

abstract
Synthetic libraries are a major source of human-like antibody (Ab) drug leads. To assess the similarity between natural Abs and the products of these libraries, we compared large sets of natural and synthetic Abs using “CDRs Analyzer,” a tool we introduce for structural analysis of Ab-antigen (Ag) interactions. Natural Abs, we found, recognize their Ags by combining multiple complementarity-determining regions (CDRs) to create an integrated interface. Synthetic Abs, however, rely dominantly, sometimes even exclusively on CDRH3. The increased contribution of CDRH3 to Ag binding in synthetic Abs comes with a substantial decrease in the involvement of CDRH2 and CDRH1. Furthermore, in natural Abs CDRs specialize in specific types of non-covalent interactions with the Ag. CDRH1 accounts for a significant portion of the cation-pi interactions; CDRH2 is the major source of salt-bridges and CDRH3 accounts for most hydrogen bonds. In synthetic Abs this specialization is lost, and CDRH3 becomes the main sources of all types of contacts. The reliance of synthetic Abs on CDRH3 reduces the complexity of their interaction with the Ag: More Ag residues contact only one CDR and fewer contact 3 CDRs or more. We suggest that the focus of engineering attempts on CDRH3 results in libraries enriched with variants that are not natural-like. This may affect not only Ag binding, but also Ab expression, stability and selectivity. Our findings can help guide library design, creating libraries that can bind more epitopes and Abs that better mimic the natural antigenic interactions.
KEYWORDS: Antigen binding site, antibody–antigen interactions, human-like antibodies, libraries, synthetic
Abbreviations
- Ab
antibody
- mAb
monoclonal antibody
- Ag
antigen
- CDR
complementarity-determining region
- Fab
fragment antigen binding
- scFv
single chain Fv
- VH
heavy chain variable domain
- VL
light chain variable domain
- Ig
immunoglobulin
- PDB
Protein Data Bank
- SHM
somatic hypermutation
Introduction
Forty years after the introduction of the hybridoma technology in 19751 and 30 y after the first therapeutic monoclonal antibody (mAb), muromonab-CD3, was approved by the US Food and Drug Administration,2 over 30 Ab-based drugs are marketed and hundreds more are in clinical trials.3 Attempts to engineer Abs are inspired by the power of in vivo Ab generation by B cells, which is based on gene rearrangement that could potentially produce 1011 different Abs.4 Somatic hypermutations (SHMs) on selected sequences increases this diversity further. While all Abs share the same immunoglobulin (Ig) fold and use the same homologous patch for antigen (Ag) recognition,4 they recognize very different epitopes, covering virtually any patch on the Ag surface,5-7 for a variety of Ag types (heptane, peptides or proteins).8
Initially, therapeutic mAbs against a specific Ag were obtained by immunizing an animal. However, this technique fails for many proteins such as toxic or self Ags. In addition, animal-derived Abs require humanization to reduce their immunogenicity, which often hampers Ag binding. Molecular display methods cope with these challenges by using in-vitro display-and-selection systems, such as phages, to isolate binders from a library of Igs.9 These libraries may be based on Ab sequences from an immunized individual10,11 or from a naïve one.12,13
An improvement for the display technologies, modified synthetic libraries offer diversity greater than that of natural repertoire (up to 1014 clones).14 This arguably increases the chances of identifying high affinity binders.14,15 It has been shown that introducing non-random diversity into these libraries can yield synthetic Abs with improved biophysical properties such as improved expression or stability.16-19
However, although these synthetic, man-made Abs may be considered fully human by their V-D-J sequence, they are arguably different than natural Abs. In every library only a small fraction of the sequences can become effective, human-like, Abs. The rest may not fold or not express well, tend to aggregate, to be highly cross-reactive or to bind the target in a non-canonical way. Often, the Abs that emerge from these libraries are found to be immunogenic or cross-reactive with self-epitopes.20-23 In addition, many synthetic libraries are based on a single,24-26 or limited set of V region frameworks17,27 and many introduce diversification only to CDRH3.28-30 Similarly, some libraries limit the introduced diversity to only 2–4 amino acids per position.31,32
A critical question, therefore, in designing synthetic libraries is to what extent the resulting Abs are similar to natural Abs in the way they recognize and bind the Ag. Indeed, good therapeutic biomolecules do not have to mimic natural Abs. However, it is often assumed that libraries that better mimic natural Abs and natural diversity are more likely to yield better binders with better profile. Some novel approaches for library design attempt to introduce diversity that will better imitate natural diversity while also yielding Abs with improved biophysical properties. For example, the human combinatorial antibody library (HuCAL) was created to represent the most frequently used germline families and was optimized to obtain high expression and low aggregation in E. coli. The CDRs cassettes were designed to mimic the length and amino acid composition of naturally occurring Abs.17,27 Sidhu et al.19 used a single stable framework scaffold to introduce diversity to the heavy chain, based on the observed propensities of amino acids in CDRs of natural Abs. Another strategy was to amplify only the CDR sequences from naïve B cells and randomly combine these CDRs into a selected Ab framework that can be highly expressed in bacterial system.33 Further understanding of key properties of naturally existing Abs will help Ab engineering technologies to obtain more promising therapeutic Abs candidates.
Here, we compare synthetic Abs to natural Abs to assess to what extent synthetic Abs indeed mimic natural ones. This comparison allowed us to review and revise common assumptions about Ab-Ag interaction. We employ a novel computational tool we developed, “CDRs analyzer” to explore biophysical characteristics of Abs. In this analysis, natural Abs are Abs that originated from hybridoma or from immunized or naïve libraries, and synthetic Abs are Abs that were selected from a synthetic library (i.e., a library that is not naïve or immunized). We found that synthetic Abs rely on CDRH3 significantly more than natural Abs. The binding contribution of CDRH1 and CDRH2 of synthetic Abs is smaller than their contribution in natural Abs. When analyzing the binding mode, we found that epitopes of natural Abs contain many epitope residues that contact multiple CDRs, while epitopes of synthetic Abs have more residues that contact only one CDR. These results show that the current way in which synthetic libraries are designed often yields Abs that do not mimic the way in which natural Abs bind their Ags. Our analysis suggests a set of considerations for library design that will take better advantage of the binding possibilities offered by the structure of the Ab. We discuss how this can yield libraries with more effective binders and with greater diversity of paratopes.
Results
Data sets of natural and synthetic Abs
Analyzing the Protein Data Bank (PDB)34 in search of a non-redundant set of natural or synthetic Abs (Methods) yielded a total of 137 Ab-Ag complexes. Of these, 101 are natural (Table S1) and 36 are synthetic (Table S2).
