

Abstract
Interleukin-1β (IL-1β) plays a key role in autoinflammatory diseases, such as systemic juvenile idiopathic arthritis (sJIA) or cryopyrin-associated periodic syndrome (CAPS). Canakinumab, a human monoclonal anti-IL-1β antibody, was recently approved for human use under the brand name Ilaris®. Canakinumab does not cross-react with IL-1β from mouse, rat, rabbit, or macaques. The crystal structure of the canakinumab Fab bound to human IL-1β was determined in an attempt to rationalize the species specificity. The X-ray analysis reveals a complex surface epitope with an intricate network of well-ordered water molecules at the antibody-antigen interface. The canakinumab paratope is largely pre-organized, as demonstrated by the structure determination of the free Fab. Glu 64 of human IL-1β is a pivotal epitope residue explaining the exquisite species specificity of canakinumab. We identified marmoset as the only non-human primate species that carries Glu 64 in its IL-1β and demonstrates full cross-reactivity of canakinumab, thereby enabling toxicological studies in this species. As demonstrated by the X-ray structure of the complex with IL-1β, canakinumab binds IL-1β on the opposite side with respect to the IL-1RAcP binding site, and in an approximately orthogonal orientation with respect to IL-1RI. However, the antibody and IL-1RI binding sites slightly overlap and the VH region of canakinumab would sterically interfere with the D1 domain of IL-1RI, as shown by a structural overlay with the IL-1β:IL-1RI complex. Therefore, direct competition with IL-1RI for IL-1β binding is the molecular mechanism of neutralization by canakinumab, which is also confirmed by competition assays with recombinant IL-1RI and IL-1RII.
Keywords: canakinumab, cryopyrin-associated periodic syndrome, crystal structure, gouty arthritis, Interleukin-1β, mode of action, species specificity, systemic juvenile idiopathic arthritis, therapeutic antibody
Abbreviations
- Å
ångström (0.1 nm)
- CAPS
cryopyrin-associated periodic syndrome
- EMA
European Medicines Agency
- Fab
antigen-binding fragment
- FDA
US Food and Drug Administration
- IL-1β
interleukin-1β
- IL-1Ra
IL-1 receptor antagonist
- IL-1RAcP
IL-1 receptor accessory protein
- IL-1RI
type I IL-1 receptor
- IL-1RII
type II IL-1 receptor
- IL-6
interleukin-6
- KD
equilibrium dissociation constant
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PyMol
python-enhanced molecular graphics tool
- rmsd
root-mean-square deviation
- RU
resonance units
- sJIA
systemic juvenile idiopathic arthritis
- TIR
Toll-IL-1 receptor
- VH
variable domain of the heavy chain
- VL
variable domain of the light chain.
Introduction
Human IL-1β is a highly regulated, highly active pro-inflammatory cytokine that plays a key role in autoinflammatory and autoimmune diseases.1-3 IL-1β signaling requires the sequential assembly of a heterotrimeric complex comprising IL-1β and 2 transmembrane receptors, the type I IL-1 receptor (IL-1RI) and the IL-1 receptor accessory protein (IL-1RAcP). The formation of this complex brings the intracellular TIR (Toll-IL-1 receptor) domains of the 2 receptor chains in close vicinity, ultimately leading to activation of the transcription factor NF-κB and stress-activated protein kinases.4 The IL-1 receptor antagonist (IL-1Ra) and the decoy receptor IL-1 receptor type II are 2 important regulatory components of IL-1β signaling. All IL-1 family members, including IL-1β and IL-1Ra, consist of a single structural domain that exhibits the so-called β-trefoil fold, which is composed of 12 β-strands connected by loop regions.5,6 The extracellular portion of the IL-1RI comprises 3 immunoglobulin-like domains that wrap around the cytokine.7 Recently, the structures of the ternary complexes of IL-1β with IL-1RAcP and either IL-1RII or IL-1RI were elucidated, revealing a large binding interface between IL-1β and IL-1RI or IL-1RII, and a small surface of interaction with IL-1RAcP, facilitating interaction of both receptor chains.8,9 Neutralization of the IL-1β bioactivity by an antibody targeting this cytokine could in principle be achieved by interference with the binding of IL-1β to IL-1RI, or with the interaction between IL-1β and IL-1RAcP, or with the recruitment of IL-1RAcP into the IL-1β-IL-1RI complex.
Derived from genetically engineered mice carrying part of the human antibody repertoire, canakinumab (previously known as ACZ885) is a human IgG1/κ monoclonal antibody that binds and neutralizes human IL-1β with high potency and selectivity.10 Canakinumab has demonstrated efficacy and safety in pivotal clinical studies of patients suffering from cryopyrin-associated periodic syndrome (CAPS), systemic juvenile idiopathic arthritis (sJIA), and gouty arthritis.11-13 Canakinumab has received market authorization for the treatment of CAPS and sJIA in the US and EU, and for the treatment of gouty arthritis in a restricted patient group in the EU.
Like many other antibodies directed against human cytokines, canakinumab does not cross-react with rat, mouse, or rabbit IL-1β. Surprisingly, it does not bind to rhesus or cynomolgus monkey IL-1β, in spite of the very high conservation of the amino acid sequence (human IL-1β shares 96% sequence identity with the rhesus and cynomolgus monkey cytokines). However, we found that canakinumab fully cross-reacts with marmoset (Callithrix jacchus) IL-1β (also 96% sequence identity to human IL-1β), thereby enabling toxicological evaluation and preclinical development.
We present here the molecular basis for this very narrow species selectivity. We have determined the crystal structures of the canakinumab Fab in the free and IL-1β-bound states. Our X-ray analyses, corroborated by mutational and biochemical studies, show that Glu 64 of human IL-1β plays a central role in antibody-antigen recognition and reveal new structural details in comparison to a recently published structure.14 In contrast to previous inferences,14 we show that canakinumab binding to IL-1β largely obeys a lock-and-key type mechanism, with contributions by all complementarity-determining regions (CDRs) and without any large structural changes of the paratope. The canakinumab and IL-1RI binding surfaces on IL-1β overlap partially, resulting in direct competition for cytokine binding. Hence, the structural and biochemical data reported here provide a detailed understanding of the molecular interactions between canakinumab and its target antigen, and show how this antibody exerts its pharmacological effect by preventing IL-1β from binding to its receptor IL-1RI.
