

Abstract
The aggregation of biotherapeutics is a major hindrance to the development of successful drug candidates; however, the propensity to aggregate is often identified too late in the development phase to permit modification to the protein's sequence. Incorporating rational design for the stability of proteins in early discovery has numerous benefits. We engineered out aggregation-prone regions on the Fab domain of a therapeutic monoclonal antibody, bevacizumab, to rationally design a biobetter drug candidate. With the purpose of stabilizing bevacizumab with respect to aggregation, 2 strategies were undertaken: single point mutations of aggregation-prone residues and engineering a glycosylation site near aggregation-prone residues to mask these residues with a carbohydrate moiety. Both of these approaches lead to comparable decreases in aggregation, with an up to 4-fold reduction in monomer loss. These single mutations and the new glycosylation pattern of the Fab domain do not modify binding to the target. Biobetters with increased stability against aggregation can therefore be generated in a rational manner, by either removing or masking the aggregation-prone region or crowding out protein-protein interactions.
Keywords: aggregation, bevacizumab, biobetter, glycosylation, monoclonal antibody, protein engineering, spatial aggregation propensity, stability
Abbreviations used
- APR
aggregation-prone region
- CDR
complementarity-determining region
- DSC
differential scanning calorimetry
- ELISA
enzyme-linked immunosorbent assay
- Fab
fragment antigen binding
- Fc
fragment crystallizable
- KD
dissociation constant
- mAbs
monoclonal antibodies
- MD
molecular dynamics
- SEC-HPLC
size-exclusion high performance liquid chromatography
- SAA
solvent accessible area
- SAP
spatial aggregation propensity
- VEGF-A
vascular endothelial growth factor
- Φeff
effective hydrophobicity.
Introduction
Monoclonal antibodies (mAbs) are the fastest growing category of biotherapeutics with an average yearly market growth rate of 38% in 2012.1 Their use as therapeutic agents has generated unprecedented interest because these molecules can specifically target a large number of proteins implicated in disease. MAbs are often required to be in a very high dosage form (often over a hundred mg per mL for subcutaneous injection), and are often manufactured and stored for extended periods of time in a liquid solution form. In these conditions, protein stability becomes a great concern and challenge. Aggregation is one of the most prominent forms of antibody instability, and can cause issues including manufacturing failure,2 fatal immunological responses3,4 upon drug delivery, and loss of efficacy. Protein degradation is usually dealt with through appropriate manufacturing, formulation and storage conditions of the drugs.5,6 These strategies, though effective, are costly and time consuming. Another approach is to alter the protein itself, typically by conjugation with a small molecule stabilizer7,8See comment in PubMed Commons below or through substitutions of amino acids in aggregation-prone regions.9 Molecular-based and computational approaches for the rational design of these proteins, beyond trial and error, permit the determination of protein developability at an early stage.10 The incorporation of drug developability within the discovery phase should reduce the risk, time, and cost to launch drugs on the market.
Protein aggregation is a complex phenomenon with no established single mechanism. Several states of proteins (folded, partially unfolded, unfolded) can be involved in the aggregation of monomers into small multimers and then into larger oligomeric structures in a reversible or irreversible manner.11 It has been postulated that certain sequence or structural features,10 termed as aggregation-prone regions (APR), drive protein aggregation. Computational tools have been developed for the identification of APRs on proteins, but very few of them have been validated on antibody molecules.10 Sequence-based computational tools such as TANGO and PAGE,12,13 and structure-based tool such as the spatial aggregation propensity (SAP) tool14-17 allow the identification of several APRs in mAbs. Since extensive literature data is available on validation of the SAP tool on antibodies14-17 (to our knowledge, no public domain data exist on validation of TANGO or PAGE for mitigating antibody aggregation), we use the SAP method to design variants of bevacizumab with enhanced stability.
With the purpose of improving the stability of mAbs without a loss in efficacy, the SAP tool has been applied to identify the aggregation-prone regions on the surface of the antigen-binding fragment (Fab) domain of a model IgG1, the therapeutic antibody bevacizumab (Avastin®, Genentech). Bevacizumab is an anti-vascular endothelial growth factor (VEGF)-A recombinant humanized monoclonal IgG1 used in the treatment of several cancers,18,19 as well as for age-related macular degeneration.20 This mAb is an interesting molecule to start with when rationally designing biobetters with enhanced stability because bevacizumab is particularly unstable. Bevacizumab is formulated at low concentration and has previously been shown to be highly prone to aggregation,21,22 leading to not only a substantial loss of activity23 but also to large aggregates that could potentially be harmful to patients. Following the identification of aggregation-prone regions, stable variants were designed by substituting the hydrophobic aggregation-prone residues for more hydrophilic charged residues. As an alternative strategy, we also rationally designed and engineered glycosylation sites on the Fab domain to shield these APRs.
Previous work has shown that the degree of glycosylation of biologics affects both their biological and biophysical properties. In particular, it has been shown that N-glycans in antibodies have an effect on the conformational and colloidal stability of mAbs, protecting the protein from both thermal and chemical denaturation. Additionally, glycosylation can stabilize the tertiary and quaternary structures of mAbs, and in the case of the Fc domain, hydrophobic regions, which are aggregation prone, are covered by a glycosylation moiety.24-27 Glycoengineering has proved to be effective in substantially increasing the solubility of biotherapeutics,28,29 as well as reducing the aggregation propensity of mAbs. It is well accepted that N-linked carbohydrates participate in the stabilization of mAbs against aggregation by covering aggregation-prone motifs and through steric hindrance that disrupts intermolecular interactions.8,30 We adopted this strategy to stabilize bevacizumab against aggregation. Four glycosylation sites were independently introduced on the surface of the constant region of the Fab domain of bevacizumab by single-point mutation in the CH1 and CL domains. These two strategies, decreasing the Fab surface hydrophobicity while modifying the protein net charge and introducing a carbohydrate in the Fab domain, were successfully applied to bevacizumab with no loss of binding to its target.
Results
Rational design of stabilizing variants
The rational design of aggregation resistant antibodies in this work is based on the identification of key aggregation-prone residues on the surface of a mAb. For this purpose, the SAP computational tool was applied to bevacizumab, which is a humanized IgG1. For each residue, a SAP value was calculated (Supplementary Information S1), which is the sum of the effective hydrophobicity (Φeff) of all residues surrounding the residue of interest within a radius R. The effective hydrophobicity is based on both the intrinsic amino acid hydrophobicity and the extent of its surface exposure (or solvent accessible area, SAA).14 The residues with high SAP value are predicted as part of APR and thus rationally engineering these residues offers an efficient and manageable solution to mitigate protein aggregation. One engineering strategy to disrupt these APR is to mutate high SAP value residues to hydrophilic and charged residues, such as lysine, while avoiding mutations in protein-binding regions.14,31-33
Both the Fab and Fc domains of bevacizumab could drive its aggregation; however, since mutations stabilizing the Fc domain of IgG1 molecules have already been discussed in previous publications,14 in this work, we focus on mutations stabilizing the Fab domain of bevacizumab. In fact, recent work has also suggested a role of the Fab domain of bevacizumab in its aggregation via the interaction of a Fab arm with the K445 residue of the CH3 domain of a second bevacizumab.34,35 Moreover, in the case of bevacizumab, it has been shown that aggregates had a 60% potency in the binding of the mAb to its target relative to the monomer, suggesting that aggregation predominantly involves Fab domain interactions, potentially rendering about half the number of complementarity-determining region non-accessible for binding to VEGF-A.23
The SAP tool identified 10 high SAP residues (SAP (R=5 Å) > 0.15; Φeff > 0.15) on the Fab domain of bevacizumab (Fig. 1). The SAP profile on the protein surface (Fig. 1A) shows high SAP regions (i.e., potential APRs) in red and low SAP regions in blue. Among those potential APRs, 6 exposed hydrophobic residues (Fig. 1B and C) were selected for mutation based on a rational design approach.32 A great challenge when engineering biobetters could be to improve mAb stability along with antigen binding. In this work we solely focused on improving stability against aggregation. Therefore, high SAP residues involved in the binding to the antigen (i.e., V94, Y54, Y103) were excluded since a mutation of these residues has higher chances of affecting bevacizumab activity.36,37 Residue P41 was also not mutated because the mutation of proline may cause a significant perturbation in the backbone structure, and thus the stability of the molecule. A rational and previously validated approach was followed to select which amino acids were inserted as substitutes for the selected candidate residues for mutation. Most of the selected residues were mutated into lysine because this amino acid is both positively charged and hydrophilic (V110K, L154K, L201K, V5K, L180K).14,31 Increasing the number of positively charged residues on the surface of bevacizumab (pI= 8.3)38 is likely to increase the colloidal stability of the mAb and to further reduce aggregation. The introduction of a lysine residue on the Fab surface could also disrupt electrostatic interactions between the Fab domains and the tip of the CH3 domain, observed for bevacizumab dimer aggregates.34,35
Figure 1.
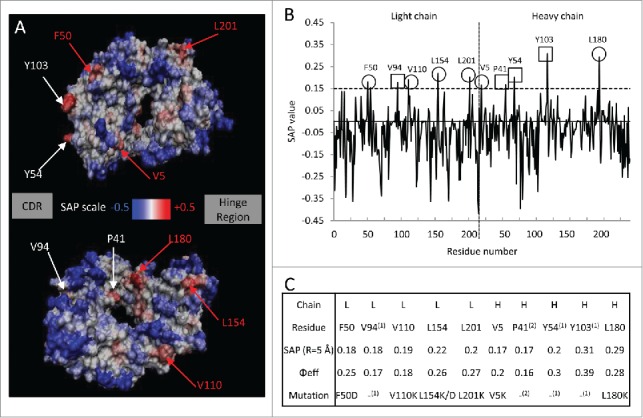
Spatial aggregation propensity (SAP) for the Fab fragment of bevacizumab. (A) The SAP values at R=5 Å for the wild-type are mapped onto the bevacizumab Fab fragment structure. The red regions represent positive SAP values (areas of high hydrophobicity, more aggregation prone) and blue regions have negative SAP values (areas of low hydrophobicity, less aggregation prone). Residues with SAP values above 0.15 and of Φeff greater than 0.15 are marked. The sites chosen for mutation are indicated in red. The residues are numbered in linear fashion. The faces on which the CDR and the hinge region are located are identified by the boxes “CDR” and “Hinge region.” The second image is the first image rotated ~180 degrees around the axis going from the CDR to hinge region. (B) SAP values at R=5 Å for the Fab fragment. ○ Peaks chosen for mutations, □ peaks not chosen for mutations. (C) Table of the residues with the highest SAP values for the Fab fragment; residues that exhibit SAP values at R=5 Å of greater value than 0.15 and of Φeff superior to 0.15, 10 of them can be considered potentially prone to aggregation. Residues were mutated into hydrophilic negatively charged residue (F50D and L154D) or hydrophilic positively charged residue, i.e., lysine. (1) Peaks not chosen for mutation are involved in binding, (2) peaks not chosen for mutation are likely involved in determining structure.
