

ABSTRACT
The antibody rilotumumab, which has been tested in multiple Phase 2 and Phase 3 trials, has been reported to neutralize hepatocyte growth factor (HGF), the ligand for the oncogene MET. However, we report that rilotumumab does not prevent HGF from directly binding to MET on conventional and primary patient-derived human gliomasphere lines, a trait driven by the HGF α-chain, which remains free to engage cell-surface glycosaminoglycans and the receptor MET. This binding induces MET phosphorylation, initiates robust AKT and ERK signaling and potentiates biological effects such as cell scattering. This partial antagonism was highly exacerbated in the presence of activated epidermal growth factor receptor, which is common in several cancers. Hence, we confirm that rilotumumab is only a partial antagonist of HGF activity, a finding that has considerable implications for the therapeutic use of rilotumumab.
KEYWORDS: Cancer, EGFR, HGF, MET, targeted therapy
Abbreviations
- BCA assay
bicinchoninic acid assay
- bFGF
basic fibroblast growth factor
- BIOrilo
biotinylated rilotumumab
- BSA
bovine serum albumin
- EGFR
epidermal growth factor receptor
- ELISA
enzyme-linked immunosorbent assay
- HGF
hepatocyte growth factor
- mAb
monoclonal antibody
- NSC
neural stem cell
- NSCLC
non-small-cell lung carcinoma
- rilo–HGF
pre-complexed rilotumumab and HGF
- RTK
receptor tyrosine kinase
- SF-BSA
serum-free medium containing 0.1% BSA
- WCL
whole cell lysate
- wt
wild-type
MET is a transmembrane receptor tyrosine kinase (RTK) implicated in the initiation and progression of several cancers, including glioma, gastric adenocarcinoma and non-small-cell lung carcinoma (NSCLC).1 An elevated level of hepatocyte growth factor (HGF), the MET ligand, is common in dysregulated MET signaling in cancer.1 Furthermore, HGF markedly reduces the anti-tumor efficacy of various targeted therapeutics, e.g., vemurafenib in melanoma patients, crizotinib in acute myeloid leukemia primary cultures, and erlotinib in NSCLC patients.2–4 Hence, neutralizing HGF's biological activity is an important node in blocking oncogenic signaling and preventing drug resistance in various cancers.
Three candidate antibodies have been developed for the purpose of neutralizing HGF, ficlatuzumab (AVEO), huL2G7 (Takeda) and rilotumumab (Amgen),5 with rilotumumab being the most advanced in clinical development. Preclinical data have shown that rilotumumab neutralizes HGF binding to the MET extracellular domain, abrogates HGF-induced MET activation in PC-3 human prostate cancer cells, and reduces human glioma xenograft size.6 However, rilotumumab in combination with the standard of care has not increased survival in 13 of 14 Phase 2 trials. The exception is a Phase 2 trial for gastric and esophageal cancer (NCT00719550),7 which was extended to the multi-institutional Phase 3 trials RILOMET-1 (NCT01697072) and RILOMET-2 (NCT02137343), which have subsequently been terminated because of increased toxicity in patients treated with rilotumumab. In light of this poor response observed in clinical trials, we investigated the binding of rilotumumab to its ligand and the downstream effects in cell lines from a variety of cancers to determine whether the antibody was a genuine full antagonist of HGF activity.
We first observed that pre-complexed rilotumumab and HGF (rilo–HGF), at a 55:1 molar excess of antibody, can still stimulate MET phosphorylation in the glioma cell line U87MG (Fig. 1A, left), the NSCLC cell line A549 (Fig. 1B) and the MET-positive patient-derived primary gliomasphere line SB2 (Fig. 1B). In U87MG, this phosphorylation was exacerbated by expression of the autoactive epidermal growth factor receptor (EGFR) mutant EGFRvIII, which is common in glioma8 (Fig. 1A, right), or by EGF-stimulation of U87MG.wtEGFR cells (Fig. 1C), which overexpress wtEGFR. We then assessed whether rilo–HGF binding to cell-surface MET exerted a prolonged functional effect (indicated in Fig. 1A), by measuring chronic MET activation. MET phosphorylation was rapid (within 7 min) after incubation with rilo–HGF and was sustained for as long as after stimulation with HGF alone in U87MG.vIII and A549 cells; however, the level of phosphorylated protein obtained after rilo–HGF stimulation was slightly lower than for HGF alone (Fig. 1D). Importantly, total MET had not been downregulated after 4 h rilo–HGF exposure, in contrast to HGF alone (Fig. 1D). Therefore, in several lines other than PC-3, despite HGF being bound by rilotumumab, it can still elicit substantial MET phosphorylation, albeit less than free HGF.
