

Abstract
The phosphatidylinositol 3-kinase (PI3K) signaling pathway regulates many important cellular functions. The functional impact of deregulating the PIK3CA gene, encoding the p110α catalytic subunit of PI3K, is validated by frequent gain of function mutations in a range of human cancers. We generated a mouse model with an inducible constitutively active form of PI3K. In this model Cre recombinase activates expression of a myristoylated form of p110α (myr-p110α). The myristoylated version of p110α brings the protein to the cytoplasmic side of the cell membrane, which mimics the normal activation mechanism for the p110α catalytic subunit and activates the PI3K enzyme. Constitutively activated PI3K signaling induced by myr-p110α in all cells of the developing mouse caused lethality during embryonic development. Transgenic Cre;myr-p110α heterozygous embryos displayed morphological malformation and poor vascular development with extremely dilated blood vessels and hemorrhage in the embryo and the extraembryonic yolk sac. Previous studies demonstrated that loss of p110α during embryonic development causes angiogenic disruption and here we show that constitutive activation of p110α by gain of function mutation during development also disrupts vasculogenesis/angiogenesis in what appears to be a similar manner. These finding demonstrate the importance of tight regulation of PI3K signaling during embryonic vasculogenesis/angiogenesis..
Keywords: PIK3CA, p110α, Cre recombinase, myristoylated, lethality, embryonic development, morphological malformation, hemorrhage, vasculogenesis, angiogenesis
1 Introduction
The phosphatidylinositol 3-kinase (PI3K) signaling pathway controls a range of fundamental cellular processes involving metabolism, growth, proliferation, and survival signaling in response to a variety of extracellular stimuli [1-3]. The PI3K isoforms in mammalian cells can be divided into class I, II, and III, based on their structural characteristics, substrate specificity, distribution, mechanisms of activation, and functions [4-6]. Based on different associated adaptors and the mechanism of PI3K activation, class I PI3Ks are further classified into class IA and IB PI3Ks [4-6]. Although signaling through all classes of PI3Ks have key regulatory roles in many cellular processes, only class IA enzymes are implicated in human cancers. Class IA PI3Ks are heterodimers of a p85 regulatory subunit and a p110 catalytic subunit [4-7].
In the basal state, the p110 catalytic subunit is bound to the p85 regulatory subunit through its N-terminus and its catalytic activity is kept at a low state [6,7]. Upon the appropriate ligand binding on the extracellular domain of cell surface receptors, such as receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs), the receptors are phosphorylated and activated [8]. The p85α regulatory subunit binds to activated RTKs, causing conformational changes that release the inhibitory interaction of p85α on p110α and lead to anchorage of the p85α/p110α complex to the cell membrane where PI3K is active [9-11]. Membrane-localized PI3K catalyzes the production of phosphatidylinositol-3,4,5-trisphosphate (PIP3) at the plasma membrane. PIP3 is an important signal for many cellular mechanisms and facilitates the recruitment of many proteins including the serine/threonine kinase AKT to the membrane via binding to their pleckstrin homology (PH) domain [1]. Membrane-bound AKT becomes fully active through phosphorylation at Thr308 and Ser473 by 3-phosphoinositide-dependent kinase (PDK1) and mammalian target of rapamycin complex 2 (mTORC2) [12,13]. Activated AKT phosphorylates a range of downstream substrates, thereby activating or inhibiting these targets, and diversifying the PI3K signal into various functional outcomes.
The binding of p85α to p110α is necessary and sufficient for stabilization and inhibition of p110α catalytic activity by sequestering p110α away from the cell membrane [6, 7, 14, 15]. In addition to regulation of p110α catalytic activity by p85α subunit, the tumor suppressor gene PTEN acts as the most important negative regulator of the PI3K signaling pathway. PTEN is a protein/lipid phosphatase that antagonizes PI3K activity by dephosphorylation of PIP3 to phosphatidylinositol-4,5-bisphosphate (PIP2). Loss of PTEN function results in the accumulation of PIP3, sustaining the activation of PI3K signaling [16].
