

ABSTRACT
From March 2014 through February 2015, the Ebola virus spread rapidly in West Africa, resulting in almost 30,000 infections and approximately 10,000 deaths. With no approved therapeutic options available, an experimental antibody cocktail known as ZMapp™ was administered to patients on a limited compassionate-use basis. The supply of ZMapp™ was highly constrained at the time because it was in preclinical development and a novel production system (tobacco plants) was being used for manufacturing. To increase the production of ZMapp™ for an uncertain future demand, a consortium was formed in the fall of 2014 to quickly manufacture these anti-Ebola antibodies in Chinese hamster ovary (CHO) cells using bioreactors for production at a scale appropriate for thousands of doses. As a result of the efforts of this consortium, valuable lessons were learned about the processing of the antibodies in a CHO-based system. One of the ZMapp™ cocktail antibodies, known as c13C6FR1, had been sequence-optimized in the framework region for production in tobacco and engineered as a chimeric antibody. When transfected into CHO cells with the unaltered sequence, 13C6FR1 was difficult to process. This report describes efforts to produce 13C6FR1 and the parental murine hybridoma sequence, 13C6mu, in CHO cells, and provides evidence for the insertion of a highly conserved framework amino acid that improved the physical properties necessary for high-level expression and purification. Furthermore, it describes the technical and logistical lessons learned that may be beneficial in the event of a future Ebola virus or other pandemic viral outbreaks where mAbs are considered potential therapeutics.
KEYWORDS: Anti-viral; ebola; manufacturability; thermal stability; ZMapp
Abbreviations
- EVD
Ebola virus disease
- mAb
monoclonal antibody
- FR1
framework 1
- mu
murine
- CHO
Chinese hamster ovary
- VL
variable light
- VH
variable heavy
- CDR
complementarity-determining region
- DHFR
dihydrofolate reductase
- GHT
glycine, hypoxanthine and thymidine
- NaCl
sodium chloride
- CEX
cation exchange chromatography
- CV
column volumes
- HIC
hydrophobic interaction chromatography
- AEX
anion exchange chromatography
- DSC
differential scanning calorimetry
- DS
drug substance
Introduction
The 2014/2015 Ebola virus epidemic in West Africa was the largest outbreak in history.1 This outbreak was caused by the Zaire species of the virus, which can have fatality rates up to 90%.2 Patients experiencing Ebola virus disease (EVD) display high viral loads that cause immune and vascular dysregulation. Major symptoms include fever, severe headache, muscle pain, weakness, fatigue, diarrhea, vomiting, abdominal pain and hemorrhaging.
There are currently no licensed vaccines or therapeutics for the treatment of EVD. Depending upon the resources available at a particular Ebola Treatment Unit (ETU), infected patients are treated with supportive-care rehydration of oral or intravenous fluids while oxygen and blood pressure levels are maintained. Therapeutic strategies targeting EVD include recombinant human activated protein, recombinant nematode anticoagulant protein c2, small interfering RNA, positively-charged phosphorodiamidate morpholino oligomers, the vesicular stomatitis virus vaccine, and monoclonal antibody (mAb) cocktails.2-11 Of these strategies, the mAb cocktail ZMapp™, in particular, has shown great promise in non-human primate studies,11 and it was compassionately administered to 9 patients during the 2014/2015 outbreak.12 ZMapp™ consists of 3 antibodies, c13C6FR1, c2G4, and c4G7, expressed in a species of tobacco, Nicotiana benthamiana. As a novel manufacturing system, infrastructure for production in this system is currently limited. However, there is substantial manufacturing infrastructure for production of mAbs in Chinese hamster ovary (CHO) cell lines.13 While manufacturing and clinical evaluation of tobacco-derived ZMapp™ continues, efforts are underway to produce the ZMapp™ anti-Ebola antibodies in CHO cells.
Antibody constant and variable domain sequences are known to affect binding specificity and biological activities such as pharmacokinetics and effector functions, but these sequences can also significantly influence antibody manufacturability, expression levels, downstream processing and formulation conditions required for stable long term storage.14,15 13C6FR1 proved to be particularly challenging with respect to CHO cell culturing and downstream processing. We thus interrogated the behavior of two 2 parental chimeric 13C6 versions. The two parental 13C6 versions, 13C6FR1 and 13C6mu, consisted of identical sequences except for framework 1 of the variable light (VL) and variable heavy (VH) domains. Framework 1 of 13C6FR1 VL contained 4 amino acid differences and the 13C6FR1 VH framework 1 contained 15 differences compared to 13C6mu. 13C6FR1 reflected the historical sequence optimization, which suggested key residues in VL and VH domains correlated with high expression in tobacco. 13C6mu was the original mouse hybridoma sequence.
Sequence analysis indicated many opportunities for engineered optimization for CHO cell expression, as well as potential improvements to antibody stability.15 However, time and resource constraints did not allow for the generation of hundreds of variants and subsequent binding and activity testing to identify improved antibodies. Therefore, only one modification was made to the C-terminal end of the VL in both parental versions as the composition was highly unusual.16 Human germline analysis indicated a conserved lysine was missing at position 148 (based on the AHo numbering scheme).17 The missing K148 shortens the linker length between the variable and constant regions and likely alters the surface charge properties of the antibody. We hypothesized that alterations of both parental versions could have an effect on stability and expression in CHO cells. Therefore, one variant was designed for each 13C6FR1 and 13C6mu with a lysine in position 148 and an arginine in position 149 of the VL. Although these modifications were distal to the complementarity-determining regions (CDRs), the paratope structure could still be altered and hence, potentially affect bioactivity.18 This study evaluated all 4 antibodies during cell culture and purification to assess the effect of sequence composition on manufacturability and product quality. Understanding the effect of sequence composition in a CHO production system could result in a molecule that is more manufacturable and would enable a quicker response to future epidemics.
Results
13C6 light chain variant
Both the parental 13C6FR1 and 13C6mu light chains have non-standard germline composition on the C-terminal end of the VL domain. Fig. 1 shows an alignment of 13C6FR1, 13C6mu, germline and the engineered variant, 13C6FR1_R148K,149R. The two amino acid positions boxed in red indicate the region of interest. The germline contains a lysine at position 148 and arginine at position 149, whereas the parental 13C6 contains an arginine at position 148 and nothing at position 149. The unusual parental composition in this region could have an effect on IgG stability, VL-VH interface interaction and expression.19 Therefore, position 148 was substituted with a lysine and an arginine was inserted at position 149 for both 13C6FR1_LC and 13C6mu_LC. The variants were named ‘13C6FR1 +K’ and ‘13C6mu +K’ due to the R148K substitution; this nomenclature is used in the sections below.