Figure 1.

Rilotumumab does not completely prevent HGF-induced MET phosphorylation in multiple cell lines. (A) MET phosphorylation detected in U87MG and U87MG.vIII cells after incubation with variable concentrations of HGF or rilo–HGF for 7 min at the indicated molar ratio and immunoprecipitation. (B) As for (A) for A549 cells or SB2 gliomaspheres with vehicle, 100 ng/mL HGF, 10 µg/mL rilotumumab or 55:1 rilo–HGF. (C) MET and EGFR phosphorylation detected in U87MG.wtEGFR cells after incubation with 100 ng/mL HGF, 100 ng/mL EGF, 10 µg/mL rilotumumab or 55:1 rilo–HGF alone or in various combinations for 7 min. MET detection was by immunoprecipitation, and EGFR detection was by standard immunoblotting. (D) MET phosphorylation detected in U87MG.vIII and A549 cells at various time points after incubation with vehicle, 100 ng/mL HGF, 10 µg/mL rilotumumab or 55:1 rilo–HGF and immunoprecipitation. Actin was used as a loading control. Data represent results from 2 or more independent experiments. Blots were imaged using an Odyssey Infrared Imaging System and software and are cropped for clarity and concision. Note that the right blot in A was exposed less than the left blot in A (to avoid overexposure of the p-Met bands) and the blot in D.
Using immunofluorescence microscopy, we confirmed that rilo–HGF can bind to the cell surface of several MET-positive human cancer cell lines, including U87MG.vIII, A549 and the gastric cancer cell line GTL-16 (Fig. 2A). The human anti-EGFR mAb panitumumab verified that bound human IgG was detectable for all cell lines tested. Flow cytometric quantification of binding showed that a high level of rilo–HGF was engaged on the surface of these cell lines and on the MET-negative cell line T-47D (human breast cancer), but not on the MET-negative Jurkat cell line (human leukemia) (Fig. 2B, top, and Supplementary Fig. S1).
Figure 2.
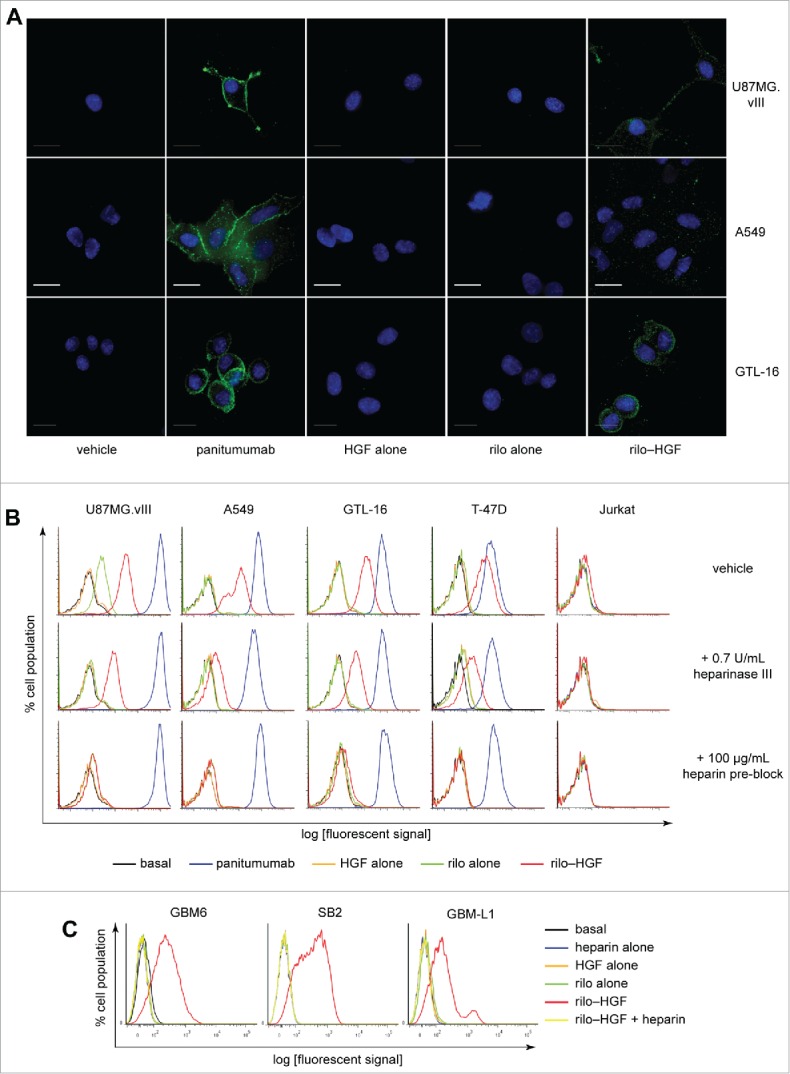
rilo–HGF engages with the cell surface in multiple different cell lines. (A) Immunofluorescence demonstrating engagement of 55:1 rilo–HGF (green) with the surface of U87MG.vIII, A549 and GTL-16 cells. Nuclei were stained with DAPI (blue). Scale bar, 50 μm. Panitumumab was included as a positive control for human IgG detection. (B, C) Flow cytometry tests demonstrating cell-surface engagement of rilo–HGF under differing conditions with conventional cell lines derived from several distinct cancers (B) or primary gliomaspheres derived from patient tissue (C). Treatment concentrations for all tests were 10 µg/mL rilotumumab, 10 µg/mL panitumumab, 100 ng/mL HGF, 100 µg/mL heparin or 55:1 rilo–HGF.
HGF has 2 chains (α and β) and binds MET via a high-affinity binding site in the NK1 domain of the α-chain.9 HGF also binds sulfonated cell-surface glycosaminoglycans such as heparan sulfate as a pre-requisite to MET binding.10 This binding occurs via another high-affinity site, which is in the N-terminus of the α-chain and is not bound by rilotumumab (which targets the β-chain).5 To test whether rilo–HGF engages MET-negative T-47D cells through cell-surface heparan sulfate, we removed this sugar enzymatically. This reduced rilo–HGF binding markedly (Fig. 2B, center), albeit not completely, as determined by flow cytometry, suggesting