again. In a control experiment, a NiP-free KPi solution was added to another blue NCNCNx suspension pre-irradiated for 30 min and the solution remained blue with no significant change in the absorption spectra. These ultralong-lived “trapped electrons” can be stored in the dark for a prolonged period of time (>30 min) in NCNCNx and transferred to NiP when available.51
Figure 5.
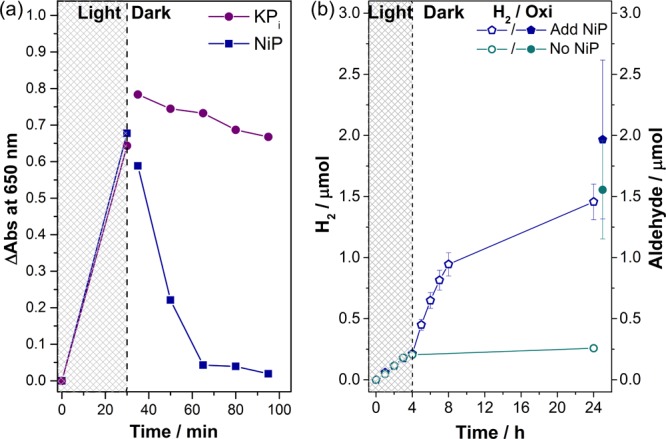
(a) Spectrophotometry of the appearance (during irradiation) and disappearance (upon addition of NiP in the dark) of photoexcited electrons in NCNCNx at λ = 650 nm. A suspension with NCNCNx (5 mg) and 4-MBA (30 μmol) in KPi (0.02 M, pH 4.5, 3 mL) was irradiated (AM 1.5G) for 30 min, followed by addition of NiP (50 nmol, 400 μL) or KPi (400 μL). (b) Two photoreactors were prepared with NCNCNx (5 mg) and 4-MBA (30 μmol) in the absence of NiP in an aqueous KPi solution (0.02 M, pH 4.5, 3 mL) and irradiated with 1 sun irradiation (AM 1.5G, 25 °C). Both of these photoreactors were taken into the dark after 4 h of irradiation, while NiP (50 nmol) was added to one of them. The pair of hollow and filled symbols of the same shape and color corresponds to H2 and aldehyde production, respectively, under the specified reaction conditions.
To demonstrate quantitative H2 evolution in the dark phase, two photoreactors were prepared using NCNCNx (5 mg) and 4-MBA (30 μmol) in KPi in the absence NiP and were irradiated under simulated solar light (AM 1.5G) for 4 h (Figure 5b). Then, both systems were protected from light, and NiP (50 nmol) was added to only one of the photoreactors; the H2 production was monitored for both (Table S8). The same effect was observed, the photoreactor kept in the dark without NiP did not show any H2 production in the 20 h dark phase, whereas the vial containing NiP showed a substantially increased H2 production. The amount of aldehyde produced after 24 h was the same as after 4 h for both of the systems (independent of the NiP) indicating that alcohol oxidation ceased as soon as the vials were removed from light. These results are in agreement with efficient alcohol oxidation by NCNCNx, using the photogenerated holes, even in the absence of an electron acceptor (Figure 2). Thus, NCNCNx can be charged during the light phase by 4-MBA oxidation and the electrons can be released in the absence of light to carry out proton reduction to H2, which enables the temporary decoupling of the oxidative and reductive half-reactions.52
Transient Absorption Spectroscopy
To gain a better understanding of the kinetics behind electron transfer in this system, transient absorption spectroscopy (TAS) measurements were carried out. As detailed below, TAS investigations confirmed that electron transfer to NiP is the rate-limiting step for the photocatalytic system (Figure 6). Titration experiments carried out by increasing the NiP concentration did not quench the excited state of the photosensitizer, NCNCNx*, eliminating the possibility for an oxidative quenching mechanism (Figures S17 and S18).
Figure 6.
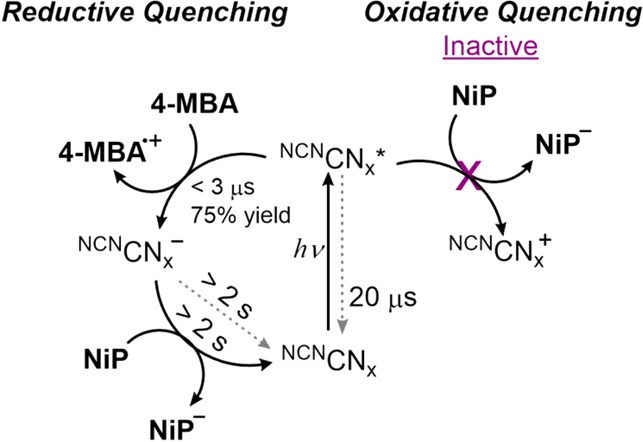
Summary of mechanism and kinetic rates from TAS measurements with suspensions of NCNCNx (1–5 mg mL–1), NiP (up to 24 μM), and 4-MBA (10 mM) in an aqueous KPi solution (0.02 M, pH 4.5, 25 °C). Representative lifetimes, calculated using 3 μs as the initial time, are indicated near reaction arrows. Recombination reactions are indicated by dashed gray arrows.