Figure 1.

Alignment of three 13C6 variable light regions, 13C6mu, c13C6FR1 and c13C6FR1_R148K_149R. The murine version is aligned to the closest murine germline whereas the human versions are aligned to the closest human germline. The C-terminal region of interest, AHo positions 148 and 149, is boxed in red.
Bioreactor production of the 13C6 variants in CHO cells
To evaluate the expression and product quality of the 13C6 variants, each antibody was transfected into a CHO DXB-11 clonal host cell line selected for growth in media lacking glycine, hypoxanthine and thymidine (-GHT media). Once viability of the –GHT selected pools reached greater than 85%, a 24-deep well plate production assay was performed to identify the highest expressing pool of each antibody. Due to time constraints, pools were not amplified with methotrexate and only the top-expressing -GHT pool of each antibody was carried forward into bioreactors (data not shown). Two liter (L) perfusion bioreactors were run in duplicate for 14 days to assess antibody titers and provide material for downstream processing. To ensure the –GHT pools did not overgrow, a temperature shift was implemented to control cell growth. Temperature shifts were based on cell density and did not occur on the same day for all the bioreactors. The 13C6FR1 parent and 13C6FR1 +K cultures were temperature shifted to 32.5° C on day 6, one day earlier than the 13C6mu and 13C6mu +K bioreactors. Despite an initial temperature shift to 32.5° C, continued growth was observed; therefore, all of the bioreactors were temperature shifted again on day 9 to 31.5° C to further suppress growth. Antibody production of each pool was monitored over the days of culture. The titer results for all of the variants are shown in Table 1 (it should be noted that these are a small number of unamplified pools and additional experimentation would be required to verify the effect of sequence on expression. Additionally, higher expressing clones would likely be selected during cell line development). All antibody sequences were verified by mass spectroscopy with 100% sequence coverage.
Table 1.
Day 14 titer and protein A pool HMW content for 13C6 variants.
| Construct | Day 14 titer (g/L) | HMW (%) |
|---|---|---|
| 13C6mu | 4.23 | 8.85 |
| 13C6mu +K | 5.79 | 7.20 |
| 13C6FR1 | 3.05 | 25.20 |
| 13C6FR1 +K | 5.13 | 6.25 |
Downstream processing
Purification of the 4 antibodies was performed to supply material for analytical characterization. In addition, the effects of the sequence modifications on downstream operations and general manufacturability were evaluated. Harvest for each of the antibodies was performed by centrifugation on day 14 of the cell culture production. Antibody capture from the harvested cell culture fluid was achieved by protein A chromatography. Polishing chromatography was performed by either HIC or CEX and the resulting pools were formulated with 20 mM acetate, 250 mM sucrose, pH 5. Typical performance was observed during protein A chromatography with all the antibodies.
The most notable differences between the antibodies were slight differences in elution pool turbidity and the high molecular weight (HMW) content of the protein A elution pool (Table 1), which ranged from 6.25% through 25.2%. 13C6FR1 had an exceptionally high HMW level, averaging 25.2%. The high HMW burden for 13C6FR1 would likely require multiple chromatography steps for aggregate reduction and result in an overall low yield. When comparing 13C6FR1 and 13C6FR1 +K, the lysine insertion significantly reduced the HMW content in the protein A pool (average of 6.25% for 13C6FR1 +K). 13C6mu and 13C6mu +K had similar HMW levels (7.2% - 8.85%). It should be noted that these are unamplified pools; clones producing lower HMW levels could potentially be selected during cell line development.
Cation exchange chromatography (CEX) is a common unit operation to reduce HMW content in mAb processes.20,21 The 13C6 variants were evaluated on the strong cation exchanger Fractogel SO3-. Fractions (0.5CV) were collected over the elution peak and analyzed for protein concentration and HMW content. A comparison of the purity plots (Fig. 2) show that under the same chromatography conditions, higher purity was achieved at higher yield for the 13C6 antibodies with the lysine insertion (13C6FR1 +K and 13C6mu +K). In contrast, the parental sequences (13C6FR1 and 13C6mu) had lower purity and recovery. It should be noted that the parental sequences both had higher starting HMW in the protein A pool compared to the lysine insertion variants. It is also likely that additional resin screening could be performed or the CEX operating conditions could be optimized to further improve yield and purity for each individual antibody.
Figure 2.
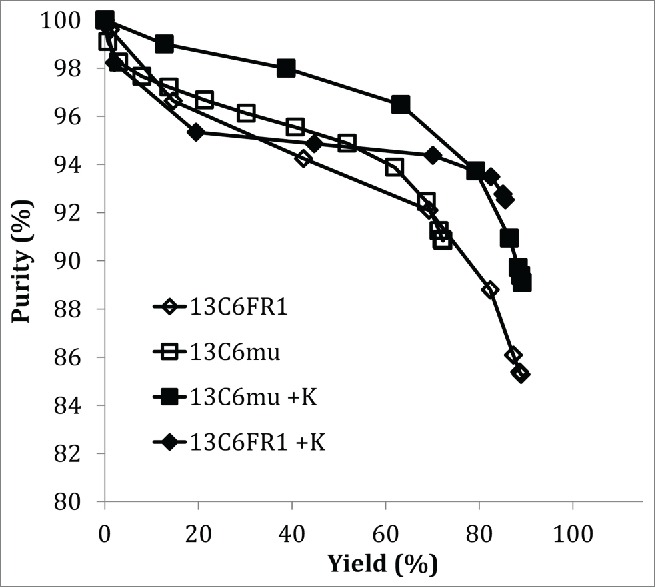
Purity plot for CEX chromatography.
Hydrophobic interaction chromatography (HIC) is also a common aggregate removal step for mAb products;20-22 therefore, the effect of the sequence changes on HIC performance was examined. Cumulative purity versus yield is shown in Fig. 3. When comparing the cumulative purity versus the cumulative yield, there was not a dramatic difference between 13C6mu and 13C6mu +K. In contrast, there was a significant difference between 13C6FR1 and 13C6FR1 +K. Higher step yield and overall purity was achieved for 13C6FR1 +K. Additionally, early eluting fractions for 13C6FR1 contained elevated HMW; if these fractions were removed from the product pool it would result in a further decrease in step yield. The HIC conditions tested were not designed for each specific molecule, and it is likely that the HIC conditions could be optimized to improve yield and purity for each antibody.
Figure 3.