Results
X-ray analysis
Overall structure of the canakinumab Fab
The crystal structure of the free canakinumab Fab was determined to 2.0 Å resolution from a monoclinic crystal with 4 Fab molecules in the asymmetric unit (Figure S1). The final model includes the complete light chain (214 amino-acids) and residues 1 to 218–224 of the heavy chain, with the exception of residues 132 to 138 in one of the 4 heavy chains (Table S1). The light and heavy chain of canakinumab originates from the human Vκ 6–21 and the human VH 3–33 germline genes, respectively.15,16 The observed conformations of the 3 light-chain CDRs matched known canonical structures (L1:2A, L2:1 and L3:1, as defined by Al-Lazikani et al17). Likewise, the H-CDR1 and H-CDR2 loops formed the expected canonical structures (H1:1 and H2:3A). The H-CDR3 loop of canakinumab is relatively short (9 residues according to the Kabat18 definition, 7 according to Chothia's nomenclature19) and exhibits the typical bulged torso structure19 while its head adopts a twisted β-hairpin conformation with a type I′ β-turn at its apex (Figure S2).
Overall structure of the Fab complex
The structure of the canakinumab Fab complex with IL-1β was determined at 2.2 Å resolution from an orthorhombic crystal with one complex per asymmetric unit (Table S1). Canakinumab recognizes an extended, discontinuous epitope on human IL-1β (Fig. 1 and Fig. S3). The binding interface is remarkably flat, extensively hydrated, and very large. The shape complementarity statistic Sc20 is typical of other protein-antibody complexes (0.707). Upon association, 1972Å2 of combined solvent-accessible surface are buried at the protein-protein interface.
Figure 1.
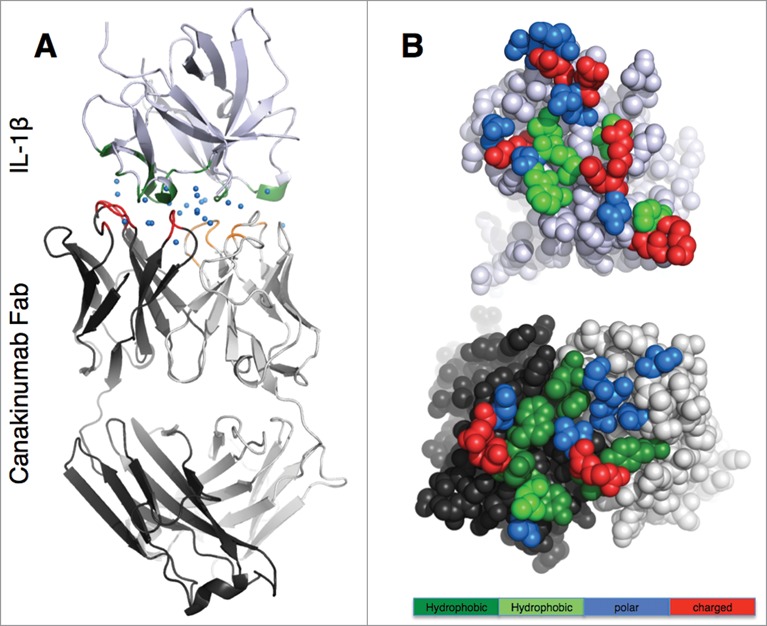
Overall view of the canakinumab Fab complex with human IL-1β (A). The complex is shown in cartoon representation with the Fab heavy and light chain in dark and light gray, respectively, and IL-1β in light violet. The paratope residues are colored red (heavy chain) and orange (light chain), and epitope residues are in green. Blue spheres represent water molecules at the antibody-antigen interface. (B). Open book view of the antibody (lower picture)- antigen (upper picture) interface. Epitope and paratope residues are colored according to their physico-chemical properties: large hydrophobic/aromatic (deep green), small hydrophobic (light green), polar (blue) and charged (red).
Canakinumab paratope
All six CDRs of canakinumab contribute to antigen binding with, in total, 19 Fab residues involved in direct intermolecular contacts (Table 1 and Fig. S4). The heavy chain contributes 2/3 of the total buried surface on the antibody. The amino acid composition of the antigen-combining site is a balanced mix of hydrophobic aromatic (25%), hydrophobic aliphatic (15%), polar (40%) and charged residues (20%). A structural overlay with the crystal structure of the free Fab shows that the canakinumab paratope is largely pre-organized with minimal induced-fit changes affecting the CDRs (Fig. 2A). In comparison to the free Fab structure, the overall root-mean-square deviation (rmsd) for all Cα atoms of the VH-VL domain (224 atoms) is only 0.60–0.80 Å.
Table 1.
Epitope and paratope residues
| IL-1 epitope |
Canakinumab paratope |
||
|---|---|---|---|
| Structural element | Residues in direct contact to canakinumab | Residues in direct contact to IL-1 | Structural element |
| β2-β3 β-hairpin | Val 19 | Tyr H53 | H-CDR2 |
| Met 20 | Trp H52, Tyr H53 | H-CDR2 | |
| Ser 21 | Trp H52, Tyr H53, Asn H57 | H-CDR2 | |
| Gly 22 | Trp H52 | H-CDR2 | |
| Pro 23 | Trp H52, Leu L94 | H-CDR2, L-CDR3 | |
| Lys 27 | Tyr H53, Asp H54, Asp H56 | H-CDR2 | |
| β3 to β4 loop | Gln 32 | Thr H28 | H-CDR1 |
| Gln 34 | Thr H28 | H-CDR1 | |
| Asp 35 | Thr H28, Val H31, Tyr H32 | H-CDR1 | |
| Glu 37 | Tyr H32, Leu H100, Arg H101 | H-CDR1, H-CDR3 | |
| Gln 38 | Val H31, Tyr H32, Leu H100, Arg H101 | H-CDR1, H-CDR3 | |
| Gln 39 | Arg H101 | H-CDR3 | |
| Val 41 | Arg H101 | H-CDR3 | |
| β5 to β6 loop | Glu 64 | Arg H101, Ser L32, Tyr L50, Ser L91 | H-CDR3, L-CDR1, L-CDR2, L-CDR3 |
| Lys 65 | Thr H102, Ser L91, Ser L92, Phe L96 | H-CDR3, L-CDR3 | |
| Asn 66 | Tyr H53, Asp H54, Ser L92, Ser L93 | H-CDR2, L-CDR3 | |
| β7 to β8 loop | Asp 86 | Ser L28 | L-CDR1 |
| Pro 87 | Ser L28 | L-CDR1 | |
| Lys 88 | Ser L28 | L-CDR1 | |
| β10 to β11 loop | Asn 129 | Tyr H53, Asp H54 | H-CDR2 |
Figure 2.