that heparan sulfate and other sulfonated glycosaminoglycans (such as dextran sulfate11 and chondroitin sulfate) engage rilo–HGF in MET's absence. Pre-blocking the high-affinity glycosaminoglycan binding site in rilo–HGF with excess heparin abolished binding to all cell types (Fig. 2B, bottom), confirming that HGF needs a free α-chain in the initial stages of cell-surface engagement. Jurkat cells, which are MET negative (Supplementary Fig. S1) and heparan-sulfate negative,12 did not bind rilo–HGF, confirming the requirement for MET and/or heparan sulfate. Robust rilo–HGF binding, abolished by heparin pre-incubation, was also observed in several primary human gliomasphere cell lines (GBM-6, SB2 and GBM-L1) (Fig. 2C), suggesting that rilo–HGF could bind tumors in glioma patients. Overall, these results confirm that rilo–HGF binds the surface of multiple conventional and primary cancer cell lines, and that this binding relies on a free HGF α-chain binding to sulfonated glycosaminoglycans and can occur in the absence of MET.
Using immunoprecipitation, we confirmed that rilo–HGF and MET directly physically interact. Incubating cells with biotinylated rilotumumab (BIOrilo) complexed with HGF led to co-immunoprecipitation of BIOrilo heavy and light chains with MET in U87MG.vIII cells, in contrast to MET-negative T-47D cells (Fig. 3A). These findings indicate that HGF within rilo–HGF physically binds to MET. Downstream signaling pathways were also activated after a 7 min exposure of cells to HGF or rilo–HGF, as indicated by robust phospho (p)-AKT and p-ERK1/2 signals in A549 WCLs (Fig. 3B). We also observed prominent activation of p-ERK1/2 in the NSCLC cell line HCC827 (Fig. 3B), which contains the EGFR-activating deletion E746–A75013 and also binds rilo–HGF by flow cytometry (Supplementary Fig. S2). Addition of HGF or rilo–HGF to the MET-positive primary human gliomasphere cell lines SB2 and RN1 also resulted in strong induction of p-AKT and p-ERK1/2 (Fig. 3C). Therefore, despite rilo–HGF triggering less MET phosphorylation than HGF (Fig. 1), signal transduction still occurs in the presence of antibody. To determine whether rilo–HGF-induced MET activation and signal transduction induce a biological effect, we used real-time xCELLigence to analyze cell scattering, a classic MET-stimulated biological effect, in 2 human cell lines documented to display this effect in response to HGF: A549 and DU 145 (prostate cancer) cells.14,15 Rilo–HGF and HGF both induced scattering profiles (Fig. 3D, left), with an immediate (A549) or delayed (DU 145) effect, shown by a cell index shift. The response to rilo–HGF, however, was lower than to HGF. The cell index changes were not due to HGF-induced cell death, as cell viability assays at the maximum scatter point for HGF (40 h post treatment of DU 145 cells) showed no decrease in cell number (Supplementary Fig. S3). These changes were, by contrast, due to direct MET stimulation, as pre-incubating the cells with 2 µM of the MET inhibitor SU11274 (Fig. 3D, center) or administering rilo–HGF pre-blocked with 100 µg/mL heparin (Fig. 3D, right) abolished the scattering phenotype. These findings confirm that rilotumumab is a partial antagonist of HGF's biological activity.
