

Abstract
Daptomycin (DAP) is a cyclic lipopeptide with in vitro activity against a variety of Gram-positive pathogens, including multidrug-resistant organisms. Since its introduction in clinical practice in 2003, DAP has become an important key front-line antibiotic for severe or deep-seated infections caused by Gram-positive organisms. Unfortunately, DAP-resistance (R) has been extensively documented in clinically important organisms such as Staphylococcus aureus, Enterococcus spp, and Streptococcus spp. Studies on the mechanisms of DAP-R in Bacillus subtilis and other Gram-positive bacteria indicate that the genetic pathways of DAP resistance are diverse and complex. However, a common phenomenon emerging from these mechanistic studies is that DAP-R is associated with important adaptive changes in cell wall and cell membrane homeostasis with critical changes in cell physiology. Findings related to these adaptive changes have offered novel insights into the genetics and molecular mechanisms of bacterial cell envelope stress response and the manner in which Gram-positive bacteria cope with the antimicrobial peptide attack and protect vital structures of the cell envelope such as the cell membrane. In this review, we will examine the most recent findings related to the molecular mechanisms of resistance to DAP in relevant Gram-positive pathogens and discuss the clinical implications for therapy against these important bacteria.
Keywords: daptomycin resistance, Bacillus subtilis, Staphylococcus aureus, Enterococcus
Introduction
Antimicrobial resistance is increasingly recognized as a major public health problem that threatens the medical care of patients worldwide. Infections caused by multi-drug resistant (MDR) organisms result in significant increases in mortality and have been associated with a large economic burden. Indeed, a recent report estimated that antibiotic resistance would be responsible for around 300 million premature deaths by 2050, with a loss of up to $100 trillion to the global economy.1 Due to this worrisome scenario, a number of governmental agencies, academic societies and international organizations have issued statements calling for action to tackle the antimicrobial resistance crisis.2, 3 To make things worse, this situation is aggravated by the lack of a robust antibiotic pipeline, resulting in emergence of infections that are almost untreatable and leaving clinicians with no reliable alternatives to treat these patients.
Daptomycin (DAP), a lipopeptide antibiotic produced by Streptomyces roseosporus has become an important option for the management of MDR infections due to Gram-positive organisms. DAP was initially examined for clinical use in the 1980s, but its development was halted due to the high frequency of muscle related toxicity in phase I and II trials.4 However, subsequent animal studies indicated that muscle toxicity could be markedly reduced by using once-daily dosing (as compared to the original twice-daily dosing regimen). Thus, in 2003, DAP was granted FDA-approval for the treatment of complicated skin and soft tissue infections caused by Gram-positive organisms, including vancomycin-susceptible E. faecalis. In 2006, DAP received approval for Staphylococcus aureus bacteremia and right-sided infective endocarditis. Similarly, DAP was approved by the European regulatory agency for the same clinical indications in 2006.
DAP is active against a wide-range of Gram-positive bacteria, including most clinically relevant MDR organisms such as methicillin-resistant S. aureus (MRSA), vancomycin-resistant enterococci (VRE), vancomycin-intermediate S. aureus (VISA) and penicillin-resistant Streptococcus pneumoniae. The clinical susceptibility breakpoints established by the Clinical Laboratory Standards Institute (CLSI) are ≤ 1 mg/L for staphylococci and ≤ 4 mg/L for enterococci. No resistance breakpoint has been officially established; hence isolates exhibiting MIC values above the susceptibility cut-off are technically labeled as “non-susceptible”. For ease of use, in this manuscript we will refer to this population as daptomycin-resistant (DAP-R). To date, the majority of Gram-positive organisms remain susceptible to DAP (DAP-S). However, the development of DAP-resistance (DAP-R) emerging during therapy or as a de novo phenomenon, has been described in several species.5 Although the mechanisms of DAP-R remain to be fully elucidated, recent work from different research groups has provided important insights into the mechanistic bases of resistance. Indeed, such mechanisms appear to be complex, diverse and mainly related to activation of the inherent bacterial self-defense processes in response to the antimicrobial “attack”. In this manuscript, we will summarize our current understanding of the molecular and biochemical basis of DAP-R. In order to organize the discussion, and considering that the resistance pathways vary among Gram-positive organisms, we will discuss the most relevant mechanisms of DAP-R in each species separately.
Mechanism of action of daptomycin
DAP is structurally and functionally related to cationic antimicrobial peptides (CAMPs) produced by the innate immune system. The DAP molecule consists of a cyclic polypeptide core of 13 amino acids attached to a lipophilic tail (a decanoyl fatty acid) (Figure 1).6 DAP exerts its bactericidal effect by altering the bacterial cell envelope homeostasis interacting with phospholipids of the cell membrane (CM), in a process that has not been fully elucidated. The bactericidal activity of DAP depends on the presence of ionized calcium. Indeed, the positive charge of the DAP-calcium complex is thought to facilitate the insertion of the antibiotic into the bacterial cell membrane. Furthermore, addition of calcium also appears to favor the formation of DAP micellar structures that have been postulated to serve as vehicles for the delivery of DAP to the bacterial CM.7, 8
Figure 1.
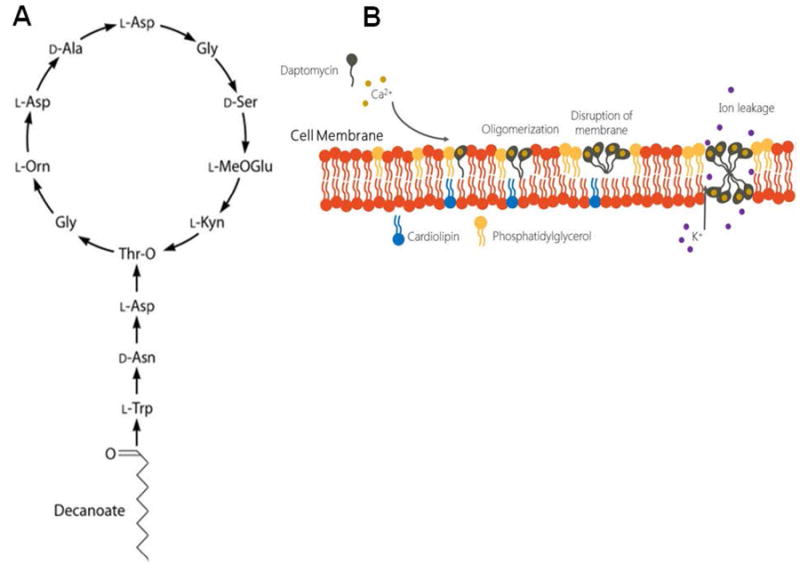
Structure of daptomycin (A) and proposed mechanism of action (B). See text for details
The mechanisms that lead to DAP-mediated bacterial cell dead are also not fully established but the antibiotic has been shown to rapidly depolarize cells of Bacillus spp. and to inhibit the active transport of amino acids.9 In addition, it has also been proposed that DAP’s mode of action includes inhibition of peptidoglycan and/or lipoteichoic acid synthesis through mechanisms that remain poorly understood.10, 11
The specific interactions between the DAP-calcium complex and the CM are still a matter of active research but several steps are thought to be important (Figure 1). In the first step (after insertion to the CM), the DAP-calcium complex oligomerize in the outer leaflet of the CM in a process that appears to be dependent on the presence of the phospholipid phosphatidylglycerol (PG). Indeed, experiments using liposomes with different composition of phospholipids have indicated that PG is crucial for DAP’s oligomerization into the CM,12, 13 which, in turn, is necessary for its bactericidal activity. Furthermore, DAP-R has been consistently associated with a decrease in PG content or increase in the conversion of PG to its positively-charged derivative lysyl-PG (L-PG) in the CM of several Gram-positive organisms such as B. subtilis, S. aureus and E. faecalis (see below). The second step involves translocation of DAP oligomers into the inner leaflet of the CM in a model in which two opposing structures (located in the outer and inner CM leaflet, respectively) result in the formation of a functional pore-like structure.14 Indeed, using fluorescence resonance energy transfer (FRET), Muraih et al. have confirmed that DAP forms oligomers on PG-containing membranes and that the oligomers contain approximately 6- 7 subunits.15 Furthermore, characterization of DAP pores show that they are cation- and size-selective with the highest permeability to Na+, K+, and alkali metal ions.14 Interestingly, the translocation of DAP oligomers from outer to inner leaflets of the CM appear to be influenced by the presence of cardiolipin (CL), a negatively charged phospholipid that plays important roles in CM homeostasis in bacteria. Indeed, enrichment of liposomes with 10% CL was sufficient to prevent translocation of DAP oligomers, disrupting the formation of pore-like structures.16 These findings suggest that increased CL concentrations might prevent, at least in part, the antibacterial effect of DAP.
Chen et. al. recently postulated an alternative model for DAP’s mechanism of action. Using microscopy imaging of giant unilamellar vesicles (GUVs) formed by different lipid compositions, the authors evaluated DAP’s activity and provided evidence that suggested that interaction of DAP with the cell membrane results in a marked alteration of phospholipid content, an effect that they designated “lipid extracting effect”. The authors showed that in the presence of low concentrations of DAP, an initial expansion of the GUV occurs due to binding of the antibiotic molecule to the surface. At higher DAP concentrations, this expansion is followed by a decrease in outer surface area of the GUVs which is associated with lipid-peptide aggregates exuding from the surface of GUVs (ie., lipids are removed from the lipid bilayer, a phenomenon designated the lipid extracting effect). This phenomenon was only seen when binding of DAP increased the membrane surface area to > 3%. Importantly, the lipid extracting effect was only observed when PG was present in the GUVs and when calcium was added to the solution.17
Although these data have increased our understanding of the molecular events that occur when DAP inserts into the CM, the specific mechanisms leading to bacterial cell death in DAP-treated organisms remain obscure. Using a Bacillus subtilis model, Pogliano et al. showed that interaction of DAP with the CM preferentially occurs at nascent septa and induces marked changes in cell shape. The exposure of B. subtilis to sublethal concentrations of DAP produces bent and patchy areas of the CM at sites of interactions of the DAP molecule with the CM target. These areas of CM “damage” appear to trigger mislocalization of cell division proteins (including the essential protein DivIVA of B. subtilis) and induce localized synthesis of peptidoglycan.18 Based on these observations, a refined model of the DAP mechanism of action is proposed where DAP binds to the bacterial CM in the presence of PG causing local alterations in membrane curvature. The changes in membrane structure are recognized by DivIVA, which incorrectly identifies this area as a potential site of division, triggering peptidoglycan biosynthesis. At high DAP concentrations, the overwhelming changes in CM homeostasis (and perhaps cell wall synthesis) leads to leakage of ions and loss of cell membrane potential.18
Mechanisms of daptomycin resistance
Through years of evolution Gram-positive bacteria have developed a myriad of sophisticated mechanisms to survive hostile environments. In order to succeed in the human host, bacteria need to adapt and, particularly, resist the attacks orchestrated by the immune system. CAMPs produced by the innate immune system are among the first potent anti-bacterial molecules and first line of defense against bacterial “invaders”. In order to respond to the CAMP challenge, bacterial pathogens have devised a cadre of very complex mechanisms to counteract this attack and prevent disruption of pivotal cellular process such as cell wall synthesis and membrane homeostasis. As it will be discussed below, resistance to DAP frequently results in decreased activities of CAMPs, suggesting convergent mechanisms. Although some environmental, non-pathogenic organisms have been found to harbor enzymes with the ability to hydrolyze and inactivate the DAP molecule, this type of resistance has not been shown in clinically important bacteria to date. However, these genes coding for DAP inactivating enzymes could be a potential source of resistance determinants if they could be captured by pathogenic bacteria, as it has been previously reported for vancomycin and β-lactam resistance determinants (van gene clusters originating in Paenibacillus spp. and CTX-M enzymes from Kluyvera spp., respectively). A summary of proposed mechanisms of DAP-R in B. subtilis, S. aureus, and enterococci is presented in Table 1.
Table 1.
