

Abstract
Background
Mitochondrial myopathy severely affects skeletal muscle structure and function resulting in defective oxidative phosphorylation. However, the major pathomechanisms and therewith effective treatment approaches remain elusive. Therefore, the aim of the present study was to investigate disease-related impairments in skeletal muscle properties in patients with mitochondrial myopathy. Accordingly, skeletal muscle biopsies were obtained from six patients with moleculargenetically diagnosed mitochondrial myopathy (one male and five females, 53 ± 9 years) and eight age- and gender-matched healthy controls (two males and six females, 58 ± 14 years) to determine mitochondrial respiratory capacity of complex I-V, mitochondrial volume density and fiber type distribution.
Results
Mitochondrial volume density (4.0 ± 0.5 vs. 5.1 ± 0.8 %) as well as respiratory capacity of complex I-V were lower (P < 0.05) in mitochondrial myopathy and associated with a higher (P < 0.001) proportion of type II fibers (65.2 ± 3.6 vs. 44.3 ± 5.9 %). Additionally, mitochondrial volume density and maximal oxidative phosphorylation capacity correlated positively (P < 0.05) to peak oxygen uptake.
Conclusion
Mitochondrial myopathy leads to impaired mitochondrial quantity and quality and a shift towards a more glycolytic skeletal muscle phenotype.
Keywords: Bioenergetics, Fat oxidation, Mitochondria, Mitochondrial cytopathy, Neuromuscular disease, Skeletal muscle phenotype
Background
Despite progress in understanding the biochemistry and genetics of mitochondrial myopathy, many of the pathophysiological mechanisms remain unclear [1]. Validated therapeutic options as well as simple and effective diagnostic tools are lacking [1–5]. So far, it has barely been investigated how muscle metabolism and morphology may be affected by mitochondrial dysfunction. Fiber type abnormalities including varying distribution of type I and II fibers as well as general or selective atrophy have been reported in patients with various mitochondrial respiratory chain dysfunctions [6–8] but not in adults with mitochondrial myopathy. In a rat model of mitochondrial myopathy [9] a transformation from type I to type II fibers was observed in response to a primary defect of the mitochondrial respiratory chain.
In addition, patients with mitochondrial myopathy have been characterised by lower peak oxygen uptake (V̇O2peak) and work capacity (Ppeak) when performing cycling exercise [10–12]. This limited oxidative capacity in mitochondrial myopathy patients is most likely related to a limited O2 extraction with reduced arterio-venous O2 difference in exercising muscles [10–13]. The limited O2 extraction, in turn, might be explained by an impaired oxidative capacity of the mitochondria itself and/or a reduction in their number [14]. Exercise intolerance can be a prominent clinical manifestation of mitochondrial myopathy, leading to relatively low levels of exertion, fast fatigablity and a sedentary lifestyle [15, 16]. A sedentary lifestyle may further restrict the capacitiy of skeletal muscle for oxidative phosphorylation by a decrease in levels of functional mitochondria [17]. Hence, it remains difficult to distinguish to what extent exercise intolerance is related to impaired mitochondrial oxidative capacity or associated with physical deconditioning. Assessments of muscle oxidative capacity and potential alterations with mitochondrial myopathy have previously only been conducted in single cases or small case series [11]. Thereby, defects of the mitochondrial respiratory chain have been identified polarographically or by enzymatic assays [18, 19], usually involving complex I or III in adults and complex IV in children [8, 20]. Poloarographic measurements have been mainly conducted in complex combinations and not independently for each complex [21]. Moreover, these global assessments of mitochondrial function were conducted in muscle homogenates or isolated fibers [19]. Mitochondrial isolation and homogenation procedures, however, may disrupt the functional network and could thereby alter innate mitochondrial characteristics [22]. Therefore, in vitro measurements of O2 consumption in permeabilised myofibers preserving innate mitochondrial function present a more promising approach to determine mitochondrial characteristics [22]. Determination of the respiratory capacity of each individual complex could lead to a better understanding of mitochondrial function and identification of potential mitochondrial-myopathy-related pathomechanisms. So far, the absence of a consistent clinical phenotype has limited the viability of clinical identification and classification criteria of mitochondrial myopathy [3, 21, 23]. However, detailed elucidation of potential pathomechanisms would be important not only for the diagnosis of mitochondrial myopathy but also for developing rational treatment options for mitochondrial myopathy.
Consequently, the primary aim of the present study was to determine mitochondrial volume density and respiratory capacity of each individual complex in permeabilised muscle fibers of mitochondrial myopathy patients by implementing high-resolution respirometry. In our set of patients with moleculargenetically verified mitochondrial myopathy and concomitant exercise intolerance, we hypothesised that mitochondrial volume density and respiratory capacity of each single complex I-V would be decreased compared to age- and gender-matched healthy controls. Furthermore, we aimed to test whether high-resolution respirometry measurements would represent a diagnostic tool leading to enhanced diagnostic and classification criteria of mitochondrial myopathy.
Methods
Aim, design and setting
The main aim of the study was to determine mitochondrial volume density and respiratory capacity of each individual complex in permeabilised muscle fibers of mitochondrial myopathy patients and age- and gender-matched healthy controls by the application of high-resolution respirometry measurements. The second aim was to determine if human mitochondrial myopathy with shift towards type II fibers. Additionally, we evaluated if the applied mitochondrial respirometric measurements would represent a diagnostic tool leading to enhanced diagnostic and classification criteria of mitochondrial myopathy.
Participants
Six patients with mitochondrial myopathy, and eight age-and gender-matched healthy controls, participated in this study (Table 1). Patients were selected from a patient base followed at the Center of Neuromuscular Disorders Zurich (Department of Neurology, University Hospital Zurich, Switzerland) and healthy controls were matched by gender, age and by physical activity patterns (assessed by interrogation). In all patients, clinical and biopsy findings and/or molecular genetic analysis of muscle mitochondrial DNA were in accordance with the diagnosis of mitochondrial myopathy. Apart from histological signs of myopathy and/or serum phosphocreatine kinase (CK) elevation, patients featured a history of muscle weakness, exercise intolerance or exercise-dependent myalgia (Table 2). Prior to the experiments, study participants were clinically examined, exclusion criteria were pregnancy as well as cardiac and/or respiratory disease interfering with exercise tolerance. Several metabolic parameters including plasma lactate, glucose, insulin, leptin, glycated hemoglobin (HbA1c), thyroid stimulating hormone (TSH) and CK were measured prio to any study examination and exercise. Cardiac and respiratory health were assessed by 24 h - electrocardiogram, cardiac ultrasound, spirometry and laboratory examination (lipid profile, glucose, precursorprotein brain natriuretic peptide (proBNP)). Patients and controls were neither completely sedentary nor highly active and none of them exhibited diabetes, coronary heart disease, peripheral vascular disease or clinically significant hyperlipidemia.
Table 1.