Figure 3.

Rilotumumab acts as a partial, not full, HGF antagonist. (A) Detection of biotinylated rilotumumab (BIOrilo) engagement of MET in U87MG.vIII and T-47D cells after pre-incubation with 10 µg/mL BIOrilo or 55:1 BIOrilo–HGF complex and immunoprecipitation. H, heavy chain; L, light chain. (B, C) Detection of AKT and ERK signaling in WCLs isolated from A549 and HCC827 cells (B) or SB2 and RN1 gliomaspheres (C) exposed to vehicle, 100 ng/mL HGF, 10 µg/mL rilotumumab or 55:1 rilo–HGF for 7 min. Blots were imaged using an Odyssey Infrared Imaging System and software and are cropped for clarity and concision. (D) Real-time xCELLigence cell scattering assays of A549 and DU 145 cells after treatments alone (PBS vehicle, 100 ng/mL HGF, 10 µg/mL rilotumumab or 55:1 rilo–HGF) or treatments after pre-incubation with 2 µM SU11274 or 100 µg/mL heparin. The data represent results from 3 or more independent experiments.
Using multiple techniques and a range of tumor cell lines, we demonstrated that rilotumumab functions as a partial, not full, antagonist of HGF activity. This arises from the HGF α-chain's retaining the ability to engage sulfonated glycosaminoglycans and to subsequently bind MET and potentiate signaling and biological effects. As we found that these effects occur in patient-derived primary gliomaspheres (Fig. 1B, Fig. 2C and Fig. 3C), it is likely that rilo–HGF complexes activate MET and trigger signaling in patient tumors.
This study clearly identifies an important shortcoming of current high-throughput platform technologies, such as surface plasmon resonance and enzyme-linked immunosorbent assays (ELISAs), used to screen for and identify antibody therapeutics that are targeted to ligands with known cell-surface co-factors. We present strong evidence that antibody therapeutics identified in such a screen require a more robust validation on live cells, using the flow cytometry/cell signaling approaches that we outline here on multiple cell lines. As these tests are relatively simple, quick and only need to be applied to leading candidates, this approach will save substantial financial outlay and effort by eliminating antibodies with seemingly good neutralization capabilities that do not translate to live cells before the preclinical and clinical trial stages.
We also found that activated EGFR enhanced MET activation in the presence of rilotumumab, suggesting that activated EGFR may exacerbate resistance to this therapeutic antibody. This is important because many tumors bear activated EGFR generated by either an activating mutation, such as in NSCLC and glioblastoma,8,16 or by overexpression of EGFR, such as in colon cancers. Hence, the presence of this commonly activated RTK in cancers may significantly impede the activity of rilotumumab as a monotherapy.
