

Summary
FACT (facilitates chromatin transcription) is a histone chaperone that facilitates transcription through chromatin and promotes histone recovery during transcription. Here, we describe a highly purified experimental system that recapitulates many important properties of transcribed chromatin and the key aspects of hFACT action during this process in vitro. We present the protocols describing how to prepare different forms of nucleosomes, including intact nucleosome, covalently conjugated nucleosome, nucleosome missing one of the two H2A/2B dimers (hexasome) and tetrasome (a nucleosome missing both H2A/2B dimers). These complexes allow analysis of various aspects of FACT’s function. These approaches and other methods described below can also be applied to the study of other chromatin remodelers and chromatin-targeted factors.
Keywords: transcription, histone chaperone, histone cross-linking, chromatin assembly
1. Introduction
Nucleosomes form strong barriers to transcribing RNA polymerase II (Pol II) in eukaryotic nuclei (1). In order to efficiently transcribe genes, these nucleosome barriers need to be properly relieved. This task is mainly conducted by various chromatin remodelers which could slide, evict, transfer or exchange core histones during transcription to allow Pol II progression along the DNA template (2).
Histone chaperones represent one type of chromatin remodelers which can alter the nucleosome structure without ATP hydrolysis (3). Various histone chaperones have high binding affinity to H2A/H2B dimer, H3/H4 tetramer and/or nucleosome. Therefore, it has been suggested that histone chaperones relieve the nucleosome barriers during transcription by transiently dissembling/reassembling the nucleosome through their chaperone activity (4). To evaluate this hypothesis, two major questions have to be addressed: (i) whether the presence of dimer and/or tetramer in a nucleosome is critical for chaperone’s activity, and (ii) whether the action of histone chaperone is indeed dependent on the disassembly/reassembly of the nucleosome. Here we describe a set of approaches to address these questions using hFACT as an exemplary histone chaperone.
FACT has histone chaperone activity, with a higher binding affinity to H2A/H2B dimer (5, 6). To study FACT’s action in facilitating Pol II transcription, we have developed a methodology to systematically investigate this process (7). It includes preparation of different forms of well-positioned nucleosomes and subnucleosomes, including intact nucleosome, nucleosomes missing the promoter-distal (–D) or -proximal (–P) H2A/H2B dimer and tetrasome (nucleosome missing both H2A/H2B dimers) assembled on the same nucleosome positioning sequence (NPS) 603 (Fig. 1). These complexes allow analysis of the effect of each histone dimer and tetramer in FACT’s action during transcription through chromatin. Our data indicate that the histone dimers play a major role in this process: removal of either distal or proximal dimer results in less pronounced stimulation of transcription by FACT, as compared with the intact nucleosome (7).
Fig. 1. Schematic diagrams of the templates for analysis of the mechanism of FACT action.

(1) intact nucleosome, (2) -D hexasome, (3) -P hexasome and (4) tetrasome. Primers used for PCR amplification of each template are shown. TspR1 site is indicated as solid gray rectangle. Position of nucleosomes/subnucleosomes on DNA are indicated by dashed lines.
The second approach described here involves analysis of the role of H2A/H2B dimer dissociation during FACT-dependent Pol II transcription through chromatin. The technique we employed to tackle this problem is covalent cross-linking of the histone octamer by dimethyl suberimidate (DMS) to prevent the dissociation of dimer(s) form nucleosome. DMS does not introduce any charges to cross-linked protein and is unlikely to cause changes in the nucleosome structure. Two different ways to preparing cross-linked nucleosome were used: (i) reconstitution of the nucleosome and subsequent DMS treatment or (ii) DMS cross-linking of purified histone octamer with subsequent nucleosome assembly. The data obtained by both protocols were consistent and demonstrated that the treatment with DMS does not affect the wrapping of DNA on histone octamer, minimally affect the ability to be transcribed by Pol II, and does not interfere with FACT function during transcription. This result suggests that H2A/H2B dimer dissociation observed during transcription is dispensable for FACT action.