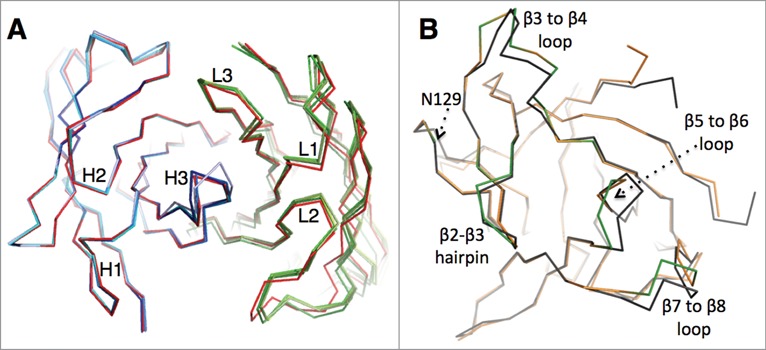
Comparison of the free and bound states of canakinumab and IL-1β (A). Structural overlay showing the Cα backbone of canakinumab's VH-VL domain, with the bound state in red and the 4 independent copies present in the asymmetric unit of the crystal of the free Fab colored in shades of blue (heavy chain) or green (light chain). (B). Structural overlay showing the Cα backbone of human IL-1β in the free (black; PDB entry 2I1B) and canakinumab-bound state (orange). Epitope loops are highlighted in green.
Residues Val H31 and Asp H56, which differ from the germline sequence, are in direct contact to the antigen. Two more amino-acid differences with the germline sequence are located within the framework β-sheet and appear to stabilize H-CDR3 (Asn H35) and H-CDR2 (Ile H50). The paratope residues that stand out in terms of number of intermolecular contacts to IL-1β are Arg H101, Trp H52, Tyr H53, Tyr H32 and Tyr L50 (Figure S4). Arg H101 of the H-CDR3 loop plays a particularly important role by forming strong electrostatic interactions with the key epitope residue Glu 64 (see infra). The sidechain of Tyr 50L of L-CDR2, which was not described as being involved in IL-1β binding,14 makes critical contacts to Glu 64, contributing favorable anion−π interactions and partially shielding the Arg H101-Glu 64 salt bridge from bulk solvent.
IL-1β epitope
The IL-1β epitope is made of 5 distinct structural elements, which form an extended, mostly planar and hydrophilic surface: the β2-β3 β-hairpin (residues 19 to 27), the α-helical region of the β3 to β4 loop (residues 32 to 39), the β5 to β6 loop (residues 63 to 66), the β7 to β8 loop (residues 86 to 88), and the β10 to β11 loop (Asn 129) (Figures S3 and S5). In sharp contrast to the canakinumab paratope, the IL-1β epitope does not include any aromatic residues or bulky hydrophobic residues (Fig. 1B). Rather, 70% (14 of 20) of all epitope residues engaged in direct contacts are either polar (1 serine, 2 asparagines, 4 glutamines) or charged amino-acids (3 lysines, 2 aspartates, 2 glutamates). These epitope residues form direct H-bonded contacts (Asp 35, Gln 34, Glu 37, Gln 38, Lys 65, Asn 129) or salt-bridge interactions (Lys 27, Glu 64) with canakinumab (Fig. 3 and Figure S6).
Figure 3.
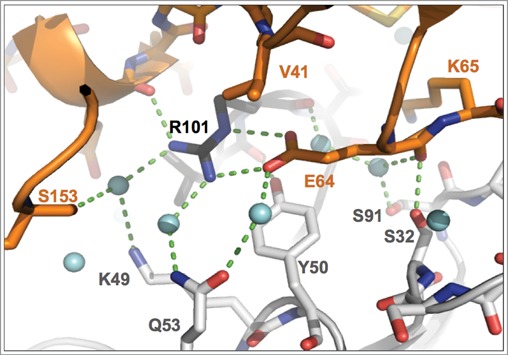
Close-up view of binding interactions involving the key epitope residue Glu 64. Heavy- and light-chain paratope residues are in black and light-gray sticks, respectively, while epitope residues are in orange. Light-blue spheres denote water molecules, dashed lines close contacts. Note the double interaction of the Glu 64 carboxylate group with Arg H101, the interaction with Tyr L50 and the water-mediated H-bonded contact to Gln L53.
The epitope residues that contribute the largest number of intermolecular contacts are Glu 64, Gln 38, Lys 65, Asp 35 and Ser 21 (Figure S5). Worthy of note, the sidechain of Glu 64, presented by the β5 to β6 loop of IL-1β, makes contacts to 4 CDRs (L-CDR1, L-CDR2, L-CDR3 and H-CDR3). Its carboxylate group is stacked onto the Tyr L50 aromatic ring and engaged in a strong, largely buried salt bridge interaction with Arg H101 of H-CDR3, involving both the Nη2 and Nε nitrogen atoms of Arg H101 (Arg H101 Nη2 – Glu 64 Oε1: 2.9 Å; Arg H101 Nε – Glu 64 Oε2: 2.8 Å) (Fig. 3 and Figure S1D). In addition, the carboxylate group of Glu 64 is also engaged in water-mediated H-bonded interactions with Arg H101 O, Tyr L50 Oη and Gln L53 Oε1 (Fig. 3).
A structural overlay of IL-1β in the free (PDB entry 2I1B) and canakinumab-bound state reveals several induced-fit changes at the binding interface (Fig. 2B). Three epitope loops undergo local conformational changes, namely the β3 to β4 loop (residues 32 to 39), the β5 to β6 loop (residues 63 to 66), and the β7 to β8 loop (residues 86 to 88). The most pronounced structural change involves the β5 to β6 loop that harbors Glu 64, Lys 65 and Asn 66. Upon canakinumab binding, the peptide bond between Lys 63 and Glu 64 is flipped, and the position of the Glu 64 Cα atom moves by 3.3Å. The positions of the following epitope residues Lys 65 and Asn 66 are also displaced by 1.5–2.0 Å. In contrast to a previous report,14 however, the orientation of the Leu 62 – Lys 63 peptide bond is not affected (Figure S7).
Hydration of the binding interface
The IL-1β epitope is almost exclusively hydrophilic, and, as a consequence, the antibody-antigen interface has a pronounced hydrophilic character. Our X-ray analysis identifies many ordered water molecules at the canakinumab / IL-1β interface that contribute to recognition and binding (Fig. 1). In total, 27 waters contribute to the H-bonded network that connects epitope and paratope residues. Among these, 13 waters are in direct H-bonded contacts to both proteins (using a distance cut-off of 3.5 Å). Examples of such water-mediated hydrogen-bonded interactions can be found in Figure 3 and Figure S6.
Mutational analysis
The experimental structure predicts key residues of human IL-1β for the interaction with canakinumab. To test whether these residue are indeed critical for the binding to canakinumab, we constructed different mutants of human IL-1β comprising key residues or loops predicted to be involved or not involved in the interaction with canakinumab. These mutants were expressed in E. coli and probed for interaction with canakinumab by Western blotting or surface plasmon resonance.