“CDRs analyzer” – a computational framework for exploring Ab-Ag interactions
The analysis utilized “CDRs Analyzer,” a computational tool we introduce for analyzing Ab-Ag interfaces. It is designed to assist Ab engineering by providing quantitative assessment of the biophysical properties of each residue and each CDR in the paratope. “CDRs Analyzer” takes as input a 3D structure of an Ab-Ag complex in a PDB format and the chain IDs of the Ab and Ag chains to be analyzed. The server provides output both at the residue and at the CDRs levels. The output includes a list of H-bonds (calculated by HBPLUS35), salt-bridges, pi-pi and cation-pi interactions, and a list of contacting residues (see Methods). Additionally, “CDRs Analyzer” calculates, for each CDR, 4 parameters to evaluate its contribution to Ag binding: (1) “Contacting residues” is the sum of the number of residues in the CDR that are in contact with the Ag and the number of residues in the Ag that are in contact with the CDR; (2) “Specific interactions” is the number of salt-bridges, pi-pi and cation-pi interaction and the number of possible H-bonds between the CDR residues and the Ag; (3) “Calculated ΔΔG” is the predicted effect on binding of mutating each CDR residue to ALA calculated using FoldX; 36 and (4) “delta relative accessible surface area (ΔRSA)” is the sum of the changes in the relative solvent accessibility of each CDR residue upon dissociation of the Ab-Ag complex calculated using DSSP.37 These 4 binding parameters were combined to give a score that assesses the contribution to Ag binding of a given CDR. This score varies from 4 (no contribution to Ag binding) to 16 (highest contribution to Ag binding; see Methods). It is a unified score that gives an equal weight for the 4 binding parameters. Ideally, as more structural data become available, the weight that each parameter should have in the final score can be explored and optimized. The binding contribution score is a combined measurement of the Ag binding portion of a given CDR relative to other CDRs of the Ab.
Additionally, “CDRs analyzer” provides the potential of a CDR to bind the Ag as peptide, based on a computational approach that was described previously.38 “CDRs Analyzer” is available online in http://www.ofranlab.org/CDRs_Analyzer.
Synthetic Abs rely heavily on CDRH3 at the expense of CDRH2 and CDRH1
CDRH3, which encompasses the V-D-J recombination site, is the most diverse component of natural Abs. As shown in Table 1, in natural Abs CDRH3 has, on average, higher values than any other CDR, for all of the 4 parameters that were assessed. Fig. 1, shows how this is translated into the binding contribution score, which is, overall, the highest for CDRH3. Notwithstanding, in natural Abs CDRH2 is a very close second, and has only slightly lower binding contribution than CDRH3. Overall, CDRH3 has an average binding contribution score of 12.69 (±0 .35), while CDRH2 has a score of 11.04 (±0 .39) (Fig. 1). CDRH1 then follows with 8.66 (±0 .36). However, the binding contribution of these 3 heavy chain CDRs is remarkably different in synthetic Ab-Ag complexes. The contribution of CDRH3 increases from 12.69 (±0 .35) to 14.31 (±0 .44), while the contribution of CDRH1 and CDRH2 drops from 8.66 (±0 .36) to 7.19 (±0 .56) and from 11.04 (±0 .39) to 9.83 (±0 .73), respectively. In addition, in the synthetic Abs, there is a small decrease in the binding contribution score of the 3 CDRs on the light chain in comparison to their contribution in natural Abs.
Table 1.
Binding parameters of CDRs from natural and synthetic Abs- Average values (standard error) of the 4 binding parameters calculated by “CDRs Analyzer” for all CDRs in natural and synthetic Abs.
| CDR | Abs | Contacting residues | Specific interactions | ΔΔG | ΔRSA |
|---|---|---|---|---|---|
| H1 | natural | 9.39(±0 .45) | 1.64(±0 .16) | 2.28(±0 .25) | 0.81(±0 .05) |
| synthetic | 9.17(±1 .05) | 0.94(±0 .18) | 2.19(±0 .41) | 0.84(±0 .12) | |
| H2 | natural | 11.76(±0 .54) | 2.5(±0 .21) | 3.7(±0 .29) | 1.1(±0 .06) |
| synthetic | 11.94(±1 .08) | 1.97(±0 .33) | 3.34(±0 .43) | 1.25(±0 .13) | |
| H3 | natural | 14.25(±0 .48) | 2.79(±0 .22) | 5.77(±0 .36) | 1.31(±0 .06) |
| synthetic | 18.97(±1 .23) | 4.39(±0 .56) | 8.2(±0 .68) | 1.83(±0 .16) | |
| L1 | natural | 6.62(±0 .43) | 0.93(±0 .13) | 1.78(±0 .19) | 0.59(±0 .05) |
| synthetic | 6.03(±0 .78) | 0.92(±0 .28) | 1.4(±0 .34) | 0.59(±0 .08) | |
| L2 | natural | 4.58(±0 .51) | 0.64(±0 .11) | 1.24(±0 .17) | 0.41(±0 .05) |
| synthetic | 4.75(±0 .85) | 0.83(±0 .27) | 1.3(±0 .33) | 0.45(±0 .09) | |
| L3 | natural | 7.74(±0 .43) | 1.35(±0 .14) | 1.76(±0 .17) | 0.62(±0 .04) |
| synthetic | 7.06(±0 .71) | 1.47(±0 .28) | 1.93(±0 .32) | 0.59(±0 .07) |
Figure 1.
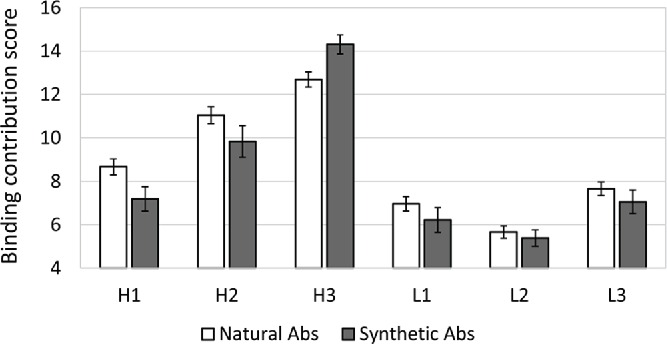
The binding contribution score of CDRs in natural and synthetic Abs –Binding contribution score (varies between 4 to 16) of each CDR in each Ab-Ag complex was calculated using the “CDRs Analyzer.” On the Y axis are the average scores of a given CDR across all of the natural (white bars) or synthetic (gray bar) Ab-Ag complexes. Error bars represent standard error.
Unlike synthetic Abs, CDRs in natural Abs specialize in specific types of contacts
“CDRs Analyzer” also provides a list of specific contacts (H-bonds, salt bridges, cation-pi or pi–pi). The distribution of each type of interaction across the 6 CDRs is shown in Fig. 2 (pi-pi interactions were excluded from the analysis due to their low occurrence, 10 interactions in synthetic Abs and 18 in natural Abs). The extreme dominance of CDRH3 in synthetic Abs emerges also from this analysis. For all types of contacts we analyzed, CDRH3 takes a larger fraction in synthetic Abs than it does in natural Abs. The most extreme is the case of the salt –bridges: In natural Ab-Ag complexes, 39.66% of the salt bridges are formed with CDRH2 and only 25.7% are with CDRH3. However, for synthetic Abs CDRH2 accounts for only 16.13% of the salt-bridges, while CDRH3 share increases to 61.29%. We also analyzed the amino acid composition of the heavy chain CDRs of synthetic and natural Abs. We found the decrease in salt-bridges from CDRH2 is accompanied with substantial decreased frequency of charged amino acids (E, D, H, K and R), from 13.13% in natural CDRsH2 to only 5.26% in synthetic CDRsH2 (Fig. S1 and Fig. S2). The percentage of salt-bridges formed by CDRH1 is also greatly affected. In natural Abs 11.17% of the salt bridges are from CDRH1 compared to only 1.61% of the salt bridges in synthetic Abs. The cation-pi and Hbond interactions are also accumulated more in CDRH3 of synthetic Abs compared to natural ones. For these contacts, we also observe a dramatic change in CDRH1, which accounts for 17.35% of the Hbonds and 22.7% of the cation-pi interactions in natural Abs. These percentages diminish to 9.96% of the Hbonds and to 10.87% of the cation-pi interactions in synthetic Abs. We also found a decreased frequency of polar amino acids in CDRH1 of synthetic Abs in comparison to that of natural ones (Fig. S2), which is consistent with the decreased share of synthetic CDRH1 in H-bonds.