Physiological characteristics of patients with mitochondrial myopathy (n = 6) and healthy controls (n = 8)
| Patients | Controls | P-value | |
|---|---|---|---|
| Age (yr) | 53 ± 9 | 58 ± 14 | 0.437 |
| Body mass (kg) | 75 ± 18 | 67 ± 12 | 0.376 |
| Height (cm) | 162 ± 7 | 167 ± 7 | 0.238 |
| BMC (kg) | 2.3 ± 0.18 | 2.4 ± 0.4 | 0.867 |
| Fat mass (kg) | 23.1 ± 16.3 | 14.2 ± 11.6 | 0.255 |
| Lean mass (kg) | 38.4 ± 13.3 | 41.4 ± 13.9 | 0.683 |
| Lean mass legs (kg) | 19.3 ± 11.1 | 19.0 ± 8.8 | 0.951 |
| Total body fat (%) | 39 ± 13 | 31 ± 6 | 0.153 |
| Relative V̇O2peak (mL min−1 kg−1) | 19.8 ± 6.8a | 32.6 ± 7.3 | <0.01 |
| P peak (W) | 78 ± 21a | 165 ± 60 | < 0.05 |
| [L−]rest (mmol L−1) | 1.64 ± 0.88 | 1.11 ± 0.62 | 0.207 |
| RER25W (-) | 0.72 ± 0.06a | 0.69 ± 0.06 | 0.447 |
| RERrel (-) | 0.78 ± 0.08a | 0.77 ± 0.05 | 0.923 |
| HRpeak(min−1) | 144 ± 28a | 158 ± 16 | 0.257 |
| tlim (s) | 278 ± 154a | 541 ± 321 | 0.118 |
| MVC (Nm) | 54.3 ± 41.9a | 80.6 ± 43.3b | 0.278 |
Values are represented as means ± SD. BMC bone mineral content; [L−], blood lactate concentration at rest; HR peak peak heart rate, MVC maximum voluntary contraction, P peak peak power; relative V̇O2peak, peak oxygen uptake per kg body weight; RER 25W respiratory exchange ratio at 25 W, RER rel respiratory exchange ratio at 50 % P peak, t lim time to exhaustion. a n = 5; b n = 7
Table 2.
Clinical and molecular characteristics of patients with mitochondrial myopathy
| Patient | Sex | Age of onset (yr) | Diagnosis | Clinical features | [CK]rest (U L−1) | [L−]rest(mmol L−1) | Genetic defect | Mutant mtDNA | Muscle biopsy |
|---|---|---|---|---|---|---|---|---|---|
| 1 | f | 20 | CPEO | ptosis, external ophthalmoplegia, facial weakness, slight general weakness | 219 | normal | mtDNA deletion 4977 bp | > 95 % | COX−, ragged blue fibers |
| 2 | m | 33 | CPEO | ptosis, external ophthalmoplegia, general weakness, exercise intolerance | 401 | normal | mtDNA deletion 4405 bp | 90 % | unremarkable |
| 3 | f | 62 | MM | mild proximal lower extremity weakness | normal | normal | mtDNA deletion 5–13 kbp | 49–71 % | COX−, ragged blue fibers |
| 4 | f | 12 | MM | exercise induced myalgia | 206 | normal | mtDNA deletion 10–13 kbp | 35 % | COX−, SDH+, ragged red fibers |
| 5 | f | 20 | MM | exercise induced myalgia | normal | normal | mtDNA deletion 6–10 kbp | 62 % | ragged red fibers |
| 6 | f | 39 | MM | exercise induced myalgia | normal | 5.3 | mtDNA deletion 8–12 kbp | 12–36 % | unremarkable |
[CK], blood creatine kinase concentration at rest; COX − cytochrome oxidase negative fibers, CPEO chronic progressive external ophthalmoplegia, [L−], blood lactate concentration at rest; MM mitochondrial myopathy, mtDNA mitochondrial DNA, SDH + succinate dehydrogenase positive fibers
Experimental design
Clinical and physiological assessments were performed. Clinical assessments consisted of a neurological examination, muscle strength examination, specific health questions (SF-36 Health Survey), fatigue severity subscale of the Checklist Individual Strength (CIS-fatigue), sensation of pain (visual analogue scale, VAS) and a skeletal muscle biopsy obtained from the vastus lateralis muscle. Physiological assessments included a scan for body composition, an incremental cycling test, a constant-load cycling test and measurement of knee extension torque.
Body composition measurement
Total body mass, bone mineral content (BMC), fat mass, percentage body fat, as well as total lean body mass and lean soft tissue mass of the leg were determined by performing dual-energy X-ray absorptiometry (DXA) measurements using a densitometer (Lunar iDXATM, GE Healthcare, Madison, WI, USA).
Exercise testing
Each study participant conducted an incremental exercise test on an electrically braked cycle ergometer until volitional exhaustion (Ergoselect 200 K, Ergoline, Bitz, Germany) to determine V̇O2peak and Ppeak. Additionally, all participants performed a constant-load cycling test at 85 % Ppeak to establish time to volitional exhaustion (tlim). Pulmonary gas exchange and ventilation were recorded during both exercise tests using an online gas collection system (Innocor™ M400, Innovision, Odense, Denmark), where flow, O2 and CO2 concentrations were recorded breath-by-breath. Flowmeter and gas analysers were calibrated prior to and after each test according to the manufacturer’s instructions. Throughout all cycling tests, heart rate was recorded (Polar S610i, Polar Electro, Kempele, Finland) and perceived exertion was assessed via a Borg CR 10 Scale. In addition, blood pressure was registered on a regular basis during the incremental cycling test. The incremental cycling test consisted of a 3 min rest phase followed by 2 min at 25 W with power increments of 25 W every 2 min until volitional exhaustion. Pedal cadences were freely chosen by the participants within a range of 60 and 80 revolutions per minute (rpm) and had to be remained constant throughout the test. V̇O2peak was determined as the highest mean over a 10 s period. The respiratory exchange ratios (RER) for submaximal power values were calculated as the mean of the last 30 s at 25 W (RER25W) and as the mean over 10 s at 50 % Ppeak (RERrel). The constant-load cycling test included a 3 min rest phase, a 3 min warm-up with 1 min at 40 % and 2 min at 60 % Ppeak, and a constant-load phase at 85 % Ppeak until volitional exhaustion.
Isokinetic dynamometry
Knee extensor maximal voluntary contraction torque (MVC) was assessed using an isokinetic dynamometer (Con-Trex MJ, Physiomed Elektromedizin, Schnaittach/Laipersdorf, Germany). Participants’ bodies were stabilised with straps and handles according to the guidelines of the manufacturer. Each participant performed three maximal knee extensions (ω = 3.14 rad s−1) separated by 1 min rest to assess MVC, whereby only the highest value out of the three trials was used for statistical analysis.
Skeletal muscle sampling
Participants were tested for coagulation abnormalities (Quick/INR, number of thrombocytes). Skeletal muscle biopsies were obtained under standardised conditions from the vastus lateralis muscle under local anesthesia (1 % lidocaine) of the skin and superficial muscle fascia, using the Bergström technique with a needle modified for suction. The biopsy was immediately dissected macroscopically free of fat and connective tissue and divided into sections for actual measurement of mitochondrial respiratory capacity, later transmission electron microscopy (TEM) and histochemistry. The part of the biopsy for the determination of mitochondrial respiratory capacity was directly placed in ice-cold biopsy preservation solution. For TEM, pieces of around 1 mm3 of each muscle biopsy were chemically fixed in 2.5 % glutaraldehyde in 0.1 M cacodylate buffer (pH 7.3), stored at room temperature (RT) for 24 h and thereafter at 4 °C until all samples were collected. Muscle tissue for immunohistochemistry was instantly mounted in an embedding medium (Tissue-Tek®, Sakura, Zoeterwoude, The Netherlands), snap frozen in isopentane cooled to −160 °C with liquid nitrogen, and subsequently stored at −80 °C until further processing.
Mitochondrial respiration measurement
Samples were prepared as described in detail previously [24]. Summarised, after mechanical fiber separation, chemical permeabilization in biopsy preservation solution and washing in mitochondrial respiration medium 05, muscle bundles were blotted dry and measured for wet weight (ww) in a balance-controlled scale (XS205 DualRange Analytical Balance, Mettler-Toledo AG, Greifensee, Switzerland). Respiration measurements were subsequently performed in mitochondrial respiration medium 06. O2 consumption of the individual muscle tissue was thereby measured at 37 °C using the high-resolution Oxygraph-2 k (Oroboros, Innsbruck, Austria). Standardised instrumental and chemical calibrations were performed as recommended by the manufacturer and described previously [24]. O2 flux was automatically calculated by the software, accounting for nonlinear changes in the negative time derivative of the O2concentration signal (DatLab, Oroboros, Innsbruck, Austria). Experiments were performed as duplicates in a hyperoxygenated environment in order to prevent any potential O2 diffusion limitation. Thereby, O2 concentration ranged between 200 and 450 nmol mL−1 within the chambers.