In addition to choosing the amino acid substitution to increase the stability of mAbs, mutations can also be selected to reduce potential immunogenicity due to the murine sequences still present in the humanized sequence. In this study, F50 was mutated to aspartic acid, a hydrophilic humanizing residue (Supplementary Information S2). Introducing hydrophilic residues, such as aspartic acid, may also be an efficient way to stabilize mAbs by locally modifying the charge on the protein surface.32,39 The variant L154D was produced as well to study the effect of adding a negatively charged residue versus a positively charged one (L154K). All single variants, listed in Figure 1C were tested for their stability via an accelerated aggregation study under heat stress and characterized by size-exclusion high performance liquid chromatography (SEC-HPLC), differential scanning calorimetry (DSC), turbidity measurement and enzyme-linked immunosorbent assay (ELISA).
Mutation of high SAP residues increases bevacizumab stability
Bevacizumab stability regarding aggregation has been studied under several conditions.35,40-43 Most studies have investigated the stability of commercial Avastin, i.e., 25 mg/mL bevacizumab formulated in 50 mM phosphate buffer (pH 6.2) containing 150 mM α,α-trehalose dehydrate and 0.4% (w/v) polysorbate 20. Under these conditions, less than 10% aggregation was observed after 3 months at 4°C or room temperature.40-42 When Avastin was incubated at 40°C, about the same amount of aggregation was observed after 5 weeks.35,43 On the other hand, when the therapeutic drug was prepared at 25 mg/mL in 50 mM phosphate buffer, pH 7.0 (samples called bevacizumab), an average of 32 ± 5% aggregation was measured after 5 weeks incubation at 40°C.34,35,43 The formulation developed by Genentech stabilized bevacizumab by 3.2-fold.
Even though aggregation might proceed through different mechanisms at low (4°C) and elevated (> 40°C) temperature, a heat stress accelerated aggregation study is a well-established way to predict protein stability at lower temperature. To accelerate bevacizumab aggregation, the incubation temperature was increased. It has previously been shown that when bevacizumab at concentrations below 2.5 mg/mL (pH 6.0-6.2) is incubated at 56°C, 65°C and 70°C, almost all of the protein aggregates within 3 days, 3 h and 10 min, respectively.21,44 To minimize unfolding of bevacizumab at such a high temperature (temperature close to Tm), a lower temperature was used for these accelerated studies. A thorough study of bevacizumab and its different fragments allowed the assignment of transition temperatures for each single domain (Tm1= 70.0°C for the CH2 domain; Tm2= 74.1°C for the Fab domain; Tm3= 82.0°C for the CH3 domain).45 A similar bevacizumab thermogram was obtained here, displaying 2 transitions with the first major transition centered around 71°C, with an increased heat capacity around 62°C and a second minor transition at 82.4°C (data not shown); Tm were extracted (Table 1). Based on this result and previous accelerated aggregation studies reported in the literature, we concluded that 52°C would be an ideal temperature at which to conduct our temperature-induced aggregation investigation (at least 10°C below the temperature leading to bevacizumab denaturation) because it is high enough to generate a substantial amount of aggregates in a relatively short time, without promoting substantial mAb unfolding other than transient perturbation to the structure. The stability of bevacizumab was studied by SEC-HPLC, which was used to determine monomer loss over time after heat stress at 52°C for up to 48 h.
Table 1.
Bevacizumab stabilization by single point mutation in its Fab domain. Wild-type (WT) bevacizumab and the variants were tested for their aggregation propensity by heat-induced accelerated aggregation study. The percentage of monomer and aggregates detectable by SEC-HPLC was measured at various time points up to 48 h of incubation at 52°C at 50 mg/mL antibody concentration (His 10 mM, pH 6.0). The percentage of monomer in the soluble fraction after 48 h of heat treatment is reported in the table. Data are the mean ± SD. (n=3 experiments with 3 different protein batches, *n=2 experiments with 2 different protein batches). The kinetic data were fitted to extract a second order rate constant (“Aggregation rate”). A stabilization factor was calculated and the “Fold increase stability” is the ratio of the percentage of WT aggregates to the percentage of variant aggregates at t=48 h. The thermostability of bevacizumab formulated in 10 mM histidine buffer (pH 6.0) was characterized by DSC to determine and attribute 3 transition temperatures, Tm1 for the CH2 domain, Tm2 which can be attributed to the Fab domain and Tm3 corresponding to the transition temperature of the CH3 domain. The transition temperatures in degrees Celsius for the WT and each variant were obtained by fitting Gaussians to each thermogram. Equilibrium dissociation constants, KD, for the WT bevacizumab and its variants binding to VEGF-A were extracted from Figure 2A. WT and engineered mAbs bound to the target VEGF with the same affinity as previously reported for bevacizumab.
| Variants | % monomer at 48 h | Aggregation rate (*10−2 mol−1.L.min−1) | Fold increase stability | Transition temperature (°C) Tm1 Tm2 Tm3 | KD (nM) |
|---|---|---|---|---|---|
| WT | 68 ± 2 | 31.3 ± 5.6 | 1.0 ± 0.1 | 70.4 71.8 82.4 | 0.85 ± 0.28 |
| V5K | 74 ± 2 | 13.9 ± 1.0 | 1.2 ± 0.1 | 70.4 71.5 82.9 | 2.57 ± 0.24 |
| F50D | 87 ± 5 | 9.6 ± 0.6 | 2.5 ± 1.0 | 71.0 72.9 83.2 | 1.19 ± 0.28 |
| V110K | 88 ± 5 | 6.3 ± 2.8 | 2.8 ± 1.2 | 71.3 72.7 83.0 | 1.32 ± 0.28 |
| L154D | 88 ± 3 | 5.9 ± 1.8 | 2.8 ± 0.8 | 71.4 72.6 83.3 | 0.62 ± 0.1 |
| L154K* | 92 ± 0 | 4.5 ± 1.2 | 4.0 ± 0.4 | 72.1 73.5 82.7 | 0.85 ± 0.02 |
| L180K | 82 ± 3 | 9.9 ± 1.7 | 1.8 ± 0.3 | 71.3 72.0 84.3 | 2.39 ± 0.13 |
| L201K* | 87 ± 2 | 3.1 ± 2.1 | 2.5 ± 0.5 | 72.2 72.6 82.8 | 0.68 ± 0.2 |
Accelerated aggregation studies were performed at several concentrations, up to 50 mg/mL, which is twice the formulation concentration of the commercial product. From these data (Supplementary Information S3), an optimal working concentration of 50 mg/mL was determined for the experimental aggregation assays. In our experimental conditions, a 50 mg/mL bevacizumab solution led to ∼25% and 32% aggregation after incubation for 24 h and 48 h, respectively, at 52°C. These findings are in relative agreement with previous results; we found ∼2.8-fold more aggregation than when commercial Avastin was incubated at 50°C for 24 h (8.8% aggregation observed).22 Given that we are working at twice the formulation concentration and at 52°C (vs. 50°C), this result was expected (a 3.2-fold difference in the amount of aggregates was observed between commercial Avastin and bevacizumab incubated at 40°C).35,43
Bevacizumab variants and wild-type monomer concentrations were measured relative to the initial 50 mg/mL by SEC-HPLC after 48 h incubation at 52°C, (see Table 1). The stability of the engineered reduced SAP variants was compared to the wild-type (WT) by calculating a “Fold of stability improvement” (ratio of aggregates observed for the wild-type versus aggregates observed for the variant) of bevacizumab upon mutation. As shown in Table 1, all mutations introduced in the Fab domain reduced the aggregation propensity compared to the wild-type. Two variants, V5K and L180K, showed a noticeable but small stabilization effect, with less than 2-fold aggregation reduction. The five other variants showed a stability improvement of 2.5-fold and up to 4-fold for the L154K variant after 2 days incubation at 52°C. With the same trend, the aggregation rate was also considerably reduced (2.3- to 10-fold slower).
Very large non-reversible aggregates cannot enter the SEC-HPLC column and be directly quantified, but are measured instead by total mass balance. Insoluble aggregates were estimated by calculating the amount of monomer loss and the amount of soluble aggregates at each time vs. the initial time of the experiment (t = 0). Bevacizumab WT showed 8.3% insoluble aggregates (Abs320nm = 0.091) after 48 h incubation at 52°C, and similar amounts were measured for variants F50D, L154D, L180K. Only variant V110K had a higher percentage of insoluble aggregates, 13%, with a corresponding increase in turbidity (Abs320nm = 0.101). No insoluble aggregates were detected by SEC-HPLC for the variants L154K and L201K, and lower absorbance at 320 nm were measured (0.012 and 0.015, respectively), indicating that these 2 mutations are the most stabilizing hydrophobic to hydrophilic charged residue substitutions. The L201K mutation stabilizes bevacizumab 3-fold against soluble and insoluble aggregates, and a 7-fold decrease in aggregates of all types was observed for our best variant containing the L154K substitution.
DSC experiments were also performed to evaluate the thermodynamic stability of our bevacizumab WT and variants (Table 1). A comparison of the thermograms shows an increase of the CH2 (Tm1) and Fab (Tm2) transition temperatures in the variants compared to the WT by up to 1.8°C, except for the V5K variant (the least stabilizing mutation). The highest increase was observed for L154K, the variant that reduced aggregation the most. The transition temperatures for the CH3 domain of the variants are less than 1°C higher than the bevacizumab WT Tm3, except for the L180K variant that exhibited a Tm3 increase of 1.9°C.
A critical aspect to creating biobetters with improved stability is that the engineered proteins must have at least the same efficacy as the innovator product. Bevacizumab is approved for the treatment of a variety of cancers and is widely used off-label to treat eye diseases such as age-related macular degeneration. It acts by blocking VEGF-A, thereby inhibiting neovascularization and blood vessel leakage. The efficacy of our engineered mAbs was estimated in vitro by measuring the affinity of the WT and variants for the target antigen, VEGF-A, through competitive ELISA. Figure 2A shows the binding curve of WT bevacizumab and the variants to VEGF-A, and their respective dissociation constants (KD) are reported in Table 1. Our competitive assay results show that our WT bevacizumab produced in house has a similar affinity for its antigen (KD = 0.9±0.3 nM) as the previously reported results (average reported values KD = 1.2±0.3 nM).46,47 All of the mutations introduced in the Fab fragment are not in the CDR and therefore, as expected, all our variants bind to VEGF-A with the same affinity as the commercial drug. These results show that a single point mutation can greatly stabilize bevacizumab against aggregation without compromising its binding to its target VEGF-A.
Figure 2.