Figure 2.
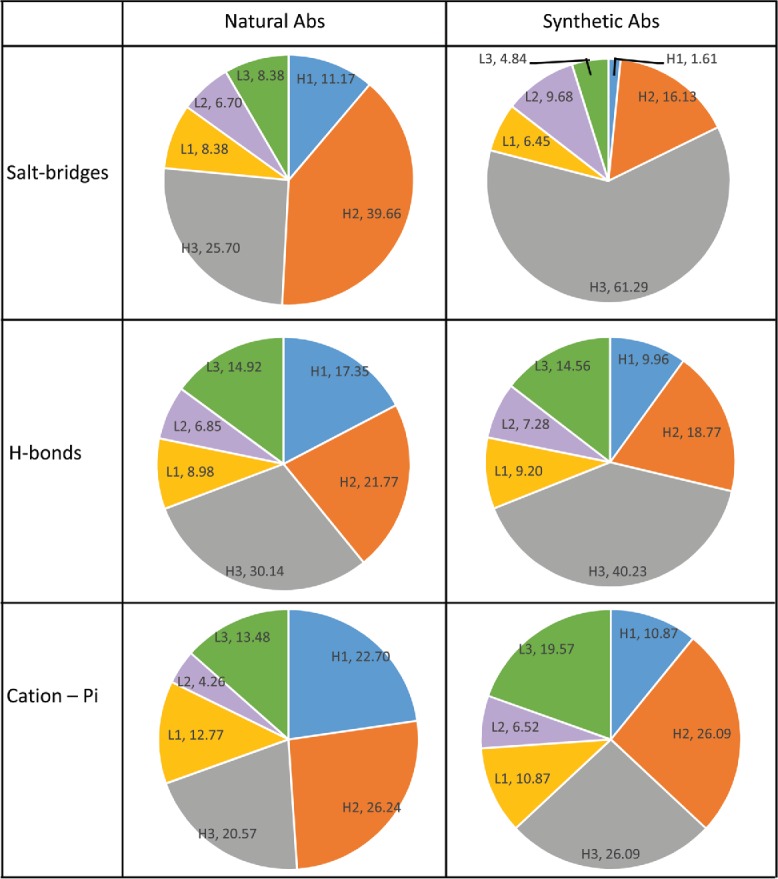
Distribution of H-bonds, salt-bridges and cation-pi across the CDRs- Percentage of salt-bridges, H-bonds and cation-pi interactions (top to bottom) that occur in each CDR in natural Abs and synthetic Abs. Labels are composed of CDR name (e.g., H1,L2) and the percentage of the specific interaction that are from residues in this CDR .
In natural Abs, each CDR on the heavy chain specializes in different types of interactions.39 As shown above, CDRH2 is responsible the largest share of salt-bridges (39.66%). CDRH3 is the main source for H-bonds (30.14%) and all heavy chain CDRs take similar parts of the cation-pi interactions (20.57%, 22.7% and 26.24% of cation-pi interactions from CDRH3, CDRH1 and CDRH2, respectively). This differentiation and specialization is lost for synthetic Abs. For the Abs that emerge from synthetic libraries, CDRH3 takes the central role in all analyzed interactions. CDRH2 has an equal share as CDRH3 only in cation-pi contacts.
The focus of synthetic Abs on CDRH3 creates interfaces that are less complex and more modular
We evaluate the complexity of Ab-Ag interaction using 2 parameters: independent epitope residue and integrated epitope residues. These parameters reflect the extent to which the 6 CDRs create an integral interface. An epitope residue on the Ag is considered an “independent residue” if it contacts only one CDR. An epitope residue that contacts 3 or more different CDRs is considered as an “integrated residue.” To assess the complexity of Ab-Ag interaction, the percentage of integrated and independent residues out of all residues that contact the paratope are calculated (note, however, that the raw output of the “CDRs Analyzer” provides this calculations as a percentage of the residues that contact a given CDR and not as a percentage of the residues that contact the entire paratope, see methods). On average, 57.49% of the epitope residues of natural Abs are independent (that is contact only one CDR). Whereas epitope of synthetic Abs are composed of 63.09% independent residues (Fig. 3A). This difference is almost exclusively accounted for by an increase in independent interactions with CDRH3 of synthetic Abs (Fig. 3B). We didn't find significant differences for the other CDRs. As for epitope residues that are integrated, their propensity drops from 12.93% for natural Abs to 8.81% for synthetic Abs (Fig. 3C). Unlike independent residues, the integrated residues are significantly decreased across all CDRs of synthetic Abs, except for CDRH1, which shows the same trend, although to lesser extent (Fig. 3D).
Figure 3.
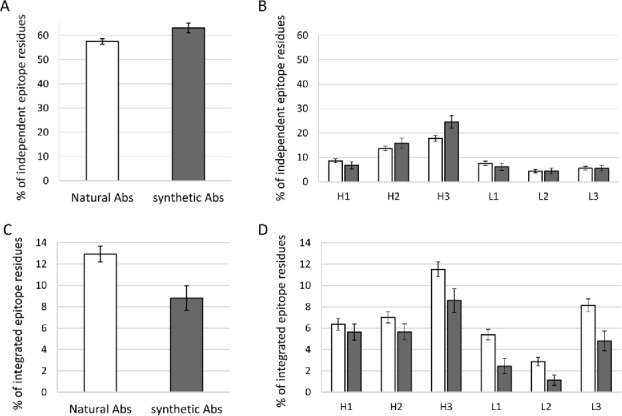
Complexity of Ab-Ag interactions in terms of independent and integrated epitope residues – The average percentage of independent residues in the epitope (Ag residues contacting only one CDR), are shown for natural and synthetic Abs (A) and for each CDR of the 2 groups (B). The average percentage of integrated residues in the epitope (Ag residues contacting at least 3 CDRs) are shown for natural Ab and synthetic Abs (C) and for each CDR of the 2 groups (D). An Integrated residue in the epitope was attributed to a certain CDR if the residue contacts that CDR and at least 2 other CDRs. Error Bars represent standard error.