The catalytic activity of p110α depends on association with the cell membrane and access to stores of the membrane-bound PIP2 substrate, and inhibition of p110α by p85α activity involves separation from the cell membrane. Therefore, the membrane localization of p110α is sufficient and necessary for its enzyme activity and associated activation of downstream kinase pathways [17-19]. Myristoylation of proteins strongly links a protein to the cell membrane [20]. It has been shown that expression of a myristoylated p110α (myr-p110α) constitutively activates the catalytic kinase activity of PI3K, similar to the active mutants of p110α [21,22]. A myristoylated p110α construct causes increased AKT phosphorylation and malignant cellular transformation [19,22-25].
The PI3K/AKT signaling pathway is important for proper development of the circulatory system [26], and the formation of blood vessels is essential for normal embryonic development [27]. The vascular endothelial growth factor (VEGF) is a primary inducer of angiogenesis. The PI3K/AKT signaling cascade plays an important role in regulating vasculogenesis and angiogenesis by mediating expression of hypoxia-inducible factor α (HIF-1α) and VEGF in tissues in response to growth factors and other signals, including hypoxia [28]. Increased signaling of PI3K and AKT increases VEGF mRNA and is sufficient to induce angiogenesis through elevating HIF-1α and VEGF expression levels [28,29]. Inhibition of the PI3K signaling pathway reduces VEGF expression and interferes with vasculogenesis and angiogenesis [30], resulting in severe defects in vascular sprouting and remodeling [31].
In order to investigate the impact of constitutively active p110α during development, we have generated a mouse strain with Cre-inducible PI3K activity by insertion of a myr-p110α as a transgene under Cre-dependent control at the Rosa26 locus. Mouse embryos that had Cre-mediated recombination of myr-p110α transgene in very early stages of embryo development show that membrane-targeted myr-p110α expression caused embryonic lethality, increased PI3K activity, and activation of downstream AKT signaling pathway despite increased active PTEN that is opposing the accumulation of the PIP3 second messenger.
2 Experimental Procedures
2.1 Generation of Cre-Inducible Rosa26-myr-p110α construct
pCAG-Myr-p110-IH plasmid, which contains a myristoylated form of murine PIK3CA (p110α) cDNA under the control of CAG promoter, was purchased from Addgene (Plasmid 15689) [32]. The activating mutation was generated by addition of the avian src myristoylation sequence (MGSSKSKPK) at the N-terminus of p110α. The NheI-XhoI linearized fragment containing myristoylated PIK3CA (myr-p110α) cDNA was inserted into the intermediary vector (pCR2.1-TOPO vector). Amplified myr-p110α cDNA NheI-XhoI cut fragment was subcloned into the corresponding sites of pBTG vector to generate PGK-Neo-tpA-myr-p110α-IRES-eGFP construct. pBTG (pBigT-IRES-GFP, DM#268), which contains a STOP cassette flanked by two loxP sites, an internal ribosome entry site (IRES), and downstream enhanced green fluorescent protein (eGFP), was purchased from Addgene (Plasmid 15037) [33]. Shuttle vectors containing PGK-Neo-tpA-myr-p110α-IRES-eGFP cassette was inserted into the PacI-AcsI linearized pRosa26 modified targeting vector (pRosa26PAm1, DM#272, Addgene plasmid 15036) [33]. The resulting myr-p110α construct in the backbone of the pRosa26 targeting vector including both 5′ and 3′ Rosa26 targeting arms as well as PGK-DTA negative selection marker was used to generate a transgenic mouse. Each construct was confirmed by restriction enzyme mapping and DNA sequencing.
2.2 Electroporation of mouse embryonic stem cells and screening for correct targeting
Mouse embryonic stem (ES) cells were cultured on mouse embryo fibroblasts in culture medium (Dulbecco’s modified Eagle medium (DMEM), supplemented with 15% Fetal Bovine Serum, 0.5 mM non-essential amino acids, 1 mM sodium pyruvate, 2 mM L-glutamine, 0.1 mM β-mercaptoethanol, and 8.3 ng/mL mouse recombinant leukemia inhibitory factor). The targeting vector was linearized by SwaI cut. Linearized DNA was purified via phenol:chloroform extraction and ethanol precipitation. 20 μg of linearized transgene cDNA construct, 5′ Rosa26-PGK-Neo-tpA-myr-p110α-IRES-EGFP-3′ Rosa26, was electroporated into the Rosa26 locus of mouse ES cells (approximately 15 × 106 cells). Transfected ES cells were grown with feeders under selection in 180 μg/ml G418 for eleven days. Antibiotic resistant ES cell colonies with good morphology were screened by PCR amplifying across the 5′ recombination junction, using three oligonucleotides: Rosa26-5F forward primer 5′-AAAGTCGCTCTGAGTTGTTAT-3′, Rosa26-3R reverse primer 5′-GGAGCGGGAGAAATGGATATG-3′, and SA-R2 reverse primer 5′-GCGAAGAGTTTGTCCTCAACC-3′. After initial denaturation at 94°C for 3 minutes, 30 cycles of PCR were performed with denaturation at 94°C for 30 seconds, annealing at 56°C for 45 seconds, and extension at 72°C for 45 seconds using a Bio-Rad S1000 Thermal cycler. The last extension was at 72°C for 7 minutes. The product size genotyped by PCR using three oligonucleotides is 602 bp in wild type and 314 bp in a targeted allele.