The β3 to β4 epitope loop (residues 32 to 39) is well conserved between human and non-human primate species. A quadruple mutant mimicking the mouse/rabbit amino-acid sequences in this region impaired the interaction significantly, but did not eliminate binding completely (Fig. 4). Close inspection of the crystal structure indicates that the observed reduction in binding affinity can mainly be ascribed to the E37N change, which eliminates favorable electrostatic interactions with Arg H98, Lys L49 and Arg H101 (Figure S6B). Mutations in the amino-terminal region (N7H, S13A, Q15L), or of residues 76 (D76G) and 77 (K77T) did not affect binding, as predicted from the lack of involvement of these residues in canakinumab binding. Replacement of the [125–130] loop had a small effect that can be explained by the N129D mutation, which maintained the H-bonded interaction to Tyr H53 but replaced the H-bonded interaction to Asp H54 by a repulsive electrostatic interaction (see Figure S6C). In sharp contrast to all previous mutations, converting the key epitope residue Glu 64 to alanine largely abrogated binding by canakinumab, underscoring the pivotal role played by this epitope residue.
Figure 4.
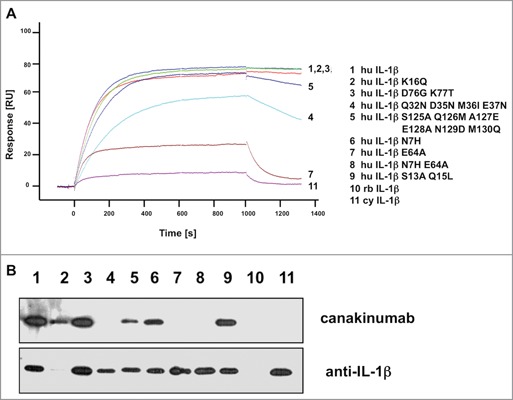
Mutational analysis of IL-1β proteins. (A) The sensorgrams of the binding kinetics of recombinant human IL-1β and muteins to immobilized canakinumab are shown. The signal values (RU) are reported as arbitrary units. (B) Western blot of E.coli expressed human, rabbit, and cynomolgus monkey IL-1β and muteins. For loading control, the blot was stripped and reprobed with a different human anti-IL-1β antibody directed against an epitope in the amino-terminus. Individual sensorgrams and lanes of the Western blot are identified by numbers which refer to the respective mutations indicated in the legend next to the sensorgrams. Abbreviations used: hu, human; rb, rabbit; cy, cynomolgus.
Reactivity of canakinumab with human, mouse, rat, rabbit, cynomolgus monkey, and marmoset IL-1β
Interaction of canakinumab with recombinant IL-1β from different mammalian species was assessed by surface plasmon resonance measurements. No binding was detected with mouse, rat, or rabbit IL-1β (data not shown). Neither was any cross-reactivity observed with cynomolgus monkey or rhesus monkey IL-1β, despite the high sequence identity (96%) to human IL-1β (Fig. 4 and Table 2). Both macaque species have all epitope residues conserved except for an alanine in position 64 of their respective IL-1β protein, which does not support a strong interaction with canakinumab. In an attempt to identify a non-human primate species neutralized by canakinumab and suitable for toxicological studies, we amplified marmoset (Callithrix jacchus) cDNA for mature IL-1β by PCR from spleen samples and produced the corresponding recombinant protein, which also shares 96% sequence identity to human IL-1β (only 5 of 133 amino acid differ; see Figure S8). Binding studies revealed full cross-reactivity with equilibrium binding constants of 30.5 pM and 22.8 pM for human and marmoset IL-1β, respectively (Table 3). To confirm full cross-reactivity to marmoset IL-1β, we tested the ability of canakinumab to neutralize natural and recombinant human or marmoset IL-1β in a cell-based bioassay. For this purpose, marmoset peripheral blood mononuclear cells were treated with lipopolysaccharide, and the IL-1β containing culture supernatant was used to stimulate IL-6 production in primary human dermal fibroblasts. IL-6 production was highly dependent on the presence of IL-1β in the unfractionated supernatant in this assay system, and addition of canakinumab repressed IL-6 production in a dose-dependent manner (Table 4).
Table 2.
Conservation of the canakinumab epitope on IL-1β across species
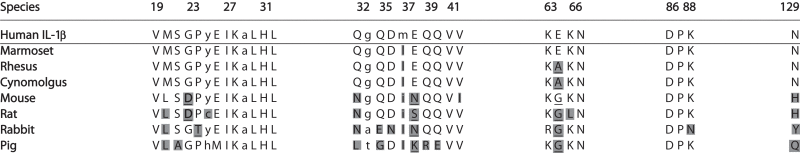 |
Conservation of the canakinumab epitope on IL-1β. The amino acid sequences of the 5 structural elements forming the epitope in human IL-1β are shown, together with the corresponding residues in several other species. Amino acid listed in bold upper case are involved in direct contacts to the antibody (3.9Å distance cut-off), while those in normal upper case have reduced solvent-accessibility upon complex formation but do not meet the 3.9Å distance cut-off. Intervening residues are shown in lower case. Non-conserved residues are shown on a gray background. Mutations which are expected to affect binding, on the basis of the X-ray structure, are shown in underlined, bold uppercase.
Table 3.
Apparent KD of canakinumab for human and marmoset IL-1β
| kon [105/Ms] | koff [10−5/s] | KD [pM] | ||
|---|---|---|---|---|
| Human IL-1β | 11.0 ± 0.24 | 3.30 ± 0.29 | 30.5 ± 2.6 | n=22 |
| Marmoset IL-1β | 15.4 ± 0.37 | 3.46 ± 0.35 | 22.8 ± 2.5 | n=20 |
Association (kon) and dissociation (koff) rate constants and equilibrium binding constant (KD) were determined by surface plasmon resonance. Values represent mean ± SD (standard deviation), n denotes the number of independent evaluations.
Table 4.
Neutralization of the biological activity of IL-1β by canakinumab
| IC50 | Canakinumab [pM] | IL-1Ra [pM] |
|---|---|---|
| Human IL-1β | 43.6 ± 5.5 | 109.2 ± 13.9 |
| Marmoset IL-1β | 40.8 ± 5.6 | 120.3 ± 15.3 |
Human dermal fibroblasts were stimulated with recombinant human IL-1β or cell derived marmoset IL-1β in the presence of canakinumab (range 0–7 nM) or recombinant human IL-1Ra (range 0–6 nM). Inhibition of IL-6 production was determined, and IC50 values are shown as mean ±SEM (standard error of mean) for n = 5 experiments.
Our site-directed mutagenesis data, combined with the observed species selectivity of canakinumab, are fully consistent with the structural results and emphasize the critical role of the epitope residue Glu 64. The comparison of the cynomolgus, rhesus and marmoset sequences demonstrates that the lack of cross-reactivity toward rhesus and cynomolgus IL-1β can be entirely ascribed to the Glu 64 change to alanine, as this is the only amino-acid difference within the canakinumab epitope (Table 2).