Respiratory titration protocol
The respiratory measurement protocol was specific to the analysis of individual aspects of respiratory capacity and coupling control efficiency during several substrate states induced via separate titrations. All titrations were added in series as presented, whereby the concentrations of substrates, uncouplers and inhibitors used were based on prior experiments [24]. The titration protocol was modified from previous protocols where they are described in detail [24]. In short, leak respiration in absence of adenylates (LN) was induced with the addition of octanoyl carnitine (0.2 mM) and malate (2 mM). Specifically, LN represents the resting O2 consumption of an unaltered and intact electron transport system (ETS) free of adenylates. Maximal electron flow through electron-transferring flavoprotein and maximal fatty acid oxidative capacity (PETF) was subsequently determined following the addition of ADP (5 mM). Electron capacity through complex I (PCI) was then induced following the additions of pyruvate (5 mM) and glutamate (10 mM). Maximal oxidative phosphorylation capacity (P) was induced with the addition of succinate (10 mM). P thereby represents respiration that results from saturating concentrations of ADP and substrate supply both for complex I and II. As an internal control for the integrity of the mitochondrial preparation, the mitochondrial outer membrane was assessed with the addition of cytochrome C (10 μM). ATP synthase was then inhibited by the titration of oligomycin (1 μM), which leads to oligomycin-induced leak respiration (LOmy). LOmy represents the corresponding leak state to P. In LOmy the chemiosmotic gradient is at maximum because of maximal substrate supply and inhibition of complex V (ATP synthase). Additionally, O2 flux is at minimum and is representative of proton leak, slip, cation cycling and overall dyscoupling. By uncoupling ATP synthase from the electron transport chain with the step-wise titration (4 × 0.5 μM) of the proton ionophore carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) phosphorylative restraint of electron transport was assessed reaching ETS capacity (E). In order to inhibit CI and to assess electron flow specific to complex II (PCII), rotenone (0.5 μM) was added. PCII is thereby not influenced by the preceding addition of FCCP, which was verified with separate protocols in our laboratory. The addition of antimycin A (2.5 μM) that inhibits complex III allows the determination and correction of residual O2 consumption (ROX), which is indicative of non-mitochondrial O2 consumption in the chamber. Finally, respiration measurements were terminated by simultaneous titration of ascorbate and TMPD to assess complex IV (COX) activity. Ascorbate and TMPD represent redox substrates that donate electrons directly to COX. Correction of O2 flux for the side reaction of auto-oxidation was conducted by chemical calibration experiments prior to the measurements. Finally, mitochondrial leak control ratios (LCR) were analysed as well. LCR allow the description of mitochondrial coupling efficiency, with a theoretical minimum of 0.0, indicating a fully coupled system, to a value of 1.0, representing a fully uncoupled system [24, 25].
Transmission electron microscopy
The chemically fixed tissue was washed 3 × in 0.1. M cacodylate buffer and consecutively post-fixed in 1 % osmiumtetroxid in 0.1. M cacodylate buffer for 2 h at RT. After 3 x washing with milliQ H2O, samples were block-stained with uranyl acetate (2 % in milliQ H2O) overnight at RT. The subsequent graded dehydration was conducted in a tissue processor (Leica EM TP, Leica Microsystems, Wetzlar, Germany) according to the following protocol: 10 min 70 % EtOH, 15 min 96 % EtOH, 4 × 30 min 100 % EtOH, 2 × 5 min and 2 × 10 min propylene oxide, 2 × 1 h 1:1 propylene oxide/Epon, 1 h 100 % Epon, 100 % Epon over night, 3 h Epon. Finally, samples were embedded in 100 % Epon in a non-oriented (isotropic) way and Epon was polymerised for 48 h at 60 °C. Ultrathin sections were cut on an ultramicrotome (Ultracut E ultramicrotome, Reichert, NY, USA) with a diamond knife (Diatome, Biel, Switzerland) and placed on 50 mesh hexagonal cupper grids (Plano GmbH, Wetzlar, Germany). Specifically, from each block three ultrathin sections (70 nm) were obtained at two depths separated by 20 μm and placed onto two grids. Finally, the grids were contrasted with lead citrate [26]. Micrographs were obtained in a FEI Tecnai G2 Spirit electron microscope (Tecnai G2 Spirit, FEI, Hillsboro, OR, USA) mounted with an Orius SC1000 charged-coupled device camera (Gatan, Pleasanton, GA, USA) and interfaced with the TEM software (TEM User Interface, FEI, Hillsboro, OR, USA). Micrographs were acquired in a uniform random systematic order. For each grid three predetermined positions were marked for further imaging. At each position an area of 222 × 146 μm2 (pixel size of 57.7 nm) was used for automated image capturing by the TEM photomontage software. A random starting point was selected for the first micrograph whereafter further micrographs were taken at fixed x-, y-intervals of 64 and 42 μm, respectively. Nine 15.9 × 10.5 μm2 micrographs (3840 × 2528 pixels) were captured in each area. Consequently, for each biopsy a total of 54 micrographs were acquired (nine micrographs per three positions per two sections per sample). Subsequently, skeletal muscle volume density of mitochondria (MitoVD) was estimated by point counting in combination with Cavalieri’s principle [27] using the Stereo-Investigator software (MBF Bioscience, Williston, ND, USA). For point counting a grid spacing of 1 μm along bot x- and y-axis was applied. Each point was assigned as either intermyofibrilar (IMFVD) or subsarcolemmal (SSVD) mitochondrial volume density, lipid droplet volume density (LDVD), skeletal muscle or “nothing”. SS mitochondria were defined as the mitochondria that were not separated by myofibrils from the sarcolemma.
Histochemistry
Consecutive transverse sections (8 μm) were cut at three depths on a microtome (Leica CM 1850, Leica Biosystems, Wetzlar, Germany) at −22 °C, mounted on glass cover slides (Superfrost Plus, Thermo Fisher Scientific Inc., Rockford, IL, USA), set to air dry and stored at -20 °C until further processing. The serial sections were then fixed in 3 % neutral buffered formalin at RT for 45 min, briefly washed and blocked with 5 % goat serum. Thereafter, the sections were firstly incubated with a primary antibody against myosin heavy chain (MyHC) isoform I (Novocastra Lyophilized Mouse Monoclonal Antibody Myosin Heavy Chain (slow), NCL-MHCs, Leica Biosystems, Wetzlar, Germany) and secondly with goat anti-rabbit IgG secondary antibody conjugated with Alexa Fluor 488 (Thermo Fisher Scientific Inc., Rockford, IL, USA). The sarcolemma of the skeletal muscle fibers were visualised by incubation with a primary antibody against laminin (Novocastra Lyophilized Mouse Monoclonal Antibody Laminin, NCL-LAMININ, Leica Biosystems, Wetzlar, Germany) in combination with goat anti-rabbit IgG secondary antibody, Alexa Fluor 647 conjugate (Thermo Fisher Scientific Inc., Rockford, IL, USA). An automated upright microscope system was used for digitizing the sections (Leica, DM5500 B, Leica Microsystems, Wetzlar, Germany). Skeletal muscle fiber type distribution was determined according to their MyHC-I and MyHC-II isoforms and classified into type I and type II fibers. For all analyses, only fibers fully encircled by adjacent fibers were evaluated using Adobe Photoshop Pro CS6 (Adobe Systems Incorporated, San Jose, CA, USA). Fiber cross-sectional area (CSA) was determined by encircling the boundaries of the muscle cells of at least 50 fibers per fiber type. Only fibers with a circularity higher than 0.7 were considered for analysis (perfect circle = 1.0).