Photoexcitation of NCNCNx produced a photoinduced absorption signal in the range of 550–1000 nm (Figure S19), which matches that observed upon irradiation of NCNCNx with 4-MBA (Figure S16). This spectral feature did not show any evolution over the decay time nor was the shape sensitive to additives, suggesting a single excited state species absorbing over this spectral range. Kinetics were probed at 750 nm in order to reduce the influence of the strong photoluminescence at wavelengths near 550 nm. The intrinsic decay of NCNCNx (Figure S20) yields a representative lifetime (τ1/2) of 21.6 μs, which is the time where 50% of the initial (at 3 μs) excited state population has decayed by half. The tens of microsecond time scale of the decay was quite comparable to those found in metal oxide photocatalysts such as hematite53 and TiO2.54 Furthermore, the same type of dispersive power law decay dynamics is found in the carbon nitrides studied as that for the metal oxides and has been attributed to bimolecular (i.e., electron–hole) recombination. The exponent of the power law is significantly smaller than unity (∼0.35), indicating that charge trapping/detrapping may play a considerable role in excited state dynamics of NCNCNx, as also indicated by the slow time scale of this recombination process.
We next performed 4-MBA titration experiments to elucidate the kinetics of 4-MBA oxidation. As the concentration of 4-MBA increases, a long-lived component of the decay becomes dominant (Figure S21), assigned to long-lived electrons formed following hole scavenging by 4-MBA. For 10 mM 4-MBA, these long-lived electrons account for about 75% of the initial amplitude (Figure 7). Assuming a bimolecular dynamic quenching process, analysis of the concentration-dependent yield and rate of formation of the long-lived electrons (see Supporting Information for details) provides a reaction rate constant of 1.43 × 107 M–1 s–1 (Figures S22 and S23). The analysis also informs us of a slow background reaction rate k0 of 8200 s–1, which we tentatively attribute to hole scavenging by small amounts of impurities or H2O and explains the nonzero absorption seen in the millisecond regime in the absence of 4-MBA. However, the shape of the decay is more indicative of quenching occurring on time scales faster than the instrument response of 3 μs, perhaps by static quenching. Specifically, the decrease in amplitude seen before 1 ms in the presence of 10 mM 4-MBA shows a τ1/2 of roughly 20 μs; that is, indistinguishable fast phase τ1/2 values are observed with or without 4-MBA. The yield of long-lived electrons plateaus for a higher concentration of 4-MBA, a possible indication that deeply trapped electrons which decay with microsecond dynamics are unable to participate in the charge transfer reaction.55 The NCNCNx/4-MBA reaction thus appears to take place faster than the ∼20 μs relaxation kinetics of NCNCNx. Upon comparison to a previously reported photocatalytic system using ascorbic acid (AA) as the sacrificial electron donor, the NCNCNx/4-MBA reaction time scale is faster than that of the reduction of an oxidized RuIII dye (RuP-TiO2/AA) and maybe comparable to the reduction time scale of the excited state of the Ru dye (RuP/AA).15
Figure 7.
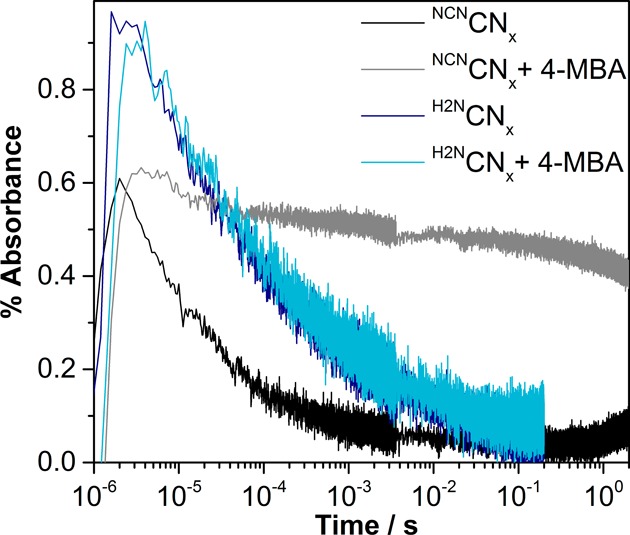
Transient decays probed at λ = 750 nm of NCNCNx (1.2 mg mL–1) and H2NCNx (1.2 mg mL–1) suspensions in aqueous KPi solution (0.02 M, pH 4.5, 25 °C) following λ = 355 nm excitation. Traces obtained following the addition of 10 mM 4-MBA are also shown.