Combining rilotumumab with anti-EGFR therapeutics may have a substantial positive effect on the efficacy of anti-HGF therapies. We have previously shown that incomplete neutralization of EGFRvIII by small molecule inhibitors, such as erlotinib, results in premature MET reactivation in the presence of rilotumumab.17 Therefore, it may be more beneficial to utilize the anti-EGFR antibody panitumumab for effective combination therapy. This approach is validated by our previous preclinical data demonstrating that co-administration of panitumumab with rilotumumab is required to inhibit glioma xenografts bearing EGFRvIII.17,18 Recently reported results of a Phase 2 trial (NCT00788957) in wild-type K-RAS metastatic colorectal cancer19 show that rilotumumab has some survival benefit when combined with the anti-EGFR antibody panitumumab, but not the anti-IGF-1R antibody ganitumumab. This finding also supports the idea of supplementing rilotumumab therapy with panitumumab. To improve clinical outcomes, patients with MET-positive and HGF-positive tumors should be stratified for EGFR activation by ligand or mutation, and combination treatment with rilotumumab and EGFR inhibitors should be explored. Additionally, several novel experimental anti-MET antagonist antibodies, such as h224G11, MM-131 and LY2875358, which target MET directly and block HGF's action without agonizing the receptor, should be investigated as alternatives in combination EGFR therapy.
Our results go some way toward explaining why Phase 2 and Phase 3 trials of rilotumumab, in combination with standard of care chemotherapy, have been disappointing in several major solid tumor types.20-23 Moreover, because the epitope of the next leading HGF-neutralizing candidate antibody, ficlatuzumab, has not yet been described, we strongly suggest that this antibody, and others such as huL2G7, be tested in the simple live cell assays that we document here before progressing to further clinical trials, to verify that it too does not bind cell-surface MET and activate signaling.
Methods and materials
Antibodies and reagents
The anti-EGFR monoclonal antibody 806 (mAb 806) and the anti-MET monoclonal antibody LMH 8524 were produced in-house at the Ludwig Institute for Cancer Research, Heidelberg, Australia. Rilotumumab and panitumumab were provided by Amgen. The rabbit anti-MET polyclonal antibody C-28 conjugated to agarose beads (catalog number, sc-161-AC) and the protein A/G agarose beads were purchased from Santa Cruz Biotechnology. The rabbit anti-p-MET (Y1234/1235) 3D7 mAb (catalog number, 3129), the mouse anti-MET mAb L41G3 (catalog number, 3148), the rabbit anti-p-EGFR (Y1173) mAb 53A5 (catalog number, 4407), the mouse anti-pan AKT mAb 40D4 (catalog number, 2920S), the rabbit anti-p-AKT (S473) mAb D9E (catalog number, 4060), the rabbit anti-p-ERK1/2 (Thr202, Tyr204) mAb D13.14.4E (catalog number, 4370S) and the mouse anti-ERK1/2 mAb L34F12 (catalog number 4696S) were purchased from Cell Signaling Technology. Recombinant human HGF (catalog number, PHG0254), Alexa Fluor 647-conjugated anti-mouse antibody (catalog number, A21235) and Alexa Fluor 647-conjugated anti-human antibody (catalog number, A21445) were purchased from Life Technologies. Heparinase III (catalog number P0737S) was purchased from New England BioLabs. Heparin was purchased from Sigma Aldrich; and streptavidin conjugated to IRDye 680, from LI-COR.
Cell lines
All cell lines were maintained at 37°C in 5% CO2. The human brain cancer cell lines U87MG, U87MG.vIII and U87MG.wt(wild-type)EGFR, all obtained from Dr Frank Furnari (Ludwig Institute for Cancer Research, San Diego, CA), and the human lung cancer cell line A549 were maintained in DMEM/F-12 medium containing 5% fetal bovine serum (FBS) and 2.5 mM GlutaMAX with the appropriate selection antibiotic if required. The human lung cancer cell line HCC827 was maintained in DMEM/F-12 medium containing 20% FBS and 2.5 mM GlutaMAX. The human acute T-cell leukemia cell line Jurkat was maintained in DMEM/F-12 medium containing 10% FBS, non-essential amino acids and 2.5 mM GlutaMAX. The human breast cancer cell line T-47D, the human gastric cancer cell line GTL-16 and the human prostate cancer cell line DU 145 were maintained in RPMI-1640 containing 10% FBS and 2.5 mM GlutaMAX. Gliomasphere lines GBM-6 (provided by Dr Paul Mischel, Ludwig Institute for Cancer Research, CA), SB2, GBM-L1 and RN1 (provided by Dr Bryan Day, QIMR Berghofer Medical Research Institute) were maintained in StemPro neural stem cell (NSC) media containing DMEM/F-12, StemPro NSC SFM Supplement, 0.02 µg/mL basic fibroblast growth factor (bFGF), 0.02 µg/mL EGF, 2.5 mM GlutaMAX and penicillin/streptomycin (Life Technologies).