Demonstrating the differences between synthetic and natural Abs
Fig. 4 demonstrates structurally the differences between synthetic and natural Abs. The residues in the figure are colored according to their binding contribution score from red (high contribution) to blue (no contribution). Fig. 4A shows the structure of the 1918 influenza virus hemagglutinin (HA) bound to the 2D1 Ab, which was isolated from a survivor of the 1918 Spanish flu.40 In this natural Ab, CDRH3 and CDRL3 are both colored in red or pink, reflecting their high contribution to binding, while CDRL1 and CDRH2 are colored in light blue, indicating moderate role in HA recognition. CDRH1 and CDRL2 are colored in blue, reflecting low or no involvement in the interaction. In this complex, 59.25% of the epitope residues are independent residues and 11.1% are integrated residues, which is very similar to the average of all natural Abs. This Ab represents typical features of natural Abs: residues in different CDRs have a major role in Ag recognition, creating a complex, integral interaction. In contrast, the E2 Ab against membrane-type serine protease 1 (MT-SP1) selected from an engineered synthetic library,41 shown in Fig. 4B, displays a different recognition pattern. Four of the CDRs, H1, L1, L2 and L3, are colored blue. This indicates a low or no influence of these CDRs on binding. Most of the contacts in this case are by CDRH3, colored in red, which is the key player of this Ab-Ag interaction. In addition, only 5.55% of the epitope residues are integrated residues and 83.33% independent residues. Notably, 61.11% of the MT-SP1 epitope residues contact residues only from CDRH3 in comparison to 46.66% of HA epitope residues. This Illustrates the way in which engineered Abs may become a mere scaffold for CDRH3, whereas natural Abs often rely on integral participation of specialized CDRs.
Figure 4.
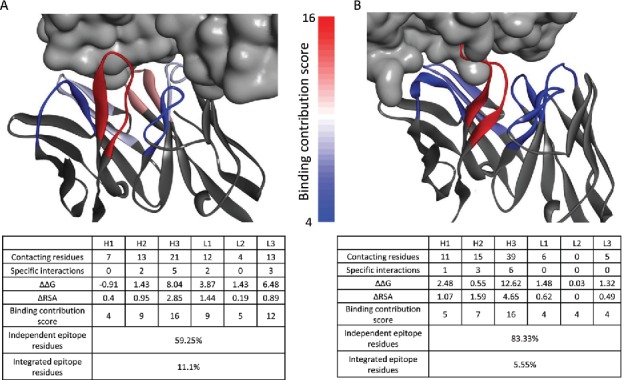
Natural and synthetic Ab-Ag interactions - The complex between hemagglutinin (HA) and the natural 2D1 Ab (PDBID: 3LZF) (A) and the complex between membrane-type serine protease 1 (MT-SP1) and the synthetic E2 Abs (PDBID: 3BN9) (B) are shown. Ags are presented in a gray surface view. Abs are presented in gray ribbon for non CDRs residues. CDRs are in ribbon representation colored according to their binding contribution score, from low contribution (blue) to high contribution (red). Images were generated using Discovery Studio Visualizer. Each Ab-Ag complex is accompanied by a table, detailing the calculated parameters, the binding contribution score and the percent of independent and integrated epitope residues.
Discussion
Synthetic libraries are clearly successful in yielding specific binders that often become successful drug leads. Here, we ask to what extent the products of these libraries mimic natural Abs. One may argue that, as long as the leads are successful, there is no need for the libraries to mimic natural Abs. However, our analysis can be important in 2 ways: first, as a basic science endeavor, it helps reveal the principles that guide natural Ab-Ag interaction. Second, revealing these principles suggests new avenues that may make synthetic libraries even more potent. While the dataset of synthetic Abs is smaller than that of the natural Abs, the data set represent a diverse collection of synthetic Abs isolated from a variety of generic (e.g., HuCAL17 or Lee et al.24) or custom-made libraries (see Table S2). The synthetic Abs in the dataset bind 30 different Ags, which are varied in their size from 51 to 915 residues. We validate that the Ag recognition occurred in different epitope in case 2 Abs bind the same Ags. Thus, the synthetic Abs data set represents the current strategies for library design. Obviously, as more synthetic Abs become available this analysis should be repeated to refine the insights and establish their significance further.
Large-scale analysis of Ab-Ag complexes can help reveal the principles that allow Igs to accommodate an exquisitely matching paratope for virtually any surface, while strictly maintaining its overall fold.5-7 The great challenge of Ab design is to make synthetic libraries that will yield Abs against a wide range of targets and epitopes. Indeed, in vivo Ab development relies on a more complex process, and hence may yield Abs with improved properties. This complex process includes gene rearrangement, somatic hyper mutations, clonal selection, both through positive selection for Ag recognition and negative selection for self-binding. We aimed to identify the differences between the Ag binding mechanism of synthetic Abs and natural Abs, which may help improve library design to yield more natural-line Abs. It also allowed us to revisit common assumptions about the role of CDRH3 in Ag recognition.
Obviously, some individual natural Abs and some individual synthetic Abs may be exceptions to the rule. Yet, our results reveal consistent differences between natural and synthetic Abs. The focus of synthetic libraries on engineering CDRH3 creates CDRH3 loops that participate in Ag recognition above the average of CDRH3 in natural Abs. As a result, CDRs H1 and H2 of synthetic Abs contribute less to Ag binding. CDRH3 loops encompass the V-D-J junction, hence this region displays the largest diversity among the 6 CDRs of the Abs in terms of length, sequence, and structure.42-44 CDRH3 is also located at the center of the binding site and is the CDR loop that undergoes the most significant conformational changes upon binding.45 Thus, it is commonly assumed that CDRH3 accounts for the ability of Abs to recognize and bind specific epitopes. Understandably, Ab engineering methods often focus on CDRH3. For example, Fellouse et al. designed phage display libraries with diversity of 104 to 1022 in CDRH3 and diversity of 32 to 896 in other CDRs.25 In the initial HuCAL libraries,17 diversity beyond the 49 chosen frameworks was introduced only to CDRH3 and CDRL3. In other studies, specific Abs were obtained from libraries with introduced diversity only to CDRH3.28-30
However, the relative importance of CDRH3 compared to other CDRs has been recently revisited in numerous studies. Large scale analyses39,46 of Abs have assessed the role of CDRH3. It has been demonstrated that CDRH2 may be as important as CDRH346 in its contribution to the binding free energy of the Ab-Ag complex. In addition, in 93% of the Ab-Ag complexes, CDRH2 contained at least one residue with high energetic contribution (ΔΔG>0.8kcal/mol) in comparison to 90% of the complexes with such residues from CDRH3. In another study, CDRH3 was found to be responsible for 30.6% of the energetically important Ag-binding residues.39 That is, most of the energetically important Ag-binding residues come from other CDRs. This has been shown also for specific examples like the interaction between HyHEL-10 and lysozyme, in which CDRH2 and CDRL1 display a dominant role, while CDRH3 shows very low binding contribution.38 The fact that CDRH3 is not necessary for the versatility of Abs was ultimately demonstrated by a study that has shown that synthetic libraries can yield specific Abs against different Ags with diverse CDRL3 and fixed CDRH3.47 In another study, the introduction of diversity into the sequence of anti ErbB2 Ab only at CDRH3 did not result in affinity enhanced variant, while beneficial mutants could be obtained by engineering one of the other contacting CDRs (CDRH1,H2,L1 or L3).48 This emphasizes that the importance of CDRH3 differ between Abs.
The reliance of synthetic Abs on CDRH3 may take a toll on the diversity of the epitopes that the library can bind, which may be referred to as the effective diversity of the library (as opposed to its actual diversity, represented by the number of unique sequences). Existing synthetic libraries tend to yield Abs with CDRH3 dominance. The typically fixed length and sequence of the other loops does not allow for paratopes with other binding topologies. It is therefore possible that, while the number of variants in the library may be higher than the number of variants in natural repertoires, these synthetic Abs represent only a small subset of the possible Abs that would be represented in a much smaller natural set of Ab sequences.