Proposed mechanisms and genes associated with daptomycin resistance in B. subtilis, S. aureus, and enterococci.
| Organism | Gene | Predicted Function | Proposed Mechanism of Resistance | Ref. |
|---|---|---|---|---|
| B. subtilis | pgsA | Phosphatidylglycerol synthase | Repulsion | 24, 34 |
| liaFSR | Three-component regulatory system as part of the cell envelope stress response | 24, 28, 34 | ||
|
| ||||
| S. aureus | dltABCD | Proteins that are involved in d-analylation of cell wall teichoic acids | Repulsion | 63-68 |
| mprF | A bifunctional enzyme that catalyzes the lysinylation of phosphatidylglycerol and translocation of lysyl- phosphatidylglycerol from the inner to the outer leaflet of the cell membrane | 46-62 | ||
| yycG | Two-component regulatory system of cell wall synthesis and homeostasis | 57, 75, 76 | ||
| cls | Cardiolipin synthase | 74, 77-80 | ||
| pgsA | Phosphatidylglycerol synthase | 80 | ||
|
| ||||
| E. faecalis | liaFSR | Three-component regulatory system as part of the cell envelope stress response | Diversion | 89, 91, 94 |
| cls | Cardiolipin synthase | 89, 91, 97 | ||
| gdpD | Glycerophosphoryl diester phosphodiesterase | 89, 91 | ||
|
| ||||
| E. faecium | liaSR | Histidine kinase and response regulator of three-component regulatory system | Repulsion | 89, 98, 100, 105, 106, 126 |
| cls | Cardiolipin synthase | 89, 101-105, 109 | ||
| yycFG | Two-component regulatory system of cell wall synthesis and homeostasis | 101, 104, 109 | ||
DAP resistance in B. subtilis
Although B. subtilis is not a common human pathogenic species, an important amount of work has been performed in this organism as a model for studying the Gram-positive cell envelope stress response (CESR). Therefore, it deserves to be discussed in detail. The CESR of B. subtilis is mediated by two-component regulatory systems (TCS) and extra-cytoplasmic function (ECF) σ factors.19-21 B. subtilis response to antibiotics that target the cell wall (such as vancomycin and bacitracin, among others) and cationic antimicrobial peptides is mediated by activation of several regulatory networks. Among them, the most studied are those that involve σM, the TCS LiaSR and BceRS (for bacitracin) and σW (for vancomycin) .21, 22 In the case of DAP, exposure to the antibiotic appears to induce a general response mediated by ECF σ factors σV, σM, σW, and σX (which are overexpressed by ~ 3-fold) and a specific response that involves the TCS LiaSR.23 Indeed, expression of LiaSR appears to be upregulated >400-fold in the presence of DAP, a value that is several order of magnitude higher than that of any other regulatory systems.23, 24 Interestingly, molecules that are structurally similar to DAP, such as the cyclic lipopeptide friulimicin B did not trigger the same response as seen with DAP suggesting that activation of LiaSR maybe a specific signature of DAP CM disruption.23 Additionally, LiaSR has been described as part of a complex general network that orchestrates the cell envelope response to a wide range of stresses, including detergents, ethanol, organic solvents and antibiotics.25
LiaSR (for lipid-II-interacting antibiotics) is named because it was initially identified after exposing B. subtilis to antibiotics that affect lipid II and cell membrane undecaprenol cycles (e.g. bacitracin, nisin, ramoplanin, and vancomycin), which are crucial steps in cell wall synthesis.21 The liaRS genes are clustered with an additional gene designated liaF, which encodes a transmembrane protein that appears to strongly inhibit LiaR-dependent gene expression.26 The LiaFSR three-component regulatory system is well conserved in Gram-positive pathogens with low G + C content (Figure 2).25, 26
Figure 2. The lia genes in some Gram-positive bacteria with low G + C content.
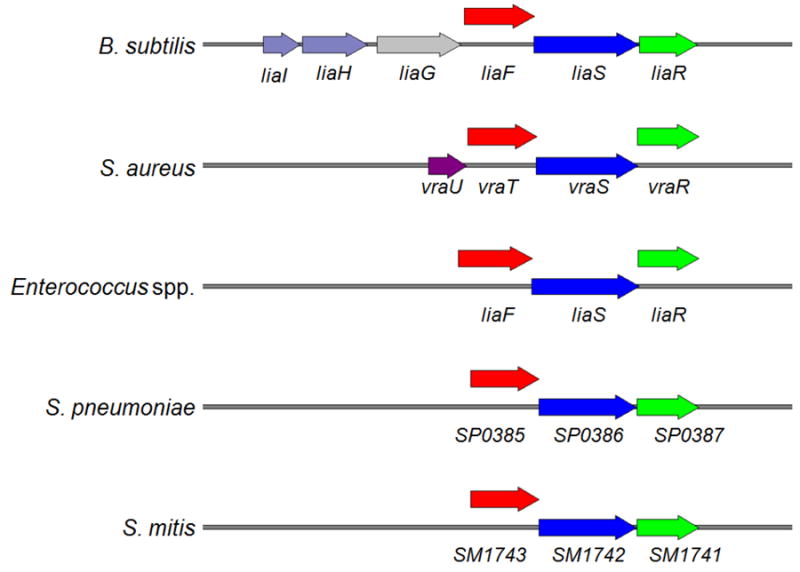
The loci are drawn to scale. Gene names follow NCBI entries of the published genome sequences.
Characterization of the lia locus of B. subtilis indicates that it consists of six genes, designated liaIH-liaGFSR, which are differentially expressed.26 In un-induced conditions, low-level expression of liaGFSR is driven by a weak constitutive promoter (PliaG) located upstream of liaG. Upon activation (i.e., exposure to antibiotics) high expression of the liaIH operon is observed by induction of the promoter PliaI (upstream of liaIH-liaGFSR), a phenomenon that is strictly dependent on the presence of the response regulator LiaR.21, 25-28. Indeed, LiaR not only induces liaH expression but also upregulates its own expression by read-through of the entire liaIH-liaGFSR locus.21, 26, 27 Furthermore, gene expression studies and proteomic analysis have shown that liaIH seems to be the only relevant target locus of LiaR.28
Stoichiometry studies of the LiaFSR system indicates that different amounts of the three proteins are produced under uninduced conditions at a ratio of 18:4:1 for LiaF, LiaS and LiaR, respectively.29 As mentioned above, liaF encodes a membrane protein that acts as a specific inhibitor of the LiaRS TCS by exerting its activity through LiaS.26, 29 LiaS is a bifunctional kinase which functions as a phosphatase in uninduced conditions, preventing the phosphorylation of LiaR and hence keeping the system “off” (PliaI activity) .29 In the presence of cell envelope stress (e.g., exposure to antibiotics), alterations in the stoichiometry of LiaFSR or overexpression of LiaS switches LiaS into its kinase mode which results in phosphorylation of LiaR and strong induction of PliaI.21, 26, 29 Notably, LiaR can also be phosphorylated by acetyl phosphate (a small molecule phosphor-donor produced as part of the cellular metabolism) in the absence of its cognate LiaS or when LiaR is produced in excess, illustrating different levels of regulation in response to cell envelope stressors.
Recent data have shed light into the function of liaIH (the target of LiaR). The liaI gene encodes a small membrane protein with two transmembrane regions whose N- and C-terminus are cytoplasmic, while LiaH is a homologue of the phage-shock protein described in Gram-negative bacteria and involved in the cell membrane response to the attack by bacteriophages.30 Indeed, LiaI appears to function as a membrane anchor for LiaH through its C-terminal domain in cell envelope stress conditions. Under normal conditions (i.e., non-stressed), LiaI remains highly motile and can move about the cytoplasmic membrane in fast and random fashion, presumably to scan for perturbations. In presence of an inducing trigger (e.g., antibiotics, including daptomycin), LiaI stalls into distinct foci while recruits LiaH to the cell membrane. Interestingly, another feature of LiaH is the formation of large oligomeric ring-like structures, comparable to the structure observed in phage-shock proteins described in Escherichia coli. Of note, the genome of B. subtilis also contains a LiaH paralog, PspA, which suggests functional redundancy of these phage shock proteins. The dynamics of Lia system via LiaIH highly resembles the Psp system described in E. coli and Yersinia enterocolitica.31, 32 The Psp system is a highly conserved system that functions as an adaptive mechanism to respond to conditions that can affect the cell envelope adversely.31-33
The strong induction of the Lia system by DAP appears to protect B. subtilis against stress caused by this antibiotic. Deletion in the liaIH operon (liaH, or liaIH) increased B. subtilis susceptibility to DAP while deletion of liaF appeared to have no effect on DAP MIC.24, 28 Since B. subtilis is a model organism, the mechanism of DAP resistance has only been studied in laboratory settings after in vitro serial passage.34 DAP-R B. subtilis displayed increased expression of the LiaRS TCS and its operon, including LiaH, but was not associated with mutations in any of its component.34 Phenotypically, development of in vitro DAP-R in B. subtilis has been associated with aberrant septum placement, thickened cell wall and cross-resistance to vancomycin, moenomycin, and bacitracin. DAP-R B. subtilis cells also displayed reduced binding of the antibiotic molecule (compared to its susceptible counterpart) (Figure 3). Interestingly, DAP-R B. subtilis was found to have a marked decrease in the concentration of PG in the cell membrane.
Figure 3. Proposed mechanisms of daptomycin resistance in Gram-positive organisms.
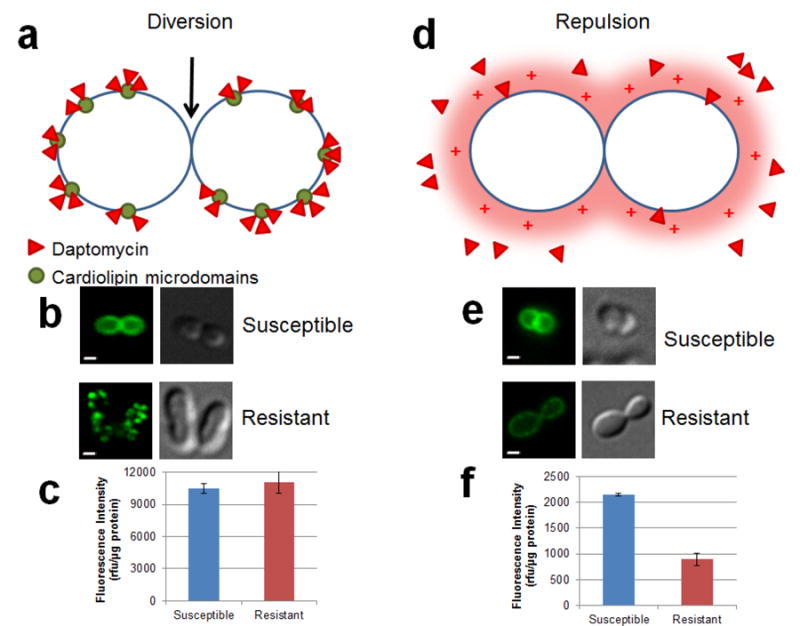
Two main mechanisms of resistance have been postulated in enterococci. The first is diversion (Enterococcus faecalis only) of the antibiotic from the preferential binding site of DAP at the septum (black arrow) resulting in ineffective binding of DAP (panel a). Images of cells treated with BODIPY-labeled DAP (a fluorescent derivative of DAP) demonstrate binding of the antibiotic to the septum in DAP-S. When exposed to the same concentration, DAP binding does not appear to occur at the septum in a DAP-R isolate (panel b). Panel c evaluates the amount of DAP bound to cell membrane of enterococci by measurement of fluorescence intensity normalized to protein content. As shown, no change in fluorescence intensities were noted between DAP-S and -R, indicating similar binding of the antibiotic molecules to the cell membrane. The second mechanism, seen in Bacillus subtilis, Staphylococcus aureus, and Enterococcus faecium, is electrostatic repulsion of the positively charged DAP-Ca2+ complex from the cell membrane (panel d). Binding of BODIPY-labeled DAP is decreased in DAP-R isolate compared to its –S counterpart (panel e), demonstrated by E. faecium. Lower fluorescence intensity is also noted in DAP-R versus DAP-S (panel f) as described in E. faecium. DAP – daptomycin; R –resistant; rfu – relative fluorescence unit; S – susceptible. Bar – 1 μm.
Another gene that has been implicated in DAP-R in B. subtilis is mprF, which encodes an enzyme with ability to modify membrane phospholipids by lysinylation of PG (leading to formation of L-PG) increasing the amount of positively charged PLs (similar to S. aureus, see below). Deletion and overexpression of mprF both led to changes in DAP susceptibility in B. subtilis.24, 35 However, changes in mprF were not found in adapted DAP-R B. subtilis. Rather, a mutation was found in the gene encoding the PG synthase (pgsA) and genetic reconstruction of such pgsA mutation demonstrated that this gene contributes significantly to the development of DAP-R in B. subtilis leading to a 20-fold increase in DAP MIC. Mutation in other genes, including mreB (cell shape-determining protein) and relA were also seen in a DAP-R derivative. However, their roles appear to be compensatory rather than major determinants of the resistance phenotype.
DAP-R in staphylococci
Although DAP-R in staphylococci is uncommon in clinical practice, development of this phenomenon during therapy has been widely described. Cases of DAP-R staphylococci are generally seen in high-inoculum infections (e.g. infective endocarditis and abscesses) and when lower doses of the drug have been used (i.e. ≤ 6 mg/kg/day).36-38 In addition, the prior use of vancomycin associated with development of the vancomycin-intermediate S. aureus (VISA) phenotype has also been linked with increased resistance to DAP during therapy.36, 38-46 The mechanisms of DAP-R in S. aureus are yet to be completely understood. A prevailing phenomenon that mechanistically links all the pathways of DAP-R in staphylococci appears to be “repulsion” of the DAP antibiotic molecule from the cell surface, which is generally associated with an overall change in the net charge of the bacterial surface (towards a more positive CM) (Figure 3). However, as it will be discussed below, the repulsion hypothesis does not explain the emergence of DAP-R in all S. aureus isolates.
MprF and the repulsion theory
One of the genes most consistently implicated in the development of DAP-R in S. aureus both in vivo and in vitro is mprF, which codes for a bifunctional enzyme (MprF, for multiple peptide resistance factor) that catalyzes the lysinylation of PG and the translocation of L-PG from the inner to the outer leaflet of the CM. MprF is composed of a cytosolic C-terminal domain that uses lysyl-tRNA as substrate to add lysine residues to negatively charged PG resulting in lysyl-PG (L-PG). The positively charged nature of L-PG appears to be a major contributor of the change of surface charge that helps repel the DAP antibiotic molecule from the surface.47-54 In order to alter surface charge, L-PG must be transferred from the inner to the outer leaflet of the CM, an activity that appears to be catalyzed by the first 6 - 8 (out of a total of 14) hydrophobic transmembrane domains in the N-terminal domain of the protein (flipase activity).55, 56 Thus, an increase in L-PG in the outer leaflet of the CM (and subsequent increase in net positive charge) appears to be of paramount importance to prevent the binding of the DAP-calcium complex to the CM.