The same spectral feature is seen with and without the addition of 4-MBA, and the oxidation of 4-MBA consumes holes, which confirms the assignment of the blue-colored solution deriving from photogenerated electrons in the NCNCNx (Figure S16). The same observation was also made with MeOH as a hole scavenger (Figure S19). An analogous increase of the photogenerated electron lifetime in TiO2 has been observed upon the addition of MeOH as a hole scavenger.56 Therefore, we may directly monitor the population of electrons in the NCNCNx. This further implies that formation of separated charge carriers in NCNCNx occurs within 3 μs.
We attempted to quantify the rate of the electron transfer reaction from NCNCNx to NiP, but the time scale of this reaction was too long for direct observation by TAS. Both in the absence or presence of 4-MBA, addition of NiP did not affect the relaxation kinetics of NCNCNx (Figures S17 and S18). The upper time limit of our measurements was ∼2 s due to settling of the heterogeneous NCNCNx dispersion. We thus conclude that the lifetime of long-lived electrons in NCNCNx is longer than 2 s, consistent with the persistent blue color observed upon irradiation of NCNCNx/4-MBA (see above). Similarly, the reaction lifetime for NCNCNx electron transfer to NiP is also longer than 2 s, in line with the disappearance of the blue color that takes place over 30 min upon NiP addition (Figure 5a). By taking into account the NiP concentration, the upper bound of the bimolecular rate constant is thus set to 2 × 105 M–1 s–1.
We have compared the TAS results of NCNCNx and the unfunctionalized H2NCNx to gain insights into the significant differences in efficiency. Surprisingly, for the studied time scale, we observed slower decay kinetics (τ1/2 = 60.2 μs) for H2NCNx dispersions compared to the photocatalytically more efficient NCNCNx (Figures 7 and S24). Instead of increasing the lifetime of reactive charges, the increased efficiency of cyanamide-functionalized carbon nitride seems to stem from higher hole transfer reactivity toward the oxidation reaction. In particular, we have observed no changes in the decay kinetics of H2NCNx upon the addition of 4-MBA, supporting an inefficient alcohol oxidation step also observed in the bulk photocatalysis experiments. Negligible change in the Fermi levels rules out the change in band energies as the reason for the enhanced photocatalytic activity for NCNCNx, eliminating the possibility of differences in driving force from the electron transfer side.42 Rather, it is plausible that oxidation of 4-MBA by NCNCNx is aided by more favorable hydrogen bonding interactions between the anionic cyanamide moiety on the surface of the material and the benzyl alcohol, in addition to ion–dipole forces originating from the cyanamide groups, increasing the preassociation and effectiveness of static quenching and/or effectively increasing the basicity of the carbon nitride and the driving force for reaction.57 These observations highlight the necessary consideration of both charge photogeneration and separation as well as complex electron–proton transfer processes in photocatalyst reactivity.
It is well-known that proton reduction by NiP occurs via successive multiple electron and proton transfers.44,58,59 By taking this and the results obtained from the characterization of the system into account, it is suggested that the alcohol oxidation also occurs via multiple electron and proton transfer steps.60,61 A stepwise mechanism is proposed in which irradiation of NCNCNx results in light absorption and charge separation. The photogenerated hole in the material is first reductively quenched by the 4-MBA for selective alcohol oxidation. The photoexcited electrons accumulated in the NCNCNx are then transferred to the NiP, which carries out reduction of aqueous protons. The rate of photogenerated electron transfer from NCNCNx to NiP is slower than the rate of reductive quenching, giving rise to an intense blue color. Studies are ongoing to develop a better understanding of the full mechanism of this coupled alcohol oxidation and proton reduction hybrid system.
Conclusions
In summary, we report a closed redox system for simultaneous photocatalytic alcohol oxidation and proton reduction, utilizing the cyanamide functionalized carbon nitride, NCNCNx, and a hydrogenase-inspired molecular H2 evolution catalyst, NiP. The hybrid system consists of earth abundant materials and functions in a purely aqueous solution and at room temperature in the absence of sacrificial reagents. Under optimized conditions, a remarkable EQE of 15% (λ = 360 ± 10 nm), a H2 production rate (TOFNiP) of 76.3 ± 7.6 h–1, and a TONNiP of 408.8 ± 40.9 were achieved, with a 4-MBA conversion yield of 83.0 ± 8.3% with quantitative selectivity for 4-MBAd.