The effective diversity of a library is not the number of unique Ab sequences it has, but the number of different epitopes they can bind. This is defined by how many of the variants are expressed and fold into Abs with paratopes that are very different from each other. Our results suggest that tampering only with CDRH3 may not be a good way to obtain diverse paratopes. Based on the results presented here, one can propose approaches for improving Ab engineering. Building libraries that allow for higher diversity in all CDRs may result in Abs that have binding modes that are more similar to those of natural Abs, which might increase the effective diversity of the library and culminate in higher success rates. Of note is the degeneration of CDRH2 and CDRH1 in synthetic Abs, most remarkably in the percentage of salt-bridges coming from these CDRs and H-bonds and cation-pi interactions from CDRH1. To correct for this and create better libraries, the amino acid composition in these CDRs should be corrected to favor these types of interactions. This could be achieved by elevating the propensity of charged amino acids in CDRH2 and CDRH1 to produce more salt bridges or elevating the propensity of aromatic, positively charges or polar amino acids in CDRH1 to produce more cation-pi and H-bonds. It is also possible that the frameworks that are commonly used in synthetic libraries are suitable for producing interactions that rely on CDRH3. Considering additional frameworks may, therefore, be beneficial.
A novel approach for the design of synthetic libraries is based on the diversity of natural Ig repertoire (naïve, memory and plasma B cells), which can be characterized using next-generation sequencing (NGS).49,50 Glanville et al.49 analyzed ˜105 sequences of Ab variable fragments from 654 healthy human donors and, consistent with our finding, reported a substantial contribution to total diversity from somatically mutated residues in CDRs 1 and 2. Based on these results, a synthetic Ab library was constructed by introducing a diversity at positions across the 6 CDRs while the amino acid usage in each position was designed to mimic the natural repertoires usage.50 The 3D structures of the Ab-Ag complexes that were selected by these modern libraries are still not available. We expect that the relative binding contribution of the different CDRs in these synthetic Abs will better mimic the natural Ab binding mechanism than the synthetic Abs analyzed in the current study.
Although there are many available tools for the automated analysis of Abs sequences,51-56 the development of tools for the structural analysis of Ab-Ag complexes is still in its infancy. Two existing tools that provide comprehensive structural analysis of Abs are ABangle, for calculating the orientation between the VH and the VL,57 and the “AgAbDb dataset,” which contains interaction profiles of ˜500 Ab-Ag complexes in the PDB.58 In the “AgAbDb,” the data is available only for the curated PDBs and most of the output is at the atoms or residues level and not at CDRs level, similarly to tools analyzing general protein-protein interactions.59,60 “CDRs Analyzer” is designed to assist Ab engineering protocols by providing quantitative assessment of the biophysical properties both at the loop level – by assessing the contribution of each CDR - and at the residue level by identifying specific positions of interest within interface. Here, we used “CDRs Analyzer” to explore the differences between natural and synthetic interactions. This tool can be used to analyze Abs against pathogenic Ags or human-self Ags (see Table S1 for the origin of the Ags in our analysis), to explore the theory that V-genes are evolutionarily pre-configured to recognize common motifs in Ags from pathogenic source. “CDRs Analyzer” can also be applied to characterize other sets of immunological interactions. For example, it allows evaluation of the differences in binding properties of peptide-binding Abs and protein-binding Abs, or the differences between different families of Abs or even differences between Abs against different Ags. However, the most straightforward way to use “CDRs Analyzer” is for the analysis of individual Abs. It is applicable for experimentally solved Ab-Ag complexes as well as to computational models of such complexes. The output of “CDRs Analyzer” can assist different Ab engineering protocols. The contacting residues list and the specific interactions list can guide choosing specific positions for Ab affinity enhancement, decreasing aggregation or for deimmunization. The CDRs binding contribution may be an important consideration for CDR grafting, Ab humanization, design of 2-in-one Abs and for identifying CDR-derived peptides.38
Methods
Construction of natural Ab–Ag complexes data set
To build a large non-redundant set of natural Abs, a previously published non-redundant dataset of 196 Ab-Ag complexes61 was further filtered to create the current study data set of natural Ab-Ag complexes. The “CDRs Analyzer” cannot analyze single-chain variable fragment (scFv) Abs, Abs that contain disorder residues in the CDRs or non-standard amino acids, complexes that were solved by NMR and complexes composed of more than 25000 atoms. Complexes that met these conditions were deleted from the original dataset. In addition, complexes that included synthetic Abs were moved to the synthetic Ab-Ag data set. Finally, complexes that contain Ag with length of ≤30 amino acids were also removed. The resulting dataset contained 101 natural Ab-Ag complexes that are listed in Table S1.
Construction of synthetic Ab–Ag complexes data set
A synthetic Ab-Ag complexes dataset was constructed using both the PDB34 and SAbDab.62 The PDB query search was used to curate manually synthetic Ab-Ag complexes. The PDB query type was set to “PubMed abstract” and search words were “phage display antibody” and “library antibody.” In addition, the sequences of the light chain, the heavy chain or the full variable domain of a representative synthetic Ab (PDBID:2H9G) was used to search the SAbDab database using the framework region only option. The retrieved PDB entries were considered synthetic Ab-Ag complexes if the library from which it was isolated included variable domains sequences that were not obtained from a natural repertoire. Two Ab-Ag complexes were considered redundant in case the 2 Abs bound the same Ag at a similar epitope. Redundancy was removed according to this criterion. We removed from the data set complexes that contain scFvs, Ag length ≤30 amino acids, Abs that contains disordered residues in the CDRs or non-standard amino acids, complexes with resolution ≤3.6° and complexes that are composed of more than 25000 atoms. The final synthetic Ab-Ag complexes dataset contained 36 non-redundant PDB entries (Table S2).
Analyzing Ab-Ag complexes using “CDRs analyzer”
CDRs analyzer takes as an input an X-ray structure of Ab-Ag complex in a PDB file format. It automatically identifies the CDRs residues and calculates a set of parameters for all 6 CDRs. The output is an HTML page presenting the calculated parameters (described below) for each of the CDRs, a list of contacting residues and list of specific interactions. “CDRs Analyzer” was implemented in Perl and Python. The front end of the server is designed in HTML and XML.
Description of CDRs analyzer
CDRs Identification
The CDRs are identified using Paratome.53,63 An Ag-contacting residue within ±/5 residues from the Ag binding region boundaries as defined by Paratome is added to the nearest CDR. An Ag-contacting residue is a residue on the Ab that has at least one non-hydrogen atom within 5Å from a non-hydrogen atom in the Ag.
Number of contacting residues
The number of “contacting residues” is the number of residues in a CDR that are in contact with the Ag and the number of residues in the Ag that are in contact with the CDR.