In an in vitro passage experiment that exposed S. aureus to ascending sub-lethal concentrations of DAP, changes in MprF were one of the first genetic changes identified in DAP-R derivatives.57 A number of mprF mutations have been described in at least 12 different loci, but the exact role of each mutation has not been established. Nonetheless, mutations appear to localize in “hot spots” of the enzyme (mainly in the central transmembrane domains, but also one in the C-terminal cytosolic domain) and are associated with a gain of function.58, 59 Bayer et al. recently analyzed over 30 DAP-S and DAP-R S. aureus and demonstrated that although mprF single-nucleotide polymorphisms were not uncommon in DAP-S bacteria, none of them were localized in the “hot spots”. Conversely, all DAP-R isolates with mprF mutations harbored the changes within the already identified “hot spots”. Furthermore, only mutations in these specific locations were correlated with the expected gain of enzymatic function and with an increase in the positive charge of the CM. Of note, these mutational changes in MprF have also been correlated with a decrease in PG content.58 Also of interest, using a model of S. aureus prosthetic joint infections in rabbits, Mishra et al. recovered DAP-R isolates harboring mutations in the same MprF “hot spots” from animals not exposed to any antibiotics, and suggested that this phenotype was presumably driven by the exposure to endogenous host defense antimicrobial peptides.53
The role of MprF in the development of DAP-R has been further supported by experimental evidence showing that mprF-deletion mutants exhibited increased susceptibility to DAP and other CAMPs.60, 61 Likewise, translation blockade using an antisense strategy was able to reverse DAP-R in a strain harboring mprF mutations.62 Furthermore, trans-complementation of mprF deletion mutants with mutated alleles was followed by an increase in DAP MICs.46 Interestingly, some data suggest that the main function of MprF might not be specifically related to an increase in the positive net charge of the CM since not all DAP-R isolates with mprF mutations exhibit changes in cell surface charge.40, 63 It has been postulated that MprF may be controlling the concentration of PG (by converting it to L-PG) and, thus, altering the interaction of DAP and CAMP with their target cell membrane.46 Thus, the reduction in PG content (as a result of conversion to L-PG) may impair the ability of DAP to oligomerize in the outer leaflet of the membrane compromising its antibacterial activity.
Another strategy used by S. aureus to change the surface charge and, therefore, alter DAP activity is by overexpression of the dlt operon.63-68 The dlt genes are involved in the introduction of the positively charged amino acid D-alanine to cell wall teichoic acids. Thus, the alanylation of these surface-exposed structures results in an increase in the net positive charge of the CM, similar to what is observed in DAP-R strains with mutated mprF alleles.
Vancomycin non-susceptibility and development of DAP-resistance in S. aureus
Several lines of research have suggested a connection between the molecular pathways leading to the development of the VISA phenotype and DAP-R in S. aureus. First, a number of studies have demonstrated that an important number of VISA strains exhibit DAP MICs above the susceptibility breakpoint, with percentages of DAP-R as high as 80%.39 Furthermore, development of DAP-R in vivo was documented in a S. aureus VISA strain that failed vancomycin therapy and was never exposed to DAP.45 Second, several independent studies have described that DAP-R S. aureus strains frequently exhibit phenotypic changes that parallel those typically observed in VISA isolates; the most important, is a marked increase in the thickness of the cell wall. Furthermore, the degree of thickness of the cell wall has been positively correlated with the increase in DAP MIC in VISA isolates.41, 42 Third, transcriptional analysis studies have shown that one of the most consistent findings after DAP exposure is the up-regulation of the cell wall “stimulon”, similar to what is observed with the response to vancomycin and other cell envelope-acting drugs.46, 69, 70 Likewise, comparison of DAP-S and DAP-R S. aureus strain pairs have located changes in genes that have been associated with the VISA phenotype indicating that genes involved in cell wall synthesis and/or homeostasis play an important role in both vancomycin and DAP-R.66
Among the most relevant group of genes involved in VISA and DAP-R phenotypes, two gene clusters encoding TCS (vraSR and yycFG [walKR]) are the most studied. Of note, VraSR is the ortholog of LiaSR in other Gram-positive organisms (Figure 2). Both systems play an important role in cell envelope homeostasis and have been consistently implicated in the development of the VISA phenotype along with an additional regulatory system designated GraSR.71, 72 Mehta et al. showed that construction of a vraRS-null mutant resulted in reversion of DAP-R (and decrease in cell-wall thickness) in S. aureus. Trans complementation with vraSR was able to restore resistance to DAP with MICs restored to the values determined in the original resistant strain.73 In addition, up-regulation of vraSR expression and its regulon has been reported in DAP-R S. aureus selected in vitro.74
YycFG (WalKR) is an essential two-component regulatory system of cell-wall synthesis and homeostasis that has also been implicated in DAP-R enterococci (see below). Indeed, analysis of in vitro derived DAP-R S. aureus mutants revealed changes affecting the histidine kinase of the system (YycG).57, 75 Furthermore, Howden et al. demonstrated that the YycFG system plays a major role in the in vivo evolution of the VISA phenotype and DAP-R in S. aureus.76 Several mutations in different locations of the yycFG operon (or in its regulatory genes yycH and yycI) have been reported, although the exact contribution to DAP-R is unclear. However, a single amino acid change in YycG (K208R) was sufficient to increase the DAP MIC from 0.5 to 2 mg/L, a value that is sufficient for clinical DAP resistance.76
Role of genes involved in phospholipid metabolism
Other genes strongly implicated in the development of DAP-R in S. aureus are those encoding enzymes involved in phospholipid metabolism (other than MprF). In bacteria, cardiolipin synthases (Cls) is the critical enzyme for the synthesis of CL often using two molecules of PG as substrate.77 Changes in this enzyme(s) are likely to play a role in DAP-R by altering the pool of PG/CL in the CM (see “Mechanism of DAP action”, above). Importantly, S. aureus (and other Gram-positive organisms) harbor two (or more) cls genes (cls1 and cls2) and their differential expression is thought to vary depending on the presence of different stress conditions.78, 79 Using a whole genome approach, Peleg et al. analyzed clinical and laboratory-derived strain pairs of S. aureus and reported that three clinically derived DAP-R S. aureus harbored amino acid changes (F60S, A23V and L52F) in Cls2, all of which were found in conjunction with mutations in mprF (Figure 4). Similarly, Cls2 was also found to have an amino acid substitution (T33N) in two laboratory-derived DAP-R mutants, but in this case, the mutation was the only genetic change reported and its presence was sufficient to increase the DAP MIC from 0.5 mg/L to ≥ 2 mg/L (above the clinical breakpoint).80 The role of Cls in DAP-R was further supported by a transcriptomic analysis of isogenic strains obtained after DAP passage in which the expression of cls was significantly down-regulated in the resistant mutant as compared to its susceptible parental strain.74
Figure 4. Schematic representation of cardiolipin synthase Cls.
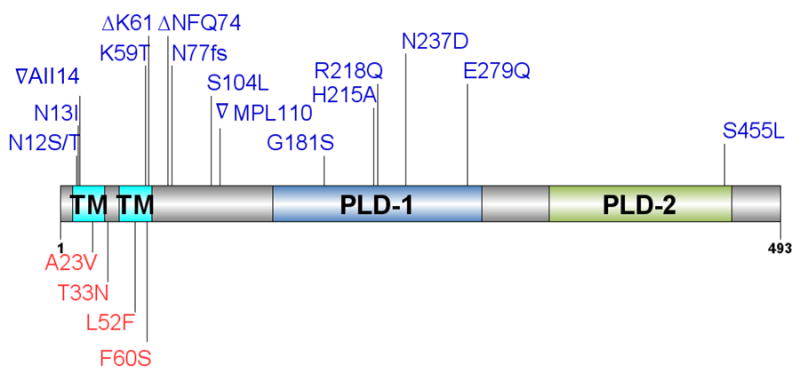
Predicted N-terminal transmembrane (TM) domains and phospholipase (PLD) domains are indicated. Lines refer to positions of amino acid changes associated with daptomycin resistance in Enterococcus spp. (blue) and Staphylococcus aureus (red).
Another gene involved in phospholipid metabolism and implicated in the DAP-R phenotype is pgsA, which encodes a CDP-diacylglycerol-glycerol-3-phosphate 3- phosphatidyltransferase that is involved in the production of PG. As mentioned above, PgsA has also been reported as an important determinant of DAP-R in B. subtilis (see above). In the genomic analysis performed by Peleg and colleagues, mutations in pgsA were often observed in DAP-R laboratory derivatives but not in DAP-R clinical isolates.80 Of note, one particular amino acid change (A64V) was seen in two different isolates and its presence was sufficient to increase the DAP MIC above the established breakpoint. The role of the mutations is unclear but they likely impair or abolish the enzymatic activity, decreasing the overall pool of PG.
Other genes and phenotypic changes associated with DAP-R in S. aureus
As mentioned, the most consistent phenotypic changes reported in DAP-R strains of S. aureus are an increase in the positive charge of the CM and a thickened cell wall, which have been found in clinical pairs of DAP-S/DAP-R MRSA and methicillin-susceptible S. aureus isolates 64, 67. The net increase in positive charge has been related to an increase in L-PG (generally associated with a gain in function of MprF, see above) or to the alanylation of cell wall teichoic acids, which is secondary to the over expression of the dlt operon. This electrostatic change results in a decreased binding of DAP to the CM, reducing its antibacterial properties. On the other hand, the increase in cell-wall thickness has been correlated with higher levels of expression of tagA, a gene that is directly involved in the early steps of the synthesis of cell-wall teichoic acids.64, 67 However, other phenotypic changes have also been reported, albeit less frequently. For instance, changes in membrane fluidity have been correlated with the development of DAP-R S. aureus strains. Interestingly, strains that developed DAP-R in vivo have been found to have membranes with increased fluidity, whereas mutants obtained in vitro had more rigid membranes.75, 81 Therefore, it has been suggested that an optimal amount of fluidity might be necessary for specific CAMPs to exert their action and that changes in any direction (higher or lower fluidity) could play an important role in resistance to these compounds.46
Another important component of S. aureus CM associated with DAP-R is staphyloxanthin, a carotenoid pigment which gives S. aureus their iconic golden color. A study by Liu et al. demonstrated that blockage of staphyloxanthin synthesis resulted in colorless bacteria with increased susceptibility to H2O2 and killing by human whole blood. In addition, inhibition of staphyloxanthin synthesis was associated with rapid clearance of S. aureus by the innate immune system.82 Mishra et al. provided evidence implicating the role of carotenoid in DAP-R. Indeed, when carotenoid production was increased by plasmid induction in a methicillin-susceptible S. aureus, the DAP MICs also increased. 83 In a follow-up study, reduced carotenoid content in an MRSA was associated with DAP-R.5, 84 Thus, it is postulated that carotenoid content influences CM order (rigidity) and fluidity, a phenomenon that may affect susceptibility to a variety of CAMPs. Further clarification of the role of staphyloxanthin and CM fluidity is the object of active investigation.
Finally, a recent publication analyzed the differences in physiologic fitness and metabolic pathways between 6 DAP-R/DAP-S isogenic strain pairs. Utilizing a metabolomic approach, Gaupp and colleagues found that DAP-R strains exhibit a decrease in the tricarboxylic acid cycle activity and a preference for the pentose phosphate pathway and purine/pyrimidine metabolism. The latter pathways are associated with cell wall teichoic acid and peptidoglycan biosynthesis.85
DAP resistance in enterococci
Enterococci are less susceptible to DAP than staphylococci and streptococci and exhibit higher MICs (e.g. the current CLSI breakpoint is 4-fold higher than that of S. aureus). Despite this difference, most enterococcal isolates remain susceptible to the antibiotic.86 However, the emergence of DAP-R enterococci during and after therapy has been extensively documented in both E faecalis and E. faecium. Interestingly, DAP-R has also been reported in isolates recovered from patients without any exposure to DAP or any other lipopeptide.87, 88
Most genes implicated in DAP-R in enterococci can be grouped into two broad categories, i) genes encoding regulatory systems that orchestrate cell- envelope homeostasis and stress-response, and ii) genes coding for enzymes involved in the metabolism of CM phospholipids. Although some of the genes implicated in resistance in E. faecalis and E. faecium code for proteins of similar function, there seem to be important differences in the genetic and biochemical routes leading to DAP-R among enterococcal species. Furthermore, it is also likely that multiple different pathways to resistance exist within a same species, and that specific isolates demonstrate a preference for one resistance pathway over the other.