The experimental system for analysis of the mechanism of FACT action described below consists of highly purified core yeast Pol II (yPol II) and precisely positioned mononucleosomes assembled using purified DNA and X. laevis recombinant histones. This “minimal” experimental system faithfully recapitulates many important properties of chromatin transcribed in vivo (8–13), including effects of histone and Pol II mutations on transcription through chromatin (10, 14), and action of various elongation factors (including FACT) (6, 12, 15–17), ATP-dependent chromatin remodelers (18) and multiple complexes of Pol II (11) during this process.
In summary, the protocols described here present some useful strategies for preparation of various nucleosomes/subnucleosomes for analysis of chromatin structure/dynamics during Pol II transcription and other chromatin remodeling processes in vitro.
2. Materials
2.1. DNA template preparation
Plasmid pGEM-3Z-603, a single copy of 603 sequence introduced into pGEM-3Z vector.
-
Oligonucleotides:
Uplong1: 5′TCTCAAATTTTATGGCACTGGGCGAGACATACACGAATATGGCGTTTTCCTAGTACAAATCACCC-3′;
603F tsp dim: 5′-AAGCGACAGCTTTGGCACT GGGTCACTCGGGCTTCTAAGTACG-3′;
603R: 5′-ACCCCAGGGACTTGAAGTAATAAG-3′
and 603 hexR: 5′-CTTTCAACATCGATGCACG GTG -3′.
γ-[32P] ATP (6000 Ci/mmol).
T4 polynucleotide kinase.
TspR1 enzyme (New England Biolabs, NEB).
Phenol and Chloroform (Sigma-Aldrich).
Gel Extraction kit (QIAquick, Qiagen, or comparable).
TB buffer: 20 mM Tris–HCl (pH 8.0), 5 mM MgCl2, 2 mM β-mercaptoethanol, and various concentrations of KCl. For instance, TB40 contains 40 mM KCl.
TE buffer: 10 mM Tris-HCl (pH 8.0) and 1 mM EDTA.
2.2. Nucleosome reconstitution
Core Reconstitution Buffers (CRB) 1–6: all six buffers contain 10 mMTris-HCl (pH 8.0), 0.2 mM EDTA, 5 mM β-mercaptoethanol, 0.1% NP-40, and NaCl at the following concentrations: buffer 1, 2 M; 2, 1.5 M; 3, 1 M; 4, 0.75 M; 5, 0.5 M; and 6, 0.01 M.
Purified histones and histone octamer (see reference (19) for detail).
Salmon sperm dsDNA.
2.3. Histone cross-linking
Dimethyl suberimidate dihydrochloride, DMS.
Cross-linking Buffer: 0.1 M sodium borate (pH 10), 0.1 M NaCl, 0.2 mM EDTA.
Stop Buffer: 25 mM Tris-HCl (pH 6.9), 1 mM EDTA.
Storage Buffer: 10 mM Tris-HCl (pH 7.5), 10 mM NaCl, 0.1% NP40, 0.2 mM EDTA, 5 mM β-mercaptoethanol.
Neutralization Buffer: 20 mM Tris-HCl (pH 7.5), 1 mM EDTA, 1 mM β-mercaptoethanol and 50 mM NaCl.
2.4. Transcription assays
Low adhesion microcentrifuge tube.
ssDNA oligonucleotide 50 T: 5′-GGTGTCGCTTGGGTTGGCTTTTCGGGCTGTCCCTCTCG ATGGCTGTAAGT-3′.
9-nt RNA primer: 5′-AUCGAGAGG-3′.
Non-template DNA strand 59NT: 5′-ACTTACAGCCATCGAGAGGGACACGGCGAAAAGCCAACCCAAGCGACACCGGCACTGGG-3′.
Purified yeast Polymerase II (see reference (20) for detail).
Purified FACT (see reference (6) for detail).
Ni2+-NTA agarose (Qiagen) or comparable His affinity resin.
Acetylated BSA.
Imidazole.
T4 DNA ligase.
Polyethylene glycol (PEG)-8000.
α-[32P] GTP (6000 Ci/mmol).
Ultrapure NTPs.