Canakinumab competes with IL-1RI and IL-1RII for binding IL-1β
Inspection of the crystal structure of the human IL-1β complex with the soluble type I IL-1 receptor7 revealed some, albeit limited, overlap with the canakinumab epitope on IL-1β. In particular, several amino acids of the first (Lys 27), second (Gln 32, Gln 34, Asp 35, Gln 38) and third (Asn 129) structural components of the IL-1β epitope are also involved in direct contacts to IL-1 RI (Figure S9). Moreover, the superposition of these 2 IL-1β complexes provided evidence for severe steric hindrance between the VH domain of the antibody and the D1 domain of IL-1 RI, which would preclude simultaneous binding to the cytokine (Fig. 5). Hence, canakinumab directly competes with IL-1RI for IL-1β binding. Structural overlays with the trimeric complexes of IL-1β with IL-1RAcP and IL-1RI8 or IL-1RII9 reveal that canakinumab binds on the opposite side of IL-1β with respect to the IL-1RAcP binding surface (Fig. 5). Hence, the antibody does not interfere with the binding of IL-1RAcP to the IL-1β binary complex with either IL-1RI or IL-1RII. The predicted interference with binding of IL-1β to IL-1RI was tested in a competitive binding assay, which confirmed that recombinant soluble IL-1RI and IL-1RII effectively compete with canakinumab for binding to IL-1β (Fig. 6).
Figure 5.
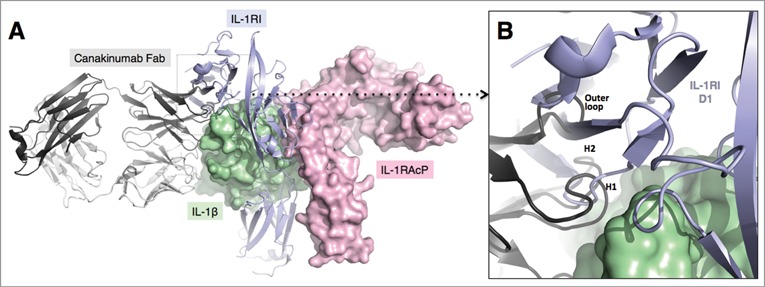
Mechanism of IL-1β neutralization by canakinumab. Structural overlay of the IL-1β - canakinumab complex with the IL-1RI – IL-1RAcP – IL-1β ternary complex (PDB entry 4DEP). (A). IL-1β (green) and the IL-1RAcP (light magenta) are shown in surface representation. The IL-1RI extracellular domains (light violet) and the canakinumab Fab (dark and light-gray) are shown in cartoon representation. (B). Close-up view showing the steric hindrance between the D1 domain of IL-1RI and the VH domain of canakinumab, notably the H-CDR1, H-CDR2 and outer loop regions.
Figure 6.

Canakinumab competes with IL-1RI and IL-1RII for the binding to human IL-1β. Human IL-1β (1nM) was incubated with the indicated concentrations of recombinant human soluble IL-1RI (solid line) or IL-1RII (dashed line). Binding of human IL-1β to immobilized canakinumab was determined by surface plasmon resonance measurement. Shown is the standard error of the mean obtained from 4 independent measurements.
Discussion
The crystal structures of the canakinumab Fab in the free and IL-1β-bound states reported here were determined from crystal forms that were distinct from those recently reported.14 Nevertheless, the refined structures are remarkably similar overall, as indicated by an rmsd of only 0.72 Å when the 2 IL-1β complexes with the canakinumab VH-VL domain are compared (371 equivalent Cα atoms), and of only 0.55–0.76 Å when the variable domain of the published free Fab structure (PDB entry 4G5Z; 224 equivalent Cα atoms) is compared to each of the 4 independent copies present in the asymmetric unit of our free Fab crystals (pairwise comparisons of the latter show an rmsd range of 0.39–0.61 Å). Importantly, the main conclusions regarding the mode of neutralization of IL-1β are in agreement. Our X-ray analyses, however, reveal new, significant structural details and do not confirm previous inferences regarding dramatic conformational changes of the canakinumab paratope upon IL-1β binding. Rather, the structural overlay presented here proves that the canakinumab VH-VL domain binds its antigen essentially as a rigid body, which is typical of affinity-matured antibodies.21 To elucidate the reason for this discrepancy, we superimposed the published free Fab structure (PDB entry 4G5Z) onto the published Fab complex (PDB entry 4G6J) and found no evidence for the reported dramatic conformational changes, with an rmsd for all Cα atoms of the VH-VL domain (224 atoms) of only 0.66Å instead of the reported value of 3.12 Å,14 indicating that the previous analysis was misled by a flawed superposition.
Furthermore, our work shows that L-CDR2 is involved in antigen binding, with Tyr L50 contributing to the recognition and binding of the critical epitope residue Glu 64. This important interaction is also present in PDB entry 4G6J but was apparently not recognized in the description of the structure. The refined conformation of Arg 101H presented here reveals the engagement of 2, rather than only one,14 guanidinium nitrogen atoms in short electrostatic contacts with the Glu 64 carboxylate group. Electron-density maps calculated with PDB entry 4G6J and the deposited structure factors show residual difference peaks consistent with our interpretation. Taken together, these new structural details account better for the pivotal role played by Glu 64, which is clearly identified as a hot spot of the canakinumab-IL-1β interface by our mutagenesis data and by the cross-reactivity profile of this therapeutic antibody toward closely-related monkey IL-1β orthologues. In this context, the pronounced induced-fit of the “Glu 64 loop” (β5 to β6 loop) is undoubtedly an intriguing, salient feature of IL-1β recognition by canakinumab. A change in the conformation of this loop upon canakinumab binding has already been noted,14 but the extent of the change and how it affected the position of Glu 64 was not fully clear. The conformation reported here is identical to that observed in PDB entry 4G6J except for the orientation of the Leu62-Lys63 peptide bond. Electron-density maps calculated with PDB entry 4G6J and the associated X-ray data support our model, which has this peptide linkage in approximately the same orientation as in free IL-1β.