This colloidal NCNCNx–NiP system is straightforward to prepare and provides a novel platform to produce a valuable organic chemical and a H2 fuel simultaneously, and the products are also spontaneously separated in the solution and gaseous phase, respectively. The degree of substrate oxidation in the system can be controlled by tuning the strength and selectivity of the electron acceptor. The suspension system also benefits from functioning in a stirred bulk solution where radial diffusion to the dispersed carbon nitride occurs, whereas conventional PEC cells can suffer from diffusion limitations at the electrode surface. The lifetime of this hybrid system is limited by the stability of the molecular H2 evolution catalyst NiP, whereas the NCNCNx maintained its activity for at least 50 h of irradiation, providing a significant advantage over similar systems based on photodegrading molecular dyes.15,25,28 Taken together, the results demonstrate that identification and design of photostable molecular catalysts could enhance the long-term photocatalytic activity of the hybrid system. The reported system also provides a potential pathway for extension into a broad range of other useful substrate oxidation reactions, as well as applications into CO2 and other reduction processes,55 by altering the molecular catalyst used.
TAS results confirmed that the cyanamide groups on the surface of the material play a vital role in providing enhanced catalytic activity toward selective substrate oxidation, which outperforms possible charge recombination reactions. The material can accumulate ultralong-lived “trapped electrons” during irradiation and effectively transfer these charge carriers to a catalyst in a dark phase. Work is currently in progress to further characterize the ultralong lived trapped electrons and to develop viable systems and devices that are not limited by the diurnal availability of sunlight by temporarily and spatially decoupling light absorption and oxidation from fuel generation.
Experimental Section
Materials and Synthesis
All reagents for the synthetic part of the work were purchased from commercial suppliers and used directly without any further purification. The buffer solutions were prepared using analytical grade reagents and titrated to the desired pH with a pH meter (Mettler Toledo; SevenEasy). H2NCNx was prepared by heating melamine to 550 °C for 12 h under Ar following a published procedure.20 The yellow solid obtained was then thoroughly ground using a pestle and mortar prior to further analysis and applications. NCNCNx was prepared from ground H2NCNx and KSCN (weight ratio of 1:2; dried overnight at 140 °C under vacuum) and heated at 400 °C for 1 h and then at 500 °C for 30 min under Ar as previously reported.42 After cooling to room temperature, the residual KSCN was removed by washing with water, and the product was dried under vacuum at 60 °C.42 Platinized H2NCNx and NCNCNx (containing 8 wt % Pt) were prepared following a published procedure,42,62 by in situ photodeposition from H2PtCl6 (10 μL aqueous solution corresponding to 8 wt % Pt loading) in a phosphate buffer (KPi) solution (3 mL, 0.02 M, pH 4.5) containing 4-MBA. NiP was synthesized and characterized as previously described.15
Characterization of NCNCNx
Samples of NCNCNx were pressed onto indium foil for XPS, and the spectra were collected on an Axis Ultra (Kratos Analytical, Manchester) XPS instrument with charge neutralization. The spectra were processed using the software CasaXPS 2.3.16 and referenced with the adventitious carbon 1s peak at 284.80 eV. Binding energies were compared with the NIST Standard Reference Database 30 unless otherwise specified. Attenuated total reflectance FTIR spectra were collected with a PerkinElmer UATR Two spectrometer equipped with a diamond crystal. UV−vis difference spectra of NCNCNx were recorded on an Edinburgh Instruments FS5 spectrofluorometer equipped with an integrating sphere. Synchronous scans (λex = λem) were run for samples after irradiating for 30 min with a solar light simulator equipped with an AM 1.5G filter. The absorption spectra were generated by comparing samples before and after irradiation using Fluoracle software supplied with the FS5 instrument.
Photocatalytic Experiments
The simultaneous 4-MBA oxidation and proton reduction photocatalytic experiments were performed and studied as follows: Polymeric NCNCNx (5.0 mg) was placed in a borosilicate glass photoreactor (total volume 7.74 mL) equipped with a magnetic stir bar. A KPi solution containing 4-MBA and NiP was prepared (total volume 3 mL) and added to the photoreactor, which was then tightly sealed with a rubber septum. The light-protected suspension was sonicated for 10 min at room temperature. The vials were then purged for 15 min with N2 containing 2% CH4, an internal gas chromatograph (GC) standard, before irradiation.
All photocatalytic experiments were carried out using a Newport Oriel Solar Light Simulator (100 mW cm–2) equipped with an AM 1.5G filter. IR irradiation was removed from all the experiments using a water filter, and UV irradiation was eliminated using a 400 nm cutoff filter (UQG), whenever specified. The photoreactor was stirred and kept at a constant temperature (25 °C) with continuous water circulation through a water-jacketed reservoir during the light experiments.
Quantification of Products
The amount of H2 was quantified using an Agilent 7890A Series GC equipped with a 5 Å molecular sieve column against an internal CH4 standard. The GC oven was kept at 45 °C, and N2 was used as the carrier gas at a flow rate of approximately 3 mL min–1. Samples of the headspace gas (approximately 20 μL) were taken from the photoreactor for analysis using an airtight Hamilton syringe at regular time intervals.