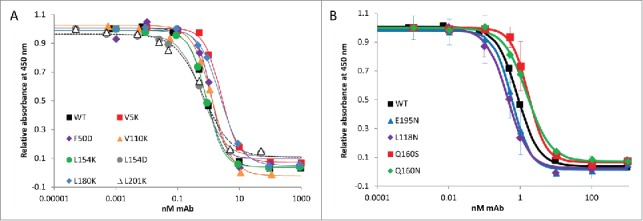
Competitive ELISA of WT bevacizumab and single variants for VEGF-A. Bevacizumab and engineered proteins binding to VEGF were measured using a VEGF Human ELISA Kit in which the VEGF protein (1500 pg/mL) was pre-incubated and shacked for 60 min at 37°C with bevacizumab or the variants at various concentrations (0.5 pM to 1 μM). The pre-mixes were then used in the ELISA, which allows the quantification of antibodies bound to the VEGF by reading absorbance at 450 nm. The more bevacizumab (or variants) binds to VEGF, the less VEGF can bind to the anti-VEGF IgG1 adhered onto the ELISA plate, and the lower the signal at 450 nm. Each assay was run at least in duplicate. The WT and variants bound to the target VEGF with the same affinity, as previously reported for bevacizumab. (A) Affinity of WT bevacizumab and the high SAP single point mutation variants for VEGF-A. (B) Affinity of WT bevacizumab and hyperglycosylated variants for VEGF-A.
Rational design of stabilizing hyperglycosylated variants
An alternate approach to increasing the stability of bevacizumab is to introduce glycosylation sites near the identified APRs to mask them. To introduce these new sites, residues near each aforementioned APR were considered. Residues close to the antigen-binding site were not considered to prevent any chance of altering bevacizumab activity. Therefore, we did not investigate the introduction of a carbohydrate moiety masking either the V5 or F50 high SAP value residues. To reduce the aggregation propensity of bevacizumab, we intended to mask residues V110, L154, L180 and L201 with a glycosylation motif. Each of these residues have shown, upon mutation to hydrophilic and charged residues, to increase the stability of bevacizumab.
To generate a list of potential N-glycosylation sites, i.e., NXS or NXT (X is any amino acid but P), close enough for the sugar group to mask those residues of interest, all of the serine, threonine and asparagine (residue i) within 10 Å of these high SAP value residues within the constant domains of the Fab region of bevacizumab (CH1 and CL domains) were identified. The neighboring residues (residues i-2 and i+2) were then considered as candidates for mutation to generate a foreseeable glycosylation site. All of the residues that can potentially be mutated to generate glycosylation sites are listed in Table 2 in the column “Variants to generate glycosylation site.” For example, the L154 high SAP value residue has been shown to be involved in aggregation (Table 1). The residue (i) T197 is located at a distance less than 10 Å from L154 and is highly surface exposed. If the residue (i-2) E195 is mutated into an asparagine, it will create the sequence NXT197 (instead of EXT), making it a potential glycosylation site (Table 2). The carbohydrate introduced in vivo during mAb expression in position N195 is expected to mask the L154 residue (and its surrounding residues) and could potentially reduce the aggregation propensity of bevacizumab. A large set of residues were identified and further selective parameters were applied to ensure that feasible and efficient glycosylation would take place. Figure 3 summarizes the different criteria that were used (details in the Materials and Methods section) to select these glycosylation sites. After dismissing mutations leading to glycosylation site motifs that would not undergo glycosylation, as well as mutations potentially affecting protein structure, together with high SAP value residues, 8 residues were identified for site-directed mutagenesis to create potential glycosylation sites. Four different possible variants have been identified that will introduce a glycosylation site to mask the V110 high SAP value residue, whereas the 4 other mutations identified should permit the masking of 3 residues (L154, L180 and L201). With the aim of making our experiments more efficient and ensuring that we chose pertinent glycosylation sites, we did not introduce any glycosylation sites to mask the high SAP value residue V110. The V110 residue has been shown to be involved in aggregation, based on the observation that its mutation into lysine resulted in a 2.8-fold stabilization of bevacizumab against aggregation. Nevertheless, this reduction in aggregation is similar to that observed for variants L154D and L201K, and V110 has one of the lowest SAP scores among the above listed high SAP value residues, making its masking by a carbohydrate moiety less attractive.
Table 2.
List of residues for glycosylation site engineering. Identification of variants which are likely to be glycosylated in the vicinity of high SAP regions. Residues in gray were not selected for the reasons described in the footnotes.
| High SAP Residues masked | Ser/Thr/Asn within 10 Å of the high-SAP value residues | Variants to generate glycosylation site | SAA of side-chain atoms of Asn or residue to be mutated to Asn (Å2) |
|---|---|---|---|
| L:L154 (øeff = 0.26) | L:N152 | L:L154S2 | 106.8 |
| L:S156 | L:L154N2 | 84.2 | |
| L:N158 | L:Q160S | 53.22 | |
| L:S159 | L:G157N1, 4 | 12.5 | |
| L:S177 | L:L175N2, 4 | 7.6 | |
| L:T197 | L:E195N | 33.9 | |
| L:T206 | L:P204N1 | 56.9 | |
| L:V110 (øeff = 0.18) | L:T109 | L:K107N | 107.7 |
| L:N138 | L:Y140S | 67.8 | |
| L:S171 | L:K169N | 160.1 | |
| L:T172 | L:D170N | 57.3 | |
| L:S202 | L:G199N1, 4 | 12.3 | |
| L:L201 (øeff = 0.27) | L:S114 | L:A112N3 | 39.2 |
| L:N137 | L:F139S4 | 8.4 | |
| L:T197 | L:E195N | 33.9 | |
| L:S202 | L:G199N1, 4 | 12.3 | |
| L:S203 | L:L201N2 | 85.2 | |
| L:N158 | L:Q160S | 53.2 | |
| L:S159 | L:G157N1, 4 | 12.5 | |
| H:L180 (øeff = 0.28) | L:S162 | L:Q160N | 28.5 |
| L:T178 | L:S176N4 | 1.56 | |
| L:T180 | L:T178N4 | 6.6 | |
| H:T120 | H:L118N | 28.2 | |
| H:S122 | H:T120N4 | 10.3 | |
| H:S182 | H:L180N2 | 87.7 | |
| H:S183 | H:Q181N4 | 12.0 | |
| H:S187 | H:L185N5 | 34.8 |
Reasons for rejections:
1. Residue to be mutated is either a glycine or a proline.
2. Residue to be mutated is a high SAP value residue. Only the effect of masking the aggregation-prone region was to be investigated, therefore, potential aggregation-prone residues must not be mutated.
3. Residue X of N-X-S/T is a proline. NPS/T are not substrates for glycosylases.
4. SAA of the side chain of asparagine or of the residue to be mutated in asparagines is less than 15 Å2 and not accessible to glycosylases.
NXS/T motif to be generated is oriented on a different face relative to the high SAP value residue.5.
Figure 3.
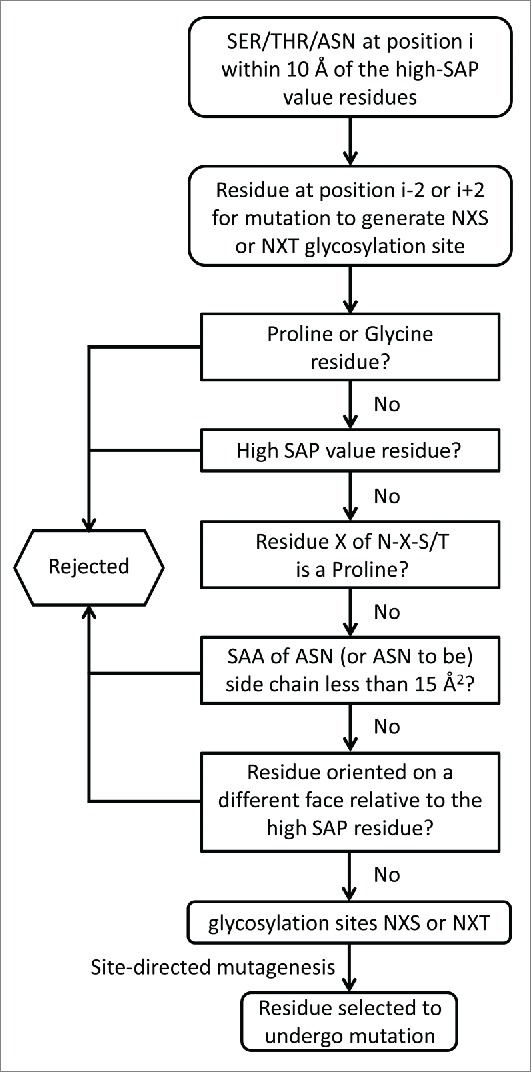
Rational selection of residues for mutation to introduce glycosylation sites on the bevacizumab Fab domain. The five different questions can be asked in any particular order.
As a proof of principle, hyperglycosylated variants were generated to cover high SAP residues L154, L180 and L201. Interestingly, there is an overlap of the potential glycosylation sites (Table 2). L154 could be masked by a glycosylation motif introduced by the mutations Q160S and E195N, which generate, respectively, glycosylation sites NSS160 and NVT197 in bevacizumab light chain. These two mutations generate a site with glycosylation moieties that should mask not only residue L154 but also residue L201. The high SAP value residue L180 could be masked by a glycosylation motif introduced by either the substitution of the residue Q160 to asparagine, generating here the glycosylation motif NES162 or by the mutation of the residue L118 to asparagine, introducing the glycosylation site NVT120 on the heavy chain of bevacizumab.
Glycoengineered proteins for increased stability against aggregation
The four variants L118N, E195N and Q160N/S (like the WT and reduced SAP variants) were produced in HEK293 human embryonic kidney cells, which are able to carry out the original post-translational modifications and should produce mAbs with glycosylation at the engineered sites. To test whether the introduction of an N-glycosylation site near a high SAP region led to an overall reduction in the aggregation propensity of bevacizumab, the hyperglycosylated variants were expressed, purified and characterized by DSC, turbidity evaluation and SEC-HPLC. The incorporation of N-glycan on the Fab domain of our bevacizumab variants was verified by reducing SDS-PAGE. The HC of L118N and the LC of E195N and Q160N/S have clearly higher molecular weights than those of the WT, corresponding to the addition of an N-glycan (data not shown). All variants were tested by DSC for their thermal stability. Three transition temperatures were extracted from the obtained thermograms (Table 3). The introduction of the N-glycan motif on the Fab domain of bevacizumab does not affect the transition temperature of the CH3 domain (Tm3), which changed by less than 0.7°C. The Fab domain shows a higher thermal stability when hyperglycosylated at one of the 4 positions we tested (Tm2 increased by 1.6°C to 2.3°C). Surprisingly, the transition temperature Tm1 attributed to the CH2 domain45 is also affected. Glycosylation of the sites N195, N160 and N158 increased the CH2 domain stability by 1.6°C to 2.1°C, whereas glycosylation of N118 reduced the CH2 domain thermal stability by 2.3°C.