Statistical analysis
All data are presented as mean ± SD in text and figures. The statistical analysis was conducted using the software SPSS Statistics 22.0 (SPSS, Chicago, IL, USA). After verification of normal distribution of the data, two-tailed unpaired samples t-tests were conducted to test the null hypothesis stating no difference between patients with mitochondrial myopathy and healthy controls. For all statistical analyses, a value of P < 0.05 was considered significant. To evaluate the effect sizes of the important findings Cohen’s d were calculated, whereby d < 0.3 is considerd a small effect, d > 0.3 and < 0.8 a medium effect and d > 0.8 a large effect. Spearman’s rank correlation coeffiencts were calculated to determine potential dependences of several variables. The number of patients and controls included in each data set are always indicated and can vary due to inability of cycling above a power output of 0 W (one case) or technical problems with the devices (two cases).
Results
All participants completed the skeletal muscle biopsy procedure and DXA measurement. Except from one patient who was not able to cycle against a certain resistance (25 W), all participants conducted the incremental and the constant-load cycling exercise test. Due to technical reasons, one patient and one control could not perform the isokinetic dynamometry test. There were no group differences for age, body composition, resting blood lactate concentration, tlim and MVC (Table 1).
Exercise performance
Ppeak was 53 % lower (P < 0.05, d = 1.8) in patients with mitochondrial myopathy. In accordance, V̇O2peak was 45 % lower (P < 0.05, d = 1.8) in mitochondrial myopathy patients than in healthy controls, with a broad range in both groups (Table 1). However, RER at the same absolute and relative power output did not differ between groups, suggesting similar metabolic regulations in patients and controls at submaximal work rates. Additionally, tlim in the constant-load test did not differ between groups.
Skeletal muscle mitochondrial volume density and respiratory capacity
Maximal mitochondrial respiration specific to each individual mitochondrial complex (mass-specific respiration, respiration per mg ww) was lower (P < 0.05) in patients with mitochondrial myopathy vs. healthy controls (Fig. 1a). In particular, difference in mass-specific respiratory capacity with mitochondrial myopathy amounted to 50 (PCI, d = 2.3), 46 (PCII, d = 1.8), 45 (P, d = 2.3), 48 (COX, d = 1.8) and 47 % (ETS, d = 2.0). Considering the range of P for example, patients show a markedly lower maximal mitochondrial oxidative capacity (44.10 – 81.94 pmol O2 mg−1 s−1) than controls (86.54 – 127.89 pmol O2 mg−1 s−1; P < 0.01). Additionally, MitoVD was on average 23 % lower (P < 0.05, d = 1.5) in patients, which was related to the lower IMFVD (Table 3, d = 1.4). Moreover, after normalizing mitochondrial respiratory capacity to MitoVD all respiratory states specific to the five mitochondrial complexes were lower (P < 0.05, Fig. 1b) in patients (28 (PCI, d = 1.9), 24 (PCII, d = 1.5), 23 (P, d = 1.8), 28 (COX, d = 1.3) and 24 % (ETS, d = 1.4)), suggesting quantitative and qualitative alterations in mitochondrial function with mitochondrial myopathy. In contrast, LCR did not differ (P = 0.501) between groups (mitochondrial myopathy 0.42 ± 0.07; controls 0.44 ± 0.05).
Fig. 1.
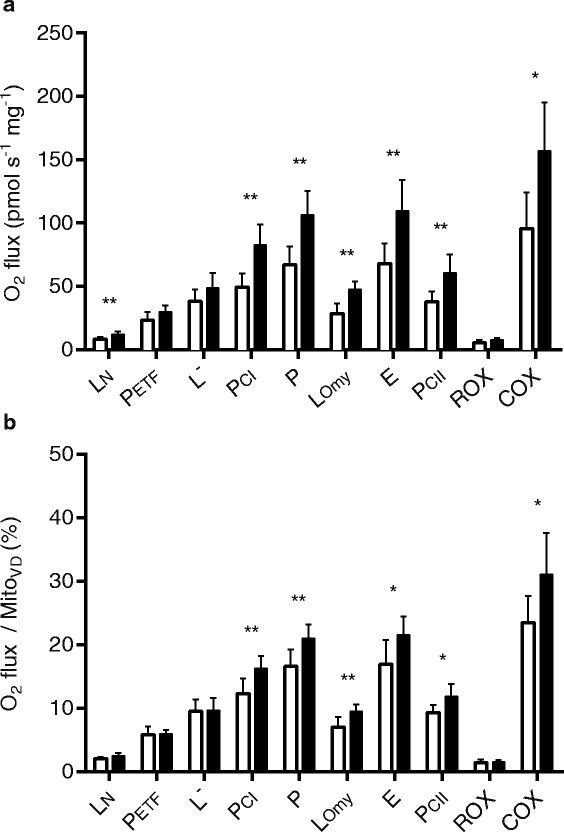
Mass-specific mitochondrial (a) and mitochondrial-specific (b) respiratory capacity (normalised to MitoVD). Respiratory capacities are presented for patients with mitochondrial myopathy in white bars and healthy controls in black bars) LN, leak respiration without adenylates; PETF, fatty acid oxidative capacity; PCI, respiratory capacity of complex I; P, oxidative phosphorylation capacity; LOmy, oligomycin-induced leak respiration; E, electron transport system capacity; PCII, respiratory capacity of complex II; ROX, residual oxygen consumption; COX, respiratory capacity of complex IV. Values are mean ± SD. *P < 0.05; **P < 0.01. nPatients = 6, nControls = 8
Table 3.
Skeletal muscle properties of patients with mitochondrial myopathy (n = 6) and healthy controls (n = 8)
| Patients | Controls | P-value | |
|---|---|---|---|
| Type I fibers (%) | 34.8 ± 3.6 | 55.7 ± 5.9 | < 0.001 |
| Type II fibers (%) | 65.2 ± 3.6 | 44.3 ± 5.9 | < 0.001 |
| CSA total (μm2) | 3,438 ± 915 | 4,698 ± 1,328 | 0.070 |
| CSA type I fibers (μm2) | 3991 ± 1211 | 5234 ± 1566 | 0.133 |
| CSA type II fibers (μm2) | 3,178 ± 852 | 4,359 ± 1,592 | 0.127 |
| MitoVD (%) | 4.0 ± 0.5 | 5.1 ± 0.8 | < 0.05 |
| IMFVD (%) | 3.4 ± 0.4 | 4.1 ± 0.5 | < 0.05 |
| SSVD (%) | 0.6 ± 0.3 | 1.0 ± 0.5 | 0.118 |
| LDVD (%) | 0.4 ± 0.1 | 0.4 ± 0.3 | 0.660 |
Values are represented as means ± SD. CSA cross sectional area; IMF VD intermyofibrilar mitochondrial volume density, LD VD lipid droplet volume density, Mito VD mitochondrial volume density, SS VD subsarcolemmal mitochondrial volume density
Skeletal muscle fiber type distribution
Mitochondrial myopathy patients demonstrated a higher (P < 0.001, d = 4.2) ratio of type II to I fibers and a tendency (P = 0.070, d = 1.1) towards lower CSA, indicating potential skeletal muscle atrophy (Table 3). In accordance with these results, the distribution of type II fibers in patients ranged from 60.4 – 69.0 % in contrast to 32.8 – 48.8 % in healthy controls. However, CSA specific to type I and II fibers did not differ (P = 0.133; P = 0.127) between patients and controls (Table 3).
