

Abstract
Retinoblastoma (RB) is the most common malignant intraocular tumor in children. In the last decade, basic research has led to a better understanding of events after two hits in RB susceptibility gene (RB1), molecular mechanism of tumor growth, the cell of origin of RB, etc. This would pave way to identify biomarkers and molecular targeted therapy for better treatment option in the future. Furthermore, improvement in molecular techniques has led to enhanced diagnostic methods for early diagnosis, genetic counseling, and prevention of the disease. This review will help to understand the essence of basic research work conducted in recent times and its implication in the management of RB in the future.
Keywords: Genetics, pRB, RB1, retinoblastoma
Retinoblastoma (RB) is the most common malignant intraocular tumor in children with a reported average incidence of 1 in every 15,000–1 in every 18,000 live births.[1] It is second only to uveal melanoma in the frequency of occurrence of malignant intraocular tumors. There is no racial or gender predisposition in the incidence of RB. RB is bilateral in about 25–35% of cases.[2] The average age at diagnosis is 18 months, unilateral cases being diagnosed at around 24 months and bilateral cases before 12 months.[2] However, the average age at diagnosis in India and other developing nations has been 6 months later than those in the Western population.[3] Pawius was the first to describe RB in 1597.[4] Our understanding of the disease in the form of its etiopathogenesis and genetics has improved immensely since then. Early diagnosis and newer treatment modalities have improved the prognosis for survival; with more than 90% surviving the disease.[5] The treatment approach has changed from enucleation in 1970s to chemoreduction followed by sequentially aggressive focal therapy. Molecular characterization of the disease with improved molecular techniques has helped enormously in understanding the disease and its management. The availability of molecular diagnosis and genetic counseling has helped in preventing the disease in the community.
Etiology and Types
RB is a genetic disease; inactivation of both alleles of RB susceptibility gene (RB1) (OMIM 180200) predisposes an individual to the disease.[6] The disease can be categorized as hereditary (25–35%) and nonhereditary or sporadic RB (65–75%). The hereditary RB is an autosomal dominant disease with germline mutation; it accounts for approximately 6% of the newly diagnosed RB.[2] The rest of inherited RB is without familial transmission and occurs due to the inactivation of the first RB1 allele at the time of conception. In hereditary type, 85% of tumors are early onset, bilateral, and multifocal with an average of five tumors per eye, the distribution being random between the two eyes. In nonhereditary or sporadic RB both the RB1 alleles are inactivated somatically in the retinal cells. Sporadic RB results in late-onset, unilateral, and unifocal tumors. Some children with the familial form of RB develop pinealoblastoma (trilateral RB).[7] The pinealoblastoma has embryological, pathological, and immunological similarities to RB. The other clinical variant, retinoma, is an uncommon benign form of RB. It is considered as spontaneously arrested RB.
RB1 mutation and development of retinoblastoma
Knudson proposed the two hit hypothesis model explaining the development of RB tumors.[6] The RB1 gene is located in chromosome 13 at q arm region 14. It spans for about 180 kb in length, having 27 exons. Transcription of RB1 results in a 4.8 kb mRNA that encodes an 110 kDa ubiquitously expressed nuclear phophoprotein, pRB containing 928 aminoacid residues. The tumor is initiated by mutations in both copies of the RB1 gene. The first allele is inactivated by an intragenic mutation in the germline (hereditary RB) or a somatic cell (sporadic RB). Around 10% of the RB1 germline mutations may be present in only a fraction of the cells of the proband, which could be due to mosaicism of RB1 mutations.[8] Mosaicism becomes obvious only when a parent with more than one affected child with RB does not show the same mutant allele present in the children. The second allele is lost by one of the mechanisms involved in the first mutation, but most commonly lost by processes involving chromosomal mechanism such as mitotic nondisjunction with the loss of the wild-type chromosome or duplication of the mutant chromosome, mitotic recombination between the RB1 locus and the centromere, or gene conversion and deletion, leading to loss of heterozygosity (LOH) at the RB1 locus.[9] LOH represents 50–70% of the second hit in RB.[10] In unilateral RB, silencing of the RB1 gene due to methylation of the promoter region is also a known mechanism of one of the two hits.