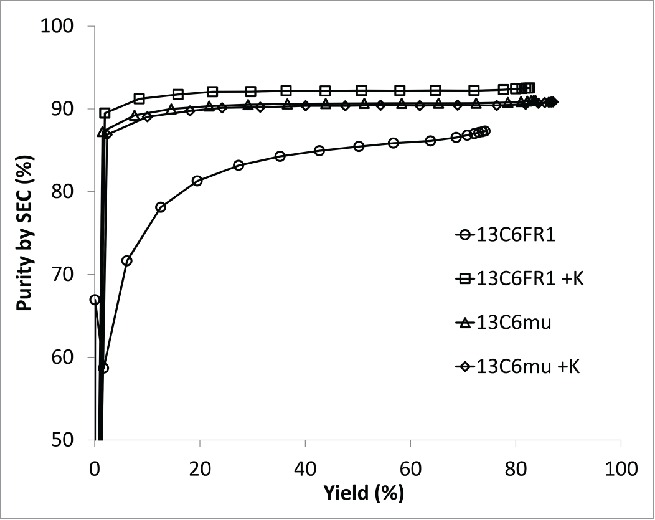
Purity plot for HIC chromatography.
In addition to step yield and chromatographic performance, the solution stability of the 13C6 variants was examined over a range of conditions that are relevant for downstream processing. In these experiments, the pH and salt strength of the protein A pool were varied from pH 5 to pH 7.5 and 0 mM to 200 mM NaCl by the addition of stock solutions. Light scattering measurements were taken at 405 nm in a microtiter plate on a Tecan plate reader after 24 hours. Contour plots derived from the model fits are shown in Fig. 4. The highest turbidity was observed with 13C6FR1. For 13C6FR1, as the pH increased and the salt concentration decreased, there was an increase in turbidity. The highest turbidity observed for the 13C6FR1 variant was ~0.2 AU at ≥pH 6 and ≤50 mM sodium chloride. While, in this system, turbidity has not been directly correlated to filter capacity, this level of precipitation could create column backpressure or filtration challenges in a manufacturing setting. As a result of the precipitation, there is also a smaller operating space for 13C6FR1, which could limit purification options (e.g., anion exchange chromatography (AEX)). In contrast, 13C6FR1 +K had A405 values ≤0.075 AU across the entire range tested. 13C6mu and 13C6mu +K both had A405 values ≤0.125 AU, and there did not appear to be a difference between the two 2 antibodies.
Figure 4.
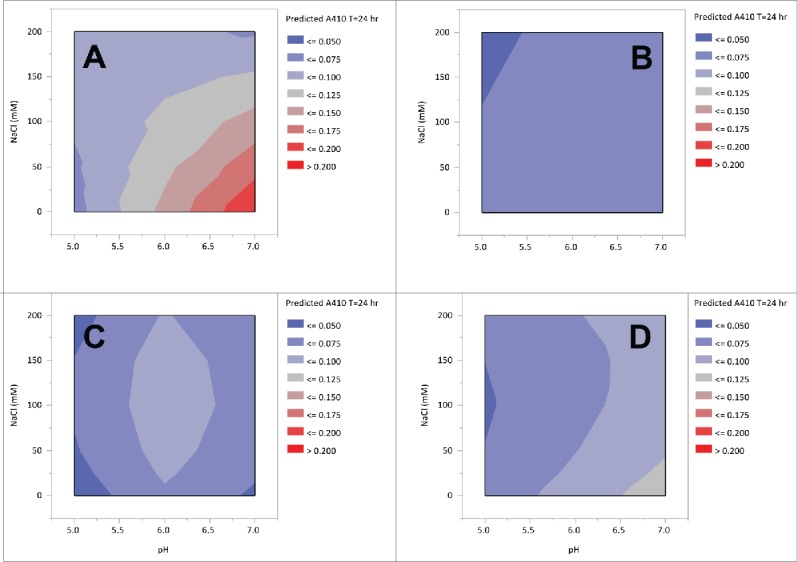
Contour plots of 13C6 variants protein A pool turbidity of pH and NaCl strength. Panel A: 13C6FR1, B: 13C6FR1 +K, C: 13C6mu, D: 13C6mu +K.
The effects of pH and ionic strength on aggregation were also evaluated with the 13C6 variants. In this study, the pH and salt strength of the protein A pool was varied from pH 5.0 to 7.5 and 0 to 200 mM NaCl by the addition of stock solutions. Samples were analyzed for HMW species by size-exclusion chromatography (samples were analyzed within 24 hours of preparation). Contour plots derived from the model fits are shown in Fig. 5. The highest HMW content was observed with 13C6FR1, and this aggregate content was strongly influenced by the pH. As the pH increased there was an increase in the HMW level, with an overall range of 11.6% through 19.7%. Salt strength had only a minor effect on 13C6FR1 HMW levels. The sensitivity to pH (and NaCl concentration) was eliminated with the lysine insertion (13C6FR1 +K). 13C6mu and 13C6mu +K were not sensitive to pH or NaCl concentration within the ranges tested. Aside from the absolute aggregate level, the most noticeable difference observed between the different sequences was the sensitivity to operating conditions. There were few limits relative to solution stability for variants 13C6FR1 +K, 13C6mu, and 13C6mu +K resulting in freedom to operate over a wide range of pH and salt strength when selecting downstream process options. In contrast, 13C6FR1 had a dramatic increase in HMW when the pH was increased, which would limit the downstream operating space.
Figure 5.

Contour plot of 13C6 variants protein A pool HMW as a function of pH and NaCl strength. Panel A: 13C6FR1, B: 13C6FR1 +K, C: 13C6mu, D: 13C6mu +K. White test indicates range of HMW measured across entire space.
The thermal stability of the different 13C6 variants in relevant downstream buffer systems was measured by a high throughput extrinsic fluorescence assay using SYPRO orange.23 The thermal transition temperatures measured by this method are shown in Table 2. Overall, 13C6FR1 had the lowest unfolding temperature in the downstream buffer conditions at ≤ 60.8°C under all conditions tested. The lysine insertion sequence (13C6FR1 +K) had a significantly higher unfolding temperature than 13C6FR1 for all buffer systems screened. Although higher than 13C6FR1, 13C6mu also had a fairly low unfolding temperature, which improved with the lysine insertion (13C6mu +K). There were no obvious trends for the parental sequences (13C6FR1 and 13C6mu) with respect to solution conditions. In contrast, the lysine insertion sequences (13C6FR1 +K and 13C6mu +K) both showed an increase in unfolding temperature as the solution pH increased (in downstream buffers). There was also a decrease in unfolding temperature as the NaCl concentrations increased; however, the difference was small. The relative differences in unfolding temperature can be indicative of solution stability (such as aggregation propensity in formulated drug substance or drug product) and susceptibility to stresses during downstream processing, such as surface mediated unfolding or solution dependent aggregation.23-26
Table 2.