Correlations
The mutation load in the 6 patients ranged from 12 to 95 %. There was no relationship between percentage mutation and any of the other determined variables. V̇O2peak was positively correlated (P < 0.05) to P (ρ = 0.626), MitoVD (ρ = 0.577) and P normalised to MitoVD (ρ = 0.648). Moreover, P, MitoVD, P/MitoVD as well as lean mass were all related to CSA of type I fibers (ρ = 0.653, ρ = 0.600, ρ = 0.618, ρ = 0.578; P < 0.05).
Discussion
The main finding of the present study is that skeletal muscle mitochondrial function in mitochondrial myopathy patients is lower than in healthy controls due to reduced mitochondrial volume density as well as diminished intrinsic mitochondrial function. Furthermore, patients exhibited a shift in skeletal muscle fiber type towards more type II fibers and tended to an atrophic muscle phenotype. Moreover, V̇O2peak and Ppeak in patients with mitochondrial myopathy were lower than in healthy controls. Mitochondrial volume density and maximal oxidative phosphorylation capacity were positively correlated to V̇O2peak. Thus, mitochondrial myopathy is associated with decreased mitochondrial quantity and quality as well as diminished capacity for whole-body maximal O2 uptake.
This study determined skeletal muscle mitochondrial volume density and respiratory capacity of each single complex I-V in permeabilised fibers from mitochondrial myopathy patients in comparison to age- and gender-matched healthy controls. Thereby, not only MitoVD but also the mass-specific (per mg ww) and mitochondrial-specific (normalised to MitoVD) respiratory capacity of each individual complex were affected by mitochondrial myopathy, indicating an evident impairment of innate mitochondrial function. This lower mitochondrial function may lead to greater disturbances in homeostasis in skeletal muscle, higher reliance on muscle glycogen, increased lactate production and, therewith, lower exercise capacity compared to healthy individuals [28]. Moreover, these alterations could limit the skeletal muscle’s ability to extract and/or oxidize O2 from the blood [14, 28], potentially resulting in an impaired V̇O2peak. Particularly, the lower O2 extraction capacity of the skeletal muscle is likely to lead to a higher systemic O2 delivery relative to O2 utilization, i.e. a hyperkinetic circulation, causing typical symptoms of mitochondrial myopathy such as metabolic acidosis, exertional dyspnea, exercise intolerance, low exercise performance and consecutively reduced quality of life [10–13, 29].
Hence, the present bioenergetic impairments at the cell organelles’ level result in abnormal cardiac and ventilatory responses to increased O2 requirements as in response to physical activity and exercise [29–31], and might impair exercise capacity in patients with mitochondrial myopathy. Noteworthy, exercise capacity in patients with mitochondrial myopathy was lower compared to healthy controls and varied broadly, reflecting the heterogenous clinical manifestation and severity of this disease. In addition, V̇O2peak correlated with measures of mitochondrial quantity and quality, indicating an association with reduced skeletal muscle O2 uptake capacity, which has been indicated by previous studies in our laboratory before [32, 33]. In support, diminished V̇O2peak may be entirely explained by a lower systemic a-vO2 diff [10–13], since peak cardiac output (Q̇peak) has been reported not to differ between mitochondrial myopathy and healthy controls [11]. Although these metabolic adaptations in skeletal muscle are critical for physical activity, especially for submaximal exercise performance, V̇O2peak is limited by O2 transport capacity in healthy human individuals whithout metabolic disturbances [34–36].
To our knowledge, this is also the first study demonstrating skeletal muscle fiber type transformation from type I to type II fibers in adult mitochondrial myopathy patients. This finding contrasts to the histochemically described type I fiber predominance as a compensation for impaired energy production and mitochondrial function in children with mitochondrial myopathy [8]. As type I fibers usually contain a higher proportion of mitochondria than type II fibers [37], an enhanced ratio of type I as compared to type II fibers could increase the number of mitochondria and lead to a potentially greater capacity for energy production [8]. In turn, decreased oxidative capacity could also be compensated by a switch from an oxidative to a more glycolytic phenotype in order to partially restore muscle strength and energy production, yet leading to higher lactic acid production, which is characteristic for this disease [10, 38]. Lactate in turn represents an important fuel for oxidative metabolism and has been reported to rather enhance energy production than inducing fatigue in patients with mitochondrial myopathy [39]. In support of our findings, Venhoff et al. [9] reported a fiber type shift from type I to type II fibers in rodent models of mitochondrial myopathy. The lower MitoVD in patients with mitochondrial myopathy, which was related to lower IMFVD, may thereby reflect the decreased type I to type II fiber ratio and the potentially higher reliance on glycogen metabolism. SSVD in the present study did not differ and only half of the mitochondrial myopathy patients demonstrated ragged-red fibers (RRF, Table 2). Together these findings contrast previous reports of SS mitochondrial proliferations and simulatenously occurring RRF with mitochondrial myopathy [8, 40, 41]. Moreover, mouse models of mitochondrial myopathy demonstrated RRF and increased skeletal muscle mitochondrial volume density, particularly in the SS region, wheras respiratory chain enzyme activites were decreased [42]. Therefore, the authors suggested the higher mitochondrial volume density to partly compensate for the respiratory chain deficiency and the redcued mitochondrial ATP production being not as critical for the pathophysiology of mitochondrial myopathy as previously thought [42]. Accordingly, recent studies displayed altered Ca2+ handling in mitochondrial myopathy [43, 44], stimulating the discussion of progressive muscle weakness rather than energy deficiency as the potential dominating pathomechanism [44]. Collectively, these partially divergent findings may once more reflect the heterogenous character of this disease and the possible detrimental effect of physical inactivity in mitochondrial myopathy patients, which in turn emphasizes the need for further studies also on alternate pathophysiological mechanisms.
Discriminating exercise intolerance caused by defective mitochondria from that of disease-associated inactivity and hence physical deconditioning remains challenging. Exercise intolerance inevitably results in reduced levels of habitual physical activity and causes physical deconditioning, which in turn leads to a vicious cycle of further deconditioning and progressive exercise intolerance [45]. Similar total lean mass and lean mass of the lower limbs in mitochondrial myopathy patients and healthy controls may suggest that the diminished mitochondrial volume density, as well as the reduced function and exercise performance are possibly associated with the disease itself rather than with inactivity alone. Moreover, MVC and tlim did not significantly differ between patients with mitochondrial myopathy and healthy controls, which contradicts different levels of physical activity. In support, mice with mitochondrial myopathy showed a decrease in ATP levels even with exercise, which was less than in sedentary diseased mice but dropped to 30 % of wild-type after 10 month [46]. Nonetheless, in spite of matching patients and controls according to age, gender and, as far as possible, to reported physical activity levels, the lower V̇O2peak and Ppeak in patients with mitochondrial myopathy could be due to sustained physical inactivity associated with the disease-related exercise intolerance. Thereby, the present lower MitoVD and mitochondrial respiratory capacity in patients with mitochondrial myopathy could indeed also simply be a reflection of this vicious cycle. Disuse and physical inactivity, respectively, have been reported to lead to decreases in mitochondrial volume density and hence mitochondrial function [47, 48]. In line herewith is the lower IMFVD with mitochondrial myopathy, as IMF mitochondria represent a specialization towards energy production for contractile activity [49]. Reduced contractile activity due to physical inactivity in patients with mitochondrial myopathy could have resulted in lower IMFVD and secondary impaired mitochondrial function. In support, muscle homogenates of mice with mitochondrial myopathy due to a COX deficiency exhibited increased mitochondrial mass, oxidative capacity and exercise performance after an endurance exercise training intervention in comparison to sedentary diseased mice [46]. However, although the enhanced physical activity led to a preservation of oxidative capacity of 50 – 60 % of wild-type levels in comparison to the drop to 10 – 40 % in sedentary diseased mice in 3 month duration, oxidative capacity still remained reduced compared to wild-type controls. Moreover, mitochondrial COX activity was not affected by the exercise intervention [46]. The oxidative capacity in muscle homogenates, which was sustained only partially, together with the persistent COX defect at the mitochondrial level in exercised diseased mice indicates increased mitochondrial quantity but not quality with exercise [46]. Therefore, lower MitoVD but not impaired intrinsic mitochondrial function could be explained by physical inactivity secondarily to exercise intolerance. Congruently, two weeks of physical inactivity induced by one leg immobilization led to reduced mitochondrial respiratory capacity due to changes in mitochondrial content but not intrinsic mitochondrial function [47]. Moreover, considering the shift towards more type II fibers and the trend to general fiber atrophy, physical inactivity should be considered as a potential cause. However, muscle disuse affects mainly type I fiber diameter [50, 51], which did not differ between patients and controls. This is further supported by a rat model of mitochondrial myopathy harborating a predominate type II fiber atrophy [9]. Conclusively, mechanisms related to mitochondrial dysfunction and the disease-causing mutations itself could have induced the present pathologic findings. However, physical inactivity could secondarily have led to further aggraviation of decreased exercise capacity and mitochondrial function. Therefore, the present study cannot definitively clarify whether the findings are related to the disease per se or in addition to inacitivity. Another limitation of the present study is the small sample size, which hovewer, according to the preceding power calculation and the significant differences of the main results was sufficient. The small number of participants can be explained by the large expected contrasts in MitoVD and mitochondrial respiratory capacity between patients with mitochondrial myopathy and healthy controls and the small coefficients of variation for the TEM and high-resolution respirometry. The present findings in this small cohort with a rather inhomogenous degree of heteroplasmy are further strengthend by the high effect sizes.
