

Abstract
The use of liquid chromatography – mass spectrometry (LC-MS) for the characterization of proteins can provide a plethora of information related to their structure, including amino acid sequence determination and analysis of posttranslational modifications. The variety of LC-MS based applications has led to the use of LC-MS characterization of therapeutic proteins and monoclonal antibodies as an integral part of the regulatory approval process. However, the improper use of an LC-MS system, related to intrinsic instrument limitations, improper tuning parameters, or poorly optimized methods may result in the production of low quality data. Improper system performance may arise from subtle changes in operating conditions that limit the ability to detect low abundance species. To address this issue, we systematically evaluated LC-MS/MS operating parameters to identify a set of metrics that can be used in a workflow to determine if a system is suitable for its intended purpose. Development of this workflow utilized a bovine serum albumin (BSA) digest standard spiked with synthetic peptides present at 0.1% to 100% of the BSA digest peptide concentration to simulate the detection of low abundance species using a traditional bottom-up workflow and data-dependent MS2 acquisition. BSA sequence coverage, a commonly used indicator for instrument performance did not effectively identify settings that led to limited dynamic range or poorer absolute mass accuracy on 2 separate LC-MS systems. Additional metrics focusing on the detection limit and sensitivity for peptide identification were determined to be necessary to establish system suitability for protein therapeutic characterization by LC-MS.
Keywords: system suitability, protein therapeutics, liquid chromatography, mass spectrometry, quality control
Abbreviations
- ADC
analog-to-digital converter
- AGC
automatic gain control
- ANOVA
analysis of variance
- BSA
bovine serum albumin
- CID
collision induced dissociation
- CV
coefficient of variation
- EIC
extracted ion chromatogram
- ETD
electron transfer dissociation
- LC
liquid chromatography
- LOD
limit of detection
- MS
mass spectrometry
- MS1
single stage mass spectrometry (precursor ion scan)
- MS2 or MS/MS
tandem mass spectrometry
- m/z
mass-to-charge ratio
- S/N
signal-to-noise ratio
- TDC
time-to-digital converter
Introduction
Mass spectrometry (MS) has become a vital tool in characterization of protein therapeutics and monoclonal antibodies because it can provide details of amino acid sequence and posttranslational modifications at the molecular level. Characterization is important in all stages of the product life cycle, ranging from early product development to routine quality control.1,2 Although proteins can be directly characterized intact or with partial enzymatic digestion on high resolution mass spectrometers,2-6 most routine MS analyses of protein therapeutics and monoclonal antibodies use a bottom-up proteomics approach for peptide mass mapping. Typically, proteins are enzymatically digested into small peptides and subjected to online liquid chromatography (LC) separation prior to introduction into the mass spectrometer. Often multiple enzymes with different but complementary specificity are used to maximize the sequence coverage for complete assessment of the protein.7-9 While some peptides can be directly resolved chromatographically, additional separation based on their mass-to-charge ratios (m/z) in the mass spectrometer significantly increases sequence coverage. In many applications, tandem mass spectrometry (MS/MS or MS2) is used to increase the confidence of the peptide identification. For a MS2 experiment, individual m/z values are selected, isolated and subjected to ion activation such as collision induced dissociation (CID) or electron transfer dissociation (ETD) to cause fragmentation, which can provide information on the peptide sequence. The complexity as well as the large number of spectra generated from MS2 experiments necessitates the use of peptide identification algorithms and software packages in order to quickly identify proteins based on the primary sequence analysis of their corresponding peptides.
An important component of the characterization of protein therapeutics and monoclonal antibodies involves the detection and relative quantitation of low abundance species present in a sample that may include impurities, degradation products, sequence variants or post-translationally modified forms. Reliable detection and quantitation of these species may be necessary in order to demonstrate manufacturing control as a part of the approval process. However, the complexity of each of the many stages of a bottom-up LC-MS/MS analysis,10-14 including sample processing (e.g., variability in enzymatic digestion), separation techniques (e.g., reproducibility of LC separation), MS analysis (e.g., MS method settings), and data processing (e.g., parameters in data processing and database search software), creates substantial challenges for the evaluation of data quality.
A common practice used to evaluate the LC-MS system performance for proteins is to analyze a standard protein digest and report the corresponding sequence coverage obtained. For peptide mass mapping experiments, near complete protein sequence coverage is required for verification of the amino acid sequence of the product, which may require the use of multiple proteases. Many laboratories determine the sequence coverage of bovine serum albumin (BSA) tryptic digest in order to rapidly evaluate instrument performance because it is sensitive to method settings in both MS1 and MS2 acquisition modes. However, measurement of system performance in this manner may not reflect many other important components of LC-MS analysis, such as the detection limit, dynamic range, and peak area precision.
Other comprehensive approaches for LC-MS experiment quality evaluation have been reported for a variety of applications, with different focuses ranging from sample preparation to instrument performance.15-19 For example, a full set of metrics covering a wide range of aspects, such as chromatography, ionization, mass accuracy, signal strength, dynamic sampling and peptide identification, have been proposed to evaluate system performance and quality of datasets in discovery proteomics where the goal is to identify large numbers of proteins in a complex mixture.15 Similar efforts have been made in defining performance metrics to benchmark instrument performance for targeted protein quantitation in biological matrix.17,18 Although some of the metrics have much in common and can be applied to LC-MS of protein therapeutics, many of them are either too comprehensive or do not directly address specific issues for complete assessment of a high purity protein sample, particularly for defining the detection limit of low abundance impurities. Therefore, it is beneficial to study the experimental variables, and design a test procedure that will directly demonstrate system suitability of mass spectrometer components specifically for LC-MS-based protein therapeutic characterization that can be performed directly prior to sample analysis in order to assist in the optimization of instrument settings and demonstrate to regulatory reviewers that a particular LC-MS system is fit for its intended purpose.
While a variety of applications that use LC-MS/MS for the analysis of protein therapeutics exist, this work focuses on the 2 most common applications: sequence identification (peptide mapping) and impurity testing (detection and relative quantitation of sequence variants, degradants and other forms). In this study, a protein digest standard spiked with peptides at different concentrations, simulating a protein therapeutic product containing impurities or other low abundance species, was used to develop and identify metrics that can be used to evaluate mass spectrometer performance. Peptide standards were spiked into a BSA digest at known concentrations in order to explicitly define the sensitivity and relative detection limits of the method. A set of instrument parameters that could significantly influence system performance were selected and systematically varied on 2 different commonly used LC-MS systems, a quadrupole-orbitrap (Q-Exactive) and a quadrupole-time-of-flight mass spectrometer (QTOF). While these instruments are commonly used in our and many laboratories, the results obtained on either platform should not be construed to represent all available Q-Orbitrap or QTOF platforms, as new entries to the market may feature instrumental improvements in several areas, including scan speeds and mass accuracy. The data acquired under different instrument conditions led to different results in regard to detection and relative quantitation of the spiked peptides due to changes in instrument performance.
The goal of the study is to use the experimental data as a foundation for evaluating the effectiveness of the proposed metrics. Because the mass spectrometer parameters are the focus of this study, other possible causes of experimental variability were minimized by using a pre-digested protein standard with constant LC gradient settings and an identical data processing routine. No comparison between the qualities of the instruments was intended or attempted. The instruments used in this study were selected based on laboratory availability, and are representative of 2 types of instruments commonly used for peptide mass mapping by LC-MS. The metrics presented here are generic recommendations based on the experimental data obtained in this study. The general methodology can be adapted on a case-by-case basis with standards for demonstrating system suitability and fulfilling the regulatory requirements for specific applications specific to the product of interest. The samples and the metrics discussed here are presented as model systems and proposed workflows, and are not intended to reflect a regulatory requirement.