DAP resistance in E. faecalis
Phenotypically, DAP-R in E. faecalis has been associated with thickened cell wall, aberrant septal placement, increase in relative cell surface positive charge and reduction of the ability of DAP to depolarize the CM.89 These findings parallel those of S. aureus and other Gram-positive species (see above). Analysis of CM phospholipid content of a clinical strain-pair of DAP-S and DAP-R E. faecalis revealed a significant decrease in PG content accompanied by an increase in negatively charged glycerophosphoglycolipid (glycerophospho-diglycodiacylglycerol [GP-DGDAG]), without changes in CL or positively charged amino-phospholipids.90
An important consideration is that the mechanism of resistance to DAP in E. faecalis appears to be distinctively different from that of S. aureus. Indeed, using bodipy-FL-labeled DAP, it has been shown that repulsion of DAP from the cell surface is not the main strategy used by this organism to withstand DAP bactericidal effect. Instead, it appears that E. faecalis has developed a sophisticated strategy to “divert” DAP from its principal septal target towards other CM areas (Figure 3). This diversion of the antibiotic is associated with redistribution of CL microdomains away from the septum.91 Although the specific biochemical steps mediating this strategy are not fully understood, the LiaFSR system seems to play a very prominent role in the steps leading to CM adaptation.
In E. faecalis (and E. faecium, see below), the LiaFSR system has similar components as described above for B. subtilis and other Gram-positive organisms (Figure 2).25, 26 LiaF is a transmembrane protein thought to be a negative regulator of the system; LiaS and LiaR are the histidine kinase and response regulator of the system, respectively. Using a quantitative experimental evolutionary approach, Miller and colleagues demonstrated that alterations in liaFSR were the initial pivotal step leading to DAP-R in E. faecalis.92 Furthermore, genomic analyses of a clinical strain-pair of DAP-S and DAP-R E. faecalis recovered from the bloodstream of a patient treated with DAP found a single deletion of an isoleucine at position 177 of LiaF in the DAP-R strain. Introduction of the altered liaF allele into the DAP-S E. faecalis isolate increased the DAP MIC from 1 to 4 μg/ml. More importantly, this amino acid change was sufficient to abolish DAP’s in vitro bactericidal activity.89, 93 Of note, the single introduction of the mutated liaF allele was sufficient to produce CL microdomain redistribution, suggesting that the LiaFSR system plays an important role in CM homeostasis.
To further implicate the critical role of LiaFSR in DAP-R, a non-polar deletion of liaR (encoding the response regulator of the system) was obtained in a clinical DAP-R strain of E. faecalis. The deletion resulted in reversion of DAP resistance and restoration of the normal distribution of CM CL-rich microdomains.94 Interestingly, deletion of liaR also resulted in a marked increase in the activity of several unrelated cationic antimicrobial peptides (CAMPs) and a parallel decrease in the MIC of telavancin (a CM-acting antibiotic available clinically). Moreover, a liaR knockout mutant generated in an E. faecalis laboratory strain (OG1RF, DAP-susceptible) resulted in hypersusceptibility to DAP with a marked decreased in the MIC, supporting the important role of LiaR in DAP-R.94
Recently, Davlieva et al. used a combination of structural and biophysical experiments to understand the molecular basis of LiaR-mediated DAP resistance. Indeed, activation of the response regulator LiaR (by phosphorylation or mutations in LiaR that mimic the phosphorylated state) triggers a transition of dimer to tetramer that appears to significantly increase the affinity of the protein for the target DNA (Figure 5).95 With this approach, it was confirmed that activated LiaR appears to bind to its own (LiaFSR) promoter but also regulates a cluster of genes that encode a putative soluble protein of 533 amino acid (designated LiaX) and two transmembrane proteins of 107 (LiaY) and 118 (LiaZ) amino acids, respectively. Although the function of these proteins remains to be elucidated, it is interesting to note that a frameshift mutation on LiaX has been shown to cause DAP-R in vitro.92
Figure 5.
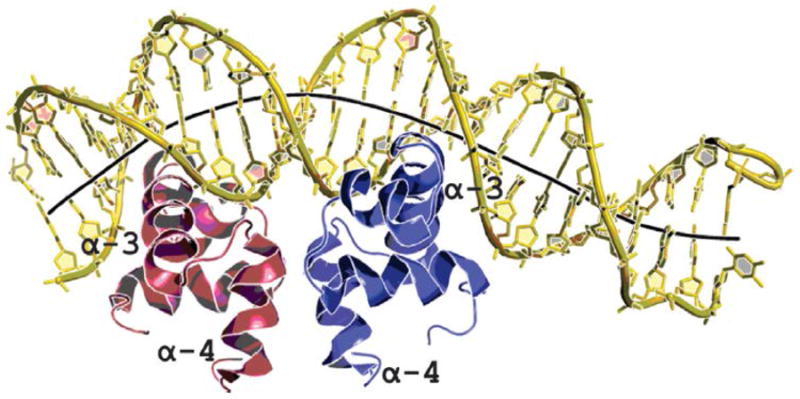
Crystal structure of the DNA binding domain LiaR of Enterococcus faecalis bound to DNA sequence upstream of its target genes. The α4 helices form part of the molecular recognition surface responsible for formation of the functional dimer required for DNA binding. The α3 DNA-recognition helices in the dimer are positioned to create a large electropositive DNA-binding surface. The LiaR-DNA complex structure shows a strong bend in the DNA, as shown by its helical axis (gray). Adapted from Davlieva M, et al. 2015. Nucleic Acids Res; 43(9):4758-73.
A second group of genes implicated in E. faecalis DAP-R code for enzymes involved in the metabolism of CM phospholipids. Among them, the two most prominent enzymes associated with the DAP-R phenotype are cardiolipin synthase (Cls, which synthesizes CL) and a glycerol-phosphodiester phosphodiesterase (GdpD) that is involved in glycerol turnover for phospholipid biosynthesis. Indeed, mutagenesis experiments confirmed the additive effect of gdpD and cls mutations in the development of DAP-R in a genetic background of a strain with changes in LiaFSR.89 Interestingly, a mutation in gdpD did not affect DAP susceptibility on its own but, when accompanied by a LiaF substitution resulted in marked increases of DAP MICs suggesting a synergistic effect of these changes. It has been postulated that mutations in genes involved in phospholipid metabolism are likely to occur in later stages of development of DAP-R that occur after the LiaFSR system is activated (triggering the cell envelope stress response).96 Thus, changes in phospholipid homeostasis are likely to “potentiate” the resistance phenotype by completing CM adaptation to the antimicrobial peptide attack.
Cls is a membrane-bound enzyme harboring two putative N-terminal transmembrane domains (Figure 4) and two phospholipase domains (PLD1 and PLD2) that catalyze the formation of CL from two PG molecules. Changes in different domains of the Cls protein (transmembrane, linker and PLDs domains) have been associated with DAP resistance but the biochemical consequences of these alterations and their contribution to the DAP-R phenotype remains unclear.89, 92, 97 Of note, overexpression in trans of a mutated cls allele harboring an amino acid change in the PLD1 domain resulted in a 4-fold increase in the DAP MIC and was sufficient to yield a DAP-R phenotype (from 4 to 64 mg/L).97
DAP-R in E. faecium
E. faecium is the most drug-resistant and recalcitrant of the enterococcal species and DAP has become a key antibiotic to treat infections caused by these organisms. Description of DAP-R in E. faecium is becoming a serious clinical problem since DAP has become a first-line option to treat severe E. faecium infections. Most worrisome is the scenario of DAP-tolerance (lack of bactericidal activity) in strains reported “susceptible” by standard susceptibility testing, which has been noted in DAP-S E. faecium clinical strains with MICs close to the breakpoint (3-4 μg/mL).98 Development of DAP-R in this species has also been associated with increased surface charge, increased cell wall thickness, and decreased depolarization after DAP exposure.90, 99 However, in contrast to what it was described above for E. faecalis, the development of DAP-R in E. faecium appears to be similar to that of S. aureus, in which “repulsion” of the antibiotic from the cell surface seems to be the predominant mechanism of resistance (Figure 3). Additionally, and also different from E. faecalis, DAP-R in E. faecium is not associated with redistribution of CM CL microdomains, despite the fact that similar genes (i.e. liaFSR) appear to be involved in the resistance phenotype.100, 101 This mechanistic difference suggests independent evolutionary trajectories in two species within the same bacterial genus.
In terms of phospholipid content of the CM, the most relevant change associated with DAP-R in E. faecium strains (where PLs have been examined) appears to be a marked decrease in the content of PG without major alterations of CL content.90 Genomic analyses of several clinical strain-pairs of E. faecium have shed light into the genetic determinants of DAP-R in this organism,102-104 although the specific role of the majority of genes involved remains to be fully elucidated. In a recent genomic analysis of 19 unrelated E. faecium strains with different DAP MICs (3 to 48 μg/mL) in which mutations in 43 genes previously associated with DAP-R were investigated, Diaz et al. showed that mutations in liaFSR were the most common changes observed in these strains.105 Among the identified changes, substitutions in LiaS (T120A) and LiaR (W73C) were the most frequent changes observed. Interestingly, these two amino acid substitutions were always found together, suggesting that they might have co-evolved. Furthermore, the same LiaSR substitutions were also found in E. faecium isolates recovered from the bloodstream of patients exhibiting DAP MICs in the higher range of susceptibility (between 3 and 4 μg/mL), but were absent in isolates with DAP MIC ≤ 2 μg/mL.98 More importantly, these mutations were sufficient to abolish the in vitro bactericidal activity of DAP and were associated with DAP failure in a neutropenic patient that presented with VRE bacteremia caused by a DAP-S isolate (MIC 3 μg/mL).106 Moreover, deletion of liaR reversed DAP resistance in E. faecium isolates independent of the genetic background or changes in LiaFSR supporting the notion that LiaR is the master regulator of the cell envelope stress response and plays a universal role in enterococci regardless of the genetic paths of DAP-R.107
Another TCS that has been found to be involved in DAP-R in E. faecium is the YycFG system and accessory proteins (YycHIJ), as described in S. aureus.108 In E. faecium, mutations in yycFG (or accessory genes) have been found in both DAP-R and –tolerant (lack of in vitro DAP bactericidal activity) strains.104, 105 Interestingly, a single mutation in YycG was not sufficient to rise DAP MICs, unless accompanied by changes in genes encoding phospholipid enzymes.101, 109 YycFG is an essential regulatory system involved in cell wall homeostasis in S. aureus, however its role in DAP-R in enteroccci has not yet been fully characterized.57, 75, 108, 110-112
Similar to what it has been found in E. faecalis, mutations in cls have also been commonly associated with DAP-R in E. faecium (Figure 4). Biochemical characterization of the impact of two independent PLD1 mutations (R218Q [also described in E. faecalis] and H215R) in Cls function showed that both changes resulted in an increase enzymatic activity (“gain of function” mutation).113. Of note, using an allelic replacement strategy, Tran et al. demonstrated that introduction of a mutated cls (resulting in R218Q substitution) associated with DAP-R did not affect the DAP MIC suggesting that Cls changes alone are not sufficient to mediate resistance in E. faecium.104
DAP-R in other Gram-positive pathogens
Although DAP-R is well described in S. aureus and enterococci, it remains bactericidal against a wide range of other Gram-positive pathogens. As such, DAP has been increasingly used for treatment of infections caused by Gram-positives especially in cases of severe or deep-seated infections. Thus, reports of DAP-R in other clinically relevant Gram-positive species have emerged in recent years. In this section, description of DAP-R in two pathogens, Corynebacterium spp. and Streptococcus mitis will be discussed.
Reports of DAP-R in Corynebacterium spp. is limited to three cases, one of C. jeikeium and two of C. striatum. DAP-R C. jeikeium was recovered from the blood of a neutropenic patient who underwent cord blood transplantation for secondary acute myeloid leukemia.114 C. striatum strains were recovered from 2 patients, one patient with a left ventricular assist device who presented with bacteremia and another case with native valve endocarditis.115, 116 All Corynebacterium isolates exhibited DAP MICs ≥ 256 μg/ml. Intriguingly, all patients were exposed to DAP prior to isolation of Corynebacterium spp., including two (C. jeikeium and one C. striatum case) patients who had been receiving DAP therapy for infections caused by other Gram-positive pathogens. These cases serve as cautionary tales for the selection of DAP-R in a genus that are considered organisms of low pathogenicity and generally regarded as human commensals. Of note, McElvania Tekippe et al. described the emergence of DAP-R in 7 of 12 clinical isolates of C. striatum after only 24 h of DAP exposure with MICs > 256 μg/ml.115 C. striatum also display heterogeneous high-level DAP resistance in one case described by Tran et al.116 The two DAP-R derivatives did not show any alterations in cell surface charge but exhibited decreased CM depolarization induced by DAP. As mentioned, the emergence of DAP-R Corynebacterium spp. appears to be related to prolonged courses of DAP. No further evaluation of the genetic basis of DAP-R in this genus has been reported.
Despite earlier studies, which reported a low prevalence of DAP-R in streptococci,117, 118 certain isolates have been shown to have the ability to adapt and rapidly develop high-level DAP-R.119-122 This phenomenon was initially described in two patients. The first was a case of breakthrough bacteremia caused by DAP-R S. anginosus in a patient who were treated with DAP for a previous MRSA infection.121 Another case, involved a patient with native valve S. oralis endocarditis whose DAP-R isolate emerged during treatment with DAP.122 An independent finding by another research group found emergence of DAP-R within 24 hr of antibiotic exposure in viridans-group streptococci exhibiting high level of resistance (MIC ≥256 μg/ml).120 Therapy with DAP alone in a rabbit endocarditis model also produced DAP-R after 48 h of exposure. Similarly, DAP exposure in a simulated endocardial vegetation model corresponded to a rapid development of high-level DAP-R after only 24 h at high concentration, up to 8X MIC.119 Like studies in Corynebacterium, the mechanism of DAP-R in streptococci remains to be elucidated.