2.3 Genomic DNA Southern blot
Genomic Southern blot hybridization was performed on genomic DNAs from ES cells digested with EcoRI. Digested genomic DNA was separated on a 0.8% agarose gel at 34mV overnight and alkaline transferred to a positively charged nylon membrane (Roche, 11417240001). A 593 bp DIG-labeled DNA probe targeting the Rosa26 locus of the 5′ side of the targeted insertion site was amplified using the PCR DIG Probe Synthesis Kit (Roche, 11630090910) and a set of primers: 5′-ACGAATATTTGGAATTTGGTATTTG-3′ and 5′-ACACTAATGAACTTTAAGTCCTGTGAA-3′, and used for Southern blot analysis. The 5′ probe was used to detect a 15.6 kb wild type band and a 4.3 kb targeted band, due to the presence of an extra EcoRI site in the targeted allele. All procedures for the DIG application system (Roche, 11585762001) were performed according to the manufacturer’s recommendations.
2.4 Generation of transgenic mice and genotyping by PCR
The selected ES colonies were expanded and injected into C57BL/6 mouse blastocysts. Embryos were implanted into the uterus of pseudopregnant CD1 females. Chimeric mice were generated and mated with C57BL/6 wild type mice. Transgenic mice carrying the targeted allele were identified by inspecting genomic DNA purified from tail using PCR with three primers: Rosa26-5F forward primer, Rosa26-3R reverse primer, and SA-R2 reverse primer. The animal study protocol was reviewed and approved by the institutional animal care and use committee (IACUC) of Geisel Medical School at Dartmouth.
2.5 Genotyping and phenotypic analysis of mouse embryo
To determine the effect of global embryonic expression of myr-p110α, CMV-Cre mice (BALB/c-Tg(CMV-cre)1Cgn/J, The Jackson Laboratory) were mated with transgenic mice harboring myr-p110α. Timed pregnant females were euthanized, and the uterine horns carrying the embryos were removed. Each embryo and its respective placenta were separated and dissected from the uterus, and put into small tissue culture dishes containing sterile phosphate-buffered saline (PBS). Yolk sac membranes were removed for genotype analysis by PCR. The presence of Cre recombinase gene (Cre) in embryos was detected by PCR amplification of a 100 bp fragment using a pair of oligonucleotides: Cre forward primer 5′-GCGGTCTGGCAGTAAAAACTATC-3′ and Cre reverse primer 5′-GTGAAACAGCATTGCTGTCACTT-3′. The transgene excision mediated by Cre recombinase was screened by PCR analysis to amplify 503 bp using a pair of primers: Rosa26-5F forward primer 5′-AAAGTCGCTCTGAGTTGTTAT-3′ and myr-p110α-R2 reverse primer 5′-ATGGTCGTGGAGGCATTCTA-3′ or to amplify 364 bp using a pair of primers: Rosa26-5F-3 forward primer 5′-GAGTTCTCTGCTGCCTCCTG-3′ and myr-p110α-R1 reverse primer 5′-CTCTTGCTGCTCCCCATAAG-3′. After initial denaturation at 94°C for 3 minutes, 30 cycles of PCR were performed with denaturation at 94°C for 30 seconds, annealing at 56°C for 45 seconds, and extension at 72°C for 45 seconds, followed by the last extension at 72°C for 3 minutes. Pictures of each embryo were taken using the LEICA MZ16 stereomicroscope and QCapturepro software. The GFP image of whole embryo body was analyzed using Zeiss Stemi SV11 Apo upright fluorescence stereomicroscope equipped with fluorescent source and captured using a Zeiss AxioCam MRm camera and AxioVision Rel 4.4 image program. Whole embryos were fixed in 10% buffered formalin in PBS (Electron Microscopy Sciences, 15740-01) and then paraffin-embedded or cryopreserved for histopathological examinations. The Cre;myr-p110α embryos referred to as myr-p110α heterozygotes are expressing both myr-p110α and Cre.