Results
Using spiked peptides in BSA digest with systematic change of critical instrument parameters to develop system suitability metrics
A BSA digest spiked with 11 “Intra-scan” and “Inter-scan” peptides at known concentrations was used as a reference sample to benchmark the performance of the system under different operation conditions. “Intra-scan” peptide standards in the sample consisted of 4 pairs of unlabeled and isotopically labeled peptides that would co-elute. Intra-scan peptides are used to simulate the detection of one species in the presence of a strong interference in order to examine intra-scan dynamic range. Isotopically labeled peptide pairs were chosen because the peptides in each pair are expected to have essentially the same ionization efficiency; thus, the ratios between peptide signals would directly reflect the concentration ratios. The four labeled (heavy) peptides were added into the BSA digest at a signal normalized concentration corresponding to ∼100% or 10% of the BSA peptide concentration in order to adjust for differences in ionization efficiency between different peptides (see Supplemental Material). Each unlabeled (light) peptide in the 4 intra-scan peptide pair was spiked at signal normalized concentrations of 0.1%, 1%, 10%, or 100% of the corresponding labeled (heavy) peptide concentration. “Inter-scan” peptide standards in the sample consisted of a set of synthetic bivalirudin sequence variants spiked in signal normalized concentrations of 100%, 10%, 1%, or 0.1% relative to the response of the most abundant BSA peptides eluting during the same range of retention times. The inter-scan peptides are used to simulate label-free quantitation of sequence variants at different concentrations for evaluating the inter-scan dynamic range. A representative LC chromatogram of the BSA digest spiked with the intra-scan and inter-scan peptide standards is shown in Figure 1. The relative quantitation of intra-scan peptides was determined by the peak area ratio of the light to heavy isotopically labeled peptide pairs. The relative quantitation of inter-scan peptides was determined by the peak area ratio of the inter-scan peptide at 10%, 1%, or 0.1% to the inter-scan peptide at 100%. The limit of detection (LOD) was defined as signal-to-noise ratio (S/N) larger than 3 from the extracted ion chromatogram (EIC) for the target m/z value of a given spiked peptide. Additional details of the sample are included in the Method section and the Supplemental Material.
Figure 1.
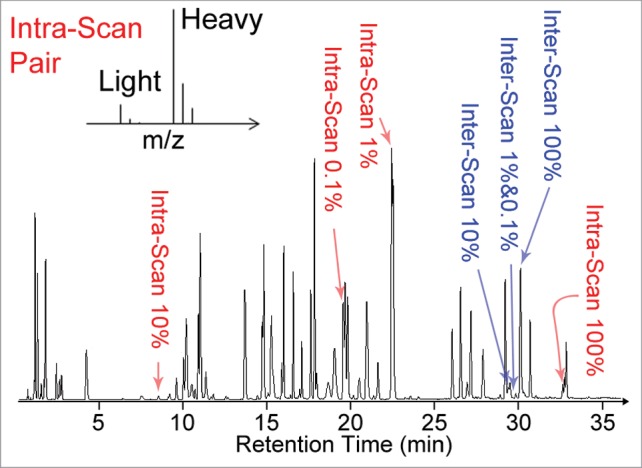
Representative total ion chromatogram of the BSA digest spiked with synthetic peptides used in this study for evaluation of system suitability. The intra-scan peptides (highlighted in red) were spiked in pairs consisting of one light peptide and one heavy isotope labeled peptide (schematic mass spectrum representation shown in insert). The light peptides were present at signal normalized concentrations corresponding to 0.1% to 100% of the heavy peptide concentrations in each pair. The inter-scan peptides (highlighted in blue) are closely related synthetic peptides eluting at similar retention times around 30 min. The other peaks in the chromatogram correspond to BSA peptides. The signal from the inter-scan peptides present at signal normalized concentrations of 1% and 0.1% are too low to be directly observed on the total ion chromatogram.
Four MS parameters (source voltage, MS1 scan time, MS2 scan time, MS2 precursor selection threshold) were selected and varied systematically to create 9 different data-dependent MS2 acquisition methods (M1 to M9 in Table 1). An additional method M0, featuring only MS1 scans (no MS2) was also evaluated. Source voltage settings were selected to optimize ion transfer at the front end of the mass spectrometers. MS1/MS2 scan time was defined by the automatic gain control (AGC) / ion injection settings on the Q-Exactive, and the scan speed (number of scans per second) on the QTOF. The MS2 selection threshold specified the minimum signal for an ion to be selected for CID. Although the 10 methods in this study cannot possibly probe every single situation for all applications, they cover several key parameters that are often empirically chosen and varied across different analyses. Improper settings would lead to system underperformance as highlighted in the following results. The experimental results from these methods were used to examine how well the performance of the LC-MS systems can be defined by the proposed system suitability metrics.
Table 1.
Experimental method settings with systematic variation of several parameters. “+” sign indicates high source voltage; long MS1/MS2 scan time/accumulation; or high MS2 precursor selection threshold. “−” sign corresponds to lower settings. M0 is a MS1 only method, thus the MS2 parameters are listed as “NA” (not applicable)
| Method ID | Source Voltage | MS1 Scan Time | MS2 Scan Time | Selection Threshold |
|---|---|---|---|---|
| M0 | − | + | NA | NA |
| M1 | − | − | + | − |
| M2 | + | − | + | − |
| M3 | − | + | + | − |
| M4 | − | − | − | − |
| M5 | − | − | + | + |
| M6 | − | + | − | − |
| M7 | − | + | + | + |
| M8 | − | − | − | + |
| M9 | − | + | − | + |
Protein sequence coverage provides limited assessment of system suitability
Despite its widespread use, sequence coverage of a digested protein standard provides only limited information in regards to the suitability of a MS system for protein therapeutic analysis. Figure 2 summarizes the BSA sequence coverage based on identified peptides with high-confidence peptide-to-spectrum assignments (high-confidence peptides) from experimental data acquired on a Q-Exactive and a QTOF mass spectrometer using each of the 9 data-dependent MS2 acquisition methods. A high confidence threshold was used to filter the peptide identifications in order to stringently evaluate the MS2 performance of the systems (see Method section for details). Reasonable sequence coverage values near 70% were achieved on both systems, with some methods showing better identification performance as reflected by slightly higher sequence coverage values (M1-M3, M5, M7 on Q-Exactive; M1, M3, M7 on QTOF). For a protein digest of relatively low complexity (such as BSA digest standard), acquisition parameters could be adjusted further to reduce the number of MS2 scans collected in each experiment without affecting the overall coverage (M7 vs. M1-M3 on Q-Exactive; M7 vs. M1, M3 on QTOF), as only a small portion of the MS2 spectra generated led to high-confidence peptide identification in most methods. Variation of the data-dependent MS2 triggering condition did not necessarily compromise the overall sequence coverage for BSA. This result allows for the flexibility to tune and optimize data-dependent MS2 settings for specific applications. For example, reducing the number of MS2 scans can increase the number of MS1 scans to better define peak shapes for relative quantification based on peak areas. However, this result also implies that sequence coverage is not sufficiently sensitive to changes in other MS performance parameters, such as the number of MS2 scans when working with simple systems. More importantly, sequence coverage only reports on the ability to detect major protein species (BSA) in the sample and does not inform on the instrument sensitivity for the detection of low abundance species, as further discussed in the following sections. As sequence coverage can also be impacted by experimental factors (i.e., differences in proteolytic peptides formed may cause some to lie outside the m/z range of the experiment, causing complete coverage to be impossible), a standard protein should be selected on a case by case basis to properly reflect the environment of which the target analyte will experience. Sequence coverage of a standard protein alone is thus an inadequate metric for complete assessment of system suitability for protein therapeutic characterization.
Figure 2.