Table 3.
Bevacizumab stabilization by hyperglycosylation of the Fab domain. The amount of monomer and soluble aggregates detectable by SEC-HPLC was measured at various time points up to 48 h of incubation at 52°C of 50 mg/mL (His 10 mM, pH 6.0) mAbs. The monomer percentage of the WT and variants in the soluble fraction after 48 h heat is reported in the table. Data are the mean ± SD (n=3 experiments with 3 different protein batches, *n=2 experiments with 2 different protein batches). The kinetic data were fitted to a simple equation to extract a second order rate constant (“Aggregation rate”). The monomer percentage at 48 h for each variant was compared to the WT through the “Fold increase stability.” For each hyperglycosylated variant, a MD simulation was performed and a SAP score (the sum of all positive SAP values) at R=5 Å was computed in addition to the standard errors. The transition temperatures in degrees Celsius for the WT and each hyperglycosylated variant were obtained by fitting 3 Gaussians to each thermogram. Equilibrium dissociation constants, KD, for the WT bevacizumab and its hyperglycosylated variants binding to VEGF-A were extracted from Figure 2B.
| Variants | % monomer at 48 h | Aggregation rate (*10−2 mol−1.L.min−1) | Fold increase stability | SAP Score | Transition temperature (°C) Tm1 Tm2 Tm3 | KD (nM) |
|---|---|---|---|---|---|---|
| WT | 68 ± 2 | 31.3 ± 5.6 | 1.0 ± 0.1 | 164 ± 1.04 | 70.4 71.8 82.4 | 0.85 ± 0.28 |
| L118N | 85 ± 3 | 9.4 ± 5.5 | 2.2 ± 0.5 | 149 ± 0.41 | 68.1 74.1 83.1 | 0.45 ± 0.06 |
| Q160N* | 79 ± 4 | 14.8 ± 8.7 | 1.5 ± 0.3 | 156 ± 1.21 | 72.5 73.4 82.9 | 1.56 ± 0.13 |
| Q160S* | 90 ± 3 | 4.2 ± 0.9 | 3.2 ± 1.1 | 159 ± 1.05 | 72.3 73.4 83.0 | 1.72 ± 0.15 |
| E195N | 89 ± 4 | 5.5 ± 2.9 | 2.9 ± 1.2 | 155 ± 0.66 | 72.0 73.4 82.5 | 0.59 ± 0.04 |
Hyperglycosylated variants were tested for their stability through accelerated aggregation studies at an elevated temperature. As with the reduced SAP variants, 52°C was chosen as the temperature to accelerate aggregation, and monomer concentrations were monitored over a 48-h period by SEC-HPLC (Fig. 4). Our SEC-HPLC data are the measure of the monomer concentration at various time points, and Figure 4 represents the average of 3 independent experiments (i.e., 3 different protein production batches) unless stated otherwise. We observed the presence of soluble aggregates in all samples from the beginning of the experiment, which was certainly due to the high concentration formulation (50 mg/mL) that was close to the solubility limit of bevacizumab. The wild-type bevacizumab sample contained 8.5% soluble aggregates at t=0, whereas the variants Q160N, Q160S and E195N contained less than 4% soluble aggregates at t=0. After 48 h incubation at 52˚C, we observed 32% soluble aggregates for the WT, whereas our best variant displayed less than 10% aggregates, representing over a 3-fold increase in stabilization. The four hyperglycosylated variants are all more stable than the WT bevacizumab against heat-induced aggregation, having a 1.5 to 3.2-fold increase in stability (Table 3). A second-order rate constant was extracted from the fitting of the monomer loss measured over time for each variant (Table 3). The WT and the Q160N variant display a fast loss of monomer in the first 16 h of incubation at 52˚C, while the variants L118N, Q160S and E195N stay relatively stable during this incubation period. They are also the 3 variants with the most stabilizing effect, and their aggregation rate is reduced by a factor 3.3 to 7.4 (Table 3 and Fig. 4).
Figure 4.
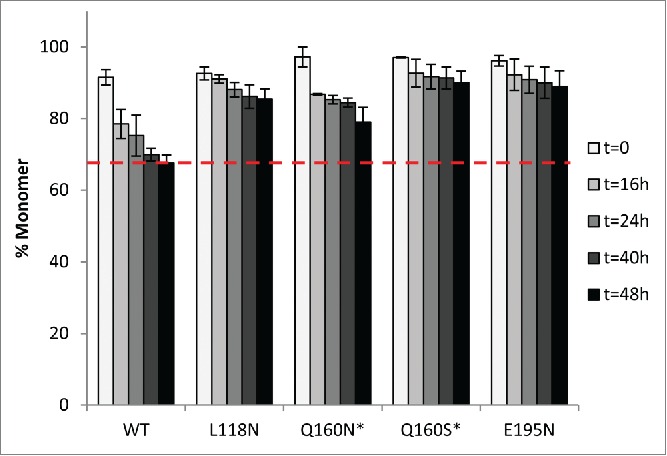
Stability comparison of WT bevacizumab and variants by SEC-HPLC. Monomer loss for WT and hyperglycosylated variants (50 mg/mL in histidine buffer, pH 6.0) was measured at various time points upon heat stress at 52°C for 48 h. Data are the mean ± SD. (n=3 experiments with 3 different protein batches * n=2 experiments with 2 different protein batches).
Using SEC-HPLC data and by comparing the amount of monomer measured at each time point (t=16 h, t=24 h, t=48 h) to the amount of monomer at t=0, we could estimate the amount of insoluble aggregates that did not enter the separation column. Our best hyperglycosylated variants, Q160N/S and E195N exhibited reduced insoluble aggregates (3-5%) compared to the WT (∼8%), whereas L118N exhibited nearly double the amount of insoluble aggregates (15%), clearly making the variant L118N the least effective hyperglycosylated variant overall (1.3-fold stabilization). This result was confirmed by turbidity measurements: Abs320nm=0.014 to 0.052 for our 3 best variants, Abs320nm=0.216 for L118N versus Abs320nm=0.091 for WT.
As described above, the engineered mAbs must have at least the same affinity to the target as the WT. The binding efficacy of our hyperglycosylated engineered bevacizumab was estimated in vitro via a competitive ELISA developed in-house, allowing measurement of the affinity of the WT and variants for the antigen, VEGF. Figure 2B shows the binding curve of WT bevacizumab and the hyperglycosylated variants to VEGF-A. Our mAbs were preincubated with VEGF, which were then incubated with anti-VEGF IgG1 for further detection by ELISA. A low absorbance at 450 nm indicates a low amount of VEGF bound to the ELISA plate and a high amount of our mAb bound to VEGF. The corresponding dissociation constants (KD) are reported in Table 3. These competitive assay results show that all of the mutations introduced in the Fab fragment, far from the CDR, produce hyperglycosylated variants that bind to VEGF with the same affinity as the commercial drug, our WT bevacizumab and our reduced SAP variants described above. Our results indicate that, when glycosylation site locations are chosen carefully, N-glycans can stabilize mAbs without compromising their activity in vitro.
To gain insights into the effect of glycosylating the bevacizumab Fab domain on aggregation, we performed molecular dynamics simulations of the Fab domain of the WT and the 4 hyperglycosylated variants. Although the nature of the glycan structures (32 different glycoforms) is of high importance, the heterogeneity and control of glycosylation pattern are complex topics48 and the identification of the N-glycosylation modification was not investigated in this study for our hyperglycosylated variants. The glycosylation of mAbs is highly dependent on the culture conditions;49 therefore, we assumed here the same glycosylation pattern observed previously in our laboratory for mAbs produced in the same conditions as our hyperglycosylated variants.30 Molecular dynamic simulations were performed based on the crystal structure of the bevacizumab Fab domain (1BJ1)37 and assuming a G0 glycosylation pattern. Figure 5 shows snapshots of typical simulation results obtained for our 4 variants. Typically, L118N and Q160N were engineered to introduce a carbohydrate that masked L180, and both variants have a comparable effect on bevacizumab stability. Interestingly, the carbohydrates behave very differently. On one hand, the Q160N carbohydrate consistently covers a wide region of residues through its interaction with the Fab surface, but at the cost of masking L180, with a negligible effect on its SAP value (Table 3). On the other hand, the L118N carbohydrate completely masked L180, whose SAP value dropped dramatically (decreased to a fifth of its original value), and made one of the most hydrophobic regions nearly hydrophilic, with a decrease in Fab SAP score of ∼15 units. Interestingly, the L118N carbohydrate was forced to point away from the surface of the Fab domain and does not reliably cover many other residues other than L180. The variant Q160S bears a carbohydrate interacting partially with the Fab domain surface and pointing away from it as well. In this case, the carbohydrate does not cover the neighboring high SAP residues L154 and L201, thereby reducing the SAP score to a smaller extent. A similar orientation, i.e., pointing away from the surface, was observed for the variant E195N carbohydrate. There was a reduction in the SAP score of the E195N variant in comparison to the wild-type, and to the same extend observed for the variants Q160N and Q160S. However, the reduction in SAP score did not correlate with reduction in aggregation propensity of the variants. As expected, L118N and Q160N glycosylation motifs are covering the targeted residues (Figs. 5A-B), while the carbohydrate of variants Q160S cover other residues, and the carbohydrate of variant E195N covers no residues.
Figure 5.
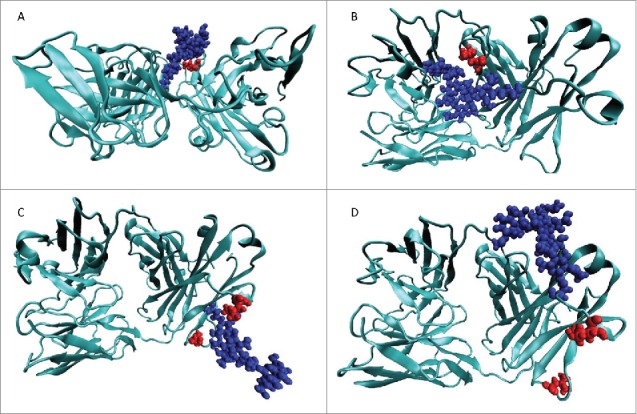
Representative structures from the MD for the 4 MD simulations of the hyperglycosylated variants. In all cases, the left of the image is the CDR and the right side is the hinge region. The protein structure is shown in teal, the glycosylation moiety is shown in blue, and the residues to be masked by the glycosylation motif are shown in red. (A) L118N hyperglycosylated variant designed to cover L180. (B) Q160N hyperglycosylated variant designed to cover L180. (C) E195N hyperglycosylated variant designed to cover L154 and L201. (D) Q160S hyperglycosylated variant designed to cover L154 and L201.