Clinical Implications of DAP resistance and tolerance
From a clinical perspective, DAP is a potent and bactericidal antibiotic that, in the case of VRE, is one of the last-resort drugs to treat these organisms. Therefore, developing of resistance during therapy is a serious threat because it leaves clinicians without options for these severe infections. Our current knowledge on the mechanism of action, resistance and pharmacological aspects of DAP is now at a stage that innovative approaches may be considered to preserve the activity of DAP, until new molecules become available. We have discussed above that developing of DAP-R during therapy involves important adaptive responses that are triggered by a variety of environmental cues that do not solely depend on the presence of DAP (bacterial cell envelope adaptation). Thus, it would be wise to consider that development of DAP resistance is a natural process and would be likely under therapy since bacterial pathogens have “learned”, through evolution, how to respond to the antibiotic challenge. Therefore, devising therapeutic strategies to prevent development of resistance by weakening the bacterial CM adaptive response would be an innovative approach. This strategy could not only preserve the activity of DAP but may also enhance the activity of other cell envelope-acting agents that have become obsolete in the treatment of Gram-positive infections. Additionally, it may contribute to the clearance of bacteria by the innate immune system.
A plethora of molecular and pharmacological data indicate that DAP is a concentration-dependent antibiotic. Neutropenic murine thigh models have demonstrated that the area under the concentration-time curve (AUC)/MIC and maximum concentration (Cmax)/MIC ratios are important the pharmacodynamic parameters to determine the in vivo activity of DAP against Gram-positive organisms.123 Indeed, the amount of active DAP molecules available to bind to the CM at any given point is of paramount importance for the bactericidal effect of the antibiotic. The two types of mechanisms of resistance discussed above (i.e., “repulsion” of the antibiotic from the cell surface and “diversion” of DAP from the septum) could potentially be overcome by increasing concentrations of the antibiotic, although the amount of drug necessary in these scenarios may be limiting due to toxicity. Using a simulated endocardial vegetation (SEV) model, Rose and colleagues compared DAP monotherapy in doses equivalent to 6 and 10 mg/Kg (the former is the FDA approved dose for S. aureus bacteremia) and demonstrated that the use of higher concentrations of DAP was able to prevent the appearance of resistant mutants in S. aureus.124 Similarly, two studies using the same SEV model to treat vancomycin-resistant E. faecium and E. faecalis showed that the only regimen that did not select for DAP-R mutants was 12 mg/kg.125, 126 Furthermore, Sakoulas et al. utilized a rat IE model and performed population analysis of bacterial isolates recovered from vegetations. They were able to show that higher DAP doses (equivalent to a human dose of 6 mg/kg vs. 4 mg/kg) were able to prevent the emergence of DAP heteroresistance in MRSA isolates in vivo.127 Therefore, it appears that optimizing the amount of drug delivered to the target site may improve its activity and also prevent, at some level, development of resistance
Another interesting observation is that once Gram-positive organisms become DAP-R, they appear to markedly increase the susceptibility to other cell-wall targeting antibiotics such as the β-lactams (the so called “see-saw” effect).128-132 This observation is not unique to DAP and has been previously documented with development of the VISA phenotype (low-level resistance to vancomycin). Similar to DAP, as the cells decrease susceptibility to vancomycin, there is a concomitant increase of susceptibility to β-lactams (even if the isolate is fully resistant in vitro to these compounds).133-137 Several in vitro and in vivo experiments support the use of the combinations of DAP and β-lactams to treat recalcitrant Gram-positive infections and achieve therapeutic success and prevent the development of resistance.105, 106, 128, 138-160 Yang et al. utilized a rabbit model of aortic IE to evaluate the combination of DAP plus oxacillin against MRSA clinical-strain pairs of DAP-S and DAP-R derivatives recovered from patients who failed DAP therapy. They showed that the combination was highly effective against DAP-R strains and were able to demonstrate the “see-saw” effect with DAP and oxacillin.128 Moreover, the combination of DAP and β-lactams has been used successfully in several cases of recalcitrant S. aureus bacteremia failing DAP and other therapies. For instance, a recent report document 24 cases (20 MRSA, 2 MSSA and 2 VISA strains) in which the combination of DAP plus ceftaroline was successfully used to clear the bloodstream of patients who had failed other therapeutic strategies (i.e. high-dose DAP monotherapy or vancomycin).140
The combination of DAP plus β-lactams has also shown promising results in recalcitrant enterococcal infections. For example, the addition of sub-inhibitory concentrations of ampicillin prevented development of DAP-R in an in vivo model of experimental endocarditis.159 Additionally, the addition of ceftriaxone to DAP (6 mg/kg) in a SEV model was able to prevent the appearance of DAP-R isolates of both vancomycin-resistant E. faecalis and E. faecium.141 With enterococci, the two β-lactams that appear to be the best in obtaining synergism are ampicillin and ceftaroline.105, 138, 152, 158 Importantly, the additive affect with ampicillin was observed regardless of the presence of high-level ampicillin resistance158 and the addition of ceftaroline has been shown to restore DAP susceptibility in DAP-R E. faecium and E. faecalis.139, 152, 161 The evidence for the use of other β-lactam compounds is less abundant, but Smith and colleagues recently performed time-kill assays with DAP plus several members of the beta-lactam family and concluded that cefepime, ceftriaxone, and ertapenem were also synergistic against a DAP-R E. faecium strain, while cefazolin and cefotaxime were not.139 In terms of clinical evidence, there are a handful of cases in which the addition of ampicillin106, 145, 158, 162 or ceftaroline152 was successfully used as salvage therapy in patients that were otherwise failing DAP treatment.
The mechanistic bases of the DAP-β-lactam synergistic effect are not well understood. It has been postulated that exposure to β-lactams results in a decrease in the net positive charge of the bacterial surface which results in an increase of DAP binding to the CM target.148, 152, 158 Of note, two important caveats of this synergistic effect deserve to be discussed. First, Berti et al. provided evidence showing that the additive effect of β-lactams might not be a drug class effect, but rather compound-specific. Furthermore, they observed that the synergistic effect was mostly seen with β-lactam molecules that target PBP-1 in S. aureus and suggested that the specific PBP activity profile of each compound would play an important role in the ability to potentiate DAP’s antibacterial effect.142 Second, recent reports by Diaz et al. and Hindler et al. independently showed that the synergistic effect of the ampicillin-DAP combination was only observed against DAP-R/tolerant E. faecium isolates harboring liaFSR mutations, but no such effect was described in the presence of changes in the YycFG system (and absence of liaFSR mutations).105, 138 Thus, synergistic activity may depend on the genetic pathway leading to DAP-R in each particular strain. Interestingly, ampicillin enhanced the activity of LL-37 (a human CAMP) against DAP-R E. faecium, regardless of the genetic pathway.138
Finally, evidence to support the use of combination therapy with DAP and other antimicrobials is scarce. Steed et al. analyzed the use of DAP plus trimethoprim sulfamethoxazole in a SEV model and reported synergism against DAP-R MRSA, including hVISA and VISA strains.146, 163 In addition, the same combination was used to manage two cases of vertebral osteomyelitis caused by a DAP-R VISA isolate.163 Lastly, a case of DAP-R MRSA bacteremia was successfully treated with DAP plus rifampin after failing DAP monotherapy (6 mg/kg).164
Concluding Remarks
Studies of mechanisms of action and resistance of DAP have uncovered an intricate relationship between Gram-positive bacterial adaptive processes and host innate immune response. The development of DAP-R appears to be a “built- in” response in these bacteria to the presence of CAMPs, including DAP. Although our understanding of DAP has evolved in the last several years, data related to the molecular mechanism(s) of resistance, treatment of DAP-R infections and alternative strategies to overcome DAP-R remain limited. As DAP usage continues to increase in clinical settings, especially in severe infections caused by Gram-positive pathogens, the threat of DAP-R will only likely escalate. Studies on the mechanistic bases of DAP-R are likely to yield novel insights into bacterial cell membrane adaptation and lead to development of innovative therapeutic strategies to combat MDR Gram-positive infections
References
- 1.Review on Antimicrobial Resistance. Antimicrobial resistance: Tackling a crisis for the future health and wealth of nations. [March 11 2015];2014 http://amr-review.org/
- 2.World Health Organization. Antimicrobial resistance: global report on surveillance 2014. [March 4 2015];2014 http://www.who.int/drugresistance/documents/surveillancereport/en/
- 3.Centers for Disease Control and Prevention. Antibiotic resistance threats in the United States. [March 9 2015];2013 http://www.cdc.gov/drugresistance/threat-report2013/index.html.
- 4.Cosgrove SE, Corey GR. A balancing act: microbe versus muscle. Clin Infect Dis. 2009;49:181–183. doi: 10.1086/600040. [DOI] [PubMed] [Google Scholar]
- 5.Munita JM, Murray BE, Arias CA. Daptomycin for the treatment of bacteraemia due to vancomycin-resistant enterococci. Int J Antimicrob Agents. 2014;44:387–395. doi: 10.1016/j.ijantimicag.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Humphries RM, Pollett S, Sakoulas G. A current perspective on daptomycin for the clinical microbiologist. Clin Microbiol Rev. 2013;26:759–780. doi: 10.1128/CMR.00030-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho SW, Jung D, Calhoun JR, et al. Effect of divalent cations on the structure of the antibiotic daptomycin. Eur Biophys J. 2008;37:421–433. doi: 10.1007/s00249-007-0227-2. [DOI] [PubMed] [Google Scholar]
- 8.Scott WR, Baek SB, Jung D, et al. NMR structural studies of the antibiotic lipopeptide daptomycin in DHPC micelles. Biochim Biophys Acta. 2007;1768:3116–3126. doi: 10.1016/j.bbamem.2007.08.034. [DOI] [PubMed] [Google Scholar]
- 9.Allen NE, Alborn WE, Jr, Hobbs JN., Jr Inhibition of membrane potential-dependent amino acid transport by daptomycin. Antimicrob Agents Chemother. 1991;35:2639–2642. doi: 10.1128/aac.35.12.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mengin-Lecreulx D, Allen NE, Hobbs JN, et al. Inhibition of peptidoglycan biosynthesis in Bacillus megaterium by daptomycin. FEMS Microbiol Lett. 1990;57:245–248. doi: 10.1016/0378-1097(90)90074-z. [DOI] [PubMed] [Google Scholar]
- 11.Canepari P, Boaretti M, Lleo MM, et al. Lipoteichoic acid as a new target for activity of antibiotics: mode of action of daptomycin (LY146032. Antimicrob Agents Chemother. 1990;34:1220–1226. doi: 10.1128/aac.34.6.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muraih JK, Harris J, Taylor SD, et al. Characterization of daptomycin oligomerization with perylene excimer fluorescence: stoichiometric binding of phosphatidylglycerol triggers oligomer formation. Biochim Biophys Acta. 2012;1818:673–678. doi: 10.1016/j.bbamem.2011.10.027. [DOI] [PubMed] [Google Scholar]
- 13.Muraih JK, Pearson A, Silverman J, et al. Oligomerization of daptomycin on membranes. Biochim Biophys Acta. 2011;1808:1154–1160. doi: 10.1016/j.bbamem.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Zhang T, Muraih JK, MacCormick B, et al. Daptomycin forms cation- and size-selective pores in model membranes. Biochim Biophys Acta. 2014;1838:2425–2430. doi: 10.1016/j.bbamem.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 15.Muraih JK, Palmer M. Estimation of the subunit stoichiometry of the membrane-associated daptomycin oligomer by FRET. Biochim Biophys Acta. 2012;1818:1642–1647. doi: 10.1016/j.bbamem.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 16.Zhang T, Muraih JK, Tishbi N, et al. Cardiolipin prevents membrane translocation and permeabilization by daptomycin. J Biol Chem. 2014;289:11584–11591. doi: 10.1074/jbc.M114.554444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen YF, Sun TL, Sun Y, et al. Interaction of daptomycin with lipid bilayers: a lipid extracting effect. Biochemistry. 2014;53:5384–5392. doi: 10.1021/bi500779g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pogliano J, Pogliano N, Silverman J. Daptomycin mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J Bacteriol. 2012;194:4494–4504. doi: 10.1128/JB.00011-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao M, Wang T, Ye R, et al. Antibiotics that inhibit cell wall biosynthesis induce expression of the Bacillus subtilis sigma(W) and sigma(M) regulons. Mol Microbiol. 2002;45:1267–1276. doi: 10.1046/j.1365-2958.2002.03050.x. [DOI] [PubMed] [Google Scholar]