More recent studies also show that RB tumors may differ in the mutagenic pathway they take from normal to the malignant cell, for example, Most of the RB is caused by RB1 mutation, while some are caused by amplification of the MYCN gene. Less is known about the development of the MYCNamp tumors beyond the initiating amplification of the MYCN oncogene.[11] Is MYCN amplification the only genomic event driving malignancy of these tumors and do these skip bypass RB1 mutation? Are these tumors different pathologically? These questions remain unanswered and, therefore, require further study.
What determines the phenotype variability in retinoblastoma?
RB has contributed tremendously to the understanding of cancer. It has provided the classic “two-hit model” for oncogenesis and has helped to identify the first tumor suppressor gene RB1. RB is characterized by extensive phenotype variability: (i) RB1 inactivation may result to the genesis of malignant or benign (retinoma/retinocytoma) tumors.[12,13] In some, malignant tumors can undergo spontaneous regression[2] and retinomas occasionally get reactivated and develop into malignant tumors.[12] (ii) Tumors may be unilateral or bilateral, unifocal, or multifocal. Bilateral and multifocal tumors are usually associated with a de novo or inherited germline mutation while unilateral tumors are usually secondary to somatic mutations. This phenotypic variation is attributed to the presence of an inactive RB1 allele in all retinal precursor cells in both eyes of the patients with germline mutations, resulting in a higher possibility of a peri- or post-natal second mutational event of the other RB1 allele being taken up by more cells. The germline RB mutation is homogenous across all retinal cells, in spite of this, why tumor event occurs in few and not in all retinal cells remains a puzzle. On the other hand, in a sporadic form of the disease, both hits occur during the postnatal period with a lower likelihood of the event affecting more than a subset of precursor cell population in one eye.[6] (iii) RB is commonly present as discrete tumors but occasionally diffuse and infiltrate. Phenotypic variations in RB offer numerous clues to disease pathogenesis. Understanding the molecular and biological basis of the phenotypic variation will provide insight into the mechanisms underlying tumor progression.
Recently, studies have shown that RB1 gene inactivation alone is insufficient to induce tumorigenesis and that there are additional genetic and stochastic events that underlie uncontrolled retinal precursor cell proliferation.[14] Comparative genomic hybridization and gene expression studies have facilitated the probing of genes controlling basic events in cellular development viz., proliferation, differentiation, and apoptosis. Knowledge of these additional mechanisms is essential, as it will aid better management and possible prevention of the disease.
What makes retinoblastoma proliferate?
Recurrent genetic changes in addition to RB1 inactivation happens in human RB tumors. The minimal regions most frequently gained in the chromosomes of RB were shown to be 1q31 (52%), 6p22 (44%), 2p24–25 (30%), and 13q32–34 (12%) and the most commonly lost was reported as 16p22 (14%).[15,16] Few candidate genes responsible for some of these chromosomal imbalances have been proposed; these include the leukemic oncogene DEK and the transcriptional factor E2F3.[16] MYCN gene amplification in the 2p24–25 regions, and cadherin II loss in the 16q22 region are other candidate genes proposed to affect RB development and progression.[16] Analysis of the gain site in 1q31–32 regions by quantitative multiplex polymerase chain reaction (PCR) and quantifying the genes in the site has revealed KIF14 (a kinesin gene) as a candidate oncogene with an increased expression of more than two orders of magnitude.[17] Studies have shown that patients with older age at diagnosis had a significantly higher expression of KIF14 compared to early diagnosed patients.[18]
Micro array-based expression analysis found E2F3 to be overexpressed in RB. Later, these findings were also confirmed by real-time PCR. Integration of data from genomic gain analysis and expression analysis in this study indicates E2F3 gene as a promising candidate for 6p22 gain rather than KIF13A as previously suggested.[19] E2F3 in conjunction with its dimerization partner regulates genes that play a role in DNA replication, such as H2 folate reductase,[20] DNA polymerase a,[21] histone H2A9 and proliferating cell nuclear antigen.[22] They also found that tumors with complete gains at loci on 6p were diagnosed significantly later with a median Age at diagnosis (AAD) compared to children whose tumors showed no or partial 6p gains.[19] Chromosome 6p gain is also common in bladder cancer and is associated with an elevated risk of progression of bladder cancer.[23] Through real-time PCR, we demonstrated a very high expression of E2F3 in a large cohort of human RB, which clearly demonstrates the role of this gene in RB proliferation.[24]
Recent evidence point out the importance of p16INK4A in RB progression. p16INK4A is a senescence protein capable of arresting proliferative cell at G1 phase of cell cycle. RB1 inactivation in the retinal cells causes genomic instability which triggers proto-oncogenes (such as KIF14 and E2F3) to cause an increase in proliferation. During the early stages of abnormal proliferation of retinal cells, p16INK4A gets overexpressed and prevents further proliferation that may result in the occurrence of retinoma. Indeed, retinoma can remain quiescent throughout life without progereing to a malignant invasive tumor. Rare cells may escape the induction of senescence, either by inactivation of p16INK4A or no activation of p16INK4A in the first place and go on to clonally progress to RB with activation of oncogenes through genomic instability. However, clinical evidence strongly suggests that the rare retinoma that remains stable and unchanging under observation can sometimes later progress to an active, malignant RB, perhaps by the failure of senescence.[25]
How is retinoblastoma proliferation controlled?