Discussion
In the work reported here, we successfully engineered biobetters of bevacizumab and designed mAbs with substantially reduced aggregation propensity while maintaining high affinity to the target antigen. Two approaches were investigated to improve stability against aggregation: 1) reducing surface hydrophobicity by single point mutation of identified aggregation-prone residues, and 2) introducing an N-glycan moiety on the Fab surface to shield identified APRs. These two strategies were both successful in improving bevacizumab stability without reducing antigen affinity.
The first step of this rational design approach is the identification of APRs on the surface of the mAb. This identification was done by applying the SAP tool to the structure of the Fab domain of bevacizumab. Ten high SAP value residues were identified, and 6 fit the criteria to further undergo single point mutation into more hydrophilic residues. Residues L154, V110, L201 and F50 were identified as the most important residues in the Fab domain of bevacizumab for improving its stability. Overall, the 2 best variants identified were L154K and L201K for which no insoluble aggregates were observed and the rate of aggregation decreased by a factor up to 10. Out of our 5 best variants, 2 bear a negative charge mutation and 3 bear a positive charge mutation.
It has been previously reported that the insertion of both positive and negative charge residues promotes mAbs stabilization, but a preference was noted for negatively charged residues, and in particular aspartic acid, as a very good substitute for hydrophobic aggregation-prone residues to stabilize proteins.32,39,50,51 Negatively charged amino acids are suggested to locally modify the charge of the protein, enhancing protein-protein repulsion. The addition of aspartic residues in the bevacizumab sequence slightly decreased the protein charge at pH 6.0 from 14 to 12 units (as calculated with PROPKA 3.0)52 present on the mAb Fab domain surface, whereas the introduction of lysine increased the charge by 2 units. In this study, the mutation of a hydrophobic residue to a negatively charged residue (F50D and L154D) was on average as efficient as mutations to positively charged residues. The one direct comparison that can be done here is between the variants L154D and L154K. Interestingly, the mutation to lysine stabilized bevacizumab 4-fold vs. 2.8-fold for the mutation to aspartic acid. It seems that the local environment of L154 in bevacizumab Fab domain is more favorably affected by the insertion of a positively charged amino acid than a negatively charged residue. It has also been shown that the more positively charged the mAb scaffold the more stabilizing the effect for a positively charged residue versus a negatively charged one.53 The net charge of one Fab domain is 7, potentially making the insertion of a lysine more effective in increasing the total net charge of bevacizumab, which would also increase the colloidal stability. The introduction of a lysine amino acid in place of a leucine also slightly increases the pI of bevacizumab (∼0.2), which correlates with increased solubility and stability.54
Several APRs identified herein are consensus residues, which are also identified in several other chimeric and humanized therapeutic mAbs, such as rituximab, cetuximab and trastuzumab. These APRs consist of L154, as well as L180, and have been investigated in our group for rituximab.32 One could expect a similar stabilization enhancement for consensus APRs. Nevertheless, we previously reported32 that the mutations L153K and L178K in the Fab domain of rituximab (corresponding to L154 and L180 for bevacizumab) were destabilizing, and the variants were poorly expressed. Mutations in aspartic acid and serine were moderately more successful (<1.5-fold stability increase). Only multiple site mutations permitted a substantial increase (3-fold) in rituximab stability. To the contrary, single point mutations L154K/D are sufficient to reduce over 2.8-fold the amount of aggregates for bevacizumab (only 1.8-fold for L180K), showing that the Fab-Fab interactions and, in particular, the implication of L154 and L180, are more important in the aggregation mechanism of bevacizumab compared to rituximab. These results demonstrate that the SAP tool allows for the prediction of accurate aggregation-prone regions. Nevertheless, one cannot predict the extent of the implication of theses APRs in the aggregation mechanism(s) for a specific mAb. Two mAbs presenting several APRs in common may follow very different aggregation pathways and may respond differently to mutations at these APRs.
Another potential way to reduce aggregation would be to mask these high SAP value residues with a carbohydrate motif to prevent protein-protein interactions. Carbohydrate groups participate in the integrity of most biologics by reducing aggregation propensity, increasing solubility, stabilizing the native conformation, protecting against various degradation pathways (hydrolysis, oxidation) and overall stabilizing the macromolecules.28,55 This functionality is particularly relevant for antibodies as all IgGs are naturally N-glycosylated at position N297 in each of the CH2 chains of the Fc domain. It has been demonstrated on several occasions that this N-glycan participates in the stabilization of mAbs against aggregation caused by various stresses.8,24,26,27,56,57 Natural IgGs present in human serum are all glycosylated in the Fc domain whereas only less than a third are glycosylated in the Fab domain.55 N-glycans are found attached to the variable region of the LC, the HC or both. It is estimated that ∼20% of the variable region of mAbs bear an N-glycosylation site motif.54 The natural occurrence of Fab glycosylation supports the idea that a glycoengineered antibody might be viable as a biotherapeutic as well.58,59 For example, cetuximab, a chimeric therapeutic mAb bears oligosaccharides in its VH region,60 and Fab domain glycoengineering has already proved to be successful for solubility improvement54 and aggregation prevention.29
In this study, we took advantage of the capacity of N-glycans to mask APRs to reduce the aggregation of our model therapeutic mAb, bevacizumab. The N-glycosylation sites NXS/T were carefully chosen to fit a series of criteria and 4 glycosylation sites were independently engineered: L118N, Q160N, Q160S and E195N. The introduction of an N-glycan at position N160 in the Fab domain, which masked the high SAP residue L180, reduces the aggregation propensity (2.1-fold reduction in aggregation rate). The L180 high SAP value residue can also potentially be masked by a carbohydrate motif introduced in position 118, or could be mutated in lysine to reduce its SAP score. Both strategies are slightly more successful at reducing aggregation: the L180K mutation reduced bevacizumab aggregation rate 3.2-fold, while the L118N hyperglycosylated variant reduced the aggregation rate by 3.3-fold. This suggests that Q160N hyperglycosylated variant did not mask the APR as well as L118N. This trend can also be seen in their average SAP scores, where L118N is lower than Q160N, which is lower than the wild-type.
Two leucine residues with high SAP value, L154 and L201, are on the same face of the Fab domain of bevacizumab, toward the hinge region. Interestingly, 2 residues have been identified as candidates for mutation to generate glycosylation substrate sites that could mask both L154 and L201: Q160S, generating the motif NSS160 and E195N introducing the NVT197 glycosylation site (Table 2). Both of the hyperglycosylated variants present the highest degree of stabilization. Interestingly, comparable results were obtained for reduced SAP variants that also removed the APRs containing L154 and L201 (see L154K, L154D, and L201K in Table 1). The similar performance of the hyperglycosylated variants and the reduced SAP variants for mutations targeting the same APRs suggests that the hyperglycosylated variants did cover the targeted APR. However, this performance of the hyperglycosylated variants does not correlate with the SAP score of the variants. This suggests that masking of the APR is not the only stabilizing mechanism, and that steric hindrance preventing protein-protein interactions cannot be dismissed and may be part of the protective effect of the N-glycans.
The added glycan is a large moiety that could prevent another molecule from interacting with the nearby residues, thus reducing their role in aggregation. The simulation of the hyperglycosylated Fab domain shows a difference in the behavior of the added carbohydrate moieties, whereas the structure of the Fab domain is only minimally perturbed by the addition of the glycans, as the root mean squared displacements (RMSD) of the proteins to the wild-type are comparable to that of the wild-type during its simulation. The masking of hydrophobic residues was the original goal for the addition of glycosylation sites, and it is present in all of the simulations. While masking is present in all simulations, it is present to varying degrees, in particular when considering the masking of the targeted residues. Typically, the Q160N carbohydrate covers a large region of the Fab domain surface, while the L118N carbohydrates is forced to point away from the surface. In fact, many of the terminal sugars spent a significant amount of time being fully solvated, suggesting that the L118N carbohydrate might also act as an exclude solute or stabilize bevacizumab through steric hindrance (Fig. 5A). The same orientation of the carbohydrate was observed for the variants Q160S and E195N, which do not interact with the Fab surface. The glycans spend the vast majority of the simulation in solution. While they do not mask the high SAP residues L154 and L201, they prevent large molecules from interacting with these APRs. These results point to the possibility that crowding plays a major role in the stability of the E195N and Q160S mAbs.
The reduced SAP score of the hyperglycosylated variants compared to the WT correlates well with the change in the transition temperature of the Fab domain. While DSC data can be a convolution of unfolding and aggregation processes, it is assumed here, that irreversible aggregation is negligible below Tm and that the transition temperature is a good estimate of the unfolding of each mAb domain and of their conformational stability.61 The correlation between SAP score and Tm supports the idea that reducing the hydrophobicity of the Fab surface increases its conformational stability, which can be predicted through the SAP score. However, this stabilization is not the only factor affecting the protein aggregation propensity. The best biobetters against aggregation do not have the lowest SAP score, as seen for Q160S and E195N variants. This finding supports the idea that masking the high SAP residues participates in the stabilization of the mAb, but the colloidal stabilization through a steric effect could be a major contributor.
While the glycosylation sites were selected such that high SAP residues were not mutated, which helped distinguish the effect of high SAP mutations and hyperglycosylated variants, any mutation will affect the SAP profile of the protein. In some cases, such as Q160N, this change is minimal because glutamine and asparagine are similar residues. However, in the case of L118N, the change in hydrophobicity is more substantial and the mutation accounts for half of the change of SAP score compare to that of the wild-type. Therefore, when possible, choosing a glycosylation site that has high SAP value would greatly aid in reducing the SAP score of the all Fab domain and should be added to the guideline for selecting glycosylation sites. Furthermore, the type of the carbohydrate could also play a role in destabilizing initial aggregate formation by modifying the net charge of the Fab domain and impacting the overall SAP score of the mAb domain. Glycosylation sites must, therefore, be cautiously chosen for each individual mAb, and clinical efficacy and immunogenicity need to be carefully investigated upon mAb modification.
Overall, single point mutations of APRs of the bevacizumab Fab domain were effective at increasing the stability of the therapeutic mAb without compromising its affinity to its target. None of the single point mutations is likely to increase the immunogenicity of bevacizumab, based on the in silico immunogenicity assessment performed with the IEDB tools (Supplementary Information S4).62,63 In general, both approaches to disrupt potential aggregation-prone regions are effective strategies for improving mAb stability early on during drug discovery or for biobetter engineering.64,65 The removal and masking of APRs increases the stability of bevacizumab to the same level of the formulated drug. It is, therefore, likely that an appropriate formulation would further increase the stability of our biobetters. The masking and crowding of APRs by glycosylation motifs could potentially present further advantages over the removal of aggregation patches. The carbohydrate moiety might also stabilize the mAbs against other degradation routes such as hydrolysis, oxidation or deamidation.