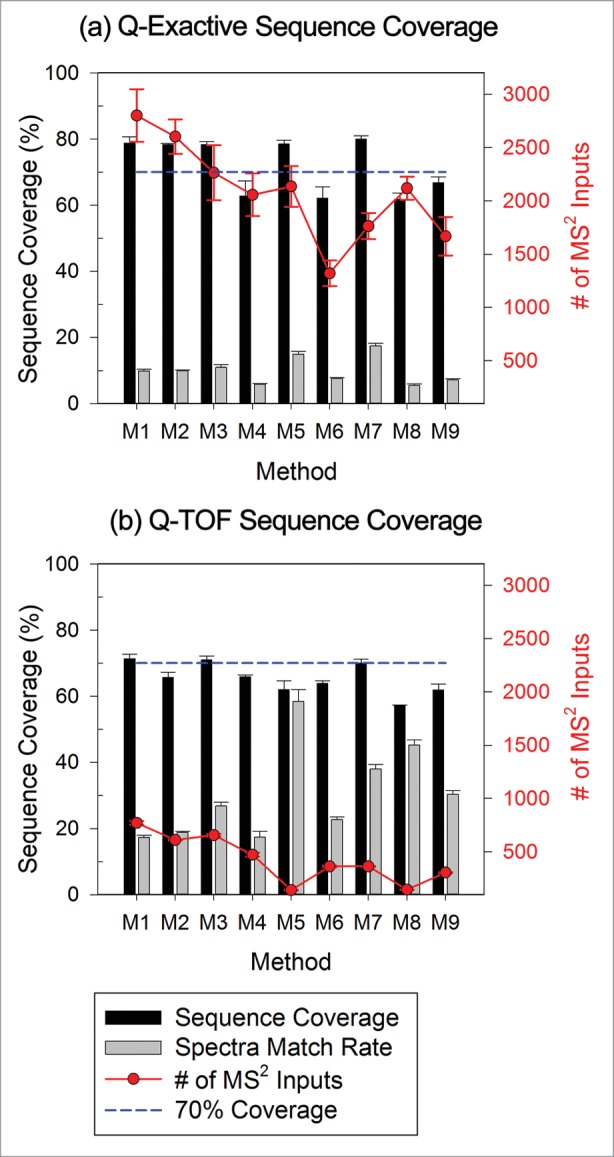
Influence of method settings on protein sequence coverage (black bars) for data from (a) Q-Exactive and (b) QTOF instrument as reported by Proteome Discoverer. Gray bars indicate the average percentage of MS2 spectra matched to peptides for each method (number of spectra associated with high-confidence peptides divided by the total number of spectra input into the search algorithm). The number of MS2 spectra inputs into the search algorithm is also included as line plots following the y-axis on the right.
Using spiked peptides to evaluate MS2 quality and sensitivity
As protein sequence coverage only assesses the ability of a mass spectrometer to identify major peptide species in a sample, other metrics are needed to evaluate system performance. In order to probe the quality of MS2 spectra for a single target peptide, a normalized MS2 score was defined for the spiked peptide standards as:
The normalization reduced the differences in score values resulting from differences in charge states, instruments or search parameters. The Sequest module in Proteome Discover (Thermo, San Jose, CA) was used to generate the cross correlation (XCorr) scores that were used as the peptide score. The high-confidence cutoff score was manually determined by choosing the cutoff score around the highest score of false positives (random match to other non-BSA proteins), without losing significant true positives by visually inspecting the identified BSA peptides (alleged true positives) at low scores. The cutoff score was chosen for both the Q Exactive and QTOF data so that the overall false discovery rate was below 0.5% (more details in Supplemental Material 1.3).
Figure 3 shows the normalized MS2 score for inter-scan peptides spiked at a signal normalized concentration of 10% on both instruments across all 9 methods. A score of zero means that the peptide was not identified by MS2 using the current method settings (e.g., MS2 was not triggered experimentally or spectral quality was too low to generate an identification). Scores between 0 and 1 indicate that an identification was made but did not meet the high confidence peptide identification criteria. The methods acquired on the Q-Exactive were split into 2 groups (M1, M2, M3, M5, M7 vs. M4, M6, M8, M9). The second group of methods had lower MS2 scores that reflected the shorter MS2 scan time (lower MS2 AGC setting) used in those methods; for these methods, the total signal accumulated for each MS2 spectrum was reduced. Lower signal accumulation resulted in lower quality MS2 identifications, reflected in the lower MS2 scores. Several methods (M4, M5, M6, M8, M9) on the QTOF platform failed to consistently identify the spiked peptides with high confidence in all 3 triplicate runs as a result of either high selection threshold settings (observed for M5 and M8), or short MS2 scan times causing poor MS2 spectra quality (observed for M4, M6, and M9). These data demonstrate that changes in acquisition parameters can cause a high degree of variability, resulting in the generation of poor quality data that can easily lead to false negative MS2 identifications without proper method optimization, even in the face of acceptable sequence coverage of a protein digest standard.
Figure 3.

MS2 scores for inter-scan peptide standards at signal normalized concentrations of 10% for Q-Exactive and QTOF systems. Higher scores were associated with higher confidence in identification. Diamonds indicate the confidence interval, with the center line indicating the mean and the vertical span representing the 95% confidence interval.
Analysis of MS2 scores was also used to determine a relative detection limit and dynamic range for MS2 methods (i.e., identification of low concentration spiked peptides in the presence of other strong ions) for inter-scan and intra-scan spiked peptides. While the spiked peptides were detected by most methods in MS1 scans, many of the methods did not generate MS2 spectra of sufficient quality for low abundance spiked peptides (≤1 % for inter-scan peptides and ≤10 % for intra-scan peptides). A common reason why the spiked peptides were not selected for MS2 could be attributed to the large number of other ions present within the same scan. This observation implies that peptide identifications made based solely on MS2 spectra may feature limited sensitivity and dynamic range in relation to methods using only MS1 detection as a result of acquisition and data processing parameters. When the MS2 acquisition has not been demonstrated to be sensitive enough to identify species at the lowest desired concentration, peptides identified based only on mass matching from MS1 spectra should be considered in order to minimize false negative identification of low abundance species.
Mass accuracy needs to be experimentally evaluated to minimize negative influences from method settings
The identification of peptides based on either MS1 or MS2 data depends on the accurate measurement of m/z for each analyte ion. The intrinsic mass accuracy for a given scan mode on a given MS system is usually well-defined by qualification specifications from the instrument manufacturer when using a valid calibration profile. However, significant shifts in mass accuracy can occur due to either temperature fluctuation or method settings. The effect of temperature on mass accuracy has been reported in literature,20 and was also observed in this study (data not shown). While laboratory temperature could be controlled or monitored appropriately to minimize the related error, the influence of method settings on mass accuracy can be easily overlooked. In order to evaluate the relationship between peptide mass error and a high confidence peptide identification, the precursor masses of BSA peptides identified with high confidence by MS2 were averaged to yield a measure referred to as the average absolute ppm error for a given set of conditions, based on output from Proteome Discoverer.
The average absolute mass error for the high-confidence BSA peptides using the 9 different acquisition methods on both MS platforms is shown in Figure 4. Most methods across both systems featured an average absolute mass error below 3 ppm. While no significant overall difference in mass error was observed among the QTOF methods, the Q-Exactive methods could be divided into 2 groups: one with average error ∼0.6 ppm and the other ∼1.6 ppm. Although the overall error was relatively low for both groups, this nonetheless indicates the acquisition parameters can alter the mass accuracy of a MS system.
Figure 4.

Average absolute mass error associated with each acquisition method on either MS platform. ANOVA analysis identifies significant differences among the Q-Exactive methods (P< 0.0001), but not for the QTOF methods (p = 0.2097). A pairwise t-test, however, suggests that significant differences in mass error between some of the methods on QTOF exist.
Further analysis of the data confirmed that the changes in mass accuracy were caused by changes in the MS1 scan time (Fig. 5a), consistent with recent reports.21 Comparison of different methods using a t-test indicated that the experiments with long MS1 scan times (high MS1 AGC) were significantly different from others acquired on the Q-Exactive. The MS1 scan time on the QTOF was also shown to influence the average mass accuracy of a MS system, with longer MS1 scans yielding smaller mass errors (Fig. 5b). In contrast to ion trap instruments, no trapping and accumulation mechanism is associated with each transient in a TOF measurement. Increasing the MS1 scan time increased the number of transients summed for a given spectrum, but did not change the ion population at each transient within a given mass scan range. Therefore, the decrease in mass error was most likely from the result of signal accumulation over a longer period of time. The data suggest that acquisition settings affect mass accuracy even for a mass spectrometer with valid calibration profile, especially for ion traps where the mass accuracy is more sensitive to the ion population.