Unfolding temperature (°C) as a function of pH and NaCl concentration.
| Construct | NaCl (mM) | pH 5 | pH 6 | pH 7.5 |
|---|---|---|---|---|
| 13C6FR1 | 0 | 60.8 | 60.6 | 58.6 |
| 50 | 59.2 | 59.6 | 59.0 | |
| 200 | 58.6 | 59.8 | 59.2 | |
| 13C6FR1 +K | 0 | 64.6 | 67.4 | 69.0 |
| 50 | 63.4 | 66.6 | 68.8 | |
| 200 | 62.8 | 65.6 | 68.4 | |
| 13C6mu | 0 | 62.2 | 62.6 | 61.6 |
| 50 | 61.0 | 61.8 | 61.4 | |
| 200 | 61.0 | 61.8 | 61.6 | |
| 13C6mu +K | 0 | 64.8 | 67.4 | 69.0 |
| 50 | 63.8 | 66.4 | 68.6 | |
| 200 | 62.2 | 66.0 | 68.4 |
Analytics/biophysical characterization
To better understand the differences in thermal stability, the different 13C6 variants were analyzed by differential scanning calorimetry (DSC) to evaluate thermal transition temperature (Tm) of the individual antibody domains (CH2, CH3, and Fab domains). The resulting DSC thermograms for 13C6FR1 and 13C6mu displayed a profile that suggested the Fab domain was the first domain to unfold, with transition temperatures of ∼66°C and 69°C, respectively (Fig. 6 and Table 3). 13C6FR1 +K and 13C6mu +K showed a significant shift in the thermogram profiles, in contrast to 13C6FR1 and 13C6mu. The addition of the lysine to both constructs resulted in an increase in the thermal stability of the Fab domain. The Tm for the Fab domain for 13C6FR1 +K and 13C6mu +K was between 8° C and 10° C higher than 13C6FR1 and 13C6mu. As a result of the increased Fab domain stability, the first unfolding event shifted from the Fab to the CH2 domain. Furthermore, the addition of lysine resulted in highly similar DSC profiles for 13C6FR1 +K and 13C6mu +K. The Tm for the CH2 and CH3 domains were similar for all of the 13C6 antibodies.
Figure 6.
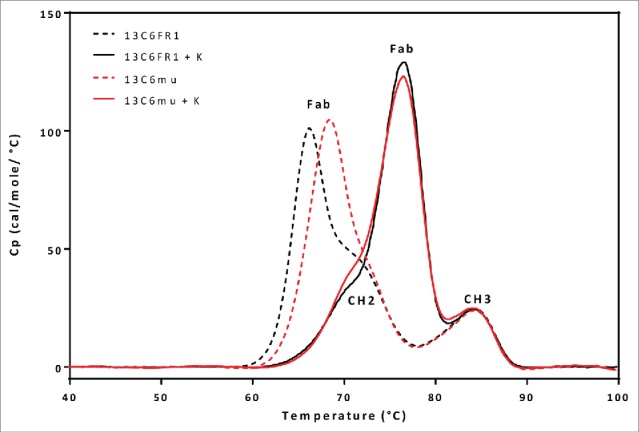
DSC comparison of 13C6FR1, 13C6FR1 +K, 13C6mu, and 13C6mu +K.
Table 3.
DSC average thermal transition temperatures.
| Tm (°C) |
|||
|---|---|---|---|
| Sample Name | Fab | CH2 | CH3 |
| 13C6FR1 | 66.0 | 71.2 | 83.8 |
| 13C6FR1 +K | 76.5 | 71.4 | 84.5 |
| 13C6mu | 68.2 | 72.8 | 83.7 |
| 13C6mu +K | 76.5 | 71.7 | 84.3 |
The Ebola Zaire binding ELISA was used to assess the binding activity of the CHO-produced 13C6 antibodies. Full dose response curves for the binding activity of the CHO produced 13C6 antibodies were compared to the binding activity of the tobacco-produced 13C6FR1 antibody currently used in ZMapp™ (tobacco c13C6FR1). The percent relative potency was calculated as a ratio of the EC50 values for the tobacco 13C6FR1 to the test sample (Table 4). 13C6FR1 compared to tobacco c13C6FR1 had a relative potency of 100%, demonstrating that the change in the expression system (tobacco vs CHO) had no effect on the binding of the molecule in the assay. 13C6FR1 +K showed a higher or increased percent relative potency compared to tobacco c13C6FR1 (121%).
Table 4.
% Relative potency for 13C6 antibodies.
| Sample Name | % Relative Potency |
|---|---|
| 13C6FR1 | 100 |
| 13C6FR1 +K | 121 |
| 13C6mu | 55 |
| 13C6mu +K | 73 |
13C6mu with and without the lysine insertion had lower percent relative potency compared to the tobacco 13C6FR1: 55% for 13C6mu and 73% for 13C6mu +K (Table 4). The reduced percent relative potency is likely due to the significant sequence differences in the variable region between the FR1 and mu variants. However, the addition of the lysine appears to increase the binding of the mu construct, but does not equate to equivalent binding to the 13C6FR1 CHO material. Whether these differences observed in the ELISA reflect changes to mAb binding to antigen or of secondary detector antibody binding to the mAb is unknown.
In silico modeling
To understand the potential mechanism of differential aggregation formation between the 13C6 variants, in silico homology models were constructed for each Fab and their exposed hydrophobicity was compared. The Spatial Aggregation Propensity algorithm27 revealed a motif that was intense in 13C6FR1, but was less intense with the lysine 148 insertion (Fig. 7). This predicted aggregation-prone region might have been sufficient to have induced aggregation formation of 13C6FR1, whereas the phenomena was remediated in the presence of lysine 148 or arginine 149. Both 13C6mu and 13C6mu +K exhibited similar aggregation behavior whether K148 and R149 was present or absent. This result is probably explained by the lack of the 13C6FR1 predicted aggregation-prone region shown in Fig. 7. The different VL residue content in framework 1 between the murine and the tobacco-optimized 13C6FR1 (Fig. 1) manifests as the lack of the intense aggregation-prone region revealed in 13C6FR1. The residue differences in framework 1 of the VL were revealed to be within the predicted aggregation-prone region shown in Fig. 7. The residue differences in framework 1 of the VL were revealed to be within the predicted aggregation-prone region shown in Fig. 7.
Figure 7.