The amounts of alcohol and aldehyde were quantified by nuclear magnetic resonance (NMR) spectroscopy as follows: The photoreactor was removed from the light source, the suspension was centrifuged (10 min at 10 000 rpm) to remove NCNCNx, and the supernatant solution was extracted with CH2Cl2 (3 × 4 mL), dried over MgSO4, filtered, and concentrated to dryness under reduced pressure. The residue was then characterized by using 1H NMR spectroscopy on a Bruker DPX 400 spectrometer at 298 K. Comparison of the methyl peaks of 4-MBA and 4-MBAd, as well as the distinctive aldehyde (COH) peak, allowed for the determination of a quantitative ratio of the starting material to product (Figure S4).
The workup procedure was confirmed to leave the ratio of starting material to product unaffected. Three vials containing different 4-MBA/4-MBAd (1:2, 1:1, and 2:1) ratios were prepared following the described procedure for the photocatalytic experiments (except the irradiation step), and the initial 4-MBA/4-MBAd ratios were preserved after working up the solutions as described above.
Determination of External Photon to H2 Quantum Efficiency (EQE)
The EQE was determined by using a solar light simulator (LOT LSN 254) equipped with a (LOT MSH 300) monochromator to irradiate the samples at λ = 360 ± 10 nm (accurate to a fwhm of 10 nm) with a light intensity (I) of I = 4.43 mW cm–2, which was measured on a power meter (ILT 1400, International Light Technologies). The EQE was calculated as follows:
where nH2 is the moles of photogenerated H2, NA is Avogadro’s constant, h is the Planck constant, c is the speed of light, tirr is the irradiation time, and A is the cross-sectional area of irradiation.
Treatment of Analytical Data
To ensure reproducibility, the solar light simulator and GC were calibrated regularly. Throughout the photocatalytic experiments, no gradual decrease in the amount of CH4 (internal GC standard) was observed, indicating that there was no significant leakage of CH4 in the vial. All analytical measurements were carried out in triplicates, and data are reported as mean value ± standard error (σ). A minimum σ of 10% was assumed in all cases.
Transient Absorption Spectroscopy
Microsecond to second transient absorption decays were acquired in diffuse reflectance mode.63 The experimental setup used an Nd:YAG laser (OPOTEK Opolette 355 II, 7 ns pulse width) as the excitation source. 355 nm light was generated from the third harmonic of the laser and transmitted to the sample through a light guide with a diameter of 0.5 cm to photoexcite the NCNCNx. Typical excitation power densities of 460 μJ cm–2 and laser repetition rates of 1.1 Hz were used. As the changes of reflectance observed are low (<1%), the transient signal is taken to be directly proportional to the concentration of excited state species.64 The probe light source was a 100 W Bentham IL1 quartz halogen lamp. Long pass filters (Comar Instruments) between the lamp and sample were used to minimize the short wavelength irradiation of the sample. A 5 cm path length cuvette filled with DI water was also placed in the beam path as an IR filter. Diffuse reflectance from the sample was collected by a 2″ diameter, 2″ focal length lens and relayed to a monochromator (Oriel Cornerstone 130) to select the probe wavelength. An additional high pass filter was positioned in front of the monochromator to reduce laser scatter. Time-resolved intensity data were collected with a Si photodiode (Hamamatsu S3071). Data at times faster than 3.6 ms were recorded by an oscilloscope (Tektronics DPO3012) after passing through an amplifier box (Costronics), while data slower than 3.6 ms were simultaneously recorded on a National Instrument DAQ card (NI USB-6251). Each kinetic trace was obtained from an average of 32–64 laser pulses. 1–5 mg mL–1 of the NCNCNx was dispersed in 20 mM KPi buffer adjusted to pH = 4.5. Samples were stirred prior to the start of each kinetic acquisition. Acquisitions were triggered by a photodiode (Thorlabs DET10A) exposed to laser scatter. Data were acquired and processed using home-built software written in the Labview environment.
The experimental setup and photoluminescence of the sample limit analyses to time scales longer than about 3 μs. From the shape of the decay, it is clear that some excited state decay occurs before 3 μs. As such, while the t50% of the NCNCNx decay is used for further kinetic analysis, a shorter value would be obtained if we could follow the entirety of the decay immediately following photoexcitation. One can thus treat the τ1/2 reported here as upper bounds.
Acknowledgments
This work was supported by the Christian Doppler Research Association (Austrian Federal Ministry of Science, Research, and Economy and the National Foundation for Research, Technology and Development) and the OMV Group (to E.R.), an Oppenheimer PhD scholarship (to B.C.M.M.), a Marie Curie Postdoctoral Fellowship (GAN 624997 to C.A.C.), a FRQNT Postdoctoral Fellowship (to R.G.), and an ERC Starting Grant (B. V. L., Grant No. 639233). We thank Dr. Mitsuharu Konuma for performing the XPS measurements, Mr. Timothy E. Rosser for help with spectrofluorometer measurements, and Mr. David Wakerley for help with 3D drawings.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b04325.