Figure 5.
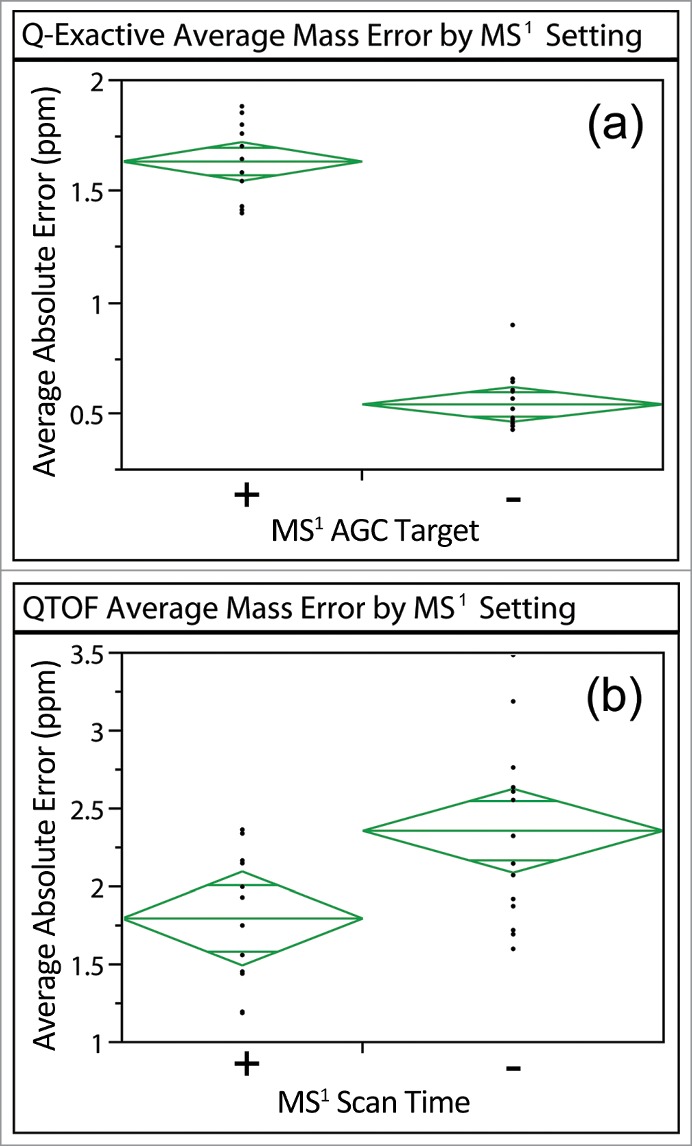
Average absolute ppm errors of high-confidence BSA peptides identified for data acquired on the Q-Exactive and QTOF sorted by differences in MS1 scan time. The (+) corresponds to a higher MS1 AGC target (Q-Exactive) or a longer MS1 scan (QTOF) than the (−). Significant differences were observed between the different MS1 settings on both systems (p < 0.0001 for Q-Exactive and p = 0.0083 for QTOF).
In addition to the MS1 scan setting, lock mass settings (an internal standard used to perform real time mass corrections) can also affect the mass accuracy. Many high-resolution MS systems now offer lock mass or internal calibrant options in order to obtain maximum mass accuracy, but incorrect usage of a lock mass can lead to increased mass error (for additional discussion, refer to Supplemental Material section 2). The results indicate that the mass accuracy of a mass spectrometer can be more appropriately defined based on actual experimental data using a known standard, rather than relying on expected instrument performance. The peptides spiked at known signal normalized concentrations can also assist in defining the mass accuracy for species at low abundance, where the mass error could potentially increase.22,23 Even for a qualified MS system with valid calibration, some acquisition settings (particularly those related to MS1 scans) can significantly affect the mass accuracy, with the extent of impact depending on the type of instrument used. Experimentally determining the mass error using a system suitability standard can eliminate the ambiguity when evaluating the confidence of peptide identifications. Similar workflows can be designed to evaluate the resolution of the system because the resolution can also change based on signal intensity (see Supplemental Material section 3). Resolution should also be evaluated using spiked peptides because the optimum extraction window size for generating EICs at the highest specificity and sensitivity is determined by the system resolution.24
Evaluating the detection limit and the accuracy of relative quantitation
The spiked intra-scan and inter-scan peptide standards were also used to evaluate the performance of the 2 LC-MS systems with the 9 acquisition methods for targeted detection and relative quantitation (Fig. 6). Missing data points in the plots indicate that the targeted species were below the LOD. Percent accuracy is defined as the experimentally determined signal normalized concentration divided by the expected signal normalized concentration. The quantitation accuracy observed on the Q-Exactive system was less than 10% for both intra-scan and inter-scan peptides present at less than a signal normalized concentration of 1%, implying that the quantitation dynamic range was quite limited for these methods on this system with this sample load. Intra-scan peptides present at signal normalized concentrations of 1% and 0.1% were not detected by some of the methods on the Q-Exactive, but all inter-scan peptides at 1% and 0.1% were detected by all methods on the Q-Exactive. The difference between intra-scan and inter-scan quantitation performance suggests that the dynamic range of MS systems can be variable depending on the intensity of other ions present within the same scan. In particular, detection of the spiked intra-scan peptide present at 1% was affected by additional interference caused by a strong co-eluting BSA peptide (stronger signal than the spiked heavy-isotope-labeled peptide), thus not accurately reflecting the scenario where an intra-scan peptide is present at a signal normalized concentration of 1%. While optimization of chromatographic conditions may circumvent this issue, this result highlights the need to understand system performance, particularly when evaluating sequence variants and low abundance modified forms.
Figure 6.

Summary of accuracy and precision results for intra-scan (left column) and inter-scan (right column) peptide pairs at different signal normalized concentration ratios on the Q-Exactive (top row) and QTOF (bottom row). Percent accuracy is defined as the experimental result divided by the expected value. The precision is represented by the standard deviation as shown with the error bars. Missing points on broken lines indicate the peptide was not detected at least twice in the triplicate runs using the associated method. Data with CV > 20% are highlighted with yellow edges on the points.
The accuracy of the relative quantitation on the QTOF was reasonably good for most of the spiked peptides. The relative quantification for the intra-scan peptide present at 0.1% featured high accuracy, similar to that observed for the intra-scan peptide present at a 100% signal normalized concentration for most methods. However, the intra-scan peptides spiked at 1% and the inter-scan peptides spiked at 0.1% were not detected with a S/N > 3 on the QTOF. Similar to what was observed using the Q-Exactive, the intra-scan peptide at 1% was not detected with S/N > 3 for the EIC due to the presence of interfering co-eluting BSA peptides. The inability to detect the inter-scan peptide present at a signal normalized concentration of 0.1% (given that the intra-scan 0.1% was detected on the QTOF platform) is likely due to the low ionization efficiency of the bivalirudin peptide. Inter-scan peptides also experience suppression from background ions, despite the fact that there were no strong interferences from other BSA peptides. The intra-scan peptides spiked at 10% did not display acceptable accuracy or sensitivity on the QTOF (35% accuracy for the 5 methods that could detect the peptide) due to the unstable signal of this peptide, as well as the relatively low concentration of the spikes (Table S1). In addition, a systematic variation of the quantitative results as a function of method was observed (e.g., M1, M2, M4 on QTOF appeared to have better results than other methods for the inter-scan peptides). This particular variation is attributed to the peak sampling and will be discussed in detail in section 6.