- 20.Cao M, Kobel PA, Morshedi MM, et al. Defining the Bacillus subtilis sigma(W) regulon: a comparative analysis of promoter consensus search, run-off transcription/macroarray analysis (ROMA), and transcriptional profiling approaches. J Mol Biol. 2002;316:443–457. doi: 10.1006/jmbi.2001.5372. [DOI] [PubMed] [Google Scholar]
- 21.Mascher T, Margulis NG, Wang T, et al. Cell wall stress responses in Bacillus subtilis: the regulatory network of the bacitracin stimulon. Mol Microbiol. 2003;50:1591–1604. doi: 10.1046/j.1365-2958.2003.03786.x. [DOI] [PubMed] [Google Scholar]
- 22.Rietkotter E, Hoyer D, Mascher T. Bacitracin sensing in Bacillus subtilis. Mol Microbiol. 2008;68:768–785. doi: 10.1111/j.1365-2958.2008.06194.x. [DOI] [PubMed] [Google Scholar]
- 23.Wecke T, Zuhlke D, Mader U, et al. Daptomycin versus friulimicin B: in-depth profiling of Bacillus subtilis cell envelope stress responses. Antimicrob Agents Chemother. 2009;53:1619–1623. doi: 10.1128/AAC.01046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hachmann AB, Angert ER, Helmann JD. Genetic analysis of factors affecting susceptibility of Bacillus subtilis to daptomycin. Antimicrob Agents Chemother. 2009;53:1598–1609. doi: 10.1128/AAC.01329-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jordan S, Hutchings MI, Mascher T. Cell envelope stress response in Gram-positive bacteria. FEMS Microbiol Rev. 2008;32:107–146. doi: 10.1111/j.1574-6976.2007.00091.x. [DOI] [PubMed] [Google Scholar]
- 26.Jordan S, Junker A, Helmann JD, et al. Regulation of LiaRS-dependent gene expression in Bacillus subtilis: identification of inhibitor proteins, regulator binding sites, and target genes of a conserved cell envelope stress-sensing two-component system. J Bacteriol. 2006;188:5153–5166. doi: 10.1128/JB.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mascher T, Zimmer SL, Smith TA, et al. Antibiotic-inducible promoter regulated by the cell envelope stress-sensing two-component system LiaRS of Bacillus subtilis. Antimicrob Agents Chemother. 2004;48:2888–2896. doi: 10.1128/AAC.48.8.2888-2896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolf D, Kalamorz F, Wecke T, et al. In-depth profiling of the LiaR response of Bacillus subtilis. J Bacteriol. 2010;192:4680–4693. doi: 10.1128/JB.00543-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schrecke K, Jordan S, Mascher T. Stoichiometry and perturbation studies of the LiaFSR system of Bacillus subtilis. Mol Microbiol. 2013;87:769–788. doi: 10.1111/mmi.12130. [DOI] [PubMed] [Google Scholar]
- 30.Dominguez-Escobar J, Wolf D, Fritz G, et al. Subcellular localization, interactions and dynamics of the phage-shock protein-like Lia response in Bacillus subtilis. Mol Microbiol. 2014;92:716–732. doi: 10.1111/mmi.12586. [DOI] [PubMed] [Google Scholar]
- 31.Brissette JL, Russel M, Weiner L, et al. Phage shock protein, a stress protein of Escherichia coli. Proc Natl Acad Sci U S A. 1990;87:862–866. doi: 10.1073/pnas.87.3.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamaguchi S, Reid DA, Rothenberg E, et al. Changes in Psp protein binding partners, localization and behaviour upon activation of the Yersinia enterocolitica phage shock protein response. Mol Microbiol. 2013;87:656–671. doi: 10.1111/mmi.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehner D, Osadnik H, Lunsdorf H, et al. The Tat system for membrane translocation of folded proteins recruits the membrane-stabilizing Psp machinery in Escherichia coli. J Biol Chem. 2012;287:27834–27842. doi: 10.1074/jbc.M112.374983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hachmann AB, Sevim E, Gaballa A, et al. Reduction in membrane phosphatidylglycerol content leads to daptomycin resistance in Bacillus subtilis. Antimicrob Agents Chemother. 2011;55:4326–4337. doi: 10.1128/AAC.01819-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salzberg LI, Helmann JD. Phenotypic and transcriptomic characterization of Bacillus subtilis mutants with grossly altered membrane composition. J Bacteriol. 2008;190:7797–7807. doi: 10.1128/JB.00720-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Julian K, Kosowska-Shick K, Whitener C, et al. Characterization of a daptomycin-nonsusceptible vancomycin-intermediate Staphylococcus aureus strain in a patient with endocarditis. Antimicrob Agents Chemother. 2007;51:3445–3448. doi: 10.1128/AAC.00559-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dortet L, Anguel N, Fortineau N, et al. In vivo acquired daptomycin resistance during treatment of methicillin-resistant Staphylococcus aureus endocarditis. Int J Infect Dis. 2013;17:e1076–1077. doi: 10.1016/j.ijid.2013.02.019. [DOI] [PubMed] [Google Scholar]
- 38.van Hal SJ, Paterson DL, Gosbell IB. Emergence of daptomycin resistance following vancomycin-unresponsive Staphylococcus aureus bacteraemia in a daptomycin-naive patient--a review of the literature. Eur J Clin Microbiol Infect Dis. 2011;30:603–610. doi: 10.1007/s10096-010-1128-3. [DOI] [PubMed] [Google Scholar]
- 39.Patel JB, Jevitt LA, Hageman J, et al. An association between reduced susceptibility to daptomycin and reduced susceptibility to vancomycin in Staphylococcus aureus. Clin Infect Dis. 2006;42:1652–1653. doi: 10.1086/504084. [DOI] [PubMed] [Google Scholar]
- 40.Pillai SK, Gold HS, Sakoulas G, et al. Daptomycin nonsusceptibility in Staphylococcus aureus with reduced vancomycin susceptibility is independent of alterations in MprF. Antimicrob Agents Chemother. 2007;51:2223–2225. doi: 10.1128/AAC.00202-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakoulas G, Alder J, Thauvin-Eliopoulos C, et al. Induction of daptomycin heterogeneous susceptibility in Staphylococcus aureus by exposure to vancomycin. Antimicrob Agents Chemother. 2006;50:1581–1585. doi: 10.1128/AAC.50.4.1581-1585.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cui L, Tominaga E, Neoh HM, et al. Correlation between reduced daptomycin susceptibility and vancomycin resistance in vancomycin-intermediate Staphylococcus aureus. Antimicrob Agents Chemother. 2006;50:1079–1082. doi: 10.1128/AAC.50.3.1079-1082.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mariani PG, Sader HS, Jones RN. Development of decreased susceptibility to daptomycin and vancomycin in a Staphylococcus aureus strain during prolonged therapy. J Antimicrob Chemother. 2006;58:481–483. doi: 10.1093/jac/dkl256. [DOI] [PubMed] [Google Scholar]
- 44.Sy CL, Lee SS, Wu KS, et al. Emergence of a strain of methicillin-resistant Staphylococcus aureus with decreased susceptibility to vancomycin 7 months after treatment with glycopeptide antibiotics. J Microbiol Immunol Infect. 2013 doi: 10.1016/j.jmii.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 45.Mwangi MM, Wu SW, Zhou Y, et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc Natl Acad Sci U S A. 2007;104:9451–9456. doi: 10.1073/pnas.0609839104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bayer AS, Schneider T, Sahl HG. Mechanisms of daptomycin resistance in Staphylococcus aureus: role of the cell membrane and cell wall. Ann N Y Acad Sci. 2013;1277:139–158. doi: 10.1111/j.1749-6632.2012.06819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang SJ, Mishra NN, Rubio A, et al. Causal role of single nucleotide polymorphisms within the mprF gene of Staphylococcus aureus in daptomycin resistance. Antimicrob Agents Chemother. 2013;57:5658–5664. doi: 10.1128/AAC.01184-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rubio A, Moore J, Varoglu M, et al. LC-MS/MS characterization of phospholipid content in daptomycin-susceptible and -resistant isolates of Staphylococcus aureus with mutations in mprF. Mol Membr Biol. 2012;29:1–8. doi: 10.3109/09687688.2011.640948. [DOI] [PubMed] [Google Scholar]
- 49.Andra J, Goldmann T, Ernst CM, et al. Multiple peptide resistance factor (MprF)-mediated resistance of Staphylococcus aureus against antimicrobial peptides coincides with a modulated peptide interaction with artificial membranes comprising lysyl-phosphatidylglycerol. J Biol Chem. 2011;286:18692–18700. doi: 10.1074/jbc.M111.226886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ernst CM, Peschel A. Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol Microbiol. 2011;80:290–299. doi: 10.1111/j.1365-2958.2011.07576.x. [DOI] [PubMed] [Google Scholar]
- 51.Nishi H, Komatsuzawa H, Fujiwara T, et al. Reduced content of lysyl-phosphatidylglycerol in the cytoplasmic membrane affects susceptibility to moenomycin, as well as vancomycin, gentamicin, and antimicrobial peptides, in Staphylococcus aureus. Antimicrob Agents Chemother. 2004;48:4800–4807. doi: 10.1128/AAC.48.12.4800-4807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Staubitz P, Neumann H, Schneider T, et al. MprF-mediated biosynthesis of lysylphosphatidylglycerol, an important determinant in staphylococcal defensin resistance. FEMS Microbiol Lett. 2004;231:67–71. doi: 10.1016/S0378-1097(03)00921-2. [DOI] [PubMed] [Google Scholar]
- 53.Mishra NN, Yang SJ, Chen L, et al. Emergence of daptomycin resistance in daptomycin-naive rabbits with methicillin-resistant Staphylococcus aureus prosthetic joint infection is associated with resistance to host defense cationic peptides and mprF polymorphisms. PLoS ONE. 2013;8:e71151. doi: 10.1371/journal.pone.0071151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang SJ, Xiong YQ, Dunman PM, et al. Regulation of mprF in daptomycin-nonsusceptible Staphylococcus aureus strains. Antimicrob Agents Chemother. 2009;53:2636–2637. doi: 10.1128/AAC.01415-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Slavetinsky CJ, Peschel A, Ernst CM. Alanyl-phosphatidylglycerol and lysyl-phosphatidylglycerol are translocated by the same MprF flippases and have similar capacities to protect against the antibiotic daptomycin in Staphylococcus aureus. Antimicrob Agents Chemother. 2012;56:3492–3497. doi: 10.1128/AAC.00370-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ernst CM, Kuhn S, Slavetinsky CJ, et al. The lipid-modifying multiple peptide resistance factor is an oligomer consisting of distinct interacting synthase and flippase subunits. MBio. 2015;6 doi: 10.1128/mBio.02340-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Friedman L, Alder JD, Silverman JA. Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrob Agents Chemother. 2006;50:2137–2145. doi: 10.1128/AAC.00039-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bayer AS, Mishra NN, Sakoulas G, et al. Heterogeneity of mprF sequences in methicillin-resistant Staphylococcus aureus clinical isolates: role in cross-resistance between daptomycin and host defense antimicrobial peptides. Antimicrob Agents Chemother. 2014;58:7462–7467. doi: 10.1128/AAC.03422-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bayer AS, Mishra NN, Chen L, et al. Frequency and distribution of single nucleotide polymorphisms within mprF in methicillin-resistant Staphylococcus aureus (MRSA) clinical isolates: Role in cross-resistance between daptomycin and host defense antimicrobial peptides. Antimicrob Agents Chemother. 2015 doi: 10.1128/AAC.00970-15. AAC.00970-00915. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cameron DR, Mortin LI, Rubio A, et al. Impact of daptomycin resistance on Staphylococcus aureus virulence. Virulence. 2015;6:127–131. doi: 10.1080/21505594.2015.1011532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ernst CM, Staubitz P, Mishra NN, et al. The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS pathogens. 2009;5:e1000660. doi: 10.1371/journal.ppat.1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rubio A, Conrad M, Haselbeck RJ, et al. Regulation of mprF by antisense RNA restores daptomycin susceptibility to daptomycin-resistant isolates of Staphylococcus aureus. Antimicrob Agents Chemother. 2011;55:364–367. doi: 10.1128/AAC.00429-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mishra NN, Bayer AS, Weidenmaier C, et al. Phenotypic and genotypic characterization of daptomycin-resistant methicillin-resistant Staphylococcus aureus strains: relative roles of mprF and dlt operons. PLoS One. 2014;9:e107426. doi: 10.1371/journal.pone.0107426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bertsche U, Yang SJ, Kuehner D, et al. Increased cell wall teichoic acid production and D-alanylation are common phenotypes among daptomycin-resistant methicillin-resistant Staphylococcus aureus (MRSA) clinical isolates. PLoS One. 2013;8:e67398. doi: 10.1371/journal.pone.0067398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cafiso V, Bertuccio T, Purrello S, et al. dltA overexpression: A strain-independent keystone of daptomycin resistance in methicillin-resistant Staphylococcus aureus. Int J Antimicrob Agents. 2014;43:26–31. doi: 10.1016/j.ijantimicag.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 66.Fischer A, Yang SJ, Bayer AS, et al. Daptomycin resistance mechanisms in clinically derived Staphylococcus aureus strains assessed by a combined transcriptomics and proteomics approach. J Antimicrob Chemother. 2011;66:1696–1711. doi: 10.1093/jac/dkr195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bertsche U, Weidenmaier C, Kuehner D, et al. Correlation of daptomycin resistance in a clinical Staphylococcus aureus strain with increased cell wall teichoic acid production and D-alanylation. Antimicrob Agents Chemother. 2011;55:3922–3928. doi: 10.1128/AAC.01226-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang SJ, Kreiswirth BN, Sakoulas G, et al. Enhanced expression of dltABCD is associated with the development of daptomycin nonsusceptibility in a clinical endocarditis isolate of Staphylococcus aureus. J Infect Dis. 2009;200:1916–1920. doi: 10.1086/648473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Utaida S, Dunman PM, Macapagal D, et al. Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology. 2003;149:2719–2732. doi: 10.1099/mic.0.26426-0. [DOI] [PubMed] [Google Scholar]