Our analysis also reveals the important role played by water at the canakinumab-IL-1β interface. The importance of water molecules located at protein-protein interfaces has long been recognized. These contribute to the shape and physico-chemical complementarity of the 2 protein partners, improving the close-packing of atoms at the protein interface and mediating polar intermolecular interactions.22 Their influence on the thermodynamic profile of antibody-antigen complexes has also been pointed out: the solvation at the protein-protein interface weakens hydrophobic effects, enhances the enthalpic component of the association equilibrium and thus also the selectivity of the binding.23
The structural insights provided by our X-ray analyses were further corroborated by site-directed mutagenesis of selected epitope residues. For example, mutations within the β3 to β4 epitope loop (Q32N D35N M36I E37N) led to a significantly reduced, albeit not abolished interaction with canakinumab (Fig. 4). On the basis of the crystal structure, this effect could be ascribed mainly to the E37N change, which led to the loss of beneficial electrostatic interactions to H-CDR3 and L-CDR2, while the other 3 mutations are expected to have little, if any, influence on IL-1β binding. In contrast, the replacement of Glu 64 by an alanine almost completely abrogated binding by canakinumab, again in line with the X-ray analysis showing that the Glu 64 sidechain plays a central role in canakinumab binding: among all epitope residues, Glu 64 contributes the largest number of intermolecular contacts, mainly to Arg H101 of H-CDR3 and Tyr L50 of L-CDR2. Worthy of note, these paratope residues are also the top contributors, in terms of intermolecular contacts, of the antibody heavy- and light-chain, respectively. By cutting the Glu 64 sidechain down to an alanine sidechain, the strong, largely shielded electrostatic interactions to Arg H101 of H-CDR3 and the anion-π interactions with Tyr L50 are lost.
The lack of binding to the Ala 64 mutant is most likely explained by the loss of these strong interactions, rather than by a change in the loop conformation which would, in turn, affect the position of neighboring epitope residues, notably Lys 65 that forms H-bonded interactions to L-CDR3 and H-CDR3. In support of this view, it should be noted that human and mouse IL-1β differ in their amino-acid sequence within the β5 to β6 epitope loop region only at position 64, where a glycine is found in the murine cytokine instead of glutamate in the human orthologue. Two independently-determined crystal structures of mouse IL-1β are available,24,25 and structural overlays with the human form clearly show that the removal of the Glu 64 sidechain does not alter the loop conformation in any way. Glu 64 is a relatively unique residue in the otherwise well conserved IL-1β from different species, and it is not part of the IL-1RI or IL-1RAcP binding sites.10 Interestingly, only the large primates, chimpanzee, gorilla, orangutan, and some smaller non-human primate species, including marmoset, share Glu 64, while most other mammalian species have alanine or glycine in this position. In line with our binding data on the E64A mutant of human IL-1β, canakinumab does not recognize cynomolgus monkey IL-1β, which has all epitope residues except Glu 64 conserved, compared to human IL-1β. Canakinumab exhibits full cross-reactivity and neutralizing capacity toward marmoset IL-1β, as all critical contact residues are identical to human IL-1β.
The crystal structure in conjunction with the binding data provided a solid rationale and argument for the selection of marmoset as the non-human primate species for toxicological evaluation of canakinumab. Marmoset is not a well-explored species for toxicological evaluation of antibodies, and, in particular, reproductive and immuno-function toxicological studies are more difficult to conduct in marmosets compared to the well-established macaque toxicological model. Though several candidate antibodies, some of which had cross-reactivity to macaque IL-1β, were derived from the immunization of genetically engineered HuMab mice, canakinumab was selected as a clinical candidate based on its high affinity. While the determination of the molecular structure of the target-antibody complex is not required for selecting a species for toxicological evaluation of therapeutic antibodies, insight into features of the target epitope greatly facilitate the selection of potential cross-reactive species by simple database searches. Final proof for cross-reactivity must then come from biochemical and cellular assays. The equilibrium binding constants and on- and off-rates for canakinumab toward recombinant human and marmoset IL-1β are similar, as predicted by the X-ray analysis. In a cell-based assay system, not only recombinant marmoset IL-1β derived from PCR-based molecular cloning (data not shown), but also IL-1β produced from marmoset immune cells were neutralized with a similar IC50 as human recombinant IL-1β (Table 4), demonstrating that marmoset monkey is indeed a valid toxicological species for the assessment of preclinical safety for canakinumab.
Regarding the mechanism of action, our results indicate that canakinumab inhibits the first step in the assembly of the IL-1 receptor ternary complex, the association of IL-1β with IL-1RI.9 The other potential mechanisms of action, interference with the interaction of IL-1RAcP with either IL-1β or IL-1RI are not supported by the X-ray structure.
Materials and Methods
Recombinant proteins and cDNA expression
Human, mouse and rat recombinant IL-1β were purchased from R&D Systems (#s 201-LB-025/CF, 401-ML-025, 501-RL-050, respectively). Cynomolgus monkey, rabbit and marmoset IL-1β cDNA were isolated from spleen RNA by PCR. A synthetic cDNA for human IL-1β cloned into the pET17 expression vector served as a template for site-directed mutagenesis, which was conducted using the QuikChange site-directed mutagenesis kit, according to the manufacturer's instructions (Stratagene). Recombinant IL-1RI and IL-1RII were purchased from R&DSystems (#s 269–1R-100/CF and 263–2R-050/CF, respectively).
Purification of human, rabbit, cynomolgus monkey, and marmoset IL-1β
Recombinant IL-1β was expressed in E. coli BL21(DE3) cells using a pET17 vector (Novagen # 69664–3). Cell pellets were lysed with a French press in 50 mM Tris-HCl pH 8.0, 5 mM benzamidine-HCl, 5 mM EDTA, 5 mM DTT. The lysate was spun for 40 min at 48,000 g at 4°C, and after 2-fold dilution with 50 mM Tris-HCl pH 8.5, 5 mM DTT, the supernatant was loaded on a XK26/10 Q-Sepharose HP column. The flow-through peaks were pooled and subjected to ammonium sulfate fractionation. Following dialysis against 20 mM MES pH 5.7, 5 mM DTT, the material was sterile filtered, loaded on a SP-Sepharose 26/10 column, and eluted with a 0.0–1.0M NaCl gradient. The IL-1β fractions were identified by HPLC analysis, pooled and further purified over a Superdex 75 (26/60) size-exclusion column in PBS buffer, pH 7.2.
Purification of the canakinumab Fab
The Fab fragment of canakinumab was prepared by papain digestion of the full-length monoclonal antibody in 100 mM sodium acetate, pH 5.5, 2 mM EDTA, 0.3 mM cysteine. The digestion was performed with 0.5% (w/w) papain (Roche Applied Sciences, #10108014001) for 23 hours at 37°C and stopped with 50 μM of the papain inhibitor E64 (Roche Applied Sciences # 1087452300). The Fab was separated from the Fc fragment by Protein A chromatography and was then further purified by SEC on a 10/30 Superdex-75 column in 10 mM Tris-HCl pH 7.4, 25 mM NaCl.