Immunoprecipitation and western blotting
Cells were washed twice in ice cold PBS after the indicated treatments and then lysed in TXC lysis buffer [1% (v/v) Triton X-100, 30 mM HEPES, 150 mM NaCl, 10 mM NaF, 2.5 mM activated Na3VO4 and protease/phosphatase inhibitor cocktails (Thermo Scientific), pH 7.4] for 20 min at 4°C. After centrifugation (21,000g, 4°C, 20 min), the protein concentration of the supernatant was determined by bicinchoninic acid (BCA) assay (Sigma), and whole cell lysates (WCLs) were subjected to immunoprecipitation and/or western blotting. Rilotumumab was biotinylated using EZ-Link Sulfo-NHS-LC-biotin (Thermo Scientific) for 2 h at 4°C and then dialyzed overnight in a 2,000-fold excess of ice-cold TBS. For the immunoprecipitation of MET, the anti-MET antibody C-28 conjugated to agarose beads was added to 50–200 µg total protein from WCLs and incubated overnight at 4°C with rotation. The immunoprecipitates were washed 5 times in excess TXC buffer and then boiled in 1× reducing LDS sample buffer. For western blotting, WCLs were prepared in 1× reducing or non-reducing LDS sample buffer. All samples were separated on 4–12% Bis-Tris SDS-PAGE gels, which were then equilibrated before transfer to polyvinylidene difluoride membranes using the iBlot Dry Blotting system (Life Technologies). The remainder of the procedure was as previously described18,25 using the appropriate primary and secondary antibodies (see Antibodies and reagents).
Immunofluorescence of rilo–HGF binding
Rilotumumab was complexed with HGF in SF-BSA for 1 h at room temperature at a 55:1 rilo:HGF molar ratio. This large molar excess of rilotumumab was used so that all of the available HGF would be bound by rilotumumab. Complete binding was also promoted by the strong affinity of rilotumumab for HGF (approximately 25 pM).
Cells were plated in 8-well confocal chamber slides (iBidi) at 5,000 cells per chamber and incubated overnight at 37°C, 5% CO2. Cells were washed once in serum-free medium containing 0.1% bovine serum albumin (SF-BSA medium) and then exposed to serum starvation in the same medium for 16 h. The pre-complexed rilo–HGF in SF-BSA at a 55:1 molar ratio or separate controls (including 10 µg/mL panitumumab, 10 µg/mL rilotumumab alone, 100 ng/mL HGF alone and vehicle) were added to cells at 4°C for 3 h, and the cells were then washed in ice cold PBS and fixed in 4% paraformaldehyde. Fixed cells were incubated with Alexa Fluor 647-conjugated anti-human antibody, diluted 1:100 in SF-BSA medium, for 1 h at 22°C. The cells were then washed in PBS and mounted in Duolink In Situ Mounting Medium with DAPI. Subsequent image acquisition was conducted using a DeltaVision deconvolution microscope on the relevant channels, and images were processed using ImageJ software.
Flow cytometry
For each test, 1 × 105 cells were stained for 3 h at 4°C with the appropriate primary antibody diluted in ice-cold SF-BSA medium. For cell-surface MET analyses, the cells were stained with 5 µg/mL LMH 85.24 For cell-surface EGFR studies, 10 µg/mL panitumumab was used as a positive control for human IgG detection. For rilo–HGF engagement studies, ice-cold pre-complexed rilo–HGF in SF-BSA medium at a 55:1 molar ratio or separate controls were added to the cells. For heparinase III pre-treatment, cells were incubated for 24 h with 0.7 U enzyme in SF-BSA medium. For heparin pre-blocking experiments, pre-formed rilo–HGF complex was incubated with 100 µg/mL heparin for 1 h. The cells were then washed 3 times in ice-cold SF-BSA medium, and anti-mouse Alexa Fluor 647-conjugated antibody (MET studies) or anti-human Alexa Fluor 647-conjugated antibody (rilotumumab studies) was added for 1 h at 4°C. After 3 further washes, the cells were analyzed on a FACSCanto II flow cytometer (BD Biosciences), and histograms were generated using FlowJo software.
xCELLigence-based cell scattering assay
A549 and DU 145 cells were seeded at 5,000 cells per well in 0.5% FBS-supplemented medium in 96-well E-Plates for 30 min before coupling to the xCELLigence apparatus (Roche) and carrying out 20 initial 2-min sweeps and then 100 30-min sweeps to monitor cell adhesion and growth. After 32 h, the treatments were added, and 60 1-min sweeps were carried out, followed by 999 30-min sweeps. Cell growth was normalized to growth at the time point immediately before treatment addition. Cell index curves were analyzed using xCELLigence software (Roche).