Even though experimental high throughput screening methods are being developed to identify developable mAbs,66,67 computational predictive approaches remain attractive and competitive due to their low cost and no requirement for materials.68 There is a clear incentive for the development of new in silico platforms for high throughput screening of the developability and aggregation propensity of proteins with high accuracy, such as the sequence-based statistical model used in Lonza's aggregation prediction tool.69 Their implementation early on during the discovery phase allows the reduction of costs, easier manufacturing, and the formulation of higher concentrations, opening the door for new delivery routes or reduced dose administration, all to the advantage of the patients and practitioners, with the benefits of a potentially safer drug and lower treatment costs.
Materials and Methods
Molecular simulation
Several Fab MD simulations were performed, one of the wild-type of bevacizumab and one of each of the hyperglycosylated Fab variants (L118N, Q160N, Q160s and E195N). All three simulations were based on the crystal structure of bevacizumab, which was obtained from the RCSB PDB (PDB ID: 1BJ1).37 Hydrogen atoms were added to this structure at pH 7 using the PSFGEN plugin of the VMD.70 Topology and structure files for the hyperglycosylated Fab variants were generated using the Glycam website,71 assuming a G0 glycosylation pattern (GLYCAM notation: DGlcpNAcb1-2DManpa1-6[DGlcpNAcb1-2DManpa1-3]DManpb1-4DGlcpNAcb1-4DGlcpNAcb1). For all simulations, the AMBER12SB72 and Glycam0673 force fields were used for the protein and glycosylation, respectively. A molecular-dynamics (MD) simulation was performed on these all-atom structures of the Fab domain with an explicit TIP3P water model.74 Each Fab domain was solvated in a cubic box with periodic boundary conditions in all 3 directions. The dimensions of the water box were adjusted such that the surface of the Fab domain is at least 10 Å away from any side of the box. The solvated system was made charge neutral by adding chlorine ions. The system temperature and the pressure were maintained at 300 K and 1 atm, respectively, by the Berendsen coupling scheme.75 The GROMACS76 package was used to perform the MD simulations in the NPT ensemble. The systems were initially minimized and then equilibrated for 20 ns. Production runs of 80 ns were then performed and frames were extracted at every 0.1 ns for further analysis. The average SAP values of each residue at 5 Å and 10 Å were computed14 over the 80 ns of the MD production runs. The average effective hydrophobicity (Φeff) value of each residue77 was also computed over these MD trajectories.
The NCBI/IGBLAST webserver78 was used to identify the closest human germline sequence to the bevacizumab heavy chain and light chain Fv sequence. The germline sequence IGKV1 shares 87% sequence identity with the LC Fv of bevacizumab. For the residues selected for mutation, we identified corresponding residues in the sequence 1GKV1. Residue F50 can be humanized via mutation into an aspartic acid residue.
Identification of glycosylation sites to engineer
To identify potential glycosylation sites in the CH1 and CL domains of bevacizumab, we identified all of the high SAP residues in these domains as described above. Using the last frame from the MD simulation, we then identified all serine, threonine and asparagine residues that are within 10 Å of these high-SAP residues (the distance is the minimum distance between all atom-pairs of 2 residues) and belong to the CH1 and CL domains. For all of the selected S/T/N residues, we chose a neighboring residue (in the sequence) for mutation to generate an NXS or NXT glycosylation motif (see Table S1). We rejected all of the variants in which a GLY or PRO needs to be mutated because mutation of these residues may cause a signification perturbation in the structure of the domain. Furthermore, we also rejected all of the mutations that lead to an N-P-S or N-P-T motif as this motif does not undergo N-glycosylation. To be efficiently glycosylated, the side-chain of the ASN residue should be surface-exposed. Therefore, we rejected all of the mutations where the exposed surface area of the side-chain atoms (of the ASN or the residue to be mutated to ASN) is less than 15 Å2. All residues oriented on a different face relative to the high SAP value residues to be masked were dismissed as well. Lastly, we rejected all of the mutations of high SAP residues. Because we are interested in observing the effect of glycosylation on the aggregation propensity of an antibody, we generated only those variants in which the reduction of the SAP was caused only by masking high SAP regions with the carbohydrates. In this work, we generated 3 such variants: E195N, L118N and Q160N/S to test our hypothesis of whether the introduction of a glycosylation site near a high SAP region leads to an overall reduction in the aggregation propensity of the antibody.
Cloning, generation of variants, expression, and purification of mAbs
The bevacizumab genes, synthesized by Genscript (Piscataway, NJ), were codon-optimized for expression in mammalian cells and subsequently subcloned separately into the vector gWiz (Genlantis, Torreyana San Diego) using the Gibson method, resulting in the vectors gWiz-A-LC and gWiz-A-HC. Bevacizumab variants were generated by site-directed mutagenesis and confirmed by sequencing. Oligonucleotides (IDT, Coralville, Iowa) were designed to introduce single mutations on bevacizumab LC (F50D, V110K, L154K, L154D, L201K, Q160s, E195N) or on bevacizumab HC (V5K, L180K, L118N).
WT bevacizumab and variants were expressed by transient transfection of FreeStyle 293-F cells (Life Technologies, Grand Island, NY) grown in GIBCO FreeStyle 293 Expression Medium (Life Technologies). Transfections were performed using 0.5 g of each heavy-chain and light-chain mAb-expressing vector and 2 mg of polyethyleneimine (Polysciences, Inc.., Warrington, PA) per liter of 106 FreeStyle 293-F cells. After five to 6 days, the supernatant was collected and filtered (0.22 μm) prior to purification. Expressed mAbs (8 to 25 mg) were purified first by affinity chromatography (protein A sepharose from GE Healthcare, Piscataway, NJ), and then, concentrated on centrifugal filter devices AMICON YM30 (EMD Millipore - division of Merck KGaA, Darmstadt, Germany) for further purification by cation exchange. The pure proteins were buffer exchanged into 10 mM histidine (pH 6.0) and further concentrated to the desired concentration, as determined by measuring the absorbance at 280 nm.
Stability of mAbs
The accelerated aggregation studies of bevacizumab and its variants were performed within 3 days after formulation. A total of 50 mg/mL of protein in 10 mM histidine (pH 6.0) were incubated at 52°C in a Bio-Rad MyCycler Thermal Cycler (Hercules, CA). Aggregation was stopped at several time points by diluting the sample to 10 mg/mL in cold 15 mM potassium phosphate buffer (pH 6.5), followed by a 10 min incubation in ice. Part of each sample was further diluted to 1 mg/mL for turbidity measurement by assessing the absorbance at 320 nm. The extent of aggregation was measured at 22°C by SEC-HPLC performed on an Agilent 1200 LC (Santa Clara, CA) using a Tosoh TSKgel super SW3000 column (Tokyo, Japan). Samples (10 mg/mL) were centrifuged for 3 min at 6000 rpm to remove large insoluble aggregates prior to injection (5 μL) in the SEC-HPLC. The proteins were eluted with a mobile phase of 150 mM potassium phosphate buffer (pH 6.5) at a flow rate of 0.5 mL/min. Proteins were quantified by detection at 280 nm. Areas of the peaks were integrated at each time point. The ratio of the aggregates peak to the total peak area at each time provides the amount of soluble aggregates. The mass balance with the initial concentration (t = 0) allows the estimation of the amount of insoluble aggregates. Each experiment was reproduced at least in duplicate with different batches of proteins. The standard errors reported herein represent the deviation observed during the all mAb stability assessments (production, purification/formulation, accelerated aggregation study).
A propagation of error was applied to calculate standard deviations reported for the stability increase factor and the aggregation rate reduction. Aggregation rate constants were extracted from the fitting of a second order function to the monomer loss measured over time.
Turbidity was estimated by measuring the absorbance at 320 nm of 50 mg/mL samples previously stressed for 48 h at 52°C and diluted down to 1 mg/mL for measurement.
Differential scanning calorimetry
DSC was used to compare the thermal stability of bevacizumab and the variants. The bevacizumab thermogram is sensitive to the pH of the sample. Therefore, we also performed a thermostability characterization by DSC of our WT bevacizumab formulated in 10 mM histidine buffer (pH 6.0). DSC measurements were performed using a VP-DSC MicroCalorimeter (Microcal, Inc.., Northampton, MA). The samples were analyzed at a concentration of 0.8 mg/mL to 1.2 mg/mL in 10 mM histidine buffer at pH 6.0 using a scan range from 20 to 95°C and a scan rate of 1°C/min. The mAb concentration is believed to be sufficiently low to prevent aggregation to occur during the acquisition of the DSC data. Histidine buffer at pH 6.0 was used as a blank reference. Thermograms were corrected by subtracting the buffer blank scans and normalizing values to the protein concentration. Data were analyzed using Origin 7.0 software (OriginLab Corporation, Northampton, MA). The transition curves were fitted to 3 Gaussians to obtain the transition temperatures of the different antibody domains (Tm1, Tm2 and Tm3).
VEGF binding assay
Bevacizumab binding to VEGF was measured using a VEGF sandwich ELISA (VEGF Human ELISA Kit, Invitrogen, Carlsbad, CA) with the following modification. VEGF protein (1500 pg/mL) was pre-incubated and shaken for 60 min at 37°C with bevacizumab or the variants diluted in the kit incubation buffer. MAbs were studied in a concentration range of 0.5 pM to 1 μM. Each mix was then transferred into ELISA plates coated with anti-VEGF antibodies. The assay protocol was then followed according to the manufacturer's instructions. The more our mAbs bind to VEGF, the less VEGF will be captured on the ELISA plate and further detected, corresponding to a lower absorbance measured at 450 nm. VEGF standards were run with each ELISA assay as recommended by the manufacturer, and each assay was run at least in duplicate. Plates were read in an Infinite M200 reader (Tecan, Männedorf, Switzerland) at 450 nm. The equilibrium dissociation constant (KD) values were determined by fitting the data points to a sigmoid curve according to the following equation: Y = B + ((A-B)/(1+(X/C)D)), A being the highest measured fluorescence intensity, B the lowest fluorescence intensity and C the KD.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to acknowledge the financial support provided by the Singapore-MIT Alliance.
The Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems (NSF-0070319) is also gratefully acknowledged.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Aggarwal RS. What's fueling the biotech engine-2012 to 2013. Nat Biotechnol 2014; 32:32-9; PMID:24406926; http://dx.doi.org/ 10.1038/nbt.2794 [DOI] [PubMed] [Google Scholar]
- 2.Joshi V, Shivach T, Kumar V, Yadav N, Rathore A. Avoiding antibody aggregation during processing: establishing hold times. Biotechnol J. 2014; 9:1195-205; PMID:24753430; http://dx.doi.org/ 10.1002/biot.201400052 [DOI] [PubMed] [Google Scholar]
- 3.Ratanji KD, Derrick JP, Dearman RJ, Kimber I. Immunogenicity of therapeutic proteins: Influence of aggregation. J Immunotoxicol 2014; 11:99-109; PMID:23919460; http://dx.doi.org/ 10.3109/1547691X.2013.821564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg AS. Effects of protein aggregates: an immunologic perspective, AAPS J 2006; 8:E501eE507; http://dx.doi.org/ 10.1208/aapsj080359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vázquez-Rey M, Lang DA. Aggregates in monoclonal antibody manufacturing processes. Biotechnol Bioeng 2011; 108:1494-508; PMID:Can't; http://dx.doi.org/ 10.1002/bit.23155 [DOI] [PubMed] [Google Scholar]
- 6.Shire SJ. Formulation and manufacturability of biologics. Curr Opin Biotechnol. 2009; 20:708-14; PMID:19880308; http://dx.doi.org/ 10.1016/j.copbio.2009.10.006 [DOI] [PubMed] [Google Scholar]
- 7.Roque C, Sheung A, Rahman N, Ausar SF. Effect of polyethylene glycol conjugation on conformational and colloidal stability of a monoclonal antibody antigen-binding fragment (Fab'). Mol Pharm 2015; 12:562-75; PMID:25548945; http://dx.doi.org/ 10.1021/mp500658w [DOI] [PubMed] [Google Scholar]
- 8.Kayser V, Chennamsetty N, Voynov V, Forrer K, Helk B, Trout BL. Glycosylation influences on the aggregation propensity of therapeutic monoclonal antibodies. Biotechnology Journal 2011; 6:38-44; PMID:20949542; http://dx.doi.org/ 10.1002/biot.201000091 [DOI] [PubMed] [Google Scholar]
- 9.Perchiacca JM, Tessier PM. Engineering Aggregation-Resistant Antibodies. Annu Rev Chem Biomol Eng 2012; 3:263-86; PMID:22468604; http://dx.doi.org/ 10.1146/annurev-chembioeng-062011-081052 [DOI] [PubMed] [Google Scholar]
- 10.Agrawal NJ, Kumar S, Wang X, Helk B, Singh SK, Trout BL. Aggregation in protein-based biotherapeutics: computational studies and tools to identify aggregation-prone regions. J Pharm Sci. 2011; 100:5081-95; PMID:21789769 [DOI] [PubMed] [Google Scholar]
- 11.Ratanji KD, Derrick JP, Dearman RJ, Kimber I. Immunogenicity of therapeutic proteins: Influence of aggregation; K.D. J Immunotoxicol 2014; 11:99-109; PMID:23919460; http://dx.doi.org/ 10.3109/1547691X.2013.821564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castillo V, Graña-Montes R, Sabate R, Ventura S. Prediction of the aggregation propensity of proteins from the primary sequence: aggregation properties of proteomes. Biotechnol J 2011. 6:674-85; PMID:21538897; http://dx.doi.org/ 10.1002/biot.201000331 [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Das TK, Singh SK, Kumar S. Potential aggregation prone regions in biotherapeutics: A survey of commercial monoclonal antibodies. mAbs 2009; 1:254-67; PMID:20065649; http://dx.doi.org/ 10.4161/mabs.1.3.8035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chennamsetty N, Voyonov V, Kayser V, Helk B, Trout BL. Design of Therapeutic proteins with enhanced stability. Proc Nat Acad Sci 2009. 106:11937-42; PMID:19571001; http://dx.doi.org/ 10.1073/pnas.0904191106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chennamsetty N, Helk B, Voyonov V, Kayser V, Trout BL. Aggregation-Prone motifs in Human Immunoglobulin G. J Mol Biol 2009; 391:404-413; PMID:19527731; http://dx.doi.org/ 10.1016/j.jmb.2009.06.028 [DOI] [PubMed] [Google Scholar]
- 16.Voynov V, Chennamsetty N, Kayser V, Helk B, Trout BL. Predictive tools for stabilization of therapeutic proteins. mAbs 2009; 1:580-1; PMID:20068399; http://dx.doi.org/ 10.4161/mabs.1.6.9773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voynov V, Chennamsetty N, Kayser V, Wallny HJ, Helk B, Trout BL. Design and application of antibody cysteine variants. Bioconjugate Chemistry 2010; 21; 385-92; PMID:20092294; http://dx.doi.org/ 10.1021/bc900509s [DOI] [PubMed] [Google Scholar]
- 18.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, et al.. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350:2335-42; PMID:15175435; http://dx.doi.org/ 10.1056/NEJMoa032691 [DOI] [PubMed] [Google Scholar]
- 19.Los M, Roodhart JML, Voest EE. Target Practice: Lessons from Phase III Trials with Bevacizumab and Vatalanib in the Treatment of Advanced Colorectal Cancer. Oncologist. 2007; 12:443-50; PMID:17470687; http://dx.doi.org/ 10.1634/theoncologist.12-4-443 [DOI] [PubMed] [Google Scholar]
- 20.Ciulla TA, Rosenfeld PJ. Antivascular endothelial growth factor therapy for neovascular age-related macular degeneration. Curr Opin Ophthalmol. 2009; 20:158-65; PMID:19417570; http://dx.doi.org/ 10.1097/ICU.0b013e32832d25b3 [DOI] [PubMed] [Google Scholar]
- 21.Paul M, Lahlou A, Carvalho M, Blanchet B, Astier A. Thermal stability of two monoclonal antibodies: cetuximab and bevacizumab. Eur J Oncol Pharm 2008; 2:37 [Google Scholar]
- 22.Oliva A, Llabrés M, Fariña JB. Capability measurement of size-exclusion chromatography with a light-scattering detection method in a stability study of bevacizumab using the process capability indices. J Chromatogr A 2014; 1353:89-98; PMID:24786652; http://dx.doi.org/ 10.1016/j.chroma.2014.04.027 [DOI] [PubMed] [Google Scholar]
- 23.Cromwell M, Gazzano-Santoro H. Protein Aggregation and Potency, Genentech, Inc, IIR Conference on Impurities of Biomolecules, 2006 Nov 7; San Francisco, CA: Available from: www.iirusa.com/upload/wysiwyg/P1198_Images/IIR_P1198_Cromwell.pdf [Google Scholar]
- 24.Latypov RF, Hogan S, Lau H, Gadgil H, Liu D. Elucidation of Acid-induced Unfolding and Aggregation of Human Immunoglobulin IgG1 and IgG2 Fc. J Biol Chem 2012; 287:1381-96; PMID:22084250; http://dx.doi.org/ 10.1074/jbc.M111.297697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Kumar S, Buck PM, Singh SK. Impact of deglycosylation and thermal stress on conformational stability of a full length murine IgG2a monoclonal antibody: Observations from molecular dynamics Simulations. Proteins 2013; 81:443-60; PMID:23065923; http://dx.doi.org/ 10.1002/prot.24202 [DOI] [PubMed] [Google Scholar]
- 26.Li CH, Narhi LO, Wen J, Dimitrova M, Wen Z, Li J, Pollastrini J, Nguyen X, Tsuruda T, Jiang Y. Effect of pH, Temperature, and Salt on the Stability of Escherichia coli- and Chinese Hamster Ovary Cell-Derived IgG1 Fc. Biochemistry 2012; 51:10056-65; PMID:23078371; http://dx.doi.org/ 10.1021/bi300702e [DOI] [PubMed] [Google Scholar]
- 27.Zheng K, Bantog C, Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. mAbs 2011; 3:568-76; PMID:22123061; http://dx.doi.org/ 10.4161/mabs.3.6.17922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinclair AM, Elliott S. Glycoengineering: the effect of glycosylation on the properties of therapeutic proteins. J Pharm Sci 2005; 94:1626-35; PMID:15959882; http://dx.doi.org/ 10.1002/jps.20319 [DOI] [PubMed] [Google Scholar]
- 29.Pepinsky RB, Silvian L, Berkowitz SA, Farrington G, Lugovskoy A, Walus L, Eldredge J, Capili A, Mi S, Graff C, Garber E. Improving the solubility of anti-LINGO-1 monoclonal antibody Li33 by isotype switching and targeted mutagenesis. Protein Science 2010; 19:954-66; PMID:20198683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voynov V, Chennamsetty N, Kayser V, Helk B, Forrer K, Zhang H, Fritsch C, Heine H, Trout BL. Dynamic fluctuations of protein-carbohydrate interactions promote protein aggregation. PLoS One 2009; 4:e8425; PMID:20037630; http://dx.doi.org/ 10.1371/journal.pone.0008425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Prediction of aggregation prone regions of therapeutic proteins. J Phys Chem B. 2010; 114:6614-24; PMID:20411962; http://dx.doi.org/ 10.1021/jp911706q [DOI] [PubMed] [Google Scholar]
- 32.Courtois F, Schneider CP, Agrawal NJ, Trout BL. Rational design of biobetters with enhanced stability. J Pharm Sci 2015; 104:2433-40; PMID:26096711; http://dx.doi.org/ 10.1002/jps.24520 [DOI] [PubMed] [Google Scholar]
- 33.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Prediction of protein binding regions. Proteins 2011; 79:888-97; PMID:21287620; http://dx.doi.org/ 10.1002/prot.22926 [DOI] [PubMed] [Google Scholar]
- 34.Westermaier Y, Veurink M, Riis-Johannessen T, Guinchard S, Gurny R, Scapozza L. Identification of aggregation breakers for bevacizumab (Avastin®) self-association through similarity searching and interaction studies. European Journal of Pharmaceutics and Biopharmaceutics 2013; 85:773-80; PMID:23665445; http://dx.doi.org/ 10.1016/j.ejpb.2013.04.012 [DOI] [PubMed] [Google Scholar]
- 35.Veurink M, Westermaier Y, Gurny R, Scapozza L. Breaking the aggregation of the monoclonal antibody bevacizumab (Avastin®) by dexamethasone phosphate: insights from molecular modelling and asymmetrical flow field-flow fractionation, Pharm Res 2013; 30:1176-87; PMID:23412914; http://dx.doi.org/ 10.1007/s11095-012-0955-6 [DOI] [PubMed] [Google Scholar]
- 36.Magdelaine-Beuzelin C, Kaas Q, Wehbi V, Ohresser M, Jefferis R, Lefranc MP, Watier H. Structure–function relationships of the variable domains of monoclonal antibodies approved for cancer treatment. Crit Rev in Oncol Hematol 2007; 64:210-25; http://dx.doi.org/ 10.1016/j.critrevonc.2007.04.011 [DOI] [PubMed] [Google Scholar]
- 37.Muller YA, Chen Y, Christinger HW, Li B, Cunningham BC, Lowman HB, de Vos AM. VEGF and the Fab fragment of a humanized neutralizing antibody:crystal structure of the complex at 2.4 A resolution and mutational analysis of the interface. Structure 1998; 6:1153-67; PMID:9753694; http://dx.doi.org/ 10.1016/S0969-2126(98)00116-6 [DOI] [PubMed] [Google Scholar]