From a clinical point of view, the diagnosis of mitochondrial cytopathy or mitochondrial myopathy, respectively, remains challenging and requires the combined application of multiple methodologies [52]. Blood samples may exhibit increased resting lactate concentrations and elevated CK levels. However, lactic acidosis is often not present and CK levels may be normal or only mildly increased. Functional muscle testing with the subanaerobic threshold exercise test (SATET) is a very specific measure for mitochondrial pathology [53] but many mitochondrial myopathy patients do not exhibit pathological values. Morphological, biochemical and molecular studies in tissue samples allow a diagnosis by the presence of suggestive histopathological findings such as RRF and ragged-blue fibers, COX depleted fibers, variously formed mitochondria, abnormal cristae, crystalloid inclusions and the determination of disease-causing mutations in the mitochondrial and/or nuclear DNA [41, 52]. As demonstrated in two of our six patients, these typical histological signs of mitochondrial myopathy can be lacking despite presence of a mitochondrial disease [52]. Molecular genetic analysis of mitochondrial DNA in tissues may demonstrate heteroplasmic deletions and point mutations. In addition, nuclear DNA mutations need to be considered. For heteroplasmic mitochondrial DNA mutations, the degree of heteroplasmy needs to be quantified, but the pathological threshold is poorly defined, particularly in mitochondrial cytopathy patients at advanaced age. This heteroplasmy, a canonical criteria for the pathogenity, is not always given as also pathogenic homoplasmic mutations have been described [3, 54]. This aspect also causes some limitations regarding the results of the present study and their generalization for all mitochondrial myopathy patients. As we included only patients with mitochondrial DNA single deletions, the present results are only true for this spectrum of phenotype and further investigations considering patients with mitochondrial DNA point mutations and nuclear DNA mutations affecting mitochondria are needed to verify these findings.
To provide proper patient management, treatment and genetic counseling, a clear diagnosis of mitochondrial cytopathies is necessary. Our results demonstrate that high-resolution respirometry measurements could represent a promising additional tool for the diagnosis of mitochondrial cytopathies. In vitro measurements as the present high-resolution respirometry allow the estimation of the functionality of specific steps implicated in mitochondrial metabolism by the use of various substrates, uncouplers and inhibitors. Such polarographic measurements of O2 consumption provide more essential information on innate mitochondrial function, as evaluations not only of isolated mitochondria but also of permeabilised cells are possible [55]. In contrast to the isolation procedure, permeabilization of muscle fibers preserves mitochondrial morphology and integrity and allows the examination of the intact mitochondrial network [22]. However, the definition of normal ranges of complex activites in skeletal muscle remains disputed and difficult. Since a skeletal muscle biopsy is needed for this approach, this minimal-invasive procedure represents a disadvantage for the patients. Conclusively, further studies to eliminate the variations and to validate high-resolution respirometry as a diagnostic measure are needed.
Conclusion
Mitochondrial myopathy led to diminished skeletal muscle mitochondrial volume density and respiratory capacity of each individual complex and was related to reduced maximal exercise capacity. Lower mitochondrial volume density was attended by a shift in skeletal muscle phenotype from type I to type II fibers, reflecting a possible compensation for the lower mitochondrial respiratory as well as exercise capacity. Hence, the affected mitochondrial quantity and quality could be responsible for the impaired exercise capacity and could further aggravate exercise intolerance. However, it cannot be excluded that changes observed in patients are simply due to physical inactivity as a cause of the disease-related exercise intolerance. Nonetheless, the clearly reduced mitochondrial respiratory capacity in patients with mitochondrial myopathy compared to age- and gender-matched healthy controls proposes high-resolution respirometry measurements in permeabilised skeletal muscle fibers to be a promising additional diagnostic tool.
Abbreviations
BMC, bone mineral content; CIS-fatigue, checklist individual strength; CK, phosphocreatine kinase; COX, complex IV activity; CSA, cross-sectional area; DXA, dual-energy X-ray absorptiometry; E, electron transport system capacity; ETS, electron transport system; FCCP, carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone; HbA1c, glycated hemoglobin; IMFVD, intermyofibrilar mitochondrial volume density; LCR, leak control ratio; LDVD, lipid droplet volume density; LN, leak respiration in absence of adenylates; LOmy, oligomycin-induced leak respiration; MitoVD, volume density of mitochondria; MVC, maximal voluntary contraction torque; P, maximal oxidative phosphorylation capacity; PCI, complex I activity; PCII, complex II activity; PETF, fatty acid oxidative capacity; Ppeak, work capacity; proBNP, precursorprotein brain natriuretic peptide; Q̇peak, peak cardiac output; RER, respiratory exchange ratio; ROX, residual O2 consumption; rpm, revolutions per minute; RT, room temperature; SSVD, subsarcolemmal mitochondrial volume density; TEM, transmission electron microscopy; tlim, time to volitional exhaustion; TSH, thyroid stimulating hormone; VAS, visual analogue scale; V̇O2peak, peak oxygen uptake; ww, wet weight
Acknowledgments
We are grateful to all participants for their contribution to this study. Moreover, we thank Prof. Dr. med. Jean-Marc Burgunder (Swiss Huntington Disease Centre, Department of Clinical Research, University of Bern, Switzerland) for the assistance with patient recruitment.
Funding
This work was supported by grants from the Zurich Center for Integrative Human Physiology (ZIHP) and Neuromuscular Research Association Basel (NeRAB).
Availability of data and material
All data is stored in separate folder of the database of the Department of Neurology, University Hospital Zurich.
Authors’ contributions
All authors substantially contributed to the conception and design, or the acquisition, analysis and interpretation of the present data. Moreover, all authors were involved in drafting or revising the manuscript and gave their final approval of the version to be published.
Competing interests
The authors declare that they have no competing interests.
Consent fo publication
Not applicable.
Ethics approval and consent to participate
The present experimental protocol was approved by the Cantonal Ethics Committee of Zurich (KEK 2012-0363) and was conducted in accordance with the Declarations of Helsinki. Participants were fully informed about the purposes, benefits and risks associated with this study and gave their written informed consent prior to the initiation of the experiments.
Contributor Information
Saskia Maria Gehrig, Email: saskia.gehrig@uzh.ch.