Supporting tables and figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fujishima A.; Honda K. Nature 1972, 238, 37–38. 10.1038/238037a0. [DOI] [PubMed] [Google Scholar]
- Lai Y.-H.; Palm D. W.; Reisner E. Adv. Energy Mater. 2015, 5, 1501668. 10.1002/aenm.201501668. [DOI] [Google Scholar]
- Luo J.; Im J.-H.; Mayer M. T.; Schreier M.; Nazeeruddin M. K.; Park N.-G.; Tilley S. D.; Fan H. J.; Grätzel M. Science 2014, 345, 1593–1596. 10.1126/science.1258307. [DOI] [PubMed] [Google Scholar]
- Maeda K.; Higashi M.; Lu D.; Abe R.; Domen K. J. Am. Chem. Soc. 2010, 132, 5858–5868. 10.1021/ja1009025. [DOI] [PubMed] [Google Scholar]
- Joya K. S.; Joya Y. F.; Ocakoglu K.; van de Krol R. Angew. Chem. Int. Ed. 2013, 52, 10426–10437. 10.1002/anie.201300136. [DOI] [PubMed] [Google Scholar]
- Duan L.; Bozoglian F.; Mandal S.; Stewart B.; Privalov T.; Llobet A.; Sun L. Nat. Chem. 2012, 4, 418–423. 10.1038/nchem.1301. [DOI] [PubMed] [Google Scholar]
- Eckenhoff W. T.; Eisenberg R. Dalton Trans. 2012, 41, 13004–13021. 10.1039/c2dt30823a. [DOI] [PubMed] [Google Scholar]
- Lubitz W.; Ogata H.; Rüdiger O.; Reijerse E. Chem. Rev. 2014, 114, 4081–4148. 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- Wood P. M. Biochem. J. 1988, 253, 287–289. 10.1042/bj2530287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farràs P.; Di Giovanni C.; Clifford J. N.; Palomares E.; Llobet A. Coord. Chem. Rev. 2015, 304–305, 202–208. 10.1016/j.ccr.2014.10.007. [DOI] [Google Scholar]
- Brink G.-J.; Arends I. W. C. E.; Sheldon R. A. Science 2000, 287, 1636–1639. 10.1126/science.287.5458.1636. [DOI] [PubMed] [Google Scholar]
- Palmisano G.; Augugliaro V.; Pagliaro M.; Palmisano L. Chem. Commun. 2007, 3425–3437. 10.1039/b700395c. [DOI] [PubMed] [Google Scholar]
- Palmisano G.; García-López E.; Marcí G.; Loddo V.; Yurdakal S.; Augugliaro V.; Palmisano L. Chem. Commun. 2010, 46, 7074–7089. 10.1039/c0cc02087g. [DOI] [PubMed] [Google Scholar]
- Fukuzumi S.; Kishi T.; Kotani H.; Lee Y.-M.; Nam W. Nat. Chem. 2011, 3, 38–41. 10.1038/nchem.905. [DOI] [PubMed] [Google Scholar]
- Gross M. A.; Reynal A.; Durrant J. R.; Reisner E. J. Am. Chem. Soc. 2014, 136, 356–366. 10.1021/ja410592d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakadamyali F.; Reisner E. Chem. Commun. 2011, 47, 1695–1697. 10.1039/c0cc04658b. [DOI] [PubMed] [Google Scholar]
- Han Z.; Qiu F.; Eisenberg R.; Holland P. L.; Krauss T. D. Science 2012, 338, 1321–1324. 10.1126/science.1227775. [DOI] [PubMed] [Google Scholar]
- Wilker M. B.; Shinopoulos K. E.; Brown K. A.; Mulder D. W.; King P. W.; Dukovic G. J. Am. Chem. Soc. 2014, 136, 4316–4324. 10.1021/ja413001p. [DOI] [PubMed] [Google Scholar]
- Lakadamyali F.; Kato M.; Muresan N. M.; Reisner E. Angew. Chem. Int. Ed. 2012, 51, 9381–9384. 10.1002/anie.201204180. [DOI] [PubMed] [Google Scholar]
- Caputo C. A.; Gross M. A.; Lau V. W.; Cavazza C.; Lotsch B. V.; Reisner E. Angew. Chem. Int. Ed. 2014, 53, 11538–11542. 10.1002/anie.201406811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale B. C. M.; Hutton G. A. M.; Caputo C. A.; Reisner E. J. Am. Chem. Soc. 2015, 137, 6018–6025. 10.1021/jacs.5b01650. [DOI] [PubMed] [Google Scholar]
- Ding Z.; Chen X.; Antonietti M.; Wang X. ChemSusChem 2011, 4, 274–281. [DOI] [PubMed] [Google Scholar]
- Fukuzumi S.; Mizuno T.; Ojiri T. Chem. - Eur. J. 2012, 18, 15794–15804. 10.1002/chem.201202041. [DOI] [PubMed] [Google Scholar]