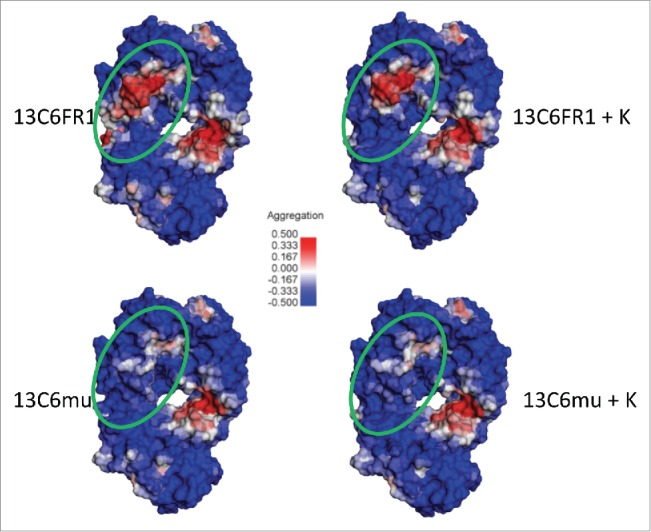
13C6 Fab models for visualization of Spatial Aggregation Propensity (SAP) (Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 4.5, San Diego: Dassault Systèmes, 2015). The SAP contour heat map consists of regions of high aggregation propensity (in red), low aggregation propensity (blue) and neutral (white). The Fabs are oriented with the complementarity-determining regions (CDRs) at the top of each model and the light chain on the left side. The green circle specifies a region that differs in SAP intensity between 13C6FR1 and 13C6FR1 +K but is similar between 13C6mu and 13C6mu +K. The major contributors of exposed hydrophobicity in this potentially aggregation-prone region are P8, L9, and P11 in the 13C6FR1 variable light domain. 13C6mu consists of less hydrophobic residues at these positions (Q8, K9, and M11). The presence of K148 is hypothesized to remediate much of the observed antibody aggregation of 13C6FR1 by dampening or modulating the exposed hydrophobicity in this SAP region.
Discussion
In early 2014, the largest outbreak of Ebola to date resulted in more deaths than all previous Ebola outbreaks combined. By fall of that year, the situation was worsening, and the trajectory of new infections in West Africa and globally was uncertain.
At that time, the worldwide focus on solutions was broad-based and included the development of new therapies such as the ZMapp™ antibody cocktail, which had displayed promising preclinical results, but was not available in sufficient quantity to address the scope of this epidemic. To meet the medical need during this large-scale Ebola outbreak, a consortium of non-profit, governmental, and industrial partners was formed to deliver sufficient anti-Ebola mAbs derived from CHO cells as rapidly as possible. The objectives of the consortium were to produce thousands of doses of anti-Ebola mAbs, with product attributes as comparable as possible to tobacco-derived ZMapp™ and initiate a clinical study by June 1, 2015.
The Ebola outbreak subsided in early 2015 and the need for large quantities of clinical doses diminished. Through the efforts of the consortium, technical advances were made in the processing of CHO-derived anti-Ebola antibodies, specifically for 13C6FR1, which may be beneficial in the event of a future Ebola virus outbreak or other pandemic viral outbreaks where mAbs are considered potential therapeutics.
This study has demonstrated that the sequence of a mAb can effect both expression and biophysical behavior, which can strongly affect manufacturability. For 13C6FR1, the insertion of a single lysine at position 148 affected aggregation, reducing HMW levels from 25% to 6%, which was similar to the HMW levels of 13C6mu and 13C6mu +K. Decreased aggregation propensity of 13C6FR1 +K compared to 13C6FR1 could be due to a structural change and improvement in thermal stability, as evidenced by the significant increase in the Tm for the Fab domain of 13C6FR1 +K. The presence of additional electropositive charge from arginine 149 could also have contributed to the observed reduction in aggregate formation.
Studies were also performed to assess the effect of sequence modification on upstream and downstream operations. From an upstream perspective, antibody titers were higher in the presence of the lysine insertion in these unamplified pools. Expression levels will most likely increase upon methotrexate amplification and after clone screening. However, due to time constraints, these experiments were not performed, and therefore whether the same titer differences will be found after more intensive cell line development is not known.
In multiple downstream conditions, 13C6FR1 was challenging due to higher initial aggregate levels as well as a higher propensity to aggregate and precipitate. As demonstrated by these data, 13C6FR1 had the most limited operating space with regard to pH and NaCl concentrations. For 13C6FR1, higher operational pH resulted in elevated levels of HMW. Additionally, at elevated pH and lower conductivity, there was an increase in product precipitation. Ultimately, the observed solution instability would limit the available operational space for 13C6FR1. For example, with 13C6FR1, operation at greater than pH 5.5 would cause unacceptable increases in HMW content and turbidity, and could result in heavy yield losses to meet drug substance (DS) product quality targets.
Insertion of the lysine in the light chain resulted in lower initial aggregate levels and decreased the susceptibility to aggregation and precipitation under downstream conditions. Additionally, the lysine insertion had a positive effect on step yield for 13C6FR1. At similar purity, 13C6FR1 +K had a 27.6% improvement in step yield over 13C6FR1 during CEX chromatography and an 8.6% improvement in step yield during HIC chromatography. Overall, the improved downstream behavior of 13C6FR1 +K resulted in lower DS HMW, higher yield, and a wider potential downstream operating space.
While this study demonstrated no negative effect on in vitro binding as measured by ELISA, data suggests 13C6 may act through effector function binding.6 Cell-based potency and other effector function assays were not employed here. Preclinical animal studies are currently being conducted to better understand the potential effect of sequence optimization on in vivo efficacy.
In the process of working through the issues of rapidly producing anti-Ebola antibodies, there were additional technical and logistical lessons learned that should be shared. First, given the high viral load of the Ebola virus among infected patients, the dose is relatively high (∼1-10 grams of antibody cocktail per cycle). For the treatment of thousands of patients, perhaps as much as 100 kg of cocktail antibodies may be needed, which could require several months of production in a multi-2000L CHO-cell bioreactor facility. Engineering antibody sequences to improve expression and purification yield is a powerful tool in reducing mass losses during manufacturing. Second, the cycle time from outbreak to resolution of an epidemic can be short (months in duration) relative to typical mAb therapeutic development timelines (years in duration) and therefore a “rapid response” approach is critical. It is not always feasible to re-engineer antibody sequences, transfect new cell lines, establish reliable production capacity, bridge results from clinical trial information, or establish product comparability between innovator and modified product in the timeframe required for a rapid response. As much as possible, these critical issues must be addressed in advance to ensure the mAb product is available to reduce the effect of a viral outbreak such as EVD. Third, the regulatory pathways for compassionate use, in the case of a worldwide threat, should be streamlined as much as possible and clearly defined in advance. Specifically, this consortium contemplated producing GMP batches with unamplified pools rather than amplified clones. To challenge conventional regulatory pathways, product quality from unamplified pool mAb production should be generated to demonstrate lot-to-lot consistency. These data could shift the current regulatory agency paradigm, at least for cell lines where these data can be established, thus enabling a more rapid response time in the event of a future Ebola type outbreak. Current regulatory guidances and pathways, as well as consistent approaches from the various agencies, are not established for a fast response to such a paradigm shift, which if left unprepared for, will no doubt produce significant delays in responding to future outbreaks.