Number of specific interactions
The number of “specific interactions” is the sum of the number of salt-bridges, pi-pi, cation-pi and possible H-bonds35 between the CDR and the Ag. A salt bridge is defined as one Asp or Glu side-chain carboxyl oxygen atom and one side-chain nitrogen atom of Arg, Lys or His that are within 4.0Å of each other. H-bonds were identified by first adding polar hydrogens atoms to the complex using Discovery Studio Visualizer and then by submitting the output file to HBPLUS program with default parameters.35 Pi-pi interactions are identified according to McGaughey et al.64 Briefly, the distance between the centroid of each pair of pi rings should be 8Å or less, at least one atom from each ring should be within 4.5Å. In addition, the angle theta between the normal of one or both rings and the centroid-centroid vector must fall between 0 and ± 60 degrees. The angle lambda between the normal of each ring must fall between 0 and ±30 degrees. A cation-pi interaction is defined if: Lys or Arg side chains cations are within 7Å from a centroid of a pi ring. The perpendicular distance between the cation and the plane of the ring is within 6Å and the angle between the cation-centroid vector and the ring plane is more than 45 degrees.
Energy calculations( ΔΔG)
The effect of in–silico mutation of each CDR residue to ALA is calculated using FoldX.36 FoldX's calculations have been previously shown to be correlated to experimentally measured results of 1030 mutants (R = 0.83).36 A recently published study curated 1100 mutations in Ab-Ag complexes and examined the performance of different energy scoring methods.65 FoldX was one of the top performers in that study, on both destabilizing (ΔΔG >1.0 kcal/mol) and stabilizing (ΔΔG < −1.0 kcal/mol) mutations.
Each PDB structure is first optimized using the FoldX RepairPDB function. Then, residues in the CDR are mutated to ALA using the BuildModel function that generated mutants and their corresponding wild-type structure models. The heavy chain and the light chain of the Ab are grouped together to calculated the energy values of the assembled Ab, and the AnalyzeComplex function is used to calculate the binding ΔG of each model. The calculated ΔΔG for each mutant is then computed by subtracting the wild-type calculated ΔG value from the mutant calculated ΔG value. The “ΔΔG” of a CDR is considered as the sum over its residues. The “CDRs Analyzer” outputs the ranking of the 6 CDRs according to the ΔΔG values.
Delta relative surface accessibility (ΔRSA)
RSA is given by dividing the solvent accessibility value by the surface area of the given amino acid.66 The solvent accessibility of the Ab residues are calculated using DSSP program.37 RSA is computed for each of the residues in the CDR, once with Ag presence (RSAbound) and once without Ag presence (RSAunbound). The ΔRSA is given by subtracting the RSAbound from the RSAunbound. The ΔRSA of a CDR is considered as the sum over its residues.
Binding contribution score
To evaluate the involvement of each CDR in Ag recognition we used an estimated calculation, which sums the 4 parameters values into a single “binding contribution score.” For each of the 4 binding parameters above, values are normalized and scored according to their quartiles: 4 points for values within the top 25% of the scores, 1 for the values within the lowest 25%. The “binding contribution score” of a given CDR is the sum of the scores over its criteria varies from 4 (no contribution to Ag binding) to 16 (highest contribution to Ag binding). The binding contribution calculation gives an equal weight for the 4 binding parameters. When more structural data becomes available, these weights should be assessed and optimized. To verify that the score is not sensitive for arbitrary cutoffs, we checked different binding contribution scores by dividing the parameters values into bins of thirds and fifths (instead of quarters). This did not change the results (data not shown).
Independent and integrated Ag residues
An “independent residue” is an Ag residue that is in contact with residues that belong to only one CDR. An “integrated residue” is an Ag residue that is in contact with at least 3 CDRs. These parameters are used by the “CDRs Analyzer” to calculate the “Independent binding score,” which measure the potential of a given CDR to bind the Ag as peptide.38 For that purpose, the percentage of independent or integrated residues for a given CDR was calculated out of Ag residues contacting that CDR. Here, we aimed to evaluate the complexity of the Ab-Ag interaction. Thus, the percentage of independent or integrated residues were calculated out of the total number of the epitope residues.
Independent binding score
The six parameters above (contacting residues, specific interactions, ΔΔG, ΔRSA, percentage of Independent and integrated Ag residues) are used to evaluate the potential of a CDR to bind the Ag as peptide.38 The values of each of the parameters are normalized and scored according to their quartiles: 4 points for values within the top 25% of the scores, 1 for the values within the lowest 25%. The “Independent binding score” of a given CDR is the sum of the scores over its 6 criteria.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed
Acknowledgments
We thank Inbal Sela-Culang (Bar-Ilan University), Sharon Fischman and Guy Nimrod (Biolojic Design, LTD) for useful comments and discussion on the manuscript.
References
- 1.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975; 256:495-7; PMID:1172191; http://dx.doi.org/ 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 2.Kung P, Goldstein G, Reinherz EL, Schlossman SF. Monoclonal antibodies defining distinctive human T cell surface antigens. Science 1979; 206:347-9; PMID:314668; http://dx.doi.org/ 10.1126/science.314668. [DOI] [PubMed] [Google Scholar]
- 3.Kinch MS. An overview of FDA-approved biologics medicines. Drug Discov Today 2015; 20:393-8; PMID:25220442; http://dx.doi.org/ 10.1016/j.drudis.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Janeway CA Jr, Travers P, Walport M, Shlomchik MJ Immunobiology: The Immune System in Health and Disease. 5th edition. New York: Garland Science; 2001. [Google Scholar]
- 5.Novotný J, Handschumacher M, Haber E, Bruccoleri RE, Carlson WB, Fanning DW, Smith JA, Rose GD. Antigenic determinants in proteins coincide with surface regions accessible to large probes (antibody domains). Proc Natl Acad Sci U S A 1986; 83:226-30; PMID:2417241; http://dx.doi.org/ 10.1073/pnas.83.2.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sela-Culang I, Kunik V, Ofran Y. The structural basis of antibody-antigen recognition. Front Immunol 2013; 4:302; PMID:24115948; http://dx.doi.org/ 10.3389/fimmu.2013.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sela-Culang I, Ofran Y, Peters B. Antibody specific epitope prediction-emergence of a new paradigm. Curr Opin Virol 2015; 11:98-102; PMID:25837466; http://dx.doi.org/ 10.1016/j.coviro.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Almagro JC. Identification of differences in the specificity-determining residues of antibodies that recognize antigens of different size: implications for the rational design of antibody repertoires. J Mol Recognit 2004; 17:132-43; PMID:15027033; http://dx.doi.org/ 10.1002/jmr.659. [DOI] [PubMed] [Google Scholar]