- 70.Muthaiyan A, Silverman JA, Jayaswal RK, et al. Transcriptional profiling reveals that daptomycin induces the Staphylococcus aureus cell wall stress stimulon and genes responsive to membrane depolarization. Antimicrob Agents Chemother. 2008;52:980–990. doi: 10.1128/AAC.01121-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gardete S, Tomasz A. Mechanisms of vancomycin resistance in Staphylococcus aureus. J Clin Invest. 2014;124:2836–2840. doi: 10.1172/JCI68834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stryjewski ME, Corey GR. Methicillin-resistant Staphylococcus aureus: an evolving pathogen. Clin Infect Dis. 2014;58(Suppl 1):S10–19. doi: 10.1093/cid/cit613. [DOI] [PubMed] [Google Scholar]
- 73.Mehta S, Cuirolo AX, Plata KB, et al. VraSR two-component regulatory system contributes to mprF-mediated decreased susceptibility to daptomycin in in vivo-selected clinical strains of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2012;56:92–102. doi: 10.1128/AAC.00432-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Camargo IL, Neoh HM, Cui L, et al. Serial daptomycin selection generates daptomycin-nonsusceptible Staphylococcus aureus strains with a heterogeneous vancomycin-intermediate phenotype. Antimicrob Agents Chemother. 2008;52:4289–4299. doi: 10.1128/AAC.00417-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mishra NN, McKinnell J, Yeaman MR, et al. In vitro cross-resistance to daptomycin and host defense cationic antimicrobial peptides in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob Agents Chemother. 2011;55:4012–4018. doi: 10.1128/AAC.00223-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Howden BP, McEvoy CR, Allen DL, et al. Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator WalKR. PLoS Pathog. 2011;7:e1002359. doi: 10.1371/journal.ppat.1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Short SA, White DC. Biosynthesis of cardiolipin from phosphatidylglycerol in Staphylococcus aureus. J Bacteriol. 1972;109:820–826. doi: 10.1128/jb.109.2.820-826.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ohniwa RL, Kitabayashi K, Morikawa K. Alternative cardiolipin synthase Cls1 compensates for stalled Cls2 function in Staphylococcus aureus under conditions of acute acid stress. FEMS Microbiol Lett. 2013;338:141–146. doi: 10.1111/1574-6968.12037. [DOI] [PubMed] [Google Scholar]
- 79.Koprivnjak T, Zhang D, Ernst CM, et al. Characterization of Staphylococcus aureus cardiolipin synthases 1 and 2 and their contribution to accumulation of cardiolipin in stationary phase and within phagocytes. J Bacteriol. 2011;193:4134–4142. doi: 10.1128/JB.00288-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peleg AY, Miyakis S, Ward DV, et al. Whole genome characterization of the mechanisms of daptomycin resistance in clinical and laboratory derived isolates of Staphylococcus aureus. PLoS One. 2012;7:e28316. doi: 10.1371/journal.pone.0028316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mishra NN, Yang SJ, Sawa A, et al. Analysis of cell membrane characteristics of in vitro-selected daptomycin-resistant strains of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2009;53:2312–2318. doi: 10.1128/AAC.01682-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu CI, Liu GY, Song Y, et al. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science. 2008;319:1391–1394. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mishra NN, Liu GY, Yeaman MR, et al. Carotenoid-related alteration of cell membrane fluidity impacts Staphylococcus aureus susceptibility to host defense peptides. Antimicrob Agents Chemother. 2011;55:526–531. doi: 10.1128/AAC.00680-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mishra NN, Bayer AS. Correlation of cell membrane lipid profiles with daptomycin resistance in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2013;57:1082–1085. doi: 10.1128/AAC.02182-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gaupp R, Lei S, Reed JM, et al. Staphylococcus aureus metabolic adaptations during the transition from a daptomycin susceptibility phenotype to a daptomycin nonsusceptibility phenotype. Antimicrob Agents Chemother. 2015;59:4226–4238. doi: 10.1128/AAC.00160-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sader HS, Farrell DJ, Flamm RK, et al. Daptomycin activity tested against 164457 bacterial isolates from hospitalised patients: summary of 8 years of a Worldwide Surveillance Programme (2005-2012) Int J Antimicrob Agents. 2014;43:465–469. doi: 10.1016/j.ijantimicag.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 87.Kelesidis T, Chow AL, Humphries R, et al. Case-control study comparing de novo and daptomycin-exposed daptomycin-nonsusceptible Enterococcus infections. Antimicrob Agents Chemother. 2012;56:2150–2152. doi: 10.1128/AAC.05918-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kelesidis T, Humphries R, Uslan DZ, et al. De novo daptomycin-nonsusceptible enterococcal infections. Emerg Infect Dis. 2012;18:674–676. doi: 10.3201/eid1804.110932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arias CA, Panesso D, McGrath DM, et al. Genetic basis for in vivo daptomycin resistance in enterococci. N Engl J Med. 2011;365:892–900. doi: 10.1056/NEJMoa1011138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mishra NN, Bayer AS, Tran TT, et al. Daptomycin resistance in enterococci is associated with distinct alterations of cell membrane phospholipid content. PLoS One. 2012;7:e43958. doi: 10.1371/journal.pone.0043958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tran TT, Panesso D, Mishra NN, et al. Daptomycin-resistant Enterococcus faecalis diverts the antibiotic molecule from the division septum and remodels cell membrane phospholipids. MBio. 2013;4 doi: 10.1128/mBio.00281-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miller C, Kong J, Tran TT, et al. Adaptation of Enterococcus faecalis to daptomycin reveals an ordered progression to resistance. Antimicrob Agents Chemother. 2013;57:5373–5383. doi: 10.1128/AAC.01473-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Munita JM, Tran TT, Diaz L, et al. A liaF codon deletion abolishes daptomycin bactericidal activity against vancomycin-resistant Enterococcus faecalis. Antimicrob Agents Chemother. 2013 doi: 10.1128/AAC.00021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reyes J, Panesso D, Tran TT, et al. A liaR deletion restores susceptibility to daptomycin and antimicrobial peptides in multidrug-resistant Enterococcus faecalis. J Infect Dis. 2015;211:1317–1325. doi: 10.1093/infdis/jiu602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Davlieva M, Shi Y, Leonard PG, et al. A variable DNA recognition site organization establishes the LiaR-mediated cell envelope stress response of enterococci to daptomycin. Nucleic Acids Res. 2015;43:4758–4773. doi: 10.1093/nar/gkv321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miller WR, Munita JM, Arias CA. Mechanisms of antibiotic resistance in enterococci. Expert Rev Anti Infect Ther. 2014;12:1221–1236. doi: 10.1586/14787210.2014.956092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Palmer KL, Daniel A, Hardy C, et al. Genetic basis for daptomycin resistance in enterococci. Antimicrobial agents and chemotherapy. 2011;55:3345–3356. doi: 10.1128/AAC.00207-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Munita JM, Panesso D, Diaz L, et al. Correlation between mutations in liaFSR of Enterococcus faecium and MIC of daptomycin: revisiting daptomycin breakpoints. Antimicrob Agents Chemother. 2012;56:4354–4359. doi: 10.1128/AAC.00509-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Steed ME, Vidaillac C, Rose WE, et al. Characterizing vancomycin-resistant Enterococcus strains with various mechanisms of daptomycin resistance developed in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother. 2011;55:4748–4754. doi: 10.1128/AAC.00084-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Panesso D, Tran TT, Munita JM, et al. Mutationsin liaSR of Enterococcus faecium are not associated with remodeling of cell membrane phospholipids. 4th ASM Conference on Enterococci; Cartagena, Colombia. March 2014.2014. [Google Scholar]
- 101.Tran TT, Panesso D, Diaz L, et al. A LiaFSR-independent pathway to daptomycin resistance in Enterococcus faecium; 53rd ICAAC; Denver, Colorado. September 2013.2013. [Google Scholar]
- 102.Humphries RM, Kelesidis T, Tewhey R, et al. Genotypic and phenotypic evaluation of the evolution of high-level daptomycin non-susceptibility in vancomycin-resistant Enterococcus faecium. Antimicrob Agents Chemother. 2012 doi: 10.1128/AAC.01318-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kelesidis T, Tewhey R, Humphries RM. Evolution of high-level daptomycin resistance in Enterococcus faecium during daptomycin therapy is associated with limited mutations in the bacterial genome. J Antimicrob Chemother. 2013;68:1926–1928. doi: 10.1093/jac/dkt117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tran TT, Panesso D, Gao H, et al. Whole-genome analysis of a daptomycin-susceptible Enterococcus faecium strain and its daptomycin-resistant variant arising during therapy. Antimicrob Agents Chemother. 2013;57:261–268. doi: 10.1128/AAC.01454-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Diaz L, Tran TT, Munita JM, et al. Whole-genome analyses of Enterococcus faecium isolates with diverse daptomycin MICs. Antimicrob Agents Chemother. 2014;58:4527–4534. doi: 10.1128/AAC.02686-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Munita JM, Mishra NN, Alvarez D, et al. Failure of high-dose daptomycin for bacteremia caused by daptomycin-susceptible Enterococcus faecium harboring LiaSR substitutions. Clin Infect Dis. 2014;59:1277–1280. doi: 10.1093/cid/ciu642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Panesso D, Reyes J, Gaston EP, et al. Deletion of liaR reverses daptomycin resistance in Enterococcus faecium independent of the genetic background. Antimicrob Agents Chemother. 2015;59:7327–34. doi: 10.1128/AAC.01073-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dubrac S, Bisicchia P, Devine KM, et al. A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol Microbiol. 2008;70:1307–1322. doi: 10.1111/j.1365-2958.2008.06483.x. [DOI] [PubMed] [Google Scholar]
- 109.Tran TT, Mishra NN, Panesso D, et al. Substitutions in YycG and cardiolipin synthase alter cell membrane phospholipids and lead to daptomycin resistance in Enterococcus faecium. 54th ICAAC; Washington, DC. September 2014.2013. [Google Scholar]
- 110.Dubrac S, I, Boneca G, Poupel O, et al. New insights into the WalK/WalR (YycG/YycF) essential signal transduction pathway reveal a major role in controlling cell wall metabolism and biofilm formation in Staphylococcus aureus. J Bacteriol. 2007;189:8257–8269. doi: 10.1128/JB.00645-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dubrac S, Msadek T. Identification of genes controlled by the essential YycG/YycF two-component system of Staphylococcus aureus. J Bacteriol. 2004;186:1175–1181. doi: 10.1128/JB.186.4.1175-1181.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Martin PK, Li T, Sun D, et al. Role in cell permeability of an essential two-component system in Staphylococcus aureus. J Bacteriol. 1999;181:3666–3673. doi: 10.1128/jb.181.12.3666-3673.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Davlieva M, Zhang W, Arias CA, et al. Biochemical characterization of cardiolipin synthase mutations associated with daptomycin resistance in enterococci. Antimicrob Agents Chemother. 2013;57:289–296. doi: 10.1128/AAC.01743-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schoen C, Unzicker C, Stuhler G, et al. Life-threatening infection caused by daptomycin-resistant Corynebacterium jeikeium in a neutropenic patient. J Clin Microbiol. 2009;47:2328–2331. doi: 10.1128/JCM.00457-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.McElvania TeKippe E, Thomas BS, Ewald GA, et al. Rapid emergence of daptomycin resistance in clinical isolates of Corynebacterium striatum… a cautionary tale. Eur J Clin Microbiol Infect Dis. 2014;33:2199–2205. doi: 10.1007/s10096-014-2188-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tran TT, Jaijakul S, Lewis CT, et al. Native valve endocarditis caused by Corynebacterium striatum with heterogeneous high-level daptomycin resistance: collateral damage from daptomycin therapy? Antimicrob Agents Chemother. 2012;56:3461–3464. doi: 10.1128/AAC.00046-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Streit JM, Steenbergen JN, Thorne GM, et al. Daptomycin tested against 915 bloodstream isolates of viridans group streptococci (eight species) and Streptococcus bovis. J Antimicrob Chemother. 2005;55:574–578. doi: 10.1093/jac/dki032. [DOI] [PubMed] [Google Scholar]