ATP-based cell viability assays
Measurement of DU 145 cell viability in response to HGF treatment was conducted using the CellTiter-Glo Luminescent Cell Viability Assay (Thermo Scientific) according to the manufacturer's instructions.
Supplementary Material
Disclosure of potential conflicts of interest
T.G.J. has received funding from Amgen.
Acknowledgments
We thank Dr D. Dadley-Moore for editing the original manuscript. This work was supported by the National Health and Medical Research Council (grant number APP1028552; T.G.J.). T.G.J. leads the Brain Cancer Discovery Collaborative, which is supported by the Cure Brain Cancer Foundation. We are grateful to the Victorian Government's Operational and Infrastructure Support Program.
References
- 1.Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer 2012; 12:89–103; PMID:22270953; http://dx.doi.org/ 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- 2.Kentsis A, Reed C, Rice KL, Sanda T, Rodig SJ, Tholouli E, Christie A, Valk PJ, Delwel R, Ngo V, et al.. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat Med 2012; 18:1118–22; PMID:22683780; http://dx.doi.org/ 10.1038/nm.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, et al.. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012; 487:500–4; PMID:22763439; http://dx.doi.org/ 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, et al.. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010; 17:77–88; PMID:20129249; http://dx.doi.org/ 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giordano S. Rilotumumab, a mAb against human hepatocyte growth factor for the treatment of cancer. Curr Opin Mol Ther 2009; 11:448–55; PMID:19649990. [PubMed] [Google Scholar]
- 6.Burgess T, Coxon A, Meyer S, Sun J, Rex K, Tsuruda T, Chen Q, Ho SY, Li L, Kaufman S, et al.. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res 2006; 66:1721–9; PMID:16452232; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3329. [DOI] [PubMed] [Google Scholar]
- 7.Iveson T, Donehower RC, Davidenko I, Tjulandin S, Deptala A, Harrison M, Nirni S, Lakshmaiah K, Thomas A, Jiang Y, et al.. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: an open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study. Lancet Oncol 2014; 15:1007–18; PMID:24965569; http://dx.doi.org/ 10.1016/S1470-2045(14)70023-3. [DOI] [PubMed] [Google Scholar]
- 8.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al.. The somatic genomic landscape of glioblastoma. Cell 2013; 155:462–77; PMID:24120142; http://dx.doi.org/ 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gherardi E, Sandin S, Petoukhov MV, Finch J, Youles ME, Ofverstedt LG, Miguel RN, Blundell TL, Vande Woude GF, Skoglund U, et al.. Structural basis of hepatocyte growth factor/scatter factor and MET signalling. Proc Natl Acad Sci U S A 2006; 103:4046–51; PMID:16537482; http://dx.doi.org/ 10.1073/pnas.0509040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubin JS, Day RM, Breckenridge D, Atabey N, Taylor WG, Stahl SJ, Wingfield PT, Kaufman JD, Schwall R, Bottaro DP. Dissociation of heparan sulfate and receptor binding domains of hepatocyte growth factor reveals that heparan sulfate-c-met interaction facilitates signaling. J Biol Chem 2001; 276:32977–83; PMID:11435444; http://dx.doi.org/ 10.1074/jbc.M105486200. [DOI] [PubMed] [Google Scholar]
- 11.Zioncheck TF, Richardson L, Liu J, Chang L, King KL, Bennett GL, Fugedi P, Chamow SM, Schwall RH, Stack RJ. Sulfated oligosaccharides promote hepatocyte growth factor association and govern its mitogenic activity. J Biol Chem 1995; 270:16871–8; PMID:7622503; http://dx.doi.org/ 10.1074/jbc.270.28.16871. [DOI] [PubMed] [Google Scholar]