Violeta Mihaylova, Email: violeta.mihayolov@usz.ch.
Sebastian Frese, frese.sebastian@googlemail.com.
Sandro Manuel Mueller, Email: sandro.mueller@usz.ch.
Maria Ligon-Auer, Email: auer.m@gmx.net.
Christina M. Spengler, Email: christina.spengler@hest.ethz.ch
Jens A. Petersen, Email: jens.petersen@usz.ch
Carsten Lundby, Email: carsten.lundby@uzh.ch.
Hans H. Jung, Phone: +41 44 255 55 45, Email: hans.jung@usz.ch
References
- 1.Di Mauro S. Pathogenesis and treatment of mitochondrial myopathies: recent advances. Acta Myol. 2010;29:333–8. [PMC free article] [PubMed] [Google Scholar]
- 2.Hassani A, Horvath R, Chinnery PF. Mitochondrial myopathies: developments in treatment. Curr Opin Neurol. 2010;23:459–65. doi: 10.1097/WCO.0b013e32833d1096. [DOI] [PubMed] [Google Scholar]
- 3.Di Mauro S. Mitochondrial medicine. Biochim Biophys Acta. 1659;2004:107–14. doi: 10.1016/j.bbabio.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Tuppen HA, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations and human disease. Biochim Biophys Acta. 1797;2010:113–28. doi: 10.1016/j.bbabio.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Larsson NG, Oldfors A. Mitochondrial myopathies. Acta Physiol Scand. 2001;171:385–93. doi: 10.1046/j.1365-201x.2001.00842.x. [DOI] [PubMed] [Google Scholar]
- 6.Gallanti A, Prelle A, Chianese L, Barbieri S, Jann S, Schiaffino S, Comini A, Scarpini E, Pellegrini G, Moggio M, et al. Congenital myopathy with type 2A muscle fiber uniformity and smallness. Neuropediatrics. 1992;23:10–3. doi: 10.1055/s-2008-1071303. [DOI] [PubMed] [Google Scholar]
- 7.Muranaka H, Osari S, Fujita H, Kimura Y, Goto A, Imoto C, Nonaka I. Congenital familial myopathy with type 2 fiber hypoplasia and type 1 fiber predominance. Brain Dev. 1997;19:362–5. doi: 10.1016/S0387-7604(97)00036-3. [DOI] [PubMed] [Google Scholar]
- 8.Enns GM, Hoppel CL, DeArmond SJ, Schelley S, Bass N, Weisiger K, Horoupian D, Packman S. Relationship of primary mitochondrial respiratory chain dysfunction to fiber type abnormalities in skeletal muscle. Clin Genet. 2005;68:337–48. doi: 10.1111/j.1399-0004.2005.00499.x. [DOI] [PubMed] [Google Scholar]
- 9.Venhoff N, Lebrecht D, Pfeifer D, Venhoff AC, Bisse E, Kirschner J, Walker UA. Muscle-fiber transdifferentiation in an experimental model of respiratory chain myopathy. Arthritis Res Ther. 2012;14:R233. doi: 10.1186/ar4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vissing J, Galbo H, Haller RG. Exercise fuel mobilization in mitochondrial myopathy: a metabolic dilemma. Ann Neurol. 1996;40:655–62. doi: 10.1002/ana.410400416. [DOI] [PubMed] [Google Scholar]
- 11.Taivassalo T, Jensen TD, Kennaway N, DiMauro S, Vissing J, Haller RG. The spectrum of exercise tolerance in mitochondrial myopathies: a study of 40 patients. Brain. 2003;126:413–23. doi: 10.1093/brain/awg028. [DOI] [PubMed] [Google Scholar]
- 12.Taivassalo T, Abbott A, Wyrick P, Haller RG. Venous oxygen levels during aerobic forearm exercise: An index of impaired oxidative metabolism in mitochondrial myopathy. Ann Neurol. 2002;51:38–44. doi: 10.1002/ana.10027. [DOI] [PubMed] [Google Scholar]
- 13.Linderholm H, Müller R, Ringqvist T, Sornas R. Hereditary abnormal muscle metabolism with hyperkinetic circulation during exercise. Acta Med Scand. 1969;185:153–66. doi: 10.1111/j.0954-6820.1969.tb07314.x. [DOI] [PubMed] [Google Scholar]
- 14.Hoppeler H, Hudlicka O, Uhlmann E. Relationship between mitochondria and oxygen consumption in isolated cat muscles. J Physiol. 1987;385:661–75. doi: 10.1113/jphysiol.1987.sp016513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taivassalo T, Haller RG. Exercise and training in mitochondrial myopathies. Med Sci Sports Exerc. 2005;37:2094–101. doi: 10.1249/01.mss.0000177446.97671.2a. [DOI] [PubMed] [Google Scholar]
- 16.Taivassalo T, Gardner JL, Taylor RW, Schaefer AM, Newman J, Barron MJ, Haller RG, Turnbull DM. Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain. 2006;129:3391–401. doi: 10.1093/brain/awl282. [DOI] [PubMed] [Google Scholar]
- 17.Iqbal S, Ostojic O, Singh K, Joseph AM, Hood DA. Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle Nerve. 2013;48:963–70. doi: 10.1002/mus.23838. [DOI] [PubMed] [Google Scholar]
- 18.Morgan-Hughes JA, Schapira AH, Cooper JM, Holt IJ, Harding AE, Clark JB. The molecular pathology of respiratory-chain dysfunction in human mitochondrial myopathies. Biochim Biophys Acta. 1990;1018:217–22. doi: 10.1016/0005-2728(90)90252-Y. [DOI] [PubMed] [Google Scholar]
- 19.Rustin P, Chretien D, Bourgeron T, Gerard B, Rotig A, Saudubray JM, Munnich A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta. 1994;228:35–51. doi: 10.1016/0009-8981(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 20.Holt IJ, Harding AE, Cooper JM, Schapira AH, Toscano A, Clark JB, Morgan-Hughes JA. Mitochondrial myopathies: clinical and biochemical features of 30 patients with major deletions of muscle mitochondrial DNA. Ann Neurol. 1989;26:699–708. doi: 10.1002/ana.410260603. [DOI] [PubMed] [Google Scholar]
- 21.Tarnopolsky MA, Raha S. Mitochondrial myopathies: diagnosis, exercise intolerance, and treatment options. Med Sci Sports Exerc. 2005;37:2086–93. doi: 10.1249/01.mss.0000177341.89478.06. [DOI] [PubMed] [Google Scholar]
- 22.Picard M, Taivassalo T, Gouspillou G, Hepple RT. Mitochondria: isolation, structure and function. J Physiol. 2011;589:4413–21. doi: 10.1113/jphysiol.2011.212712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moggio M, Colombo I, Peverelli L, Villa L, Xhani R, Testolin S, Di Mauro S, Sciacco M. Mitochondrial disease heterogeneity: a prognostic challenge. Acta Myol. 2014;33:86–93. [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobs RA, Siebenmann C, Hug M, Toigo M, Meinild AK, Lundby C. Twenty-eight days at 3454-m altitude diminishes respiratory capacity but enhances efficiency in human skeletal muscle mitochondria. FASEB J. 2012;26:5192–200. doi: 10.1096/fj.12-218206. [DOI] [PubMed] [Google Scholar]
- 25.Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int J Biochem Cell Biol. 2009;41:1837–45. doi: 10.1016/j.biocel.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 26.Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol. 1963;17:208–12. doi: 10.1083/jcb.17.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.West MJ. Estimating volume in biological structures. Cold Spring Harb Protoc. 2012;2012:1129–39. doi: 10.1101/pdb.top071787. [DOI] [PubMed] [Google Scholar]
- 28.Holloszy JO, Coyle EF. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J Appl Physiol Respir Environ Exerc Physiol. 1984;56:831–8. doi: 10.1152/jappl.1984.56.4.831. [DOI] [PubMed] [Google Scholar]