Several factors contributed to the observed limited dynamic range and inaccurate quantitation for these MS systems. The primary issue with the dynamic range came from co-eluting peptides. The response of a mass spectrometer for an analyte ion can be significantly affected by ionization efficiency and matrix suppression.25,26 Suppression during ion detection is another contributor to limited dynamic range. The acquisition methods on the Q-Exactive that experienced difficulties in detecting low abundance species (intra-scan in Fig. 6) were associated with low MS1 AGC settings (M1, M2, M4, M5 on Q-Exactive). The AGC target values for MS1 scans are sometimes tuned for optimum protein sequence coverage and dynamic range.21,27, 28 Although the optimal setting can vary for different compounds, a general recommendation is to fill less ions for optimization of mass accuracy and more ions for larger dynamic ranges.28 Other ion detection systems that use time-to-digital converters (TDCs), commonly used in TOF instruments, are also known to have limited dynamic range as a result of low ion current, 29,30 although improvements had been made using analog-to-digital (ADC) detection to obtain larger dynamic ranges.31 Accumulation of a larger number of transients (i.e., longer MS1 scans: M0, M3, M6, M7, M9 in Fig. 6c) can improve the S/N for better detection of low abundance species. The data in Figure 6 demonstrate that the accuracy and dynamic range for relative quantitation can be affected by multiple factors, including chromatographic, ionization, mass spectrometric detection, and cannot be guaranteed by simply having good mass accuracy or high resolution. Peptide standards spiked into the sample can be used to gauge the accuracy in relative quantitation experiments across a range of concentrations.
Interestingly, only one method on the QTOF, M2, did not detect the intra-scan peptide present at 0.1%. The high source voltages used in this method largely depleted the precursor ion signal of this fragile triply-charged peptide. The ion source settings affect the signal strength and stability of the ions entering the mass spectrometer. While normally optimized for good transmission of ions of interest, they must also be set low enough to avoid excessive activation of the ions. Fragile peptides can fragment and be depleted under harsh source conditions, resulting in false negatives in identification or underestimated concentration determinations. For example, the observed distribution of various glycoforms of peptides from monoclonal antibodies can change depending on the selected source voltages, with high source voltage generating more non-native glycoforms (data not shown). Spiking a fragile peptide as a reference standard that is the same as or highly similar to the type of peptide of interest in the sample can be used to probe the harshness of the selected source conditions on a given system and the suitability of those settings for a specific application.
Inadequate acquisition parameters can affect relative quantitation due to differential peak sampling
In the dataset discussed in Figure 6, several QTOF methods (M1 in particular, even though it has MS1 scans >15 across all EIC peaks) detected higher relative concentrations than those observed using the MS1 only method (M0) for the same dataset. A more in-depth examination of the data reveals that discrepancies in the determined signal normalized concentration were associated with differential EIC peak sampling depending on the intensity of the peak present. The peak sampling of the spiked (inter-scan) bivalirudin sequence variants, present at signal normalized of 1% or 10% of the bivalirudin peptide present at a signal normalized concentration of 100%, were examined (Fig. 7). To accomplish this, the intensity ratio obtained for each inter-scan peptide pair was normalized with the experimental ratio obtained in the MS1-only experiment (M0), as shown in the black plot with filled circles in Fig. 7 (Note: method M2 was excluded from this analysis as only source voltage was modified in this method). As a MS1 only method, EIC peak sampling in M0 is expected to be relatively uniform across all signal intensities, although minor AGC-based fluctuations on the Q-Exactive may be present. The presence of MS2 scans in methods M1-M9 decreases the frequency at which MS1 scans are obtained in a stochastic or data-dependent manner. To examine this trend, a sampling factor was calculated for each method, as shown in the following equation:
Figure 7.
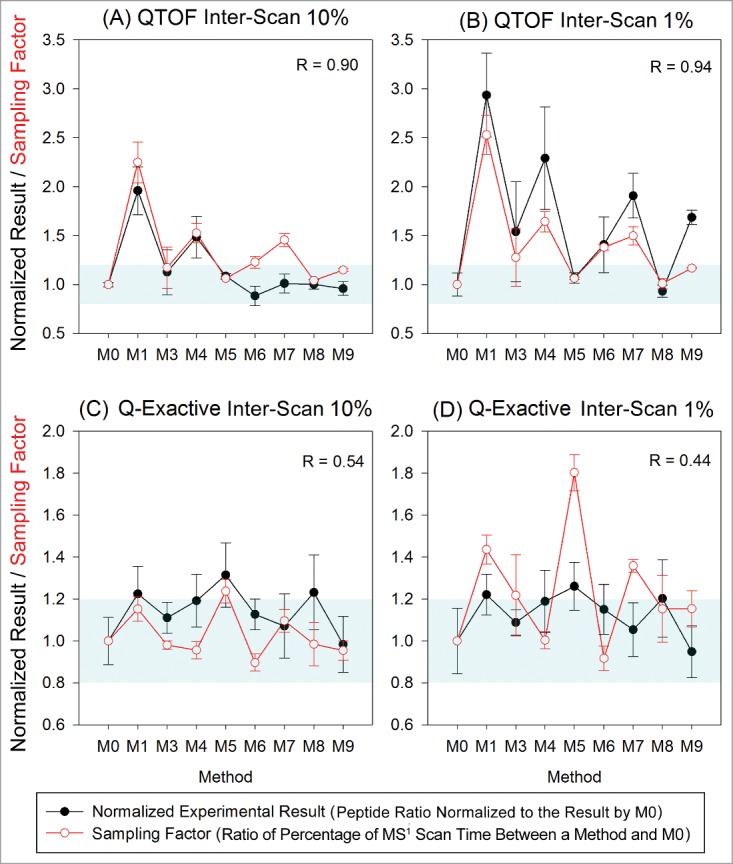
Variation in quantitation results relative to the MS1 only method (M0) due to variation in peak sampling for 2 inter-scan peptides on both the QTOF and Q-Exactive. The sampling factor featured good correlation with the QTOF data, but not with the Q-Exactive data as manifested by the correlation coefficients (R) noted in the plots. The ranges where the normalized results are 0.8∼1.2 (relative quantitation result within ± 20% of that by method M0) are highlighted in blue in each plot.
MS1 scan time refers to the amount of time spent on MS1 scans across the duration of the full width of an EIC peak. The MS1 scan time divided by the total duration of the EIC peak gives the percentage of MS1 scan time across the EIC peak. The sampling factor is the ratio of the amount of time spent on the MS1 scan compared to the full scan time for the entire EIC peak between the lower abundance peptide and the more abundant peptide in the pair, as shown above.
A sampling factor of 1 corresponds to EIC peak sampling between the 2 peptides, independent of signal concentration, while a sampling factor greater than 1 indicates more MS2 scans (and correspondingly, more scan time) were taken for the peptide at 100% than the peptides at 10% or 1%. Differential sampling across the 2 peptide EIC peaks resulted in changes in detected peak areas, thus causing different relative peak area ratios to be calculated, which would interfere with accurate quantitation. For example, the data acquired on the QTOF with M1 showed that over 80% of the time across the EIC peak was spent on the MS1 scan for inter-scan peptides at 10% and 1%, whereas the of MS1 scan time was only about 30% of the total scan time for the inter-scan peptide at 100% (i.e., more MS2 scans triggered based on the method settings). In contrast, 40–50% of the scan time was spent on MS1 scans for inter-scan peptides at all 3 concentrations (100%, 10%, 1%) analyzed with M3 on the QTOF. The final relative quantitation result by M3 was only 1.1 times higher than MS1-only method (M0), whereas M1 yielded a value 2 times higher than the result obtained using method M0. The correlation coefficients of the metric value with the normalized quantitation result were greater than 0.9 for inter-scan peptides at 10% and 1% on the QTOF, but the correlation was poor for all data acquired on the Q-Exactive. This effect likely was due to the fact that the scan time on Q-Exactive was not constant; thus, the sampling factor could not accurately represent the variability in sampling caused by the AGC settings selected. Although the overall differences between methods on the Q-Exactive were not as significant for 10% and 1% inter-scan relative concentrations (p = 0.079 and 0.040, respectively, for ANOVA) compared to the QTOF data (both p < 0.0001), several methods (M1, M5, M8) on Q-Exactive did show relative quantitation results more than 1.2 times larger than the results obtained from M0 (difference larger than 20%). To ensure proper, more accurate relative quantitation, peak sampling should be monitored by comparing the result using MS2 methods to the result obtained by a MS1 only (M0) method (shown as normalized experimental result in Fig. 7) for this type of instrument. Most intra-scan quantitation results did not show significant differences between methods (data not shown). Presumably, this behavior results from the fact that the intra-scan peptide pairs experience the same scan conditions, causing the differences in sampling to be largely normalized.