Preparation of the Fab complex with IL-1β
The complex was prepared by mixing the canakinumab Fab with a 1.5-fold molar excess of human IL-1β, followed by purification over a Superdex-75 SEC column in 10 mM Tris-HCl, 25 mM NaCl, pH 7.4.
Surface plasmon resonance analysis
Determination of kinetic binding parameters and levels of cross-reactivity as well as competition studies were done by surface plasmon resonance measurements using BIAcore 2000 (BIAcore AB). Canakinumab was coupled non-covalently via attachment to an anti-human-Fcγ antibody (Jackson Immunochemicals, #109-005-098), and sensorgrams were recorded with varying concentrations of IL-1β, ranging between 0.5 nM and 4 nM. Equilibrium binding constants were calculated from a 1:1 Langmuir binding model using the BIAevaluation 3.0 software (BIAcore AB). Competition with IL-1RI and IL-1RII was conducted with immobilized canakinumab as above, 1 nM IL-1β and concentrations of soluble receptors ranging from 0 to 12 nM. Resonance units were recorded at 1000 s, converted to % binding, and IC50 values were determined by sigmoidal curve fit.
Cellular assays
Human dermal foreskin fibroblasts were obtained from Lonza. Cells were stimulated with 100 pg/ml of recombinant human IL-1β (R&D Systems, #201 LB). Canakinumab or recombinant human IL-1Ra (R&D Systems, # 280-RA-050) at various concentrations was added to the cultures and supernatant was taken 16–17 h after stimulation. The amount of released IL-6 was determined by a fluorescence resonance energy transfer assay (Cisbio), and the IC50 was obtained by sigmoidal curve fitting with the Origin 8.0 software (OriginLab Corp).
Crystallization
Crystallization conditions were identified by screening at 19°C with the vapor diffusion in sitting drop technique in Corning 96-well microtiter plates. Crystals were subsequently optimized in Hampton VDX plates using the technique of vapor diffusion in hanging drops. Well-diffracting crystals of the free canakinumab Fab were obtained at 60 mg/ml in 10 mM Tris-HCl pH 7.4, 25 mM NaCl from 12% PEG MME 5000, 0.05 M sodium citrate pH 5.0. Initial crystallization conditions for the Fab-IL-1β complex were identified with the PACT crystallization screen (Qiagen). These conditions (0.1 M MIB buffer pH 5.0 (2:3:3 mix of malonic acid, imidazole and boric acid), 25% (w/v) PEG 1,500) were then optimized using the OptiSalts screen (Qiagen). The crystal used in this study grew from a 9:1 mix of the initial crystallization solution with 0.1 M Tris pH 8.5, 4.0 M LiCl. Protein stock was 26.8 mg/ml Fab complex in 10 mM Tris-HCl pH 7.4, 25 mM NaCl. Crystallization drops were set-up by mixing 1.0 μl protein stock with 1.0 μl reservoir solution.
Structure Determination
Free canakinumab Fab
X-ray data were collected at the Swiss Light Source, beamline XS06A, with a Mar CCD 165 mm detector. In total 465 diffraction images of 0.5° oscillation each were recorded. The diffraction extended beyond 2.0 Å, but only 2.0 Å data were recorded in order to avoid reflection overlap. The structure was determined by molecular replacement with the program AMORE26 and refined with CNX27 using cycles of torsion angle dynamics and energy minimization, interspersed with model rebuilding steps using O.28
Canakinumab Fab complex
Diffraction data were collected at 100K with a FR-E rotating anode X-ray generator equipped with a MAR 345 DTB imaging plate detector. In total, 360 images of 0.5° oscillation each were recorded at a crystal-to-detector distance of 200 mm, and processed with HKL2000.29 The structure was determined by molecular replacement with the program Phaser,30 using PDB entry 2I1B for human IL-1β4 and the structure of the free canakinumab Fab.
Initial refinement was performed with the programs CNX27 and O28 as before. Final refinement was carried out with Autobuster (BUSTER version 2.11.2, Global Phasing Ltd) and Coot.31
Solvent accessible surfaces were calculated with the program AREAIMOL of the CCP4 program suite,32 using a probe radius of 1.4Å and standard van der Waals radii. Shape complementarity was quantified with the program SC.20 Pairwise contacts were generated with the CCP4 program NCONT, using a distance cut-off of 3.9 Å. All figures were made with PyMOL (Schrödinger, LLC).
Accession number
The atomic coordinates and observed structure factors of the free canakinumab Fab and of its IL-1β complex were deposited with the Protein Data Bank under accession numbers 5BVJ and 5BVP, respectively.
Disclosure of Potential Conflicts of Interest
All authors are full-time employees of Novartis Pharma AG.
Acknowledgments
The conduct of the BiaCore experiments by Jean-François Zuber and Dr. Ute Manning is gratefully acknowledged. We thank Elke Koch for the purification of human IL-1β and Peter Schöpflin for the generation and purification of the canakinumab Fab. We thank Sylvie Lehmann for crystallization experiments. We are grateful to Patrick Graff and Francis Bitsch for mass spectrometry and N-terminal sequence analyses. Diffraction data for the free canakinumab Fab were collected at the Swiss Light Source, Paul Scherrer Institute, Villigen, Switzerland. We are grateful to the machine and beamline groups whose outstanding efforts have made this experiment possible.