Ethylenediaminetetraacetic acid (EDTA).
RNA loading buffer (RLB): 95% formamide, 10 mM EDTA, 0.1% SDS, and 0.01% bromophenol blue and xylene cyanol dyes.
2.5. Gel electrophoresis and quantification
40% (w/v) acrylamide/bis-acrylamide solution (39:1).
40% (w/v) acrylamide/bis-acrylamide solution (19:1).
30% (w/v) acrylamide/bis-acrylamide solution (29:1).
Urea.
10% SDS.
1.5 M Tris-HCl (pH8.8) and 0.5 M Tris-HCl (pH6.8)
10% (w/v) ammonium persulfate.
N,N,N′,N′-Tetramethylethylenediamine,TEMED.
5× TBE stock solution in 1 L of H2O: 54 g of Tris base, 27.5 g of boric acid and caution 20 mL of 0.5 M EDTA (pH 8.0).
3. Methods
Removal of dimer(s) from an intact, well-positioned mononucleosome was done by truncating corresponding DNA sequence (e.g., nucleosome positioning sequence NPS-603). The truncated DNA templates were generated by PCR amplification with specific primers (Fig. 1). Different truncated forms of DNA template were prepared and incubated with different amounts of various combinations of purified histones to obtain the nucleosomes, hexasomes or tetrasomes. Alternatively, cross-linking of all core histones in the nucleosome was induced by DMS; the cross-linking was conducted either before or after nucleosome reconstitution.
To obtain the nucleosomes for transcription by yPol II, DNA fragments containing NPS-603 and bearing a TspR1 restriction site upstream of the NPS were PCR amplified. After amplification and digestion with TspRI enzyme, the fragment was purified, the nucleosome was reconstituted and ligated to the preassembled Pol II elongation complex (EC) through the TspRI sticky end. In order to evaluate the quality of nucleosomes, the promoter-distal end of nucleosomal DNA was weakly labeled with γ-[32P]-ATP. This weak labeling minimally interferes with subsequent analysis of more intensely pause-labeled RNA.
3.1. Preparation of DNA templates for assembly of intact, distal (–D) or proximal (–P) dimer-truncated nucleosomes/subnucleosomes
Reverse primers 603 hexR or 603R were labeled with 0.1–0.3 μCi γ-[32P] ATP using T4 polynucleotide kinase in the TB40 buffer for 40 min at 37°C with subsequent enzyme inactivation for 20 min at 65°C.
Intact and –D DNA templates were PCR-amplified from pGEM-3Z-603 plasmid with the universal unlabeled forward primer Uplong1 (containing TspR1 site) and labeled reverse primers 603R and 603 hexR, respectively. –P DNA template was PCR amplified from pGEM-3Z-603 plasmid with unlabeled forward primer 603F tsp dim (containing TspR1 site) and labeled reverse primer 603R.
The PCR product was extracted with one volume of 1:1 (v/v) phenol:chloroform, precipitated with ethanol, washed with 70% ethanol, resuspended in 1× Buffer 4 (NEB), and digested with TspR1 at 65 °C for 4 h or overnight.
The digested 231-bp product of intact fragment, 195-bp product of –D fragment or 118-bp product of –P fragment were resolved in 2% agarose gel containing 4 M urea. The DNA fragments were excised on the trans-illuminator using long UV wavelength (see Note 1).
The DNA fragment was further purified using QIAquick Gel Extraction kit according to the manufacturer instruction and eluted with 30–50 μl of TE buffer.
DNA concentrations were determined by measuring the A260; purified DNA fragments were stored at 20°C.
3.2. Reconstitution of nucleosomes/subnucleosomes
250 ml of each of CRB1-6 buffers was cooled to 4°C.
0.5–3 μg of single end-labeled full length, –P or –D DNA fragments were dissolved in the CRB1 buffer for assembly with purified histones. Intact nucleosome can also be assembled by histone core octamer transfer (see Note 2 and Section 3.3.1).