This observation further highlights the need to examine peak sampling for data dependent MS2 acquisition methods intended for relative quantitation applications. The sampling rate is associated with MS2 scan rate and trigger settings in the method, which can be highly variable across methods depending on the specific application. In discovery proteomics, acquisition of large numbers of fast MS2 scans can maximize the number of peptide identifications.32 However, some settings for optimized discovery applications will reduce the number of MS1 scans that can be used for quantitative analysis. Insufficient sampling across a chromatographic peak can result in peak shape distortion, affecting integration. As such, the number of MS1 and MS2 scans should be balanced to provide sufficient MS2 spectra for high-confidence identifications without compromising the ability to perform relative quantitative analysis from a sufficient number of MS1 scans. A method that is identical to the proposed data-dependent MS2 method but with MS2 acquisition excluded (i.e., same LC and MS1 scan settings) can be used to evaluate the effect of dynamic sampling. The relative quantitation results for the target peptides should be expected to yield similar results for both the MS1 only method and the proposed method (e.g., the results should be similar to method M0 used in this study).
Discussion
Overall, the ability to detect and perform relative quantitative measurements on both systems featured limited sensitivity and accuracy across all methods examined in this study at the given sample load (0.2 µg on a narrow bore column). Although it is very likely that the experimental conditions (e.g., MS methods, LC methods, injection amount) can be optimized to achieve better performance on either instrument used in this study, the results highlight the necessity for careful evaluation of system suitability before analyzing any sample. Ignoring sample-specific issues (e.g., strong co-eluting species, differences in ionization efficiencies), significant variation in the detectability of low abundance peptides was observed to be a function of method parameters (e.g., MS1/MS2 scan accumulation time, MS2 triggering parameters, ion source voltages). Meanwhile, some peptides were not detected by any of the methods on a given system, reflecting intrinsic instrument sensitivity limits with the methods examined (e.g., inter-scan peptides with a 0.1% signal normalized concentration on the QTOF). Although a dynamic range of 3–4 orders of magnitude is achievable for full MS scan modes under optimized conditions,27,28 it is important to use a set of metrics to explicitly demonstrate the suitability of a system to probe the intra-scan/inter-scan LOD in order to reduce the possibility of false negative identifications caused by incorrect method settings or intrinsic instrument limitations.
MS analysis of protein therapeutics is generally not sample limited for the abundant protein species, and the concentration of the absolute LOD can be readily obtained. Sample load can be increased within the column loading capacity or detector saturation if the target analyte is below the absolute LOD of the MS system. However, higher sample load will not completely improve the dynamic range if the signal is restricted by suppression of another species in the sample for either chemical or instrumental reasons. The two categories of spiked peptides used throughout this study, inter-scan and intra-scan, can be used in combination to separately evaluate the absolute LOD and the dynamic range. The inter-scan peptides experience interference predominantly from background ions (e.g., solvent, ambient ions), and allow a relative limit of detection for a given sample load to be determined. The intra-scan peptides evaluate the dynamic range of detecting low abundance species (the non-isotopically labeled or light peptide) in the presence of a strong co-eluting species (the heavy or isotopically–labeled peptide). A similar sample load should be used for the system suitability test standard and the intended analyte sample to best mimic both the limits of relative quantification and dynamic range. The spiked peptides could also benchmark the confidence for identification of unknowns at different concentrations as discussed in the previous sections (e.g., mass error and acceptance range, MS2 quality and sensitivity).
All the system suitability metrics discussed are summarized in Table 2, and a proposed workflow is shown in Figure 8. The main purpose of these metrics is to provide an explicit measure of analytical performance for a given LC-MS system with generic parameters that can be applied across wide range of applications, so that it becomes straightforward to determine if the system is suitable for the intended purpose. Fit for purpose is a regulatory standard defined in the Guidance for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics Draft Guidance,33 as well as 21 CFR 211.165(e) and 211.194(a)(2). In a system suitability test, the standard sample should be analyzed with the proposed method to be tested on the selected LC-MS system with replicate analyses. Additionally, a separate method identical to the intended method, but with MS2 disabled, should be included to test peak sampling. The first stage is to ensure the instrument is functioning reproducibly with a stable signal. The system then needs to pass the most basic performance tests, including protein sequence coverage, average mass accuracy, resolution, and proper ion source settings. The mass accuracy and resolution should fall within the acceptable range of instrument specifications, and thus are instrument specific. Failure at this stage normally would indicate fundamental instrument or tuning problems. The protein sequence coverage, however, is primarily affected by method settings. Ion source settings need to be evaluated if the target of the analysis includes any fragile species, as discussed previously in the Results section.
Table 2.
Summary of the proposed metrics for system suitability test, with proposed acceptance criteria and possible troubleshooting targets. The thresholds of 15% for CV and +/− 20% for relative quantitation are adapted from current FDA recommendations34
| Metric | Purpose | Definition | Recommended Threshold | Possible Cause for Failure |
|---|---|---|---|---|
| CV of Peak Area | Check signal stability | %CV of peak area of EIC at 100% | <15% | Instable signal, likely an issue at the electrospray |
| Protein Sequence Coverage | Check method settings for identification capability | Determined from search algorithm based on the confidence level chosen | Near complete is ideal | Acquisition setting, intrinsic sensitivity, LC separation |
| Average Mass Error | Report overall mass accuracy | Average of absolute ppm error of all identified peptides with high confidence (or known peptides) | From system specification <5 ppm or lower (High resolution) | Temperature change, invalid calibration, incorrect method settings (e.g., lock mass, AGC) |
| Mass Resolution | Check instrument resolution | Full width at half maximum mass peak resolution at 100% concentration | Within instrument specification (instrument and operation mode dependent) | Invalid instrument tuning/qualification, instrument malfunctioning |
| Ion Source Settings | Minimize non-native species generated in source | Monitor the relative abundance of a fragile peptide in the standard | Maintain sensitivity while minimize excess activation of fragile species | Source tuning, source acceleration voltage too high |
| S/N of EIC | Defining LOD | S/N of target peptide EIC | > 3 | Bad instrument tuning, bad EIC peak shape, bad spraying condition, intrinsic sensitivity limit of system |
| MS2 Identification Score | Check MS2 quality at different concentrations | Target peptide score/cutoff threshold for intra and inter-scan | ≥1 | Acquisition setting, intrinsic sensitivity, LC separation |
| Mass Accuracy/Resolution Change | Evaluate confidence of mass measurement at low concentrations | Compare mass error/resolution at low and 100% concentration | Ideally minimal change at low concentrations | Instrument specific and can be normal to see mass error/resolution change at different signal intensities. Need to adjust data analysis procedure accordingly. |
| Accuracy of Relative Quantitation | Check accuracy and dynamic range of quantitation | Percent accuracy of the experimental result relative to the expected result | 80% –120% | Bad EIC extraction method, co-eluting species, instrument intrinsic limit |
| CV of Relative Quantitation Result | Check precision of quantitation | %CV of peak area ratios of a target peptide pair | <15% unless only intended for semi-quantitation | Low sensitivity/bad peak shape at the concentration, incorrect data processing, interfering co-eluting species |
| Differential Peak Sampling | Check potential error in quantitation due to stochastic sampling across EIC peak | Ratio of relative quantitation results between the proposed method and the MS1 only method | 0.8∼1.2 Use the MS1 only method for quantitation if exceeds the range | Improper MS2 settings such as the selection threshold, MS1 and MS2 scan rate, etc. |
Figure 8.

Proposed workflow for a system suitability test.