- 38.Kaja S, Hilgenberg JD, Everett E, Olitsky SE, Gossage J, Koulen P. Effects of dilution and prolonged storage with preservative in a polyethylene container on Bevacizumab (Avastin™) for topical delivery as a nasal spray in anti-hereditary hemorrhagic telangiectasia and related therapies. Hum Antibodies 2011; 20:95-101; PMID:22129679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dudgeon K, Rouet R, Kokmeijer I, Schofield P, Stolp J, Langley D, Stock D, Christ D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc Natl Acad Sci U S A 2012; 109:10879-84; PMID:22745168; http://dx.doi.org/ 10.1073/pnas.1202866109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bakri SJ, Snyder MR, Pulido JS, McCannel CA, Weiss WT, Singh RJ. Six-month stability of bevacizumab (Avastin) binding to vascular endothelial growth factor after withdrawal into a syringe and refrigeration or freezing. Retina 2006; 26:519-22; PMID:16770257; http://dx.doi.org/ 10.1097/01.iae.0000225354.92444.7a [DOI] [PubMed] [Google Scholar]
- 41.Paul M, Vieillard V, Roumi E, Cauvin A, Despiau MC, Laurent M, Astier A. Long-term stability of bevacizumab repackaged in 1 mL polypropylene syringes for intravitreal administration. A. Ann Pharm Fr 2012; 70:139-54; PMID:22655582; http://dx.doi.org/ 10.1016/j.pharma.2012.03.006 [DOI] [PubMed] [Google Scholar]
- 42.Liu L, Ammar DA, Ross LA, Mandava N, Kahook NY, Carpen-ter JF. Silicone oil microdroplets and protein aggregates in repackaged bevacizumab and ranibizumab: effects of long-term storage and product mishandling. Invest Ophtalmol Vis Sci 2011; 52:1023-34; http://dx.doi.org/ 10.1167/iovs.10-6431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Veurink M, Stella C, Tabatabay C, Pournaras CJ, Gurny R. Association of ranibizumab (Lucentis®) or bevacizumab (Avastin®) with dexamethasone and triamcinolone acetonide: an in vitro stability assessment. Eur J Pharm Biopharm 2011; 78:271-7; PMID:21172437; http://dx.doi.org/ 10.1016/j.ejpb.2010.12.018 [DOI] [PubMed] [Google Scholar]
- 44.Zhang A, Singh SK, Michael R, Shirts MR, Kumar S, Fernandez EJ. Distinct aggregation mechanisms of monoclonal antibody under thermal and freeze-thaw stresses revealed by hydrogen exchange. Pharm Res 2012; 29:236-250; PMID:21805212; http://dx.doi.org/ 10.1007/s11095-011-0538-y [DOI] [PubMed] [Google Scholar]
- 45.Ionescu RM, Vlasak J, Price C, Kirchmeier M. Contribution of Variable Domains to the Stability of Humanized IgG1 Monoclonal Antibodies. J Pharm Sci. 2008; 97:1414-26; PMID:17721938; http://dx.doi.org/ 10.1002/jps.21104 [DOI] [PubMed] [Google Scholar]
- 46.Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E, Pyles EA, Yancopoulos GD, Stahl N, Wiegand SJ. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 2012; 15:171-85; PMID:22302382; http://dx.doi.org/ 10.1007/s10456-011-9249-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu Y, Lee P, Ke Y, Zhang Y, Yu Q, Lee J, Li M, Song J, Chen J, Dai J, Do Couto FJ, An Z, Zhu W, Yu GL. A humanized anti-VEGF rabbit monoclonal antibody inhibits angiogenesis and blocks tumor growth in xenograft models. Plosone 2010; 5:e9072; http://dx.doi.org/ 10.1371/journal.pone.0009072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jenkins N. Modifications of therapeutic proteins: challenges and prospects. Cytotechnology 2007; 53:121-5; PMID:19003198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. The Impact of Glycosylation on the Biological Function and Structure of Human Immunoglobulins. Annu Rev Immunol 2007; 25:21-50; PMID:17029568 [DOI] [PubMed] [Google Scholar]
- 50.Lee CC, Perchiacca JM, Peter M. Tessier PM. Toward aggregation-resistant antibodies by design. Trends Biotechnol 2013; 31:612-20; PMID:23932102 [DOI] [PubMed] [Google Scholar]
- 51.Buchanan A, Clementel V, Woods R, Harn N, Bowen MA, Mo W, Popovic B, Bishop SM, Dall'Acqua W, Minter R, Jermutus L, Bedian V. Engineering a therapeutic IgG molecule to address cysteinylation, aggregation and enhance thermal stability and expression. mAbs 2013; 5:255-62; PMID:23412563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olsson MHM, Sondergard CR, Rostkowski M, Jensen JH. Consistent Treatment of Internal and Surface Residues in Empirical pKa predictions. J Chem Theory Comput. 2011; 7:525-37 [DOI] [PubMed] [Google Scholar]
- 53.Perchiacca JM, Lee CC, Tessier PM. Optimal charged mutations in the complementarity determining regions that prevent domain antibody aggregation are dependent on the antibody scaffold. Protein Engineering, Design & Selection 2014; 27:29-39 [DOI] [PubMed] [Google Scholar]
- 54.Wu SJ, Luo J, O'Neil KT, Kang J, Lacy ER, Canziani G, Baker A, Huang M, Tang QM, Raju TS, Jacobs SA, Teplyakov A, Gilliland GL, Feng Y. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng Des Sel 2010; 23:643-51; PMID:20543007 [DOI] [PubMed] [Google Scholar]
- 55.Hristodorov D, Fischer R, Linden L. With or Without Sugar? (A)glycosylation of Therapeutic Antibodies. Mol Biotechnol 2013; 54:1056-68; PMID:23097175 [DOI] [PubMed] [Google Scholar]
- 56.Hari SB, Lau H, Razinkov VI, Chen S, Latypov RF. Acid-induced aggregation of human monoclonal IgG1 and IgG2: molecular mechanism and the effect of solution composition. Biochemistry 2010; 49:9328-38; PMID:20843079 [DOI] [PubMed] [Google Scholar]
- 57.Hristodorov D, Fischer R, Joerissen H, Müller-Tiemann B, Apeler H, Linden L. Generation and Comparative Characterization of Glycosylated and Aglycosylated Human IgG1 Antibodies. Mol Biotechnol 2013; 53:326-35; PMID:22427250 [DOI] [PubMed] [Google Scholar]
- 58.Mattu TS, Pleass RJ, Willis AC, Kilian M, Wormald MR, Lellouch AC, Rudd PM, Woof JM, Dwek RA. The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fc alpha receptor interactions. J Biol Chem 1998; 273:2260-72; PMID:9442070 [DOI] [PubMed] [Google Scholar]
- 59.Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov 2009; 8:226-234; PMID:19247305 [DOI] [PubMed] [Google Scholar]
- 60.Qian J, Liu T, Yang L, Daus A, Crowley R, Zhou Q. Structural characterization of N-linked oligosaccharides on monoclonal antibody cetuximab by the combination of orthogonal matrix-assisted laser desorption/ionization hybrid quadrupole-quadrupole time-of-flight tandem mass spectrometry and sequential enzymatic digestion. Anal Biochem 2007; 364:8-18; PMID:17362871 [DOI] [PubMed] [Google Scholar]
- 61.Sanchez-Ruiz JM. Theoretical analysis of Lumry–Eyring models in differential scanning calorimetry. Biophys J 1992; 61:921-35; PMID:19431826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calis JJA, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, Sette A, Kesmir C, Peters B. Properties of MHC class I presented peptides that enhance immunogenicity. PloS Comp Biol 2013; 9:e1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett 1990; 276:172-4; PMID:1702393; http://dx.doi.org/ 10.1016/0014-5793(90)80535-Q [DOI] [PubMed] [Google Scholar]
- 64.Trout BL, Schneider CP, Agrawal NJ. Her2-and vegf-a-binding proteins with enhanced stability. U.S. Patent Application No. 61/706240 2013.
- 65.Trout BL, Schneider CP, Agrawal NJ. Cd20-and egfr-binding proteins enhanced stability. U.S. Patent Application No. 61/706242 2013.
- 66.Lavoisier A, Schlaeppi JM. Early developability screen of therapeutic antibody candidates using Taylor dispersion analysis and UV area imaging detection. mAbs 2015; 7:77-83; PMID:25514497; http://dx.doi.org/ 10.4161/19420862.2014.985544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamniuk AP, Ditto N, Patel M, Dai J, Sejwal P, Stetsko P, Doyle ML. Application of a kosmotrope-based solubility assay to multiple protein therapeutic classes indicates broad use as a high-throughput screen for protein therapeutic aggregation propensity. J Pharm Sci 2013; 102:2424-39; PMID:23712759; http://dx.doi.org/ 10.1002/jps.23618 [DOI] [PubMed] [Google Scholar]
- 68.Lauer TM, Agrawal NJ, Chennamsetty N, Egodage K, Helk B, Trout BL. Developability index: a rapid in silico tool for the screening of antibody aggregation propensity. J Pharm Sci 2012; 101:102-15; PMID:21935950; http://dx.doi.org/ 10.1002/jps.22758 [DOI] [PubMed] [Google Scholar]
- 69.Obrezanova O, Arnell A, de la Cuesta RG, Berthelot ME, Gallagher TR, Zurdo J, Stallwood Y. Aggregation risk prediction for antibodies and its application to biotherapeutic development. MAbs 2015; 7:352-63; PMID:25760769; http://dx.doi.org/ 10.1080/19420862.2015.1007828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 1996; 14:33-8; PMID:8744570; http://dx.doi.org/ 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 71.Woods Group. GLYCAM Web ; web tool Complex Carbohydrate Research Center, University of Georgia, Athens, GA; 2005. ; last accessed 2015 June 5. Available from www.glycam.org [Google Scholar]
- 72.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, et al.. AMBER 12; University of California, San Fransisco, 2012. [Google Scholar]
- 73.Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeiriño J, Daniels CR, Foley BL, Woods RJ. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J Comput Chem 2008; 29:622-55; PMID:17849372; http://dx.doi.org/ 10.1002/jcc.20820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983; 79:926-35; http://dx.doi.org/ 10.1063/1.445869 [DOI] [Google Scholar]
- 75.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984; 81:3684-90; http://dx.doi.org/ 10.1063/1.448118 [DOI] [Google Scholar]
- 76.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation 2008; 4:435-47; http://dx.doi.org/ 10.1021/ct700301q [DOI] [PubMed] [Google Scholar]
- 77.Agrawal NJ, Helk B, Trout BL. A computational tool to predict the evolutionarily conserved protein–protein interaction hot-spot residues from the structure of the unbound protein. FEBS Letters 2014; 588:326-33; PMID:24239538; http://dx.doi.org/ 10.1016/j.febslet.2013.11.004 [DOI] [PubMed] [Google Scholar]
- 78.Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 2013; 41:34-40; http://dx.doi.org/ 10.1093/nar/gkt382 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.