In case of intact nucleosome assembly from purified histones, H3 and H4 histones were added in 2-fold molar excess to DNA, while H2A and H2B were added in 3-fold molar excess; in case of –D and –P hexasome assembly, H3 and H4 histones were added in 2-fold and H2A and H2B were added in 1.5-fold molar excess; in case of tetrasome (assembly with –D DNA template), H3 and H4 histones were added in 2-fold molar excess to DNA and no H2A and H2B were added.
For intact nucleosome, –D or –P hexasome preparations, salmon sperm dsDNA was added to achieve 3-fold weight excess over the amount of the DNA fragments; for tetrasome, salmon sperm dsDNA was added to achieve 6-fold weight excess (see Note 3).
The reaction volume in all cases was adjusted to 40–100 μl to achieve 10–100 ng/ml total concentration of DNA.
The samples were subsequently dialyzed against CRB1, CRB2, CRB3, and CRB4, each for 1 h, CRB5 for 2.5 h, and CRB6 overnight at 4°C.
The resulting nucleosomes were stored at 4°C.
The quality of nucleosome reconstitution was evaluated by analysis of the samples by 4.5% (39:1 acrylamide:bis) native PAGE in 0.5× TBE buffer for 3 h at 110 V.
The gel was transferred to Whatman 3MM paper, covered with a plastic wrap, vacuum dried, and exposed to a PhosphorImager screen for 2–3 h or overnight. Assembled nucleosomes migrate as distinct bands and have a lower mobility in the gel as compared with histone-free DNA (Fig. 2 and see Note 4).
Fig. 2. Analysis of reconstituted -D hexasome and tetrasome by native PAGE.
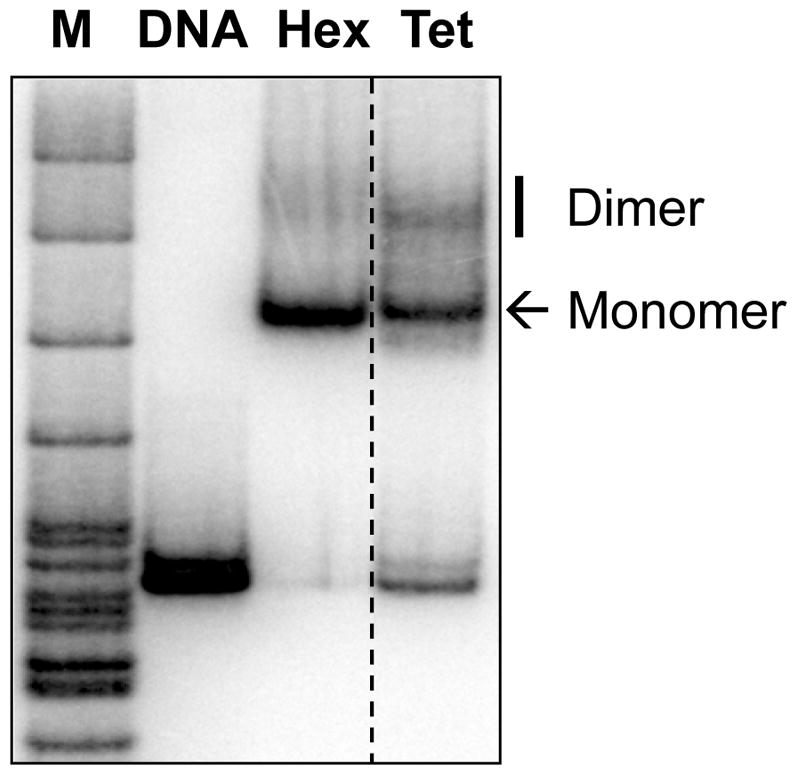
Monomer and dimer forms of nucleosomes/subnucleosomes are indicated. M: pBR322 DNA-MspI digest.
3.3. Preparation of cross-linked nucleosomes
(1) Cross-linking of nucleosomes
Intact nucleosomes were reconstituted as described above using purified histone core octamer in 4-fold molar excess relative to DNA (see Note 5).
Reconstituted nucleosome was dialyzed against the cross-linking buffer for 90 min at 4°C.