The next stage evaluates the ability of a system to detect low signals. This evaluation includes qualitative aspects such as LOD as defined by S/N of EIC, as well as MS2 sensitivity as defined by the identification score. The minimum concentrations at which spiked peptides can be detected by MS1 (peptide mass only) and MS2 (fragment ion matching by search algorithm) benchmark the MS1 and MS2 sensitivity of the system, respectively. If there is a significant change in mass accuracy or resolution at low signal intensities, it may be necessary to reconsider the data processing parameters at such intensities. This set of metrics is sufficient to demonstrate system suitability for peptide mapping experiments. For applications that involve detection and relative quantitation of sequence variants or modified forms, the quantitative aspects can be evaluated by the error, coefficient of variation (CV) of the relative quantitation results, and the differential peak sampling test for spiked peptides at a set of relative concentration values, which can define the accuracy, precision, the intra-scan and inter-scan linearity of the method. If the quantitative performance at the desired (low) concentration does not satisfy the requirements for the intended purpose, the method will need to be amended or an alternative technique should be used (such as triple quadrupole-based targeted quantitation methods).
The integrated workflow covering both the qualitative and quantitative aspects of the analysis is expected to provide a more complete assessment of system suitability, especially because it is able to address the quality and sensitivity of detection and relative quantitation of low abundance species. The experiments in this study used a BSA digest as a matrix and several spiked peptide standards with reasonably similar complexity to real samples, all of which were selected in part because they were available at the time of the study, to generate a test dataset as a proof of principle with minimal variation from other analytical processes. Different compounds may exhibit different linear dynamic ranges as a result of different responses to interference.28 In addition, differences in sample matrix will also result in different co-eluting species, even with the same LC conditions. Therefore, the use a specific standard for a system suitability test that can optimally simulate the properties for the intended sample would be beneficial, allowing the metrics values determined from the standards to be more applicable to the actual sample (e.g., using an antibody digest with spiked antibody analog peptides as a system suitability standard for analysis of antibody digests). The spiked peptides used in this study were selected based on their availability in our laboratory and used as a model system; they do not represent an optimum standard for establishing system suitability for universal applications. Any isotopically labeled peptide pairs can be used as intra-scan spikes, and any set of peptide sequence variants can be used to evaluate system suitability following the general workflow discussed in this work. While it is anticipated that more specific reference materials will be developed by various vendors and agencies to suit different applications, the system suitability metrics discussed here provide a basic framework for a workflow that would allow MS system performance to be critically and experimentally evaluated in order to facilitate the application and review processes for protein therapeutics that include LC-MS data.
Methods/Materials
MassPrep BSA digestion standard was purchased from Waters and used directly without purification. Synthetic peptides were spiked into the BSA protein digest at various concentrations (Table S1). The intra-scan peptides used were β-amyloid (1–15), angiotensin I, angiotensin II, BSA fragment 349–359. The inter-scan peptides were bivalirudin sequence variants (native, plus-Gly, des-Gly, β-Asp). LC separation was performed using a Waters Acquity UPLC BEH C18 column (2.1 × 100 mm, 1.7 µm) at 40°C using Optima LC-MS Grade Water and acetonitrile (Fisher), each containing 0.1% formic acid as mobile phase A and B, respectively. A 40 minute gradient starting at 5% B and ending at 40% B was used. The spiked BSA standard was analyzed on 2 different high resolution LC-MS systems: a Q-Exactive (Thermo Q-Exactive equipped with Accela HPLC) or a QTOF (Agilent QTOF 6520B equipped with a 1290 UHPLC), with 10 different methods used on each system as listed in Table 1 (for more details, see Tables S2 and S3). Each method was run in triplicate, and the sequence of the runs (30 runs on each instrument) was randomized. No lock mass was applied for the methods M0 to M9.
The data produced from methods M1-M9 on both MS systems were analyzed by ProteomeDiscoverer 1.4 (ThermoScientific) to obtain protein sequence coverage, average mass error based on the population of identified peptides, and peptide MS2 identification scores. The protein database including the BSA and all spiked peptide sequences was appended with a bovine protein database (a total of 1065 sequences). Protein identifications other than BSA were considered as false positives to provide a measurement of false discovery rate (FDR). The manually selected peptide high confidence filter (minimal Xcorr score 1.6 for +2 ions; 2.2 for +3; 3.2 for +4 and above) resulted in experimental FDR of 0.13% on Q-Exactive and 0.21% on QTOF each for all data acquired by 9 different methods.
EIC generation and integration were performed in Thermo Xcalibur Processing Setup (Q-Exactive) and Agilent MassHunter (QTOF). Mass extraction windows used were 20 ppm for Q-Exactive data and 100 ppm for QTOF data. The intra-scan peptide ratios at 100%, 10%, 1%, 0.1% were defined as the peak area ratio for the light isotope with respect to the heavy isotope peptide. The inter-scan peptide ratios are defined as the peak area ratio of the bivalirudin sequence variants at 10%, 1%, and 0.1%, relative to the one at 100% of endogenous BSA peptide signal to yield signal normalized concentrations. The signal normalized concentrations allowed for intensity ratios to be corrected to match expected theoretical results for these ratios using the “1:1 mix” reference standard in order to minimize the errors from differences in ionization efficiencies between different peptide sequences and any experimental errors from sample preparation. The “1:1 mix” standard contains the same spiked peptides in the BSA digest discussed earlier, but the concentrations of the spiked peptides are different: all the light intra-scan peptides have the same concentration as the heavy peptides, and all the bivalirudin peptides were spiked at 100%. Further details of the materials and experimental conditions can be found in the Supplemental Material section 1.
Supplementary Material
Disclosure of Potential Conflicts of Interest
The ideas, findings, and conclusions in this publication have not been formally disseminated by the US. Food and Drug Administration (FDA) and should not be construed to represent any agency determination or policy.
Acknowledgments
The authors acknowledge manuscript preparation assistance from Dr. Michaella Levy and Anneliese Faustino.
Funding
This study was supported in part by an appointment to the Research Participation Program at the FDA Center for Drug Evaluation and Research, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and FDA (MZ).This work was funded by the FDA Center for Drug Evaluation and Research Critical Path program (MTB).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianférani S. Characterization of therapeutic antibodies and related products. Anal Chem 2012; 85:715-36; PMID:23134362; http://dx.doi.org/ 10.1021/ac3032355 [DOI] [PubMed] [Google Scholar]
- 2.Zhang Z, Pan H, Chen X. Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom Rev 2009; 28:147-76; PMID:18720354; http://dx.doi.org/ 10.1002/mas.20190 [DOI] [PubMed] [Google Scholar]