The commercial institutions that participated in this work should be commended for their participation and collaboration. They did so with the knowledge that they could leverage their capabilities and available capacity to help with the public health threat this outbreak posed, but also with some risk to their ongoing corporate obligations. However, future response to viral outbreaks would greatly benefit from a global strategy that focuses on technologies that enable rapid development and production of improved molecules, and provides access to manufacturing capacity, experienced operations staff, cell lines, and raw materials that will result in a robust and rapid response.
Materials and methods
Material
Tobacco produced 13C6FR1 (control), CHO produced 13C6 FR1 ±K insertion, CHO produced 13C6 mu ± K insertion.
Molecular biology
Protein sequences for antibodies 13C6FR1 and 13C6mu were provided by Mapp Biopharmaceutical (San Diego, CA). The sequences were back-translated into codons that were optimized for mammalian expression using VectorNTI (Life Technologies, Carlsbad, CA). The antibody coding DNA along with the VK1|012 signal peptide were synthesized by Integrated DNA Technologies (Coralville, IA). The light chain variants were generated using the Quikchange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Both strands of the entire plasmid for each of the 6 DNA constructs were sequence confirmed using BigDye Terminator sequencing (Life Technologies, Carlsbad, CA).
Transfections
A CHO DXB11 host cell line was transfected with expression vectors containing 13C6 constructs as depicted in Figure 1. DNA was linearized with Pvu1, followed by ethanol precipitation. Four pools of each condition were transfected in 96-well plates (Bio-Rad Gene Pulser MXcell Electroporation System) following the high DNA content high-throughput electroporation method.28 Cells were selected in GHT-depleted media. Once viability of the pools reached > 85%, cells were seeded into a 24-deep well plate production assay. All four pools of both 13C6FR1 and 13C6FR1 +K were seeded into production in triplicate, whereas 13C6mu constructs took slightly longer to recover from -GHT selection and did not have as many replicates. Productions were seeded at 10 × 106 c/mL × 2mLs of cell culture medium in 24-deep well plates at 36°C, 5% CO2, shaking at 225 rpms. On days 1-3, plates were centrifuged at 1000 rpm for 5 minutes, 70% of the medium was removed and replaced with fresh medium. Samples were harvested on day 4 and analyzed for titer. The highest producing pool titer from each condition was selected and scaled-up for bioreactors.
Bioreactor experiments
Minus GHT pools from each construct were grown in shake flasks in 5% CO2, humidified Kuhner incubators (Basel, Switzerland) for the N-3 and N-2 stages. Cells were subsequently seeded into 3 L stirred-tank bioreactors for the N-1 stage and N (production). Chemically-defined medium with no animal-derived components was used throughout the growth phase and production. pH and dissolved oxygen setpoints were maintained throughout the course of production. pH was controlled with 1.0 M sodium carbonate and carbon dioxide gas. Temperature setpoint was lowered to control cell growth as cells reached high cell densities. Ex-Cell™ antifoam (SAFC) was used as needed to control foaming.
Viable cell density (VCD) and viability were determined using a ViCell XR automated cell counter (Beckman Coulter, Brea, CA). The osmolality was determined using a Model 2020 osmometer (Advanced Instruments, Norwood, MA). pH and CO2 readings were determined using a Rapidlab 1260 Blood Gas Analyzer (Siemens, Malvern, PA). Titer was determined using affinity protein A ultra-high performance liquid chromatography (Waters, Milford, MA). Glucose and lactate were measured using a Bioprofile Flex (Nova Biomedical, Waltham, Ma.). All 3 L bioreactors were manufactured by Applikon (Delft, Netherlands). Supernatant was harvested on day 14 by centrifugation for 30 minutes at 3000 rpm, and filtered through a Pall Acropak 500 capsule)
Sequence confirmation
The sequence of each antibody was confirmed using mass spectrometry.29
Size-exclusion chromatography analysis of aggregation
60 µg of each sample was separated isocratically at ambient temperature (~22° C) using a sodium phosphate mobile phase (100 mM sodium phosphate, 250 mM sodium chloride, pH 6.8) on a 4.6 × 150 mm, 1.7 µm Waters Acquity BEH200 SEC column. The column flow rate was 0.4 mL/min and detection was monitored by A280 with a method run time of 6 minutes.
High throughput screening
Downstream HTS studies were performed using a Tecan Freedom EVO™ robotic liquid handling system (Tecan US, Research Triangle Park, NC, USA). The Tecan was configured with Te-Chrom components to facilitate automated micro-chromatography. Measurements for concentration and turbidity were performed on an integrated microplate reader. Resins were acquired as pre-packed 450 µL Robocolumns from Atoll (Weingarten, Germany). Product pools were collected as fractions (1 CV/fraction) in 96-deep well plates (Qiagen Sciences, Germantown, Maryland, USA) for further analysis. Results from individual fractions were used to generate pseudo-chromatograms. Cumulative results from each fraction were used to calculate yield and mass balance.
Bench-scale purification experiments
Bench-scale chromatography experiments were conducted using an Akta Explorer 100 (GE Healthcare, Piscataway, NJ, USA). Resins were packed into 1.15 cm ID Vantage columns (EMD Millipore, Billerica, MA, USA) to a bed height of ~10–25 cm). The resins MabSelect SuRe™ and Phenyl Sepharose 6 Fast Flow hi sub were acquired from GE Healthcare (Piscataway, NJ, USA) and Fractogel SO3- was acquired from EMD Millipore (Billerica, MA, USA). Protein A chromatography was used as initial capture and purification from the harvested cell culture fluid. The second chromatography step was either CEX (Fractogel SO3-) performed in BEM or HIC (Phenyl Sepharose 6 Fast Flow hi sub) performed in FTM. For CEX, following equilibration, the column was loaded with neutralized protein A pool, washed with equilibration buffer, and eluted using a NaCl linear gradient over 10 column volumes. For HIC, the protein A pools were conditioned to the target loading conditions and then applied to the column and followed by a 6 column volume (CV) wash with equilibration buffer. The load flow through and wash volumes were collected as the process pool. Purified product pools were formulated to 20 mM acetate, 250 mM sucrose pH 5 using a 30 kD regenerated cellulose Tangential Flow Filtration membrane (EMD Millipore, Billerica, MA, USA). Polysorbate 80 was added to the final product pool to a concentration of 0.01% (v/v).23
Potency
The ELISA is designed to monitor the dose-dependent binding of anti-Ebola antibodies to the Ebola Zaire glycoprotein. The glycoprotein was coated onto a 96-well polystyrene microtiter assay plate. After coating, the plate was blocked with reagent to prevent non-specific binding of the anti-Ebola antibodies. A dilution series of the reference material (RS) and test samples were then added to the plate. The amount of bound anti-Ebola antibody was then detected by an anti-human kappa horseradish peroxidase-conjugated detection antibody, followed by the chromogenic substrate tertramethyl benzidine. The percent activity of each test sample was then calculated from the EC50 values of the test sample and the reference standard dose-response curves.