- 9.McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature 1990; 348:552-4; PMID:2247164; http://dx.doi.org/ 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- 10.Barbas CF, Kang AS, Lerner RA, Benkovic SJ. Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc Natl Acad Sci U S A 1991; 88:7978-82; PMID:1896445; http://dx.doi.org/ 10.1073/pnas.88.18.7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williamson RA, Lazzarotto T, Sanna PP, Bastidas RB, Dalla Casa B, Campisi G, Burioni R, Landini MP, Burton DR. Use of recombinant human antibody fragments for detection of cytomegalovirus antigenemia. J Clin Microbiol 1997; 35:2047-50; PMID:9230379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol 1991; 222:581-97; PMID:1748994; http://dx.doi.org/ 10.1016/0022-2836(91)90498-U. [DOI] [PubMed] [Google Scholar]
- 13.Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol 1996; 14:309-14; PMID:9630891; http://dx.doi.org/ 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- 14.Harel Inbar N, Benhar I. Selection of antibodies from synthetic antibody libraries. Arch Biochem Biophys 2012; 526:87-98; PMID:22244834; http://dx.doi.org/ 10.1016/j.abb.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 15.Hoogenboom HR. Designing and optimizing library selection strategies for generating high-affinity antibodies. Trends Biotechnol 1997; 15:62-70; PMID:9081300; http://dx.doi.org/ 10.1016/S0167-7799(97)84205-9. [DOI] [PubMed] [Google Scholar]
- 16.Presta LG. Molecular engineering and design of therapeutic antibodies. Curr Opin Immunol 2008; 20:460-70; PMID:18656541; http://dx.doi.org/ 10.1016/j.coi.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 17.Knappik A, Ge L, Honegger A, Pack P, Fischer M, Wellnhofer G, Hoess A, Wölle J, Plückthun A, Virnekäs B. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J Mol Biol 2000; 296:57-86; PMID:10656818; http://dx.doi.org/ 10.1006/jmbi.1999.3444. [DOI] [PubMed] [Google Scholar]
- 18.Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol 2005; 23:1105-16; PMID:16151404; http://dx.doi.org/ 10.1038/nbt1126. [DOI] [PubMed] [Google Scholar]
- 19.Sidhu SS, Li B, Chen Y, Fellouse FA, Eigenbrot C, Fuh G. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J Mol Biol 2004; 338:299-310; PMID:15066433; http://dx.doi.org/ 10.1016/j.jmb.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 20.Wolbink GJ, Aarden LA, Dijkmans BA. Dealing with immunogenicity of biologicals: assessment and clinical relevance. Curr Opin Rheumatol 2009; 21:211-5; PMID:19399992; http://dx.doi.org/ 10.1097/BOR.0b013e328329ed8b. [DOI] [PubMed] [Google Scholar]
- 21.Clark M. Antibody humanization: a case of the ‘Emperor's new clothes’? Immunol Today 2000; 21:397-402; PMID:10916143; http://dx.doi.org/ 10.1016/S0167-5699(00)01680-7. [DOI] [PubMed] [Google Scholar]
- 22.van Schouwenburg PA, Rispens T, Wolbink GJ. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat Rev Rheumatol 2013; 9:164-72; PMID:23399692; http://dx.doi.org/ 10.1038/nrrheum.2013.4. [DOI] [PubMed] [Google Scholar]
- 23.Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, Dijkmans BA, Tak PP, Wolbink GJ. Clinical response to adalimumab: relationship to anti-adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann Rheum Dis 2007; 66:921-6; PMID:17301106; http://dx.doi.org/ 10.1136/ard.2006.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CV, Liang WC, Dennis MS, Eigenbrot C, Sidhu SS, Fuh G. High-affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J Mol Biol 2004; 340:1073-93; PMID:15236968; http://dx.doi.org/ 10.1016/j.jmb.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 25.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, et al.. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J Mol Biol 2007; 373:924-40; PMID:17825836; http://dx.doi.org/ 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Liang WC, Dennis MS, Stawicki S, Chanthery Y, Pan Q, Chen Y, Eigenbrot C, Yin J, Koch AW, Wu X, et al.. Function blocking antibodies to neuropilin-1 generated from a designed human synthetic antibody phage library. J Mol Biol 2007; 366:815-29; PMID:17196977; http://dx.doi.org/ 10.1016/j.jmb.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 27.Rothe C, Urlinger S, Löhning C, Prassler J, Stark Y, Jäger U, Hubner B, Bardroff M, Pradel I, Boss M, et al.. The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. J Mol Biol 2008; 376:1182-200; PMID:18191144; http://dx.doi.org/ 10.1016/j.jmb.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 28.Mahon CM, Lambert MA, Glanville J, Wade JM, Fennell BJ, Krebs MR, Armellino D, Yang S, Liu X, O'Sullivan CM, et al.. Comprehensive interrogation of a minimalist synthetic CDR-H3 library and its ability to generate antibodies with therapeutic potential. J Mol Biol 2013; 425:1712-30; PMID:23429058; http://dx.doi.org/ 10.1016/j.jmb.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 29.Braunagel M, Little M. Construction of a semisynthetic antibody library using trinucleotide oligos. Nucleic Acids Res 1997; 25:4690-1; PMID:9358184; http://dx.doi.org/ 10.1093/nar/25.22.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.der Maur AA, Zahnd C, Fischer F, Spinelli S, Honegger A, Cambillau C, Escher D, Plückthun A, Barberis A. Direct in vivo screening of intrabody libraries constructed on a highly stable single-chain framework. J Biol Chem 2002; 277:45075-85; PMID:12215438; http://dx.doi.org/ 10.1074/jbc.M205264200. [DOI] [PubMed] [Google Scholar]
- 31.Fellouse FA, Li B, Compaan DM, Peden AA, Hymowitz SG, Sidhu SS. Molecular recognition by a binary code. J Mol Biol 2005; 348:1153-62; PMID:15854651; http://dx.doi.org/ 10.1016/j.jmb.2005.03.041. [DOI] [PubMed] [Google Scholar]
- 32.Fellouse FA, Wiesmann C, Sidhu SS. Synthetic antibodies from a four-amino-acid code: a dominant role for tyrosine in antigen recognition. Proc Natl Acad Sci U S A 2004; 101:12467-72; PMID:15306681; http://dx.doi.org/ 10.1073/pnas.0401786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Söderlind E, Strandberg L, Jirholt P, Kobayashi N, Alexeiva V, Aberg AM, Nilsson A, Jansson B, Ohlin M, Wingren C, et al.. Recombining germline-derived CDR sequences for creating diverse single-framework antibody libraries. Nat Biotechnol 2000; 18:852-6; PMID:10932154; http://dx.doi.org/ 10.1038/78458. [DOI] [PubMed] [Google Scholar]
- 34.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res 2000; 28:235-42; PMID:10592235; http://dx.doi.org/ 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J Mol Biol 1994; 238:777-93; PMID:8182748; http://dx.doi.org/ 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 36.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol 2002; 320:369-87; PMID:12079393; http://dx.doi.org/ 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 37.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983; 22:2577-637; PMID:6667333; http://dx.doi.org/ 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 38.Burkovitz A, Leiderman O, Sela-Culang I, Byk G, Ofran Y. Computational identification of antigen-binding antibody fragments. J Immunol 2013; 190:2327-34; PMID:23359499; http://dx.doi.org/ 10.4049/jimmunol.1200757. [DOI] [PubMed] [Google Scholar]