Freshly prepared DMS (in the cross-linking buffer) was added to nucleosomes at final concentration 0.5 mg/ml and incubate for 60 min at room temperature (RT) (see Note 6).
Sample was dialyzed against stop buffer for 90 min at RT to stop the cross-linking reaction.
Cross-linked nucleosomes were dialyzed against the storage buffer for 90 min at RT and then stored at 4°C.
Successful cross-linked nucleosome was analyzed by 8% SDS-PAGE with Coomassie blue staining. The cross-linked nucleosome appears as a slow-mobility complex on the top of gel (not shown).
The quality of nucleosomes was evaluated as described in Section 3.2.8-9 (Fig. 3A).
Fig. 3. Analysis of 603 DNA intact and DMS-cross-linked nucleosomes (Nu) by native PAGE ((17), reproduced from PNAS with permission).

Cross-linking was conducted before (A) or after (B) nucleosome assembly. The positions of mononucleosomes and dinucleosomes are indicated.
(2) Cross-linking of histone octamer before reconstitution
Incubate 10–15 μg of purified histone core octamer with freshly prepared DMS to final concentration of 0.5 mg/ml in the cross-linking buffer containing 2 M NaCl for 60 min at RT.
Add 0.25 M Tris-HCl (pH 6.9) to 50 mM final concentration to stop the cross-linking reaction.
Dialyze the sample against the neutralization buffer for 2 hours at RT.
Check cross-linked histone octamer by 8% SDS-PAGE, stain with Coomassie blue. The cross-linked histone octamer appears as a slow-mobility complex on the top of gel.
Nucleosome reconstitution was performed as described above (see Section 3.2) with cross-linked histone octamer in 5-fold molar excess relative to DNA.
The quality of nucleosomes was evaluated as described in Section 3.2.8-9 (Fig. 3B).
3.4. Transcription in the presence of FACT
The low adhesion microcentrifuge tubes are used in all steps.
200 pmol of template ssDNA oligonucleotide 50T was phosphorylated with 1 mM ATP using T4 polynucleotide kinase in TB40 buffer for 20 min at 37°C with subsequent enzyme inactivation for 20 min at 65°C (see Note 7).
RNA:DNA duplex was obtained by addition of 200 pmol of 9-nt RNA primer to 200 pmol of phosphorylated 50 T DNA oligonucleotide in 20 μl of TB40. Reaction was continued for 10 min at 45°C; then the temperature was decreased by 2°C every 2 min to the RT.
The RNA:DNA duplex was incubated with 3-fold molar excess of yeast core Pol II (purified as described in (21)) to form active EC for 10 min at RT.
Formation of the EC was completed by adding 3-fold molar excess of unlabeled non-template DNA strand 59NT for 10 min at RT.
20 μl of Ni2+-NTA agarose was washed three times with 0.5 ml of TB40, incubated in the presence of 0.5 mg/ml of acetylated BSA for 10 min, and washed two times with 0.5 ml TB40. The volume was adjusted to 25 μl (see Note 8).
Pol II EC was incubated with 25 μl of Ni2+-NTA suspension for 15 min at RT.
Immobilized EC was washed once with TB40, twice with TB300, and twice with TB40. The volume was adjusted to 25 μl.
EC was eluted from Ni2+-NTA beads in the presence of 100 mM imidazole after incubating for 5 min at RT.
The eluted EC was ligated with 100–200 ng of the nucleosomal template in the presence of 100 μM ATP, 1% PEG-8000, and 50 units of T4 DNA ligase in a volume of 50 μl at 16°C for 1–2 h.
RNA transcript was pulse-labeled and EC-5 (with active center of Pol II located 5-bp upstream of promoter-proximal nucleosomal boundary) was formed in the presence of 1 μM α-[32P] GTP and unlabeled 2 μM CTP and 10 μM ATP for 10 min at RT.
Complete formation of EC–5 was achieved by adding nonlabeled GTP to final concentration of 10 μM for 5min at RT.
Transcription was continued (EC-5 was chased) in the presence of 400 μM NTPs for 10 min at RT.