- 29.Heinicke K, Taivassalo T, Wyrick P, Wood H, Babb TG, Haller RG. Exertional dyspnea in mitochondrial myopathy: clinical features and physiological mechanisms. Am J Physiol Regul Integr Comp Physiol. 2011;301:R873–84. doi: 10.1152/ajpregu.00001.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dandurand RJ, Matthews PM, Arnold DL, Eidelman DH. Mitochondrial disease. Pulmonary function, exercise performance, and blood lactate levels. Chest. 1995;108:182–9. doi: 10.1378/chest.108.1.182. [DOI] [PubMed] [Google Scholar]
- 31.Flaherty KR, Wald J, Weisman IM, Zeballos RJ, Schork MA, Blaivas M, Rubenfire M, Martinez FJ. Unexplained exertional limitation: characterization of patients with a mitochondrial myopathy. Am J Respir Crit Care Med. 2001;164:425–32. doi: 10.1164/ajrccm.164.3.2005110. [DOI] [PubMed] [Google Scholar]
- 32.Jacobs RA, Flück D, Bonne TC, Burgi S, Christensen PM, Toigo M, Lundby C. Improvements in exercise performance with high-intensity interval training coincide with an increase in skeletal muscle mitochondrial content and function. J Appl Physiol (1985) 2013;115:785–93. doi: 10.1152/japplphysiol.00445.2013. [DOI] [PubMed] [Google Scholar]
- 33.Jacobs RA, Lundby C. Mitochondria express enhanced quality as well as quantity in association with aerobic fitness across recreationally active individuals up to elite athletes. J Appl Physiol (1985) 2013;114:344–50. doi: 10.1152/japplphysiol.01081.2012. [DOI] [PubMed] [Google Scholar]
- 34.Montero D, Cathomen A, Jacobs RA, Fluck D, de Leur J, Keiser S, Bonne T, Kirk N, Lundby AK, Lundby C. Haematological rather than skeletal muscle adaptations contribute to the increase in peak oxygen uptake induced by moderate endurance training. J Physiol. 2015;593:4677–88. doi: 10.1113/JP270250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saltin B, Strange S. Maximal oxygen uptake: “old” and “new” arguments for a cardiovascular limitation. Med Sci Sports Exerc. 1992;24:30–7. doi: 10.1249/00005768-199201000-00007. [DOI] [PubMed] [Google Scholar]
- 36.Bassett DR, Jr, Howley ET. Limiting factors for maximum oxygen uptake and determinants of endurance performance. Med Sci Sports Exerc. 2000;32:70–84. doi: 10.1097/00005768-200001000-00012. [DOI] [PubMed] [Google Scholar]
- 37.Gouspillou G, Sgarioto N, Norris B, Barbat-Artigas S, Aubertin-Leheudre M, Morais JA, Burelle Y, Taivassalo T, Hepple RT. The relationship between muscle fiber type-specific PGC-1alpha content and mitochondrial content varies between rodent models and humans. PLoS ONE. 2014;9:e103044. doi: 10.1371/journal.pone.0103044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeppesen TD, Schwartz M, Olsen DB, Vissing J. Oxidative capacity correlates with muscle mutation load in mitochondrial myopathy. Ann Neurol. 2003;54:86–92. doi: 10.1002/ana.10594. [DOI] [PubMed] [Google Scholar]
- 39.Jeppesen TD, Orngreen MC, Van Hall G, Vissing J. Lactate metabolism during exercise in patients with mitochondrial myopathy. Neuromuscul Disord. 2013;23:629–36. doi: 10.1016/j.nmd.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 40.Rollins S, Prayson RA, McMahon JT, Cohen BH. Diagnostic yield muscle biopsy in patients with clinical evidence of mitochondrial cytopathy. Am J Clin Pathol. 2001;116:326–30. doi: 10.1309/WATB-W4QV-NA53-B9MY. [DOI] [PubMed] [Google Scholar]
- 41.Di Mauro S, Bonilla E, Zeviani M, Nakagawa M, DeVivo DC. Mitochondrial myopathies. Ann Neurol. 1985;17:521–38. doi: 10.1002/ana.410170602. [DOI] [PubMed] [Google Scholar]
- 42.Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson NG. Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci U S A. 2002;99:15066–71. doi: 10.1073/pnas.232591499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aydin J, Andersson DC, Hanninen SL, Wredenberg A, Tavi P, Park CB, Larsson NG, Bruton JD, Westerblad H. Increased mitochondrial Ca2+ and decreased sarcoplasmic reticulum Ca2+ in mitochondrial myopathy. Hum Mol Genet. 2009;18:278–88. doi: 10.1093/hmg/ddn355. [DOI] [PubMed] [Google Scholar]
- 44.Gineste C, Hernandez A, Ivarsson N, Cheng AJ, Naess K, Wibom R, Lesko N, Bruhn H, Wedell A, Freyer C, Zhang SJ, Carlström M, Lanner JT1, Andersson DC, Bruton JD, Wredenberg A, Westerblad H. Cyclophilin D, a target for counteracting skeletal muscle dysfunction in mitochondrial myopathy. Hum Mol Genet. 2015;24:6580-87. [DOI] [PMC free article] [PubMed]
- 45.Taivassalo T, Haller RG. Implications of exercise training in mtDNA defects--use it or lose it? Biochim Biophys Acta. 1659;2004:221–31. doi: 10.1016/j.bbabio.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 46.Wenz T, Diaz F, Hernandez D, Moraes CT. Endurance exercise is protective for mice with mitochondrial myopathy. J Appl Physiol (1985) 2009;106:1712–9. doi: 10.1152/japplphysiol.91571.2008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Gram M, Vigelso A, Yokota T, Hansen CN, Helge JW, Hey-Mogensen M, Dela F. Two weeks of one-leg immobilization decreases skeletal muscle respiratory capacity equally in young and elderly men. Exp Gerontol. 2014;58:269–78. doi: 10.1016/j.exger.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 48.Tryon LD, Vainshtein A, Memme JM, Crilly MJ, Hood DA. Recent advances in mitochondrial turnover during chronic muscle disuse. Integr Med Res. 2014;3:161–71. doi: 10.1016/j.imr.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferreira R, Vitorino R, Alves RM, Appell HJ, Powers SK, Duarte JA, Amado F. Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics. 2010;10:3142–54. doi: 10.1002/pmic.201000173. [DOI] [PubMed] [Google Scholar]
- 50.Booth FW, Gollnick PD. Effects of disuse on the structure and function of skeletal muscle. Med Sci Sports Exerc. 1983;15:415–20. doi: 10.1249/00005768-198315050-00013. [DOI] [PubMed] [Google Scholar]
- 51.Zhong H, Roy RR, Siengthai B, Edgerton VR. Effects of inactivity on fiber size and myonuclear number in rat soleus muscle. J Appl Physiol (1985) 2005;99:1494–9. doi: 10.1152/japplphysiol.00394.2005. [DOI] [PubMed] [Google Scholar]
- 52.Milone M, Wong LJ. Diagnosis of mitochondrial myopathies. Mol Genet Metab. 2013;110:35–41. doi: 10.1016/j.ymgme.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 53.Nashef L, Lane RJ. Screening for mitochondrial cytopathies: the sub-anaerobic threshold exercise test (SATET) J Neurol Neurosurg Psychiatry. 1989;52:1090–4. doi: 10.1136/jnnp.52.9.1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, 2nd, Nikoskelainen EK. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–30. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 55.Barrientos A, Fontanesi F, Diaz F. Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr Protoc Hum Genet. 2009;Chapter 19:Unit19 13. doi: 10.1002/0471142905.hg1903s63. [DOI] [PMC free article] [PubMed] [Google Scholar]