- 39.Kunik V, Ofran Y. The indistinguishability of epitopes from protein surface is explained by the distinct binding preferences of each of the six antigen-binding loops. Protein Eng Des Sel 2013; 26(10):599-609; PMID:23754530. [DOI] [PubMed] [Google Scholar]
- 40.Xu R, Ekiert DC, Krause JC, Hai R, Crowe JE, Wilson IA. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 2010; 328:357-60; PMID:20339031; http://dx.doi.org/ 10.1126/science.1186430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Farady CJ, Egea PF, Schneider EL, Darragh MR, Craik CS. Structure of an Fab-protease complex reveals a highly specific non-canonical mechanism of inhibition. J Mol Biol 2008; 380:351-60; PMID:18514224; http://dx.doi.org/ 10.1016/j.jmb.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chothia C, Lesk AM, Tramontano A, Levitt M, Smith-Gill SJ, Air G, Sheriff S, Padlan EA, Davies D, Tulip WR, et al.. Conformations of immunoglobulin hypervariable regions. Nature 1989; 342:877-83; PMID:2687698; http://dx.doi.org/ 10.1038/342877a0. [DOI] [PubMed] [Google Scholar]
- 43.Kuroda D, Shirai H, Kobori M, Nakamura H. Structural classification of CDR-H3 revisited: a lesson in antibody modeling. Proteins 2008; 73:608-20; PMID:18473362; http://dx.doi.org/ 10.1002/prot.22087. [DOI] [PubMed] [Google Scholar]
- 44.Morea V, Tramontano A, Rustici M, Chothia C, Lesk AM. Conformations of the third hypervariable region in the VH domain of immunoglobulins. J Mol Biol 1998; 275:269-94; PMID:9466909; http://dx.doi.org/ 10.1006/jmbi.1997.1442. [DOI] [PubMed] [Google Scholar]
- 45.Sela-Culang I, Alon S, Ofran Y. A systematic comparison of free and bound antibodies reveals binding-related conformational changes. J Immunol 2012; 189:4890-9; PMID:23066154; http://dx.doi.org/ 10.4049/jimmunol.1201493. [DOI] [PubMed] [Google Scholar]
- 46.Robin G, Sato Y, Desplancq D, Rochel N, Weiss E, Martineau P. Restricted diversity of antigen binding residues of antibodies revealed by computational alanine scanning of 227 antibody-antigen complexes. J Mol Biol 2014; 426:3729-43; PMID:25174334; http://dx.doi.org/ 10.1016/j.jmb.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 47.Persson H, Ye W, Wernimont A, Adams JJ, Koide A, Koide S, Lam R, Sidhu SS. CDR-H3 diversity is not required for antigen recognition by synthetic antibodies. J Mol Biol 2013; 425:803-11; PMID:23219464; http://dx.doi.org/ 10.1016/j.jmb.2012.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu D, Hu S, Wan W, Xu M, Du R, Zhao W, Gao X, Liu J, Liu H, Hong J. Effective Optimization of Antibody Affinity by Phage Display Integrated with High-Throughput DNA Synthesis and Sequencing Technologies. PLoS One 2015; 10:e0129125; PMID:26046845; http://dx.doi.org/ 10.1371/journal.pone.0129125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, Ni I, Mei L, Sundar PD, Day GM, et al.. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci U S A 2009; 106:20216-21; PMID:19875695; http://dx.doi.org/ 10.1073/pnas.0909775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhai W, Glanville J, Fuhrmann M, Mei L, Ni I, Sundar PD, Van Blarcom T, Abdiche Y, Lindquist K, Strohner R, et al.. Synthetic antibodies designed on natural sequence landscapes. J Mol Biol 2011; 412:55-71; PMID:21787786; http://dx.doi.org/ 10.1016/j.jmb.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 51.Kaas Q, Ruiz M, Lefranc MP. IMGT/3Dstructure-DB and IMGT/StructuralQuery, a database and a tool for immunoglobulin, T cell receptor and MHC structural data. Nucleic Acids Res 2004; 32:D208-10; PMID:14681396; http://dx.doi.org/ 10.1093/nar/gkh042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ehrenmann F, Kaas Q, Lefranc MP. IMGT/3Dstructure-DB and IMGT/DomainGapAlign: a database and a tool for immunoglobulins or antibodies, T cell receptors, MHC, IgSF and MhcSF. Nucleic Acids Res 2010; 38:D301-7; PMID:19900967; http://dx.doi.org/ 10.1093/nar/gkp946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kunik V, Ashkenazi S, Ofran Y. Paratome: an online tool for systematic identification of antigen-binding regions in antibodies based on sequence or structure. Nucleic Acids Res 2012; 40:W521-4; PMID:22675071; http://dx.doi.org/ 10.1093/nar/gks480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abhinandan KR, Martin AC. Analyzing the “degree of humanness” of antibody sequences. J Mol Biol 2007; 369:852-62; PMID:17442342; http://dx.doi.org/ 10.1016/j.jmb.2007.02.100. [DOI] [PubMed] [Google Scholar]
- 55.Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 2013; 41:W34-40; PMID:23671333; http://dx.doi.org/ 10.1093/nar/gkt382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Retter I, Althaus HH, Münch R, Müller W. VBASE2, an integrative V gene database. Nucleic Acids Res 2005; 33:D671-4; PMID:15608286; http://dx.doi.org/ 10.1093/nar/gki088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dunbar J, Fuchs A, Shi J, Deane CM. ABangle: characterising the VH-VL orientation in antibodies. Protein Eng Des Sel 2013; 26:611-20; PMID:23708320; http://dx.doi.org/ 10.1093/protein/gzt020. [DOI] [PubMed] [Google Scholar]
- 58.Kulkarni-Kale U, Raskar-Renuse S, Natekar-Kalantre G, Saxena SA. Antigen-Antibody Interaction Database (AgAbDb): a compendium of antigen-antibody interactions. Methods Mol Biol 2014; 1184:149-64; PMID:25048123; http://dx.doi.org/ 10.1007/978-1-4939-1115-8_8. [DOI] [PubMed] [Google Scholar]
- 59.Tina KG, Bhadra R, Srinivasan N. PIC: Protein Interactions Calculator. Nucleic Acids Res 2007; 35:W473-6; PMID:17584791; http://dx.doi.org/ 10.1093/nar/gkm423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laskowski RA, Hutchinson EG, Michie AD, Wallace AC, Jones ML, Thornton JM. PDBsum: a Web-based database of summaries and analyses of all PDB structures. Trends Biochem Sci 1997; 22:488-90; PMID:9433130; http://dx.doi.org/ 10.1016/S0968-0004(97)01140-7. [DOI] [PubMed] [Google Scholar]
- 61.Burkovitz A, Sela-Culang I, Ofran Y. Large-scale analysis of somatic hypermutations in antibodies reveals which structural regions, positions and amino acids are modified to improve affinity. FEBS J 2014; 281:306-19; PMID:24279419; http://dx.doi.org/ 10.1111/febs.12597. [DOI] [PubMed] [Google Scholar]
- 62.Dunbar J, Krawczyk K, Leem J, Baker T, Fuchs A, Georges G, Shi J, Deane CM. SAbDab: the structural antibody database. Nucleic Acids Res 2014; 42:D1140-6; PMID:24214988; http://dx.doi.org/ 10.1093/nar/gkt1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kunik V, Peters B, Ofran Y. Structural consensus among antibodies defines the antigen binding site. PLoS Comput Biol 2012; 8:e1002388; PMID:22383868; http://dx.doi.org/ 10.1371/journal.pcbi.1002388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McGaughey GB, Gagné M, Rappé AK. pi-Stacking interactions. Alive and well in proteins. J Biol Chem 1998; 273:15458-63; PMID:9624131; http://dx.doi.org/ 10.1074/jbc.273.25.15458. [DOI] [PubMed] [Google Scholar]
- 65.Sirin S, Apgar JR, Bennett EM, Keating AE. AB-Bind: Antibody binding mutational database for computational affinity predictions. Protein Sci 2015; PMID:26473627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chothia C. The nature of the accessible and buried surfaces in proteins. J Mol Biol 1976; 105:1-12; PMID:994183; http://dx.doi.org/ 10.1016/0022-2836(76)90191-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.