2.6 Mouse embryo sex determination
The genomic DNA was extracted from yolk sacs of embryos as a lysate through proteinase K digestion at 55°C for 1 hour. Sex was confirmed by multiplex PCR for the male-specific gene SRY (sex determination region on the Y chromosome responsible for testes formation) and the autosomal gene myogenin (Myog). Primers specific for the SRY gene are mSry-F 5′-TCATGAGACTGCCAACCACAG-3′ and mSry-R 5′-CATGACCACCACCACCACCAA-3′ and primers specific for the myogenin gene are mMyog-F 5′-TTACGTCCATCGTGGACAGC-3′ and mMyog-R 5′-TGGGCTGGGTGTTAGTCTTA-3′. After initial denaturation at 94°C for 5 minutes, 30 cycles of PCR were performed with denaturation at 94°C for 30 seconds, annealing at 66°C (for SRY) or 57°C (for Myog) for 45 seconds, and extension at 72°C for 45 seconds on a Bio-Rad S1000 Thermal cycler. The last extension was at 72°C for 3 minutes. PCR products were electrophoresed on 1.5% agarose gels at 100V and visualized with ethidium bromide under UV illumination.
2.7 Hematoxylin and eosin (H&E) stain
Mouse embryos were fixed in 10% buffered formalin. Paraffin-embedded or cryopreserved tissues were sectioned at 6 μm according to standard protocols. Sections were stained with hematoxylin and eosin (H&E) following the manufacturer’s protocol and mounted with Permount (Fisher scientific, SP15-100). Images were obtained on the Olympus BX5.1 microscope, and captured using Image Pro software.
2.8 Immunohistochemistry
For PECAM-1 staining, mouse embryos were fixed in 10% buffered formalin overnight. All embryos were paraffin embedded and sectioned at 6 μm according to standard protocols. Paraffin sections were baked overnight at 60°C, deparaffinized in xylene, rehydrated, and washed with PBS. Antigen retrieval was performed with sodium citrate buffer (10mM Sodium Citrate, 0.05% Tween 20, pH 6.0) in a pressure steamer (100°C for 10 minutes). Endogenous peroxide activity was quenched with 3% hydrogen peroxide in methanol for 10 min at room temperature and incubated in matched normal sera (Vector Laboratories, Burlingame, CA). Sections were immunostained with anti-PECAM-1 antibody (Abcam, ab28364, dilution 1:50) following the manufacturer’s protocol. The biotinylated secondary antibody was added to amplify signals, sequentially followed by addition of horseradish peroxidase-avidin conjugated antibody. The Vectastain Peroxidase ABC kit (Vector Laboratories, PK-6101, PK-4002, PK-6105) was applied as described by the manufacturer for visualization of PECAM-1 antigen by the development of peroxidase substrate. Sections were counterstained with Gill’s hematoxylin (Vector Laboratories, H-3401) and mounted with Permount. PECAM-1 immunostainings are visualized as brown colors. Images were obtained and captured by using the Olympus BX5.1 microscope and Image Pro software.