- 118.Silverman JA, Oliver N, Andrew T, et al. Resistance studies with daptomycin. Antimicrob Agents Chemother. 2001;45:1799–1802. doi: 10.1128/AAC.45.6.1799-1802.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Akins RL, Katz BD, Monahan C, et al. Characterization of high-level daptomycin resistance in viridans group streptococci developed upon in vitro exposure to daptomycin. Antimicrob Agents Chemother. 2015;59:2102–2112. doi: 10.1128/AAC.04219-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Garcia-de-la-Maria C, Pericas JM, Del Rio A, et al. Early in vitro and in vivo development of high-level daptomycin resistance is common in mitis group streptococci after exposure to daptomycin. Antimicrob Agents Chemother. 2013;57:2319–2325. doi: 10.1128/AAC.01921-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Palacio F, Lewis JS, 2nd, Sadkowski L, et al. Breakthrough bacteremia and septic shock due to Streptococcus anginosus resistant to daptomycin in a patient receiving daptomycin therapy. Antimicrob Agents Chemother. 2011;55:3639–3640. doi: 10.1128/AAC.00231-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tascini C, Di Paolo A, Poletti R. Daptomycin concentrations in valve tissue and vegetation in patients with bacterial endocarditis. Antimicrob Agents Chemother. 2013;57:601–602. doi: 10.1128/AAC.01608-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Safdar N, Andes D, Craig WA. In vivo pharmacodynamic activity of daptomycin. Antimicrob Agents Chemother. 2004;48:63–68. doi: 10.1128/AAC.48.1.63-68.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rose WE, Leonard SN, Sakoulas G, et al. Daptomycin activity against Staphylococcus aureus following vancomycin exposure in an in vitro pharmacodynamic model with simulated endocardial vegetations. Antimicrob Agents Chemother. 2008;52:831–836. doi: 10.1128/AAC.00869-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hall AD, Steed ME, Arias CA, et al. Evaluation of standard- and high-dose daptomycin versus linezolid against vancomycin-resistant Enterococcus isolates in an in vitro pharmacokinetic/pharmacodynamic model with simulated endocardial vegetations. Antimicrob Agents Chemother. 2012;56:3174–3180. doi: 10.1128/AAC.06439-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Werth BJ, Steed ME, Ireland CE, et al. Defining daptomycin resistance prevention exposures in vancomycin-resistant Enterococcus faecium and E. faecalis. Antimicrob Agents Chemother. 2014;58:5253–5261. doi: 10.1128/AAC.00098-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sakoulas G, Eliopoulos GM, Alder J, et al. Efficacy of daptomycin in experimental endocarditis due to methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2003;47:1714–1718. doi: 10.1128/AAC.47.5.1714-1718.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang SJ, Xiong YQ, Boyle-Vavra S, et al. Daptomycin-oxacillin combinations in treatment of experimental endocarditis caused by daptomycin-nonsusceptible strains of methicillin-resistant Staphylococcus aureus with evolving oxacillin susceptibility (the "seesaw effect") Antimicrob Agents Chemother. 2010;54:3161–3169. doi: 10.1128/AAC.00487-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Barber KE, Ireland CE, Bukavyn N, et al. Observation of “seesaw effect” with vancomycin, teicoplanin, daptomycin and ceftaroline in 150 unique MRSA strains. Infect Dis Ther. 2014;3:35–43. doi: 10.1007/s40121-014-0023-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ortwine JK, Werth BJ, Sakoulas G, et al. Reduced glycopeptide and lipopeptide susceptibility in Staphylococcus aureus and the “seesaw effect”: Taking advantage of the back door left open? Drug Resist Updat. 2013;16:73–79. doi: 10.1016/j.drup.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 131.Lee CH, Wang MC, Huang IW, et al. Development of daptomycin nonsusceptibility with heterogeneous vancomycin-intermediate resistance and oxacillin susceptibility in methicillin-resistant Staphylococcus aureus during high-dose daptomycin treatment. Antimicrob Agents Chemother. 2010;54:4038–4040. doi: 10.1128/AAC.00533-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vignaroli C, Rinaldi C, Varaldo PE. Striking “seesaw effect” between daptomycin nonsusceptibility and beta-lactam susceptibility in Staphylococcus haemolyticus. Antimicrob Agents Chemother. 2011;55:2495–2496. doi: 10.1128/AAC.00224-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Backo M, Gaenger E, Burkart A, et al. Treatment of experimental staphylococcal endocarditis due to a strain with reduced susceptibility in vitro to vancomycin: efficacy of ampicillin-sulbactam. Antimicrob Agents Chemother. 1999;43:2565–2568. doi: 10.1128/aac.43.10.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bhateja P, Purnapatre K, Dube S, et al. Characterisation of laboratory-generated vancomycin intermediate resistant Staphylococcus aureus strains. Int J Antimicrob Agents. 2006;27:201–211. doi: 10.1016/j.ijantimicag.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 135.Dilworth TJ, Leonard SN, Vilay AM, et al. Vancomycin and piperacillin-tazobactam against methicillin-resistant Staphylococcus aureus and vancomycin-intermediate Staphylococcus aureus in an in vitro pharmacokinetic/pharmacodynamic model. Clin Ther. 2014;36:1334–1344. doi: 10.1016/j.clinthera.2014.06.027. [DOI] [PubMed] [Google Scholar]
- 136.Dilworth TJ, Sliwinski J, Ryan K, et al. Evaluation of vancomycin in combination with piperacillin-tazobactam or oxacillin against clinical methicillin-resistant Staphylococcus aureus isolates and vancomycin-intermediate S. aureus isolates in vitro. Antimicrob Agents Chemother. 2014;58:1028–1033. doi: 10.1128/AAC.01888-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Naimi TS, Anderson D, O’Boyle C, et al. Vancomycin-intermediate Staphylococcus aureus with phenotypic susceptibility to methicillin in a patient with recurrent bacteremia. Clin Infect Dis. 2003;36:1609–1612. doi: 10.1086/375228. [DOI] [PubMed] [Google Scholar]
- 138.Hindler JA, Wong-Beringer A, Charlton CL, et al. In vitro activity of daptomycin in combination with beta-lactams, gentamicin, rifampin and tigecycline against daptomycin non-susceptible enterococci. Antimicrob Agents Chemother. 2015 doi: 10.1128/AAC.05077-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Smith JR, Barber KE, Raut A, et al. β-Lactam combinations with daptomycin provide synergy against vancomycin-resistant Enterococcus faecalis and Enterococcus faecium. J Antimicrob Chemother. 2015;70:1738–1743. doi: 10.1093/jac/dkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dhand A, Sakoulas G. Daptomycin in combination with other antibiotics for the treatment of complicated methicillin-resistant Staphylococcus aureus bacteremia. Clin Ther. 2014;36:1303–1316. doi: 10.1016/j.clinthera.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 141.Hall Snyder A, Werth BJ, Barber KE, et al. Evaluation of the novel combination of daptomycin plus ceftriaxone against vancomycin-resistant enterococci in an in vitro pharmacokinetic/pharmacodynamic simulated endocardial vegetation model. J Antimicrob Chemother. 2014;69:2148–2154. doi: 10.1093/jac/dku113. [DOI] [PubMed] [Google Scholar]
- 142.Berti AD, Sakoulas G, Nizet V, et al. β-Lactam antibiotics targeting PBP1 selectively enhance daptomycin activity against methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2013;57:5005–5012. doi: 10.1128/AAC.00594-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Moise PA, Amodio-Groton M, Rashid M, et al. Multicenter evaluation of the clinical outcomes of daptomycin with and without concomitant β-lactams in patients with Staphylococcus aureus bacteremia and mild to moderate renal impairment. Antimicrob Agents Chemother. 2013;57:1192–1200. doi: 10.1128/AAC.02192-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mehta S, Singh C, Plata KB, et al. β-Lactams increase the antibacterial activity of daptomycin against clinical methicillin-resistant Staphylococcus aureus strains and prevent selection of daptomycin-resistant derivatives. Antimicrob Agents Chemother. 2012;56:6192–6200. doi: 10.1128/AAC.01525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sierra-Hoffman M, Iznaola O, Goodwin M, et al. Combination therapy with ampicillin and daptomycin for treatment of Enterococcus faecalis endocarditis. Antimicrob Agents Chemother. 2012;56:6064. doi: 10.1128/AAC.01760-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Steed ME, Vidaillac C, Rybak MJ. Novel daptomycin combinations against daptomycin-nonsusceptible methicillin-resistant Staphylococcus aureus in an in vitro model of simulated endocardial vegetations. Antimicrob Agents Chemother. 2010;54:5187–5192. doi: 10.1128/AAC.00536-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Snydman DR, McDermott LA, Jacobus NV. Evaluation of in vitro interaction of daptomycin with gentamicin or β-lactam antibiotics against Staphylococcus aureus and enterococci by FIC index and timed-kill curves. J Chemother. 2005;17:614–621. doi: 10.1179/joc.2005.17.6.614. [DOI] [PubMed] [Google Scholar]
- 148.Dhand A, Bayer AS, Pogliano J, et al. Use of antistaphylococcal beta-lactams to increase daptomycin activity in eradicating persistent bacteremia due to methicillin-resistant Staphylococcus aureus: role of enhanced daptomycin binding. Clin Infect Dis. 2011;53:158–163. doi: 10.1093/cid/cir340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Werth BJ, Barber KE, Tran KN, et al. Ceftobiprole and ampicillin increase daptomycin susceptibility of daptomycin-susceptible and -resistant VRE. J Antimicrob Chemother. 2015;70:489–493. doi: 10.1093/jac/dku386. [DOI] [PubMed] [Google Scholar]
- 150.Barber KE, Werth BJ, Ireland CE, et al. Potent synergy of ceftobiprole plus daptomycin against multiple strains of Staphylococcus aureus with various resistance phenotypes. J Antimicrob Chemother. 2014;69:3006–3010. doi: 10.1093/jac/dku236. [DOI] [PubMed] [Google Scholar]
- 151.Smith JR, Barber KE, Raut A, et al. β-Lactams enhance daptomycin activity against vancomycin-resistant Enterococcus faecalis and Enterococcus faecium in in vitro pharmacokinetic/pharmacodynamic models. Antimicrob Agents Chemother. 2015;59:2842–2848. doi: 10.1128/AAC.00053-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sakoulas G, Rose W, Nonejuie P, et al. Ceftaroline restores daptomycin activity against daptomycin-nonsusceptible vancomycin-resistant Enterococcus faecium. Antimicrob Agents Chemother. 2014;58:1494–1500. doi: 10.1128/AAC.02274-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Leonard SN, Rolek KM. Evaluation of the combination of daptomycin and nafcillin against vancomycin-intermediate Staphylococcus aureus. J Antimicrob Chemother. 2013;68:644–647. doi: 10.1093/jac/dks453. [DOI] [PubMed] [Google Scholar]
- 154.Werth BJ, Steed ME, Kaatz GW, et al. Evaluation of ceftaroline activity against heteroresistant vancomycin-intermediate Staphylococcus aureus and vancomycin-intermediate methicillin-resistant S. aureus strains in an in vitro pharmacokinetic/pharmacodynamic model: exploring the “seesaw effect”. Antimicrob Agents Chemother. 2013;57:2664–2668. doi: 10.1128/AAC.02308-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Berti AD, Wergin JE, Girdaukas GG, et al. Altering the proclivity towards daptomycin resistance in methicillin-resistant Staphylococcus aureus using combinations with other antibiotics. Antimicrob Agents Chemother. 2012;56:5046–5053. doi: 10.1128/AAC.00502-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rose WE, Schulz LT, Andes D, et al. Addition of ceftaroline to daptomycin after emergence of daptomycin-nonsusceptible Staphylococcus aureus during therapy improves antibacterial activity. Antimicrob Agents Chemother. 2012;56:5296–5302. doi: 10.1128/AAC.00797-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Garrigos C, Murillo O, Lora-Tamayo J, et al. Efficacy of daptomycin-cloxacillin combination in experimental foreign-body infection due to methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2012;56:3806–3811. doi: 10.1128/AAC.00127-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sakoulas G, Bayer AS, Pogliano J, et al. Ampicillin enhances daptomycin- and cationic host defense peptide-mediated killing of ampicillin- and vancomycin-resistant Enterococcus faecium. Antimicrob Agents Chemother. 2012;56:838–844. doi: 10.1128/AAC.05551-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Entenza JM, Giddey M, Vouillamoz J, et al. In vitro prevention of the emergence of daptomycin resistance in Staphylococcus aureus and enterococci following combination with amoxicillin/clavulanic acid or ampicillin. Int J Antimicrob Agents. 2010;35:451–456. doi: 10.1016/j.ijantimicag.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 160.Steenbergen JN, Mohr JF, Thorne GM. Effects of daptomycin in combination with other antimicrobial agents: a review of in vitro and animal model studies. J Antimicrob Chemother. 2009;64:1130–1138. doi: 10.1093/jac/dkp346. [DOI] [PubMed] [Google Scholar]
- 161.Sakoulas G, Nonejuie P, Nizet V, et al. Treatment of high-level gentamicin-resistant Enterococcus faecalis endocarditis with daptomycin plus ceftaroline. Antimicrob Agents Chemother. 2013;57:4042–4045. doi: 10.1128/AAC.02481-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Arias CA, Torres HA, Singh KV, et al. Failure of daptomycin monotherapy for endocarditis caused by an Enterococcus faecium strain with vancomycin-resistant and vancomycin-susceptible subpopulations and evidence of in vivo loss of the vanA gene cluster. Clin Infect Dis. 2007;45:1343–1346. doi: 10.1086/522656. [DOI] [PubMed] [Google Scholar]
- 163.Steed ME, Werth BJ, Ireland CE, et al. Evaluation of the novel combination of high-dose daptomycin plus trimethoprim-sulfamethoxazole against daptomycin-nonsusceptible methicillin-resistant Staphylococcus aureus using an in vitro pharmacokinetic/pharmacodynamic model of simulated endocardial vegetations. Antimicrob Agents Chemother. 2012;56:5709–5714. doi: 10.1128/AAC.01185-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ahmad NM, Rojtman AD. Successful treatment of daptomycin-nonsusceptible methicillin-resistant Staphylococcus aureus bacteremia with the addition of rifampin to daptomycin. Ann Pharmacother. 2010;44:918–921. doi: 10.1345/aph.1M665. [DOI] [PubMed] [Google Scholar]