- Su F.; Mathew S. C.; Möhlmann L.; Antonietti M.; Wang X.; Blechert S. Angew. Chem. Int. Ed. 2011, 50, 657–660. 10.1002/anie.201004365. [DOI] [PubMed] [Google Scholar]
- Kalita D.; Radaram B.; Brooks B.; Kannam P. P.; Zhao X. ChemCatChem 2011, 3, 571–573. 10.1002/cctc.201000335. [DOI] [Google Scholar]
- Zhou X.; Li F.; Li X.; Li H.; Wang Y.; Sun L. Dalton Trans. 2015, 44, 475–479. 10.1039/C4DT02945C. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Li J.; Zhang B.; Chong R.; Li R.; Yuan B.; Lu S.-M.; Li C. J. Catal. 2015, 332, 95–100. 10.1016/j.jcat.2015.08.029. [DOI] [Google Scholar]
- Singh W. M.; Pegram D.; Duan H.; Kalita D.; Simone P.; Emmert G. L.; Zhao X. Angew. Chem. Int. Ed. 2012, 51, 1653–1656. 10.1002/anie.201106494. [DOI] [PubMed] [Google Scholar]
- Song W.; Vannucci A. K.; Farnum B. H.; Lapides A. M.; Brennaman M. K.; Kalanyan B.; Alibabaei L.; Concepcion J. J.; Losego M. D.; Parsons G. N.; Meyer T. J. J. Am. Chem. Soc. 2014, 136, 9773–9779. 10.1021/ja505022f. [DOI] [PubMed] [Google Scholar]
- Harriman A. Eur. J. Inorg. Chem. 2014, 573–580. 10.1002/ejic.201301540. [DOI] [Google Scholar]
- Schneider J.; Bahnemann D. W. J. Phys. Chem. Lett. 2013, 4, 3479–3483. 10.1021/jz4018199. [DOI] [Google Scholar]
- Lotsch B. V.; Döblinger M.; Sehnert J.; Seyfarth L.; Senker J.; Oeckler O.; Schnick W. Chem. - Eur. J. 2007, 13, 4969–4980. 10.1002/chem.200601759. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wang X.; Antonietti M. Angew. Chem. Int. Ed. 2012, 51, 68–89. 10.1002/anie.201101182. [DOI] [PubMed] [Google Scholar]
- Martin D. J.; Reardon P. J. T.; Moniz S. J. A.; Tang J. J. Am. Chem. Soc. 2014, 136, 12568–12571. 10.1021/ja506386e. [DOI] [PubMed] [Google Scholar]
- Liu J.; Liu Y.; Liu N.; Han Y.; Zhang X.; Huang H.; Lifshitz Y.; Lee S.-T.; Zhong J.; Kang Z. Science 2015, 347, 970–974. 10.1126/science.aaa3145. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Lan Z.-A.; Lin L.; Lin S.; Wang X. Chem. Sci. 2016, 7, 3062–3066. 10.1039/C5SC04572J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Maeda K.; Thomas A.; Takanabe K.; Xin G.; Carlsson J. M.; Domen K.; Antonietti M. Nat. Mater. 2009, 8, 76–80. 10.1038/nmat2317. [DOI] [PubMed] [Google Scholar]
- Cao S.-W.; Liu X.-F.; Yuan Y.-P.; Zhang Z.-Y.; Fang J.; Loo S. C. J.; Barber J.; Sum T. C.; Xue C. Phys. Chem. Chem. Phys. 2013, 15, 18363–18366. 10.1039/c3cp53350f. [DOI] [PubMed] [Google Scholar]
- Song X.-W.; Wen H.-M.; Ma C.-B.; Cui H.-H.; Chen H.; Chen C.-N. RSC Adv. 2014, 4, 18853–18861. 10.1039/c4ra01413h. [DOI] [Google Scholar]
- Su F.; Mathew S. C.; Lipner G.; Fu X.; Antonietti M.; Blechert S.; Wang X. J. Am. Chem. Soc. 2010, 132, 16299–16301. 10.1021/ja102866p. [DOI] [PubMed] [Google Scholar]
- Long B.; Ding Z.; Wang X. ChemSusChem 2013, 6, 2074–2078. 10.1002/cssc.201300360. [DOI] [PubMed] [Google Scholar]
- Lau V. W.-h.; Moudrakovski I.; Botari T.; Weinburger S.; Mesch M. B.; Duppel V.; Senker J.; Blum V.; Lotsch B. V. Nat. Commun. 2016, 10.1038/ncomms12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M. L.; Stewart M. P.; Bullock R. M.; Rakowski DuBois M.; DuBois D. I. Science 2011, 333, 863–866. 10.1126/science.1205864. [DOI] [PubMed] [Google Scholar]