pRB has been found to play an important role in cell cycle exit and terminal cellular differentiation. In addition to pRB, the role of other proteins in the pathway has been extensively studied in retinal cell development. The members of Cip/Kip family of cyclin-dependent kinase inhibitors (CKIs), p27Kip1 and p57Kip2, have been shown to be important in determining the time at which the cell exits and determines the cell fate in the retina.[26,27,28,29] A member of the Cip/Kip family of CKIs, p27 has been found to be the component of an intrinsic timer which is important for retinal development.[30,31] Apart from its role in cell cycle exit, it also plays an important role in differentiation of retinal cells.[32] The role of this protein has not been studied in human RB. Studies on another CKIs - p57 in mouse retina gave the first evidence of cell cycle exit;[33] it has been detected in a subpopulation of differentiating amacrine cells confirming its role in differentiation of retinal cells.[34] It has been found in a large cohort of RB patients that the expression of p57 was either normal or less, which was also replicated at the protein level.[35]
Is retinoblastoma death resistant?
It was believed that RB arose from death-resistant cells, which escaped an intact p53 pathway.[36] However, recently a study has shown that a defective p53 pathway may be responsible for the progression of RB.[37] The study demonstrated that the p53 pathway was inactivated in RB1 deficient cells due to amplification of MDMX and MDM2. MDMX and MDM2 are structurally related proteins that act as antagonists of p53. Under normal conditions, levels of p53 protein are kept low, partly through negative regulation by MDM proteins. MDM2 is an enzyme that tags p53 with an ubiquitin molecule (Ub), thereby promoting p53 degradation. MDMX also interacts physically with p53 and inhibits its gene-regulatory activity. The study also showed that nutlin-3 (an inhibitor of the MDM2-p53 interaction) efficiently killed RB cells. An important component of the p53 tumor surveillance pathway is p14.[38] When RB activity is lost, the transcription factor E2F activates the transcription of p14.[39] p14 then inactivates MDM2,[40] leading to p53-mediated apoptosis, and exit from the cell cycle.
Model for retinoblastoma progression
The tireless efforts of scientist to understand the events that follow RB1 inactivation for tumor progression have laid a foundation to recognize the probable pathogenic signature of this tumor [Fig. 1]. As soon as both copies of RB1 are inactivated, the immature retinal transitional cells become genetically unstable and uncontrollably proliferative. During this stage, the proliferation is countered by the senescence protein p16INK4A. If the proliferating cells respond to the action of p16INK4A, the tumor stops its proliferation and gets arrested as retinoma. On the other hand, if the genetic instability in the abnormal retinal cell takes the upper hand (increase in KIF14 and E2F3 levels), it counters and overtakes the cellular senescence and become malignant. Normal or slightly lower expression of P27 and p57 may not be able to counter the proliferative capability of the very high expressing oncogenes (KIF14 and E2F3). By nullifying the activity of p53 induced cell death (overexpressing MDM2), the retinal tumor cells escape cell death and progress.
Figure 1.