Funding
Study supported by Novartis Pharma AG, Basel, Switzerland.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 2009; 27:519-50; PMID:19302047; http://dx.doi.org/ 10.1146/annurev.immunol.021908.132612 [DOI] [PubMed] [Google Scholar]
- 2.Dinarello CA, Simon A, van der Meer JWM. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discovery 2012; 11:633-52; PMID:22850787; http://dx.doi.org/ 10.1038/nrd3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunology 2010; 10:89-102; PMID:20081871; http://dx.doi.org/ 10.1038/nri2691 [DOI] [PubMed] [Google Scholar]
- 4.Sims JE, Smith DE. Regulation of interleukin-1 activity is enhanced by cooperation between the interleukin-1 receptor type II and interleukin-1 receptor accessory protein. Eur Cytokine Netw 2003; 14:77-81; PMID:12957787 [PubMed] [Google Scholar]
- 5.Priestle JP, Schär HP, Grütter MG. Crystallographic refinement of interleukin 1 beta at 2.0 A resolution. Proc Natl Acad Sci U S A 1989; 86:9667-71; PMID:2602367; http://dx.doi.org/ 10.1073/pnas.86.24.9667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schreuder HA, Rondeau JM, Tardif C, Soffientini A, Sarubbi E, Akeson A, Bowlin TL, Yanofsky S, Barrett RW. Refined crystal structure of the interleukin-1 receptor antagonist. Presence of a disulfide link and a cis-proline. Eur J Biochem 1995; 227:838-47; PMID:7867645; http://dx.doi.org/ 10.1111/j.1432-1033.1995.tb20209.x [DOI] [PubMed] [Google Scholar]
- 7.Vigers GP, Anderson LJ, Caffes P, Brandhuber BJ. Crystal structure of the type-I interleukin-1 receptor complexed with interleukin-1beta. Nature 1997; 386:190-4; PMID:9062193; http://dx.doi.org/ 10.1038/386190a0 [DOI] [PubMed] [Google Scholar]
- 8.Thomas C, Bazan JF, Garcia KC. Structure of the activating IL-1 receptor signaling complex. Nat Struct Mol Biol 2012; 19:455-7; PMID:22426547; http://dx.doi.org/ 10.1038/nsmb.2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang D, Zhang S, Li L, Liu X, Mei K, Wang X. Structural insights into the assembly and activation of IL-1β with its receptors. Nat Immunology 2010; 11:905-11; http://dx.doi.org/ 10.1038/ni.1925 [DOI] [PubMed] [Google Scholar]
- 10.Alten R, Gram H, Joosten LA, van den Berg WB, Sieper J, Wassenberg S, Burmester G, van Riel P, Diaz-Lorente M, Bruin GJM, et al.. The human anti-IL-1 beta monoclonal antibody ACZ885 is effective in joint inflammation models in mice and in a proof-of-concept study in patients with rheumatoid arthritis. Arthritis Res Ther 2008; 10:R67; PMID:18534016; http://dx.doi.org/ 10.1186/ar2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, Gitton X, Widmer A, Patel N, Hawkins PN. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med 2009; 360:2416-25; PMID:19494217; http://dx.doi.org/ 10.1056/NEJMoa0810787 [DOI] [PubMed] [Google Scholar]
- 12.Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, Brik R, McCann L, Kasapcopur O, Rutkowska-Sak L, et al.. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012; 367:2396-406; PMID:23252526; http://dx.doi.org/ 10.1056/NEJMoa1205099 [DOI] [PubMed] [Google Scholar]
- 13.Schlesinger N, Alten RE, Bardin T, Schumacher HR, Bloch M, Gimona A, Krammer G, Murphy V, Richard D, So AK. Canakinumab for acute gouty arthritis in patients with limited treatment options: results from two randomised, multicentre, active-controlled, double-blind trials and their initial extensions. Ann Rheum Dis 2012; 71:1839-48; PMID:22586173; http://dx.doi.org/ 10.1136/annrheumdis-2011-200908 [DOI] [PubMed] [Google Scholar]
- 14.Blech M, Peter D, Fischer P, Bauer MMT, Hafner M, Zeeb M, Nar H. One target-two different binding modes: structural insights into gevokizumab and canakinumab interactions to interleukin-1β. J Mol Biol 2013; 425:94-111; PMID:23041424; http://dx.doi.org/ 10.1016/j.jmb.2012.09.021 [DOI] [PubMed] [Google Scholar]
- 15.Lefranc MP. Nomenclature of the human immunoglobulin kappa (IGK) genes. Exp Clin Immunogenet 2001; 18:161-74; PMID:11549845; http://dx.doi.org/ 10.1159/000049195 [DOI] [PubMed] [Google Scholar]
- 16.Pallarès N, Lefebvre S, Contet V, Matsuda F, Lefranc MP. The human immunoglobulin heavy variable genes. Exp Clin Immunogenet 1999; 16:36-60; PMID:Can't; http://dx.doi.org/ 10.1159/000019095 [DOI] [PubMed] [Google Scholar]
- 17.Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol 1997; 273:927-48; PMID:9367782; http://dx.doi.org/ 10.1006/jmbi.1997.1354 [DOI] [PubMed] [Google Scholar]
- 18.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of proteins of immunological interest, 5th edit., NIH Publication no. 91-3242. Washington DC: US Department of Health and Human Services; 1991. [Google Scholar]
- 19.Morea V, Tramontano A, Rustici M, Chothia C, Lesk AM. Conformations of the third hypervariable region in the VH domain of immunoglobulins. J Mol Biol 1998; 275:269-94; PMID:9466909; http://dx.doi.org/ 10.1006/jmbi.1997.1442 [DOI] [PubMed] [Google Scholar]
- 20.Lawrence MC, Colman PM. Shape complementarity at protein-protein interfaces. J Mol Biol 1993; 234:946-50; PMID:8263940; http://dx.doi.org/ 10.1006/jmbi.1993.1648 [DOI] [PubMed] [Google Scholar]
- 21.Wedemayer GJ, Patten PA, Wang LH, Schultz PG, Stevens RC. Structural insights into the evolution of an antibody combining site. Science 1997; 276:1665-9; PMID:9180069; http://dx.doi.org/ 10.1126/science.276.5319.1665 [DOI] [PubMed] [Google Scholar]
- 22.Lo Conte L, Janin J, Chothia C. The structure of protein-protein recognition sites. J Biol Chem 1990; 265:16027-30; PMID:2204619 [PubMed] [Google Scholar]
- 23.Bhat TN, Bentley GA, Boulot G, Greene MI, Tello D, Dall'Acqua W, Souchon H, Schwarz FP, Mariuzza RA, Poljak RJ. Bound water molecules and conformational stabilization help mediate an antigen-antibody association. Proc Natl Acad Sci U S A 1994; 91:1089-93; PMID:8302837; http://dx.doi.org/ 10.1073/pnas.91.3.1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Oostrum J, Priestle JP, Grutter MG, Schmitz A. The structure of murine interleukin-1beta at 2.8 Å resolution. J Struct Biol 1991; 107:189-95; PMID:1807351; http://dx.doi.org/ 10.1016/1047-8477(91)90021-N [DOI] [PubMed] [Google Scholar]
- 25.Ohlendorf DH, Treharne AC, Weber PC, Wendoloski JJ, Salemme FR. A comparison of the high resolution structures of human and murine interleukin-1beta. RCSB PDB 1992; entry 8I1B; http://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?uid=58606 [Google Scholar]
- 26.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr Sect A Found Crystallogr 1994; 50:157-63; http://dx.doi.org/ 10.1107/S0108767393007597 [DOI] [Google Scholar]
- 27.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al.. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 1998; 54:905-21; PMID:9757107; http://dx.doi.org/ 10.1107/S0907444998003254 [DOI] [PubMed] [Google Scholar]
- 28.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr Sect A Found Crystallogr 1991; 47: 110-9; http://dx.doi.org/ 10.1107/S0108767390010224 [DOI] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzym 1997; 276:307-26; http://dx.doi.org/ 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 30.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr 2007; 40:658-74; PMID:19461840; http://dx.doi.org/ 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D Biol Crystallogr 2010; 66:486-501; http://dx.doi.org/ 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al.. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011; 67:235-42; http://dx.doi.org/ 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.