Differential scanning calorimetry
DSC analysis was performed using a Capillary VP-DSC (MicroCal, Northampton, Massachusetts) with a scan rate of 1°C/ min and no feedback. Immediately prior to analysis, all samples were diluted to ~0.5 mg/mL in 20 mM acetate, 250 mM sucrose, 0.01% polysorbate 80 pH 5.0. The DSC profiles were analyzed using MicroCal Origin v 7.0 software (MicroCal, Northampton, Massachusetts). All theromograms were baseline corrected and normalized to the moles of protein loaded. The Tm for each sample was determined using the MicroCal Origin Non-2State fitting algorithm with 3 peaks.
In silico modeling
Fv models for 13C6FR1 and 13C6mu were made using the Antibody Modeler in the Molecular Operating Environment (MOE, Chemical Computing Group, Montreal CA). The Amber:EHT 10 force field was used with GB/VI ranking to arrive at the working Fab model. Non-ideality (e.g. non-proline cis-peptides, atom clashes) in the model was corrected manually. Low-mode molecular dynamics was performed on the CDR3_VH loop and any non-ideality (described above) was corrected. Lysine was then inserted at position 148, which resulted in shifting R148 to become R149. Following lysine insertion, the linkers between the variable and constant region were energy minimized to allow for optimal residue interactions, followed by solid-body energy minimization to allow for optimal domain-domain interactions.The Spatial Aggregation Propensity algorithm within Discovery Studio (Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 4.5, San Diego: Dassault Systèmes, 2015) was used to calculate potential aggregation-prone regions for each of the four 4 antibodies. Each Fab was loaded into Discovery Studio and Prepare Protein was performed with the CHARMm force field applied. The Cutoff Radius parameter was set to 10 Å and all other settings were default.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank the Bill & Melinda Gates Foundation for support of this work (OPP 1126570) and guidance from Steve Hadley at the foundation. The authors wish to acknowledge Alison Moore and Maureen Halligan from Amgen for coordinating and providing resources, including lab space, equipment and supplies throughout the anti-Ebola consortium efforts. Skillful technical support was provided by Scott Freeman, Sheila Kingrey-Gebe, Kim Hardy, Bridget Sessions, Vladimir Razinkov, Lance Horton, Tim Wanek, Neeraj Agrawal, and Connie Hickey. The authors would also like to acknowledge Randal R. Ketchem, Jeff McGrew, Randal Bass, and Victor Fung for advice and critical review of the manuscript.
References
- 1.World Health Organization Ebola virus disease [Internet]. 2015; Available from: http://www.who.int/mediacentre/factsheets/fs103/en/ [Google Scholar]
- 2.Geisbert TW, Hensley LE, Jahrling PB, Larsen T, Geisbert JB, Paragas J, Young HA, Fredeking TM, Rote WE, Vlasuk GP. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet [Internet] 2003; 362:1953-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14683653; PMID:14683653; http://dx.doi.org/ 10.1016/S0140-6736(03)15012-X [DOI] [PubMed] [Google Scholar]
- 3.Feldmann H, Jones SM, Daddario-DiCaprio KM, Geisbert JB, Stroher U, Grolla A, Bray M, Fritz EA, Fernando L, Feldmann F, et al.. Effective post-exposure treatment of Ebola infection. PLoS Pathog [Internet] 2007; 3:e2. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17238284; PMID:17238284; http://dx.doi.org/ 10.1371/journal.ppat.0030002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geisbert TW, Lee AC, Robbins M, Geisbert JB, Honko AN, Sood V, Johnson JC, de Jong S, Tavakoli I, Judge A, et al.. Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference: a proof-of-concept study. Lancet [Internet] 2010; 375:1896-905. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20511019; PMID:20511019; http://dx.doi.org/ 10.1016/S0140-6736(10)60357-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hensley LE, Stevens EL, Yan SB, Geisbert JB, Macias WL, Larsen T, Daddario-DiCaprio KM, Cassell GH, Jahrling PB, Geisbert TW. Recombinant human activated protein C for the postexposure treatment of Ebola hemorrhagic fever. J Infect Dis [Internet] 2007; 196 Suppl :S390-9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17940975; PMID:17940975; http://dx.doi.org/ 10.1086/520598 [DOI] [PubMed] [Google Scholar]
- 6.Olinger GG Jr., Pettitt J, Kim D, Working C, Bohorov O, Bratcher B, Hiatt E, Hume SD, Johnson AK, Morton J, et al.. Delayed treatment of Ebola virus infection with plant-derived monoclonal antibodies provides protection in rhesus macaques. Proc Natl Acad Sci U S A [Internet] 2012; 109:18030-5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23071322; PMID:23071322; http://dx.doi.org/ 10.1073/pnas.1213709109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pettitt J, Zeitlin L, Kim H, Working C, Johnson JC, Bohorov O, Bratcher B, Hiatt E, Hume SD, Johnson AK, et al.. Therapeutic intervention of Ebola virus infection in rhesus macaques with the MB-003 monoclonal antibody cocktail. Sci Transl Med [Internet] 2013; 5:199ra113. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23966302; PMID:23966302 [DOI] [PubMed] [Google Scholar]
- 8.Qiu X, Audet J, Wong G, Pillet S, Bello A, Cabral T, Strong JE, Plummer F, Corbett CR, Alimonti JB, et al.. Successful treatment of ebola virus-infected cynomolgus macaques with monoclonal antibodies. Sci Transl Med [Internet] 2012; 4:138ra81. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22700957; PMID:22700957 [DOI] [PubMed] [Google Scholar]
- 9.Qiu X, Wong G, Audet J, Bello A, Fernando L, Alimonti JB, Fausther-Bovendo H, Wei H, Aviles J, Hiatt E, et al.. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature [Internet] 2014; 514:47-53. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25171469; PMID:25171469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu X, Wong G, Fernando L, Audet J, Bello A, Strong J, Alimonti JB, Kobinger GP. mAbs and Ad-vectored IFN-alpha therapy rescue Ebola-infected nonhuman primates when administered after the detection of viremia and symptoms. Sci Transl Med [Internet] 2013; 5:207ra143. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24132638; PMID:24132638 [DOI] [PubMed] [Google Scholar]