- 3.Mann M, Kelleher NL. Precision proteomics: the case for high resolution and high mass accuracy. Proc Natl Acad Sci U S A 2008; 105:18132-8; PMID:18818311; http://dx.doi.org/ 10.1073/pnas.0800788105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith LM, Kelleher NL. Proteoform: a single term describing protein complexity. Nat Meth 2013; 10:186-7; http://dx.doi.org/ 10.1038/nmeth.2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gucinski AC, Boyne MT. Evaluation of intact mass spectrometry for the quantitative analysis of protein therapeutics. Anal Chem 2012; 84:8045-51; PMID:22916992; http://dx.doi.org/ 10.1021/ac301949j [DOI] [PubMed] [Google Scholar]
- 6.Wang B, Gucinski AC, Keire DA, Buhse LF, Boyne Ii MT. Structural comparison of two anti-CD20 monoclonal antibody drug products using middle-down mass spectrometry. Analyst 2013; 138:3058-65; PMID:23579346; http://dx.doi.org/ 10.1039/c3an36524g [DOI] [PubMed] [Google Scholar]
- 7.Ni W, Lin M, Salinas P, Savickas P, Wu S-L, Karger B. Complete mapping of a cystine knot and nested disulfides of recombinant human arylsulfatase a by multi-enzyme digestion and LC-MS analysis using CID and ETD. J Am Soc Mass Spectrom 2013; 24:125-33; PMID:23208745; http://dx.doi.org/ 10.1007/s13361-012-0510-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiśniewski JR, Mann M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal Chem 2012; 84:2631-7; PMID:22324799; http://dx.doi.org/ 10.1021/ac300006b [DOI] [PubMed] [Google Scholar]
- 9.Gatlin CL, Eng JK, Cross ST, Detter JC, Yates JR. Automated identification of amino acid sequence variations in proteins by hplc/microspray tandem mass spectrometry. Anal Chem 2000; 72:757-63; PMID:10701260; http://dx.doi.org/ 10.1021/ac991025n [DOI] [PubMed] [Google Scholar]
- 10.Dick Jr LW, Mahon D, Qiu D, Cheng K-C. Peptide mapping of therapeutic monoclonal antibodies: Improvements for increased speed and fewer artifacts. J Chromatogr B 2009; 877:230-6; http://dx.doi.org/ 10.1016/j.jchromb.2008.12.009 [DOI] [PubMed] [Google Scholar]
- 11.Shah B, Jiang X, Chen L, Zhang Z. LC-MS/MS peptide mapping with automated data processing for routine profiling of N-glycans in immunoglobulins. J Am Soc Mass Spectrom 2014:1-13; PMID:24249043 [DOI] [PubMed] [Google Scholar]
- 12.Lowenthal MS, Liang Y, Phinney KW, Stein SE. Quantitative bottom-up proteomics depends on digestion conditions. Anal Chem 2014; 86:551-8; PMID:24294946; http://dx.doi.org/ 10.1021/ac4027274 [DOI] [PubMed] [Google Scholar]
- 13.Bakalarski C, Haas W, Dephoure N, Gygi S. The effects of mass accuracy, data acquisition speed, and search algorithm choice on peptide identification rates in phosphoproteomics. Anal Bioanal Chem 2007; 389:1409-19; PMID:17874083; http://dx.doi.org/ 10.1007/s00216-007-1563-x [DOI] [PubMed] [Google Scholar]
- 14.Addona TA, Abbatiello SE, Schilling B, Skates SJ, Mani DR, Bunk DM, Spiegelman CH, Zimmerman LJ, Ham A-JL, Keshishian H, et al.. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotech 2009; 27:633-41; http://dx.doi.org/ 10.1038/nbt.1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudnick PA, Clauser KR, Kilpatrick LE, Tchekhovskoi DV, Neta P, Blonder N, Billheimer DD, Blackman RK, Bunk DM, Cardasis HL, et al.. Performance metrics for liquid chromatography-tandem mass spectrometry systems in proteomics analyses. Mol Cell Proteomics 2010; 9:225-41; PMID:19837981; http://dx.doi.org/ 10.1074/mcp.M900223-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallien S, Bourmaud A, Domon B. A Simple protocol to routinely assess the uniformity of proteomics analyses. J Proteome Res 2014; 13:2688-95; PMID:24617767; http://dx.doi.org/ 10.1021/pr4011712 [DOI] [PubMed] [Google Scholar]
- 17.Abbatiello SE, Mani DR, Schilling B, MacLean B, Zimmerman LJ, Feng X, Cusack MP, Sedransk N, Hall SC, Addona T, et al.. Design, implementation and multisite evaluation of a system suitability protocol for the quantitative assessment of instrument performance in liquid chromatography-multiple reaction monitoring-MS (LC-MRM-MS). Mol Cell Proteomics 2013; 12:2623-39; PMID:23689285; http://dx.doi.org/ 10.1074/mcp.M112.027078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carr SA, Abbatiello SE, Ackermann BL, Borchers C, Domon B, Deutsch EW, Grant RP, Hoofnagle AN, uumlttenhain R, Koomen JM, et al.. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol Cell Proteomics 2014; 13(3):907-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jenkins R, Duggan J, Aubry A-F, Zeng J, Lee J, Cojocaru L, Dufield D, Garofolo F, Kaur S, Schultz G, et al.. Recommendations for validation of LC-MS/MS bioanalytical methods for protein biotherapeutics. AAPS J 2015; 17:1-16; PMID:25392238; http://dx.doi.org/ 10.1208/s12248-014-9685-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rochat B, Kottelat E, McMullen J. The future key role of LC–high-resolution-MS analyses in clinical laboratories: a focus on quantification. Bioanalysis 2012; 4:2939-58; PMID:23244284; http://dx.doi.org/ 10.4155/bio.12.243 [DOI] [PubMed] [Google Scholar]
- 21.Kalli A, Hess S. Effect of mass spectrometric parameters on peptide and protein identification rates for shotgun proteomic experiments on an LTQ-orbitrap mass analyzer. Proteomics 2012; 12:21-31; PMID:22065615; http://dx.doi.org/ 10.1002/pmic.201100464 [DOI] [PubMed] [Google Scholar]
- 22.Olsen JV, de Godoy LMF, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M. Parts per million mass accuracy on an orbitrap mass spectrometer via lock mass injection into a C-trap. Mol Cell Proteomics 2005; 4:2010-21; PMID:16249172; http://dx.doi.org/ 10.1074/mcp.T500030-MCP200 [DOI] [PubMed] [Google Scholar]
- 23.Makarov A, Denisov E, Lange O, Horning S. Dynamic range of mass accuracy in LTQ orbitrap hybrid mass spectrometer. J Am Soc Mass Spectrom 2006; 17:977-82; PMID:16750636; http://dx.doi.org/ 10.1016/j.jasms.2006.03.006 [DOI] [PubMed] [Google Scholar]
- 24.Zeng K, Geerlof-Vidavisky I, Gucinski A, Jiang X, Boyne M, II. Liquid chromatography-high resolution mass spectrometry for peptide drug quality control. AAPS J 2015; 17:643-51; PMID:25716148; http://dx.doi.org/ 10.1208/s12248-015-9730-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Annesley TM. Ion suppression in mass spectrometry. Clinical Chemistry 2003; 49:1041-4; PMID:12816898; http://dx.doi.org/ 10.1373/49.7.1041 [DOI] [PubMed] [Google Scholar]
- 26.Trufelli H, Palma P, Famiglini G, Cappiello A. An overview of matrix effects in liquid chromatography–mass spectrometry. Mass Spectrom Rev 2011; 30:491-509; PMID:21500246; http://dx.doi.org/ 10.1002/mas.20298 [DOI] [PubMed] [Google Scholar]
- 27.Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, Domon B. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol Cell Proteomics 2012; 11:1709-23; PMID:22962056; http://dx.doi.org/ 10.1074/mcp.O112.019802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong RL, Xin B, Olah T. Optimization of Exactive Orbitrap™ acquisition parameters for quantitative bioanalysis. Bioanalysis 2011; 3:863-71; PMID:21510760; http://dx.doi.org/ 10.4155/bio.11.37 [DOI] [PubMed] [Google Scholar]
- 29.Guilhaus M, Selby D, Mlynski V. Orthogonal acceleration time-of-flight mass spectrometry. Mass Spectrom Rev 2000; 19:65-107; PMID:10795088; http://dx.doi.org/ 10.1002/(SICI)1098-2787(2000)19:2%3c65::AID-MAS1%3e3.0.CO;2-E [DOI] [PubMed] [Google Scholar]
- 30.Chernushevich IV, Loboda AV, Thomson BA. An introduction to quadrupole–time-of-flight mass spectrometry. J Mass Spectrom 2001; 36:849-65; PMID:11523084; http://dx.doi.org/ 10.1002/jms.207 [DOI] [PubMed] [Google Scholar]
- 31.Cotter RJ. Time-of-flight mass spectrometry for the structural analysis of biological molecules. Anal Chem 1992; 64:1027A-39A; PMID:1443622; http://dx.doi.org/ 10.1021/ac00045a726 [DOI] [PubMed] [Google Scholar]
- 32.Randall S, Cardasis H, Muddiman D. Factorial experimental designs elucidate significant variables affecting data acquisition on a quadrupole orbitrap mass spectrometer. J Am Soc Mass Spectrom 2013; 24:1501-12; PMID:23913023; http://dx.doi.org/ 10.1007/s13361-013-0693-y [DOI] [PubMed] [Google Scholar]
- 33.U.S. Department of Health and Human Services, Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER), and Center for Biologics Evaluation and Research (CBER). Guidance for industry: Analytical procedures and methods validation for drugs and biologics. Silver Spring, MD: FDA; 2015 [Google Scholar]
- 34.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) and Center for Veterinary Medicine (CVM). Guidance for industry: Bioanalytical method validation. Silver Spring, MD: FDA; 2001 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.