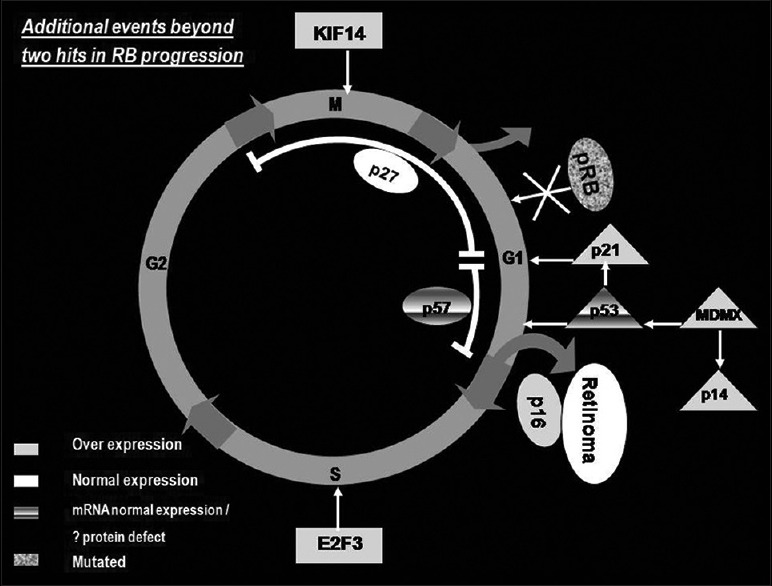
Events that leads to the progression of retinoblastoma
Cell of origin of retinoblastoma?
Identifying the cell of origin of RB may help to answer questions regarding phenotype variability and progression of the tumor. Transgenic mouse models of RB have given important clues to the cell of origin of human RB.[41] Unfortunately, RB has been developed from different types of retinal cells using animal models. A recent human study reveals that RB cells have several intrinsic features of cone photoreceptors.[42] what determines the cell of origin of RB in humans and animal models? Does the time of pRB inactivation determine the cell of origin of RB?[43] As retinal cells strictly follow the competence model for retinal development; cones cells are the first to develop during human retinal embryogenesis, lack of pRB due to RB1 inactivation at the time of conception may lead to lack of cell cycle exit and differentiation in early forming cone cells which could probably trigger RB formation. The cone phenotype of RB in human RB suggests but does not prove that the disease has arisen from cones. Early response of retinal cells to RB1 loss needs to be understood to come to a conclusion on the cell of origin of RB.[44] Despite the theories regarding cell of origin, ocular coherence tomography images of small tumors show that the tumors seem to arise within the inner nuclear layer.[45]
Future Therapy for Retinoblastoma
Improvement in our understanding on the molecular wiring of RB would result in a better treatment option in the future. The treatment modality would be targeted along the pathway deciphered in RB proliferation. This may include deactivating the overexpressed KIF14 and E2F3 and improving cell exit from the cell cycle. Further improving the P53 death signaling pathway and placing the proliferative cell in senescence may help in controlling the tumor growth at an early stage of the disease. Understanding the cell of origin may help us to intervene at the appropriate time to prevent the occurrence of the disease.
Molecular Diagnosis of Retinoblastoma
In routine clinical practice children suspected with the risk of RB are subjected to ophthalmic examination as an outpatient procedure or under anesthesia routinely at regular intervals till the clinician feels that the risk of tumor formation is negligible. A better method of screening the children with a risk of developing RB is required to replace the subjective method described earlier. Molecular diagnostics for RB can offer an accurate risk prediction and effective management. An efficient diagnostic model for RB could reduce the overall health care costs and also avoid unnecessary anxiety and worry for the family.[46] European Molecular Quality Network (EMQN) has evolved “the best practice guidelines for molecular analysis of RB” based on the reports drawn up from the workshops run by EMQN.[47] According to this model, peripheral venous blood is screened for mutation in bilateral RB and familial unilateral RB patients. In nonfamilial unilateral RB patients which warranted enucleation due to the advanced stage of the disease, tumor samples are screened for mutation and then the peripheral blood lymphocytes are examined for specific mutant alleles seen in the tumor to rule out germline mutation. The advent of next generation sequencing has tremendously improved the molecular screening of RB1 gene. The time taken and the cost to screen the entire RB1 coding region have decreased with next generation sequencing technology.