To evaluate the effect of FACT, 0.1 μM of FACT was added during the chase.
To characterize pulse-labeled RNA, the reaction was terminated by adding EDTA to final concentration of 20 mM. Pulse-labeled RNA was extracted with one volume of 1:1 (v/v) phenol:chloroform, precipitated with ethanol, washed with 70% ethanol, dried, and dissolved in 5 μl of RLB (RNA loading buffer).
Samples were boiled for 1 min and analyzed in 8% polyacrylamide gel (19:1 acrylamide:bis) containing 8M urea after electrophoresis in 0.5x TBE buffer for 1–1.5 h at 2000 V.
Gel was dried on Whatman 3MM paper and exposed to a PhosphorImager screen at RT overnight (Fig. 4).
Fig. 4. Effect of cross-linking of histone octamers on transcription through the nucleosomes in the presence or absence of hFACT ((17), reproduced from PNAS with permission).

603 nucleosomes were transcribed by Pol II at 150 mM KCl before and after cross-linking of the octamer by DMS and subsequent nucleosome assembly, followed by analysis of pulse-labeled RNA by denaturing PAGE.
Acknowledgments
This work was supported by the National Institutes of Health RO1 grant GM58650 to V.M.S. and by RAS Presidium Program “Basic research for the development of biomedical technologies”.
Footnotes
After digestion with TspR1, the fragments have 9-nt long sticky ends which could still attach to each other via base-pairing and may not completely separate in regular agarose gel. The presence of urea in the gel helps to prevent this non-covalent attachment and improves the separation of the digested fragments.
Purified histone core octamer is intact at high salt condition (2 M NaCl) without the DNA. Therefore, like donor chromatin (22), purified histone core histone octamer can also be used for nucleosome reconstitution.
The nucleosomes and hexasomes are typically obtained with 90–95% yield; however, the tetrasome yield is only ~60–70% due to competitive binding of multiple histone tetramers to the DNA fragment.
The formation of the dimer form of the nucleosome minimally affects the outcome of transcription as the dimers are not transcribed.
The purified histone octamer was used because accidental histone-histone cross-linking was observed when purified histones were used.
DMS is an imidoester cross-linker which reacts with primary amines to form amidine bonds. To ensure specificity for primary amines, imidoester reactions are best done in amine-free, alkaline conditions (pH 10), such as in the borate-containing buffer.
The efficient phosphorylation of 50T oligonucleotide is critical for the subsequent ligation step.
The treatment of acetylated BSA with Ni2+-NTA minimizes non-specific binding of the beads to microcentrifuge tube.
References
- 1.Kulaeva OI, Hsieh FK, Chang HW, Luse DS, Studitsky VM. Mechanism of transcription through a nucleosome by RNA polymerase II. Biochim Biophys Acta. 2013;1829:76–83. doi: 10.1016/j.bbagrm.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 3.Park YJ, Luger K. Histone chaperones in nucleosome eviction and histone exchange. Curr Opin Struct Biol. 2008;18:282–289. doi: 10.1016/j.sbi.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selth LA, Sigurdsson S, Svejstrup JQ. Transcript Elongation by RNA Polymerase II. Annu Rev Biochem. 2010;79:271–293. doi: 10.1146/annurev.biochem.78.062807.091425. [DOI] [PubMed] [Google Scholar]
- 5.Winkler DD, Muthurajan UM, Hieb AR, Luger K. Histone chaperone FACT coordinates nucleosome interaction through multiple synergistic binding events. J Biol Chem. 2011;286:41883–41892. doi: 10.1074/jbc.M111.301465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;301:1090–1093. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- 7.Hsieh FK, Kulaeva OI, Patel SS, Dyer PN, Luger K, Reinberg D, Studitsky VM. Histone chaperone FACT action during transcription through chromatin by RNA polymerase II. Proc Natl Acad Sci U S A. 2013;110:7654–7659. doi: 10.1073/pnas.1222198110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kireeva ML, Walter W, Tchernajenko V, Bondarenko V, Kashlev M, Studitsky VM. Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol Cell. 2002;9:541–552. doi: 10.1016/s1097-2765(02)00472-0. [DOI] [PubMed] [Google Scholar]