2.9 cDNA preparation and quantitative real-time reverse transcriptase-polymerase chain reaction (qRT-PCR)
Total RNA from mouse embryos was isolated using RNeasy kit (Qiagen) following manufacturer’s protocol. Total RNA was reverse-transcribed using the iScript™ cDNA synthesis kit for RT-PCR (Bio-Rad, 170-8891) according to the manufacturer’s instructions. The cDNA was quantified by RT-PCR on the ABI Prism 7900 Sequence Detection System (Applied Biosystems, Foster City, CA). PCR was performed using iQ™ SYBR® Green supermix reagents (Bio-Rad, 170-8882) with the following conditions: 95°C 60 seconds (1 cycle); 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 40 seconds (34 cycles). GAPDH was used as the reference control for normalization of the target genes. Each sample was run in triplicate and each PCR experiment included three non-template control wells. Quantitative real-time RT-PCR products were approximately in the range of 120–150 base pairs in length. To ensure the quality of all the primers, the dissociation curves were examined to confirm that no nonspecific bands arose with the primers. The threshold cycles (CT) were recorded for all samples for both the target gene and the reference. Relative expression values were calculated as the messenger RNA amount of each gene relative to that of GAPDH by the 2−ΔΔCT method. The gene-specific primer sequences were designed between different exons to avoid genomic DNA contamination. Primers used for the analysis were as follows: lmHIF-1α-F: 5′-GCAGCAGGAATTGGAACATT-3′, rmHIF-1α-R: 5′-GCATGCTAAATCGGAGGGTA-3′, lmHIF-1β-F: 5′-TCTCCCTCCCAGATGATGAC-3′, rmHIF-1β-R: 5′-CAATGTTGTGTCGGGAGATG-3′, lmVEGFA-F: 5′-CAGGCTGCTGTAACGATGAA-3′, rmVEGFA-R: 5′-GCATTCACATCTGCTGTGCT-3′, lmGAPDH-F: 5′-AACTTTGGCATTGTGGAAGG-3′, and rmGAPDH-R: 5′-GGATGCAGGGATGATGTTCT-3′.
2.10 Protein preparation and Western blot
Mouse embryos and cultured primary mouse embryonic fibroblasts (pMEFs) were lysed in ice-cold lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1mM NaVO4, and protease inhibitor cocktail (Roche, 11836170001) and phosphatase inhibitor cocktail (Sigma, P5726). Protein lysates were cleared by centrifugation and quantified by Bradford assay. 30 μg of protein lysates were separated on 9% SDS-PAGE gels under denaturing conditions and then transferred to Immobilon-P transfer membranes (Millipore, IPVH00010). The membranes were blocked with 5% non-fat skim milk in Tris-buffered saline (TBS)-0.1% Tween 20 (TBS-T) for 1 hour and washed according to standard protocols. The membranes were incubated with primary antibodies diluted in 5% non-fat skim milk or bovine serum albumin (for phosphorylated antibody) of TBS-T overnight at 4°C. Primary antibodies are as follows: anti-p110α (Cell Signaling technology, 4249, dilution 1:4 000), anti-AKT (anti-AKT1/2/3, Cell Signaling technology, 4691, dilution 1:5,000), anti-p-AKT Ser473 (anti-p-AKT1/2/3 Ser473, Cell Signaling technology, 4060, dilution 1:5 000), anti-p85α (Santa cruz biotech, sc-1637, dilution 1:2 000), anti-PTEN (Cell Signaling technology, 9559, dilution 1:4 000), anti-p-PTEN Ser 380/Thr 382/383 (Santa cruz biotech, sc-101789, dilution 1:4 000), anti-GFP (Santa cruz biotech, sc-9996, dilution 1:2 000), and HRP-conjugated anti-β-Actin (Abcam, ab49900, dilution 1:30 000). After rinsing with TBS-T, the membranes were incubated with anti-rabbit (Santa cruz biotech, sc-2313, dilution 1:5 000) or anti-mouse (Santa cruz biotech, sc-2060, dilution 1:5 000) secondary antibody conjugated with horseradish peroxidase diluted in 5% non-fat skim milk of TBS-T for 1 hour at room temperature. After rinsing with TBS-T several times, immunoreactive proteins were detected by using chemiluminescence ECL (Santa cruz biotech Western Blotting Luminol Reagent, sc-2048) and membranes exposed to film. Western blots were quantified using Quantity One software (Bio-Rad) and normalized with respect to β-actin expression.
2.11 Preparation of primary mouse embryonic fibroblast cells and fluorescence imaging
Mouse embryos at E12.5 day post coitum were dissected in 5 ml sterile Dulbecco’s phosphate buffered saline (DPBS) in a 100-mm tissue culture dish. Head and internal organs from the abdominal cavity were removed. After rinsing with 10 ml DPBS, the embryo was transferred to a clean 100-mm tissue culture dish and dissociated by aspirating into a 3-ml syringe through an 18-G needle. The dissociated contents were placed in tissue culture dishes containing DMEM supplemented with 10% (v/v) Fetal Bovine Serum, 1X penicillin/streptomycin, and 200 mM L-glutamine and cultured in a 37°C gassed (5% CO2) incubator. For fluorescence imaging, pMEF cells were cultured overnight on cover slips in complete DMEM medium. The coverslips were washed with PBS and fixed with 0.4% paraformaldehyde in PBS for 5 minutes at room temperature. After fixation, pMEFs on the coverslips were washed three times with PBS and permeabilized with 0.1% Triton X-100 and 0.1% sodium citrate for 5 minutes at room temperature. Then the coverslips were washed three times with PBS after permeabilization and incubated with Hoechst 33342 nucleic acid counterstain (Molecular Probes, H3570) to visualize nuclei and slides were mounted with PermaFluor (Thermo Scientific, TA-030-FM). The enhanced green fluorescent protein (eGFP) signals without enzyme-based amplification and Hoechst 33342 signals were analyzed under the fluorescence Olympus BX5.1 microscope, and images were captured using Image Pro software.
2.12 Statistical analysis
Statistical differences between control and experimental groups analyzed for real-time qRT-PCR and Western blot were determined by two-tailed unpaired Student’s t-test. Data analysis was performed using the GraphPad Prism 5.0 software. Graphs in figures denoted statistical significance of *p < 0.05, **p < 0.01, and ***p < 0.001. p > 0.05 was considered non-significant (ns). Error bars represented as standard error of the mean (SEM).
3 Results
3.1 Construction of Rosa26-myr-p110α targeting vector and generation of Cre-inducible myr-p110α mice
The homologous recombination construct used to insert Cre-activatable myr-p110α into the Rosa26 locus was generated by addition of the avian src myristoylation sequence (MGSSKSKPK) at the NH2 terminus of PIK3CA cDNA, encoding p110α. Targeted insertion in the Rosa26 locus was utilized to express the transgene ubiquitously in embryonic and adult mice [34]. The myr-p110α construct in the pRosa26 targeting vector was recombined via gene targeting into the endogenous Rosa26 locus (Figure 1A). The presence of floxed PGK-neo with its polyA site blocks expression of the transgene from the endogenous Rosa26 promoter. Cre-mediated removal of the PGK-neo-polyA cassette links Rosa26 exon 1 to myr-p110α. As the endogenous Rosa26 transcripts are not translated into protein, the first start codon in the myr-p110α is used for initiation of translation, and thus myr-p110α is expressed. The presence of IRES-eGFP in the message mediates expression of eGFP, which identifies cells that are expressing myr-p110α. Since Rosa26 is ubiquitously expressed [34] so myr-p110α should be expressed in any cell derived from a Cre-expressing progenitor. This system enables temporal and tissue specific Cre-regulated control of myr-p110α expression.
Figure 1. Generation of Cre-inducible Rosa26-myr-p110α knock-in allele.

(A) Schematic diagrams of the expression construct cloned into pRosa26 targeting vector, a portion of Rosa26 locus with restriction sites, the resulting targeted locus, and the myr-p110α allele following Cre-mediated excision. Labeled primers for PCR genotyping are indicated with blue arrows. The probe used for Southern blot analysis is shown as a light green box in the Rosa26 locus. (B) Representative result of PCR detecting 602 bp of wild-type (WT) band and 314 bp of integrated construct band. (C) Genomic Southern blotting with a 593 bp of DIG-labeled DNA probe, detecting either 15.6 kb of WT allele and 4.3 kb of targeted allele.
The correct integration of this construct into mouse embryonic stem (ES) cells was identified by PCR (Figure 1B) and genomic DNA southern blot (Figure 1C). Correctly targeted ES cells were used to establish the myr-p110α mice.
3.2 Constitutive expression of myristoylated-p110α results in embryonic lethality
To determine the phenotype of mice expressing myr-p110α beginning in early development, myr-p110α carrying mice were mated with CMV-Cre mice that express Cre recombinase in very early embryos so all or most cells have Cre-mediated genomic alterations [35]. CMV-Cre in this transgenic strain is X-linked; therefore, all female and male littermates get Cre from CMV-Cre homozygous mothers (Supplemental Figure 1). Hemizygous CMV-Cre fathers transmit Cre to all females and no male offspring (Supplemental Figure 1). Genotyping of liveborn mice from breeding of myr-p110α with CMV-Cre mice showed that except for one animal, no liveborn mice were identified with both myr-p110α and Cre (Table 1). This one exception apparently did not have strong Cre recombinase activity early during development, because it had a relatively low level of the genomic PCR band specific for the Cre-mediated transgene recombination event that activates myr-p110α (data not shown). The genotype of liveborn littermates demonstrated that myr-p110α causes embryonic lethality when it is expressed.
Table 1.
Genotype of offspring from myr-p110α heterozygote ⊥ CMV-Cre crosses.
| Cross | Number of liveborn mice |
Genotype of liveborn mice |
|||
|---|---|---|---|---|---|
| WT w/o Cre | WT w/ Cre | Myr-p110αwt/fl w/o Cre |
Myr-p110αwt/fl w/ Cre |
||
| CMV-CRE ♂ ⊥ Myr-p110αwt/fl ♀ | 54 | 12 (all ♂) | 15 (all ♀) | 26 (all ♂) | 1(♀) |
| Myr-p110αwt/fl ♂ ⊥ CMV-CRE ♀ | 62 | 0# | 62 (30 ♀, 32 ♂) | 0# | 0 |
Probability: ***p<0.0001 (χ2 test)
0#: As CMV-Cre is X-linked, littermate without Cre is not applicable from CMV-Cre homozygous mother.
To identify the stage of embryogenesis at which lethality is occurring, embryos at different stages of embryonic development from timed pregnant females were examined and genotyped. Mouse embryos were microscopically evaluated to analyze phenotype. Abnormal embryos with myr-p110α that also inherited Cre (Cre;myr-p110α) could be identified beginning at embryonic day 9.5 (E9.5). Grossly normal mice with Cre:myr-p110α were in the minority but also present at E9.5 to E11.5. By day E12.5, almost all heterozygous Cre;myr-p110α mutant embryos were grossly abnormal with developmental defects, hemorrhage or both although infrequent viable embryos were observable as late as E15.5 on mixed genetic background mice. This observation indicates that systemwide overexpression of PI3K signaling pathway disrupts normal embryonic development.
3.3 Embryonic death of Cre;myr-p110α heterozygotes involves developmental defects and hemorrhage
To evaluate gross phenotypic features, embryos in mixed genetic background were dissected and examined for morphology, blood circulation, and extravascular blood. Assessment of freshly dissected mutant Cre;myr-p110α embryos demonstrated profound growth retardation (Figures 2C, D, G, H, K, and O) compared with the wild-type (WT) controls in the left panels. In addition, Cre;myr-p110α embryos also failed to generate well-defined vitelline blood vessels in the yolk sac and thus revealed reduced and fragmented vasculature in the yolk sac (Figures 2C, G, K, O, and S), and extensive areas of hemorrhaging in the embryos (Figures 2L, P, S, and T). Some of Cre;myr-p110α embryos displayed one or more subepidermal blebs that frequently filled with blood (Figures 2D, H, and L, arrow) often involving enlargement of pericardial cavity (Figures 2D and L) with pooling of blood in the pericardial cavity (Figure 2L). We assume that lack of normal blood circulation was a major cause of intrauterine death.
Figure 2. Expression of myristoylated-p110α results in hemorrhage, vascular defects, and stunted embryos.

(A and B) Cre;WT embryos (WT embryo expressing Cre recombinase) at E10.5 showing normal phenotype. (C and D) Cre;myr-p110α embryos at E10.5 revealing defective yolk sac vasculature (C) and a shrunken body with a large subepidermal bleb filled with blood in the abdominal cavity (D, arrow). (E and F) Cre;WT embryos at E11.5. (G and H) Cre;myr-p110α embryos at E11.5 displaying defective vasculature and an unusual blood spot in the yolk sac (G), and an embryo with abnormal morphology (H). (I and J) Cre;WT embryos at E12.5. (K and L) Cre;myr-p110α embryos at E12.5 with fragmented vasculature in the yolk sac (K, arrow), and hemorrhaging areas with an enlarged pericardial cavity filled with blood (L, arrow). (M and N) Cre;WT embryos at E13.5. (O and P) Cre;myr-p110α embryos at E13.5 exhibiting poorly defined yolk sac vessels and abnormal embryo body (O), and extensive hemorrhaging areas in the embryo (P). (Q and R) Cre;WT embryos at E15.5. (S and T) Cre;myr-p110α embryos at E15.5 displaying defective yolk sac vasculature (S) and hemorrhage (T). Pictures are representative of total 434 embryos analyzed. Scale bars represent 0.5 cm.
The phenotype of Cre;myr-p110α embryos has some variability in time of death and characteristics of the phenotype. CMV-Cre is located on the X-chromosome so it was possible that, in female embryos, X-chromosome inactivation (XCI) could have modified the phenotype and thereby contributed to the phenotypic variability. To investigate this possibility, the sex of embryos showing either developmental defects or hemorrhages at different time points was tested by PCR for the male-specific marker SRY and the autosomal gene myogenin found in both sexes (Figure 3). There was no evident difference between females and males in either viability or phenotypic variability, suggesting that XCI is not a cause of phenotypic variability.
Figure 3. Variability in a phenotype of Cre;myr-p110α embryos is not associated with X-chromosome inactivation.

PCR products were generated from purified gDNA of the yolk sacs dissected from embryos harvested from CMV-Cre homozygous mother and myr-p110αwt/fl father crosses. Embryos with either developmental defects (lanes 1 to 5 at E9.5, 11 to 15 at E10.5, and 21 to 25 at E11.5) or hemorrhage (lanes 6 to 10 at E9.5, 16 to 20 at E10.5, and 26 to 30 at E11.5) were randomly chosen at E9.5 to E11.5 at which embryonic lethality is occurring. There was no evident difference between female and male in either viability or phenotypic variability. The genomic PCR products were visualized in a 1.5% agarose gel electrophoresis. Representative results of PCR detecting 441 bp of male-specific gene SRY and 245 bp of the autosomal gene myogenin (Myog) found in both males and females.
3.4 Hemorrhage in myristoylated-p110α heterozygotes is associated with vasodilation and increased VEGF
Histological analysis of embryos in mixed genetic background by staining with H&E revealed further information on defects in embryo development: abnormality in embryo body structure (data not shown) and hemorrhage to various degrees (Figures 4E, F, K, and L) in Cre;myr-p110α embryos. The head sections from hemorrhaging Cre;myr-p110α embryos clearly showed blood leaking out of blood vessels below the skin (Figures 4E and F), and other parts of the body also demonstrated areas of extravasated blood (Figures 4K and L), which suggests defective vasculogenesis/angiogenesis leading to embryonic death.
Figure 4. Cre;myr-p110α embryos have extremely dilated blood vessels with discontinuous walls.

(A) WT embryo at E15.5. (B and C) Cre;myr-p110α embryos at E15.5 revealing extensive hemorrhage. (D to F) H&E stained sagittal head sections of embryo. (D) WT embryo shown in (A), exhibiting normal histology. (E and F) Cre;myr-p110α embryos shown in (B) and (C), displaying extravasations under the skin (arrow). (G to I) Immunostaining with anti-PECAM-1 antibody of sagittal embryo head sections. (G) WT embryo (A), revealing normal blood vessels (arrow). (H and I) Cre;myr-p110α embryos (B and C), exhibiting extremely dilated blood vessels with discontinuous walls in regions of hemorrhage (arrows). (J to L) H&E stained sagittal embryo back sections. (J) WT embryo (A), showing normal histology. (K and L) Cre;myr-p110α embryos (B and C), displaying extravasations (arrows). (M to O) PECAM-1 immunostaining of sagittal embryo back sections. (M) WT embryo (A), presenting normal blood vessels (arrow). (N and O) Cre;myr-p110α embryos (B and C), exhibiting dilated vessels with discontinuous walls in regions of hemorrhage (arrows). PECAM-1 immunostainings are visualized as brown colors, demonstrating the blood vessels. Pictures of H&E staining and PECAM-1 immunostaining were taken with X400 magnification. Pictures are representative. Bar in panel (A) represents 1 cm and indicates scale for (A) to (C).
To visualize blood vessel morphology in the hemorrhaging areas of Cre;myr-p110α embryos, immunohistochemistry was used to identify cells expressing platelet endothelial cell adhesion molecule (PECAM-1, CD31), an endothelial cell-specific marker. Blood vessels of the hemorrhaging Cre;myr-p110α embryos demonstrated by PECAM-