- 12.de Parseval A, Elder JH. Binding of recombinant feline immunodeficiency virus surface glycoprotein to feline cells: role of CXCR4, cell-surface heparans, and an unidentified non-CXCR4 receptor. J Virol 2001; 75:4528–39; PMID:11312323; http://dx.doi.org/ 10.1128/JVI.75.10.4528-4539.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K, Possemato A, Nardone J, Innocenti G, Wetzel R, et al.. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A 2008; 105:692–7; PMID:18180459; http://dx.doi.org/ 10.1073/pnas.0707270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buus R, Faronato M, Hammond DE, Urbe S, Clague MJ. Deubiquitinase activities required for hepatocyte growth factor-induced scattering of epithelial cells. Curr Biol 2009; 19:1463–6; PMID:19699092; http://dx.doi.org/ 10.1016/j.cub.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fram ST, Wells CM, Jones GE. HGF-induced DU145 cell scatter assay. Methods Mol Biol 2011; 769:31–40; PMID:21748667; http://dx.doi.org/ 10.1007/978-1-61779-207-6_3. [DOI] [PubMed] [Google Scholar]
- 16.Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014; 511:543–50; PMID:25079552; http://dx.doi.org/ 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenall SA, Donoghue JF, Van Sinderen M, Dubljevic V, Budiman S, Devlin M, Street I, Adams TE, Johns TG. EGFRvIII-mediated transactivation of receptor tyrosine kinases in glioma: mechanism and therapeutic implications. Oncogene 2015; 34(41):5277–87. [DOI] [PubMed] [Google Scholar]
- 18.Pillay V, Allaf L, Wilding AL, Donoghue JF, Court NW, Greenall SA, Scott AM, Johns TG. The plasticity of oncogene addiction: implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia 2009; 11:448–58, 2 p following 58; PMID:19412429; http://dx.doi.org/ 10.1593/neo.09230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Cutsem E, Eng C, Nowara E, Swieboda-Sadlej A, Tebbutt NC, Mitchell E, Davidenko I, Stephenson J, Elez E, Prenen H, et al.. Randomized phase Ib/II trial of rilotumumab or ganitumab with panitumumab versus panitumumab alone in patients with wild-type KRAS metastatic colorectal cancer. Clin Cancer Res 2014; 20:4240–50; PMID:24919569; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin LP, Sill M, Shahin MS, Powell M, DiSilvestro P, Landrum LM, Gaillard SL, Goodheart MJ, Hoffman J, Schilder RJ. A phase II evaluation of AMG 102 (rilotumumab) in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol 2014; 132:526–30; PMID:24361733; http://dx.doi.org/ 10.1016/j.ygyno.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan CJ, Rosenthal M, Ng S, Alumkal J, Picus J, Gravis G, Fizazi K, Forget F, Machiels JP, Srinivas S, et al.. Targeted MET inhibition in castration-resistant prostate cancer: a randomized phase II study and biomarker analysis with rilotumumab plus mitoxantrone and prednisone. Clin Cancer Res 2013; 19:215–24; PMID:23136195; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-2605. [DOI] [PubMed] [Google Scholar]
- 22.Schoffski P, Garcia JA, Stadler WM, Gil T, Jonasch E, Tagawa ST, Smitt M, Yang X, Oliner KS, Anderson A, et al.. A phase II study of the efficacy and safety of AMG 102 in patients with metastatic renal cell carcinoma. BJU Int 2011; 108:679–86; PMID:21156020. [DOI] [PubMed] [Google Scholar]
- 23.Wen PY, Schiff D, Cloughesy TF, Raizer JJ, Laterra J, Smitt M, Wolf M, Oliner KS, Anderson A, Zhu M, et al.. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol 2011; 13:437–46; PMID:21297127; http://dx.doi.org/ 10.1093/neuonc/noq198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenall SA, Gherardi E, Liu Z, Donoghue JF, Vitali AA, Li Q, Murphy R, Iamele L, Scott AM, Johns TG. Non-agonistic bivalent antibodies that promote c-MET degradation and inhibit tumor growth and others specific for tumor related c-MET. PLoS One 2012; 7:e34658; PMID:22511956; http://dx.doi.org/ 10.1371/journal.pone.0034658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ymer SI, Greenall SA, Cvrljevic A, Cao DX, Donoghue JF, Epa VC, Scott AM, Adams TE, Johns TG. Glioma Specific Extracellular Missense Mutations in the First Cysteine Rich Region of Epidermal Growth Factor Receptor (EGFR) Initiate Ligand Independent Activation. Cancers (Basel) 2011; 3:2032–49; PMID:24212795; http://dx.doi.org/ 10.3390/cancers3022032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.