- 9.Kulaeva OI, Gaykalova DA, Pestov NA, Golovastov VV, Vassylyev DG, Artsimovitch I, Studitsky VM. Mechanism of chromatin remodeling and recovery during passage of RNA polymerase II. Nat Struct Mol Biol. 2009;16:1272–1278. doi: 10.1038/nsmb.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsieh FK, Fisher M, Ujvari A, Studitsky VM, Luse DS. Histone Sin mutations promote nucleosome traversal and histone displacement by RNA polymerase II. EMBO Rep. 2010;11:705–710. doi: 10.1038/embor.2010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kulaeva OI, Hsieh FK, Studitsky VM. RNA polymerase complexes cooperate to relieve the nucleosomal barrier and evict histones. Proc Natl Acad Sci USA. 2010;107:11325–11330. doi: 10.1073/pnas.1001148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bondarenko VA, Steele LM, Ujvari A, Gaykalova DA, Kulaeva OI, Polikanov YS, Luse DS, Studitsky VM. Nucleosomes can form a polar barrier to transcript elongation by RNA polymerase II. Mol Cell. 2006;24:469–479. doi: 10.1016/j.molcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Gaykalova DA, Kulaeva OI, Pestov NA, Hsieh FK, Studitsky VM. Experimental analysis of the mechanism of chromatin remodeling by RNA polymerase II. Methods Enzymol. 2012;512:293–314. doi: 10.1016/B978-0-12-391940-3.00013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang HW, Kulaeva OI, Shaytan AK, Kibanov M, Kuznedelov K, Severinov KV, Kirpichnikov MP, Clark DJ, Studitsky VM. Analysis of the mechanism of nucleosome survival during transcription. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt1120. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ujvari A, Hsieh FK, Luse SW, Studitsky VM, Luse DS. Histone N-terminal tails interfere with nucleosome traversal by RNA polymerase II. J Biol Chem. 2008;283:32236–32243. doi: 10.1074/jbc.M806636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Angelov D, Bondarenko VA, Almagro S, Menoni H, Mongelard F, Hans F, Mietton F, Studitsky VM, Hamiche A, Dimitrov S, Bouvet P. Nucleolin is a histone chaperone with FACT-like activity and assists remodeling of nucleosomes. EMBO J. 2006;25:1669–1679. doi: 10.1038/sj.emboj.7601046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsieh FK, Kulaeva OI, Patel SS, Dyer PN, Luger K, Reinberg D, Studitsky VM. Histone chaperone FACT action during transcription through chromatin by RNA polymerase II. Proc Natl Acad Sci USA. 2013;110:7654–7659. doi: 10.1073/pnas.1222198110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaykalova DA, Nagarajavel V, Bondarenko VA, Bartholomew B, Clark DJ, Studitsky VM. A polar barrier to transcription can be circumvented by remodeler-induced nucleosome translocation. Nucleic Acids Res. 2011;39:3520–3528. doi: 10.1093/nar/gkq1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004;375:23–44. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- 20.Kireeva ML, Walter W, Tchernajenko V, Bondarenko V, Kashlev M, Studitsky VM. Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol Cell. 2002;9:541–552. doi: 10.1016/s1097-2765(02)00472-0. [DOI] [PubMed] [Google Scholar]
- 21.Walter W, Kireeva ML, Tchernajenko V, Kashlev M, Studitsky VM. Assay of the fate of the nucleosome during transcription by RNA polymerase II. Methods Enzymol. 2003;371:564–577. doi: 10.1016/S0076-6879(03)71042-8. [DOI] [PubMed] [Google Scholar]
- 22.Gaykalova DA, Kulaeva OI, Bondarenko VA, Studitsky VM. Preparation and analysis of uniquely positioned mononucleosomes. Methods Mol Biol. 2009;523:109–123. doi: 10.1007/978-1-59745-190-1_8. [DOI] [PMC free article] [PubMed] [Google Scholar]