Genetic Counseling for Retinoblastoma
This inherited childhood cancer requires a proper genetic counseling for prevention, early detection and to plan a better management strategy for patients and their families. The arrival of molecular diagnostics for RB has enhanced the counseling protocol for families. The counseling schedule can be split into pre- and post-test (molecular testing) counseling. During pretest counseling, pedigree of the family is constructed based on the history. The analysis is made for potentially inherited disease in the family. The nature of the disease is elaborated. The genetics of RB is discussed, paying attention, particularly to the importance of knowing the inherited and sporadic RB. In bilateral and familial patients, the risk of transmission of the defective allele and occurrence of the disease is explained. A brief discussion on the molecular testing for RB and various results are interpreted. Presentation of risks and benefits of each option, with careful attention to patient comprehension, should be done. In posttest counseling, the explanation of the test results, the implication for further testing if needed and the management strategy based on the test report are discussed. Emotional support followed by the composition of a summary and referral letter shall be provided. Long-term support and follow-up of the affected families for proper guidance with respect to rehabilitation and research updates is a requirement.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Bishop JO, Madson EC. Retinoblastoma. Review of the current status. Surv Ophthalmol. 1975;19:342–66. [PubMed] [Google Scholar]
- 2.Shields JA, Shields CL. Intraocular Tumors – A Text and Atlas. Philadelphia, PA, USA: WB Saunders Company; 1992. [Google Scholar]
- 3.Shanmugam MP, Biswas J, Gopal L, Sharma T, Nizamuddin SH. The clinical spectrum and treatment outcome of retinoblastoma in Indian children. J Pediatr Ophthalmol Strabismus. 2005;42:75–81. doi: 10.3928/01913913-20050301-01. [DOI] [PubMed] [Google Scholar]
- 4.Albert DM. Historic review of retinoblastoma. Ophthalmology. 1987;94:654–62. doi: 10.1016/s0161-6420(87)33407-4. [DOI] [PubMed] [Google Scholar]
- 5.Chan HS, Gallie BL, Munier FL, Beck Popovic M. Chemotherapy for retinoblastoma. Ophthalmol Clin North Am. 2005;18:55–63. doi: 10.1016/j.ohc.2004.11.002. viii. [DOI] [PubMed] [Google Scholar]
- 6.Knudson AG., Jr Mutation and cancer: Statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimmerman LE, Burns RP, Wankum G, Tully R, Esterly JA. Trilateral retinoblastoma: Ectopic intracranial retinoblastoma associated with bilateral retinoblastoma. J Pediatr Ophthalmol Strabismus. 1982;19:320–5. doi: 10.3928/0191-3913-19821101-10. [DOI] [PubMed] [Google Scholar]
- 8.Munier FL, Thonney F, Girardet A, Balmer A, Claustre M, Pellestor F, et al. Evidence of somatic and germinal mosaicism in pseudo-low-penetrant hereditary retinoblastoma, by constitutional and single-sperm mutation analysis. Am J Hum Genet. 1998;63:1903–8. doi: 10.1086/302138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cavenee WK, Dryja TP, Phillips RA, Benedict WF, Godbout R, Gallie BL, et al. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature. 1983;305:779–84. doi: 10.1038/305779a0. [DOI] [PubMed] [Google Scholar]
- 10.Ramprasad VL, Madhavan J, Murugan S, Sujatha J, Suresh S, Sharma T, et al. Retinoblastoma in India: Microsatellite analysis and its application in genetic counseling. Mol Diagn Ther. 2007;11:63–70. doi: 10.1007/BF03256223. [DOI] [PubMed] [Google Scholar]
- 11.Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S, Thériault BL, et al. Characterisation of retinoblastomas without RB1 mutations: Genomic, gene expression, and clinical studies. Lancet Oncol. 2013;14:327–34. doi: 10.1016/S1470-2045(13)70045-7. [DOI] [PubMed] [Google Scholar]