- Kilgore U. J.; Roberts J. A. S.; Pool D. H.; Appel A. M.; Stewart M. P.; Rakowski DuBois M.; Dougherty W. G.; Kassel W. S.; Bullock R. M.; DuBois D. L. J. Am. Chem. Soc. 2011, 133, 5861–5872. 10.1021/ja109755f. [DOI] [PubMed] [Google Scholar]
- Palmisano G.; Yurdakal S.; Augugliaro V.; Loddo V.; Palmisano L. Adv. Synth. Catal. 2007, 349, 964–970. 10.1002/adsc.200600435. [DOI] [Google Scholar]
- Yurdakal S.; Palmisano G.; Loddo V.; Augugliaro V.; Palmisano L. J. Am. Chem. Soc. 2008, 130, 1568–1569. 10.1021/ja709989e. [DOI] [PubMed] [Google Scholar]
- Yurdakal S.; Palmisano G.; Loddo V.; Alagöz O.; Augugliaro V.; Palmisano L. Green Chem. 2009, 11, 510–516. 10.1039/b819862d. [DOI] [Google Scholar]
- Yurdakal S.; Augugliaro V. RSC Adv. 2012, 2, 8375–8380. 10.1039/c2ra20960h. [DOI] [Google Scholar]
- Shinagawa T.; Takanabe K. J. Phys. Chem. C 2015, 119, 20453–20458. 10.1021/acs.jpcc.5b05295. [DOI] [PubMed] [Google Scholar]
- Liu G.; Wang T.; Zhang H.; Meng X.; Hao D.; Chang K.; Li P.; Kako T.; Ye J. Angew. Chem. Int. Ed. 2015, 54, 13561–13565. 10.1002/anie.201505802. [DOI] [PubMed] [Google Scholar]
- Rosser T. E.; Gross M. A.; Lai Y.-H.; Reisner E. Chem. Sci. 2016, 7, 4024–4035. 10.1039/C5SC04863J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symes M. D.; Cronin L. Nat. Chem. 2013, 5, 403–409. 10.1038/nchem.1621. [DOI] [PubMed] [Google Scholar]
- Barroso M.; Mesa C. A.; Pendlebury S. R.; Cowan A. J.; Hisatomi T.; Sivula K.; Grätzel M.; Klug D. R.; Durrant J. R. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 15640–15645. 10.1073/pnas.1118326109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan A. J.; Tang J.; Leng W.; Durrant J. R.; Klug D. R. J. Phys. Chem. C 2010, 114, 4208–4214. 10.1021/jp909993w. [DOI] [Google Scholar]
- Kuriki R.; Matsunaga H.; Nakashima T.; Wada K.; Yamakata A.; Ishitani O.; Maeda K. J. Am. Chem. Soc. 2016, 138, 5159–5170. 10.1021/jacs.6b01997. [DOI] [PubMed] [Google Scholar]
- Xiao-e L.; Green A. N. M.; Haque S. A.; Mills A.; Durrant J. R. J. Photochem. Photobiol., A 2004, 162, 253–259. 10.1016/j.nainr.2003.08.010. [DOI] [Google Scholar]
- Waidmann C. R.; Miller A. J. M.; Ng C.-W. A.; Scheuermann M. L.; Porter T. R.; Tronic T. A.; Mayer J. M. Energy Environ. Sci. 2012, 5, 7771–7780. 10.1039/c2ee03300c. [DOI] [Google Scholar]
- Wilson A. D.; Shoemaker R. K.; Miedaner A.; Muckerman J. T.; DuBois D. L.; Rakowski DuBois M. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6951–6956. 10.1073/pnas.0608928104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small Y. A.; DuBois D. L.; Fujita E.; Muckerman J. T. Energy Environ. Sci. 2011, 4, 3008–3020. 10.1039/c1ee01170g. [DOI] [Google Scholar]
- Higashimoto S.; Suetsugu N.; Azuma M.; Ohue H.; Sakata Y. J. Catal. 2010, 274, 76–83. 10.1016/j.jcat.2010.06.006. [DOI] [Google Scholar]
- Megerle U.; Wenninger M.; Kutta R.-J.; Lechner R.; König B.; Dick B.; Riedle E. Phys. Chem. Chem. Phys. 2011, 13, 8869–8880. 10.1039/c1cp20190e. [DOI] [PubMed] [Google Scholar]
- Maeda K.; Wang X.; Nishihara Y.; Lu D.; Antonietti M.; Domen K. J. Phys. Chem. C 2009, 113, 4940–4947. 10.1021/jp809119m. [DOI] [Google Scholar]
- Wilkinson F. J. Chem. Soc., Faraday Trans. 2 1986, 82, 2073–2081. 10.1039/f29868202073. [DOI] [Google Scholar]
- Kessler R. W.; Krabichler G.; Uhl S.; Oelkrug D.; Hagan W. P.; Hyslop J.; Wilkinson F. Opt. Acta 1983, 30, 1099–1111. 10.1080/713821340. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.