- 11.Warren TK, Warfield KL, Wells J, Swenson DL, Donner KS, Van Tongeren SA, Garza NL, Dong L, Mourich D V, Crumley S, et al.. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat Med [Internet] 2010; 16:991-4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20729866; PMID:20729866; http://dx.doi.org/ 10.1038/nm.2202 [DOI] [PubMed] [Google Scholar]
- 12.Gallagher J. Ebola: Experimental drug ZMapp is “100% effective” in animal trials [Internet]. BBC News 2014; Available from: http://www.bbc.com/news/health-28980153 [Google Scholar]
- 13.Chartrain M, Chu L. Development and production of commercial therapeutic monoclonal antibodies in Mammalian cell expression systems: an overview of the current upstream technologies. Curr Pharm Biotechnol [Internet] 2008. [cited 2015 Aug 3]; 9:447-67. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19075685; PMID:19075685; http://dx.doi.org/ 10.2174/138920108786786367 [DOI] [PubMed] [Google Scholar]
- 14.Ó'Fágáin C. Engineering protein stability In: D Walls, Loughran ST, editors. Protein Chromatography. Springer; New York Dordrecht Heidelberg London; 2011. page 103-36. [Google Scholar]
- 15.Igawa T, Tsunoda H, Kuramochi T, Sampei Z, Ishii S, Hattori K. Engineering the variable region of therapeutic IgG antibodies. MAbs [Internet] 2011; 3:243-52. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21406966; PMID:21406966; http://dx.doi.org/ 10.4161/mabs.3.3.15234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giudicelli V, Chaume D, Lefranc M-P. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res 2005; 33:D256-61; PMID:15608191; http://dx.doi.org/ 10.1093/nar/gki010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Honegger AM, Plückthun A. Yet another numbering scheme for immunoglobulin variable domains: an automatic modeling and analysis tool. J Mol Biol 2001; 309:657-70; PMID:11397087; http://dx.doi.org/ 10.1006/jmbi.2001.4662 [DOI] [PubMed] [Google Scholar]
- 18.Janda A, Eryilmaz E, Nakouzi A, Cowburn D, Casadevall A. Variable region identical immunoglobulins differing in isotype express different paratopes. J Biol Chem [Internet] 2012; 287:35409-17. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22930758; PMID:22930758; http://dx.doi.org/ 10.1074/jbc.M112.404483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buchanan A, Clementel V, Woods R, Harn N, Bowen M a., Mo W, Popovic B, Bishop SM, Dall'Acqua W, Minter R, et al.. Engineering a therapeutic IgG molecule to address cysteinylation, aggregation and enhance thermal stability and expression. MAbs 2013; 5:255-62; PMID:23412563; http://dx.doi.org/ 10.4161/mabs.23392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yigzaw Y, Hinckley P, Hewig A, Vedantham G. Ion exchange chromatography of proteins and clearance of aggregates. Curr Pharm Biotechnol [Internet] 2009; 10:421-6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19519418; PMID:19519418; http://dx.doi.org/19519419 10.2174/138920109788488842 [DOI] [PubMed] [Google Scholar]
- 21.Lu Y, Williamson B, Gillespie R. Recent advancement in application of hydrophobic interaction chromatography for aggregate removal in industrial purification process. Curr Pharm Biotechnol [Internet] 2009; 10:427-33. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19519419; PMID:19519419; http://dx.doi.org/ 10.2174/138920109788488897 [DOI] [PubMed] [Google Scholar]
- 22.Gagnon P. Purification Tools for Monoclonal Antibodies. 1st ed. Tucson (AZ); Validated Biosystems Inc. 1996; 252p. [Google Scholar]
- 23.He F, Phan DH, Hogan S, Bailey R, Becker GW, Narhi LO, Razinkov VI. Detection of IgG aggregation by a high throughput method based on extrinsic fluorescence. J Pharm Sci [Internet] 2010; 99:2598-608. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20039384; PMID:20039384 [DOI] [PubMed] [Google Scholar]
- 24.Brader ML, Estey T, Bai S, Alston RW, Lucas KK, Lantz S, Landsman P, Maloney KM. Examination of thermal unfolding and aggregation profiles of a series of developable therapeutic monoclonal antibodies. Mol Pharm [Internet] 2015; 12:1005-17. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25687223; PMID:25687223; http://dx.doi.org/ 10.1021/mp400666b [DOI] [PubMed] [Google Scholar]
- 25.Gillespie R, Nguyen T, Macneil S, Jones L, Crampton S, Vunnum S. Cation exchange surface-mediated denaturation of an aglycosylated immunoglobulin (IgG1). J Chromatogr A [Internet] 2012; 1251:101-10. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22771262; PMID:22771262; http://dx.doi.org/ 10.1016/j.chroma.2012.06.037 [DOI] [PubMed] [Google Scholar]
- 26.Mueller M, Loh MQ, Tscheliessnig R, Tee DH, Tan E, Bardor M, Jungbauer A. Liquid formulations for stabilizing IgMs during physical stress and long-term storage. Pharm Res [Internet] 2013; 30:735-50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23143394; PMID:23143394; http://dx.doi.org/ 10.1007/s11095-012-0914-2 [DOI] [PubMed] [Google Scholar]
- 27.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci U S A 2009; 106:11937-42; PMID:19571001; http://dx.doi.org/ 10.1073/pnas.0904191106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barsoum J. Introduction of stable high-copy-number DNA into Chinese hamster ovary cells by electroporation. DNA Cell Biol [Internet] 1990. [cited 2015 Aug 5]; 9:293-300. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2112395; PMID:2112395; http://dx.doi.org/ 10.1089/dna.1990.9.293 [DOI] [PubMed] [Google Scholar]
- 29.Rogers RS, Nightlinger NS, Livingston B, Campbell P, Bailey R, Balland A. Development of a quantitative mass spectrometry multi-attribute method for characterization, quality control testing and disposition of biologics. MAbs [Internet] 2015:00-00. PMID:26186204; http://dx.doi.org/ 10.1080/19420862.2015.1069454 [DOI] [PMC free article] [PubMed] [Google Scholar]