- 12.Gallie BL, Ellsworth RM, Abramson DH, Phillips RA. Retinoma: Spontaneous regression of retinoblastoma or benign manifestation of the mutation? Br J Cancer. 1982;45:513–21. doi: 10.1038/bjc.1982.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eagle RC, Jr, Shields JA, Donoso L, Milner RS. Malignant transformation of spontaneously regressed retinoblastoma, retinoma/retinocytoma variant. Ophthalmology. 1989;96:1389–95. doi: 10.1016/s0161-6420(89)32714-x. [DOI] [PubMed] [Google Scholar]
- 14.DiCiommo D, Gallie BL, Bremner R. Retinoblastoma: The disease, gene and protein provide critical leads to understand cancer. Semin Cancer Biol. 2000;10:255–69. doi: 10.1006/scbi.2000.0326. [DOI] [PubMed] [Google Scholar]
- 15.Chen D, Gallie BL, Squire JA. Minimal regions of chromosomal imbalance in retinoblastoma detected by comparative genomic hybridization. Cancer Genet Cytogenet. 2001;129:57–63. doi: 10.1016/s0165-4608(01)00427-7. [DOI] [PubMed] [Google Scholar]
- 16.Bowles E, Corson TW, Bayani J, Squire JA, Wong N, Lai PB, et al. Profiling genomic copy number changes in retinoblastoma beyond loss of RB1. Genes Chromosomes Cancer. 2007;46:118–29. doi: 10.1002/gcc.20383. [DOI] [PubMed] [Google Scholar]
- 17.Corson TW, Huang A, Tsao MS, Gallie BL. KIF14 is a candidate oncogene in the 1q minimal region of genomic gain in multiple cancers. Oncogene. 2005;24:4741–53. doi: 10.1038/sj.onc.1208641. [DOI] [PubMed] [Google Scholar]
- 18.Madhavan J, Coral K, Mallikarjuna K, Corson TW, Amit N, Khetan V, et al. High expression of KIF14 in retinoblastoma: Association with older age at diagnosis. Invest Ophthalmol Vis Sci. 2007;48:4901–6. doi: 10.1167/iovs.07-0063. [DOI] [PubMed] [Google Scholar]
- 19.Grasemann C, Gratias S, Stephan H, Schüler A, Schramm A, Klein-Hitpass L, et al. Gains and overexpression identify DEK and E2F3 as targets of chromosome 6p gains in retinoblastoma. Oncogene. 2005;24:6441–9. doi: 10.1038/sj.onc.1208792. [DOI] [PubMed] [Google Scholar]
- 20.Fry CJ, Slansky JE, Farnham PJ. Position-dependent transcriptional regulation of the murine dihydrofolate reductase promoter by the E2F transactivation domain. Mol Cell Biol. 1997;17:1966–76. doi: 10.1128/mcb.17.4.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pearson BE, Nasheuer HP, Wang TS. Human DNA polymerase alpha gene: Sequences controlling expression in cycling and serum-stimulated cells. Mol Cell Biol. 1991;11:2081–95. doi: 10.1128/mcb.11.4.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HH, Chiang WH, Chiang SH, Liu YC, Hwang J, Ng SY. Regulation of cyclin D1, DNA topoisomerase I, and proliferating cell nuclear antigen promoters during the cell cycle. Gene Expr. 1995;4:95–109. [PMC free article] [PubMed] [Google Scholar]
- 23.Bruch J, Schulz WA, Häussler J, Melzner I, Brüderlein S, Möller P, et al. Delineation of the 6p22 amplification unit in urinary bladder carcinoma cell lines. Cancer Res. 2000;60:4526–30. [PubMed] [Google Scholar]
- 24.Madhavan J, Mitra M, Mallikarjuna K, Pranav O, Srinivasan R, Nagpal A, et al. KIF14 and E2F3 mRNA expression in human retinoblastoma and its phenotype association. Mol Vis. 2009;15:235–40. [PMC free article] [PubMed] [Google Scholar]
- 25.Dimaras H, Khetan V, Halliday W, Orlic M, Prigoda NL, Piovesan B, et al. Loss of RB1 induces non-proliferative retinoma: Increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008;17:1363–72. doi: 10.1093/hmg/ddn024. [DOI] [PubMed] [Google Scholar]
- 26.Harland R. Neural induction. Curr Opin Genet Dev. 2000;10:357–62. doi: 10.1016/s0959-437x(00)00096-4. [DOI] [PubMed] [Google Scholar]
- 27.Ebendal T, Bengtsson H, Söderström S. Bone morphogenetic proteins and their receptors: Potential functions in the brain. J Neurosci Res. 1998;51:139–46. doi: 10.1002/(SICI)1097-4547(19980115)51:2<139::AID-JNR2>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 28.Patapoutian A, Reichardt LF. Roles of Wnt proteins in neural development and maintenance. Curr Opin Neurobiol. 2000;10:392–9. doi: 10.1016/s0959-4388(00)00100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belliveau MJ, Cepko CL. Extrinsic and intrinsic factors control the genesis of amacrine and cone cells in the rat retina. Development. 1999;126:555–66. doi: 10.1242/dev.126.3.555. [DOI] [PubMed] [Google Scholar]
- 30.Durand B, Raff M. A cell-intrinsic timer that operates during oligodendrocyte development. Bioessays. 2000;22:64–71. doi: 10.1002/(SICI)1521-1878(200001)22:1<64::AID-BIES11>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 31.Durand B, Gao FB, Raff M. Accumulation of the cyclin-dependent kinase inhibitor p27/Kip1 and the timing of oligodendrocyte differentiation. EMBO J. 1997;16:306–17. doi: 10.1093/emboj/16.2.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohnuma S, Philpott A, Wang K, Holt CE, Harris WA. p27Xic1, a Cdk inhibitor, promotes the determination of glial cells in Xenopus retina. Cell. 1999;99:499–510. doi: 10.1016/s0092-8674(00)81538-x. [DOI] [PubMed] [Google Scholar]
- 33.Dyer MA, Cepko CL. p57(Kip2) regulates progenitor cell proliferation and amacrine interneuron development in the mouse retina. Development. 2000;127:3593–605. doi: 10.1242/dev.127.16.3593. [DOI] [PubMed] [Google Scholar]
- 34.Dyer MA, Cepko CL. The p57Kip2 cyclin kinase inhibitor is expressed by a restricted set of amacrine cells in the rodent retina. J Comp Neurol. 2001;429:601–14. [PubMed] [Google Scholar]
- 35.Madhavan J, Mallikarjuna K, Vikas K, George R, Bremner R, Kumaramanickavel G. CDKN1C (p57KIP2) mRNA expression in human retinoblastomas. Ophthalmic Genet. 2010;31:141–6. doi: 10.3109/13816810.2010.490544. [DOI] [PubMed] [Google Scholar]
- 36.Chen D, Livne-bar I, Vanderluit JL, Slack RS, Agochiya M, Bremner R. Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell. 2004;5:539–51. doi: 10.1016/j.ccr.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 37.Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–6. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 38.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 39.Aslanian A, Iaquinta PJ, Verona R, Lees JA. Repression of the Arf tumor suppressor by E2F3 is required for normal cell cycle kinetics. Genes Dev. 2004;18:1413–22. doi: 10.1101/gad.1196704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: Progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 41.Mills MD, Windle JJ, Albert DM. Retinoblastoma in transgenic mice: Models of hereditary retinoblastoma. Surv Ophthalmol. 1999;43:508–18. doi: 10.1016/s0039-6257(99)00047-8. [DOI] [PubMed] [Google Scholar]
- 42.Xu XL, Fang Y, Lee TC, Forrest D, Gregory-Evans C, Almeida D, et al. Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell. 2009;137:1018–31. doi: 10.1016/j.cell.2009.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madhavan J, Khetan V. Does the time of inactivation of pRb determine the cell of origin of retinoblastoma? Invest Ophthalmol Vis Sci. 2011;52:9403. doi: 10.1167/iovs.11-8843. [DOI] [PubMed] [Google Scholar]
- 44.Bremner R. Retinoblastoma, an inside job. Cell. 2009;137:992–4. doi: 10.1016/j.cell.2009.05.034. [DOI] [PubMed] [Google Scholar]
- 45.Rootman DB, Gonzalez E, Mallipatna A, Vandenhoven C, Hampton L, Dimaras H, et al. Hand-held high-resolution spectral domain optical coherence tomography in retinoblastoma: Clinical and morphologic considerations. Br J Ophthalmol. 2013;97:59–65. doi: 10.1136/bjophthalmol-2012-302133. [DOI] [PubMed] [Google Scholar]
- 46.Richter S, Vandezande K, Chen N, Zhang K, Sutherland J, Anderson J, et al. Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet. 2003;72:253–69. doi: 10.1086/345651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lohman D, Scheffer H, Gallie BL. Best practice guidelines for molecular analysis of retinoblastoma. Manchester, UK: EMQN; 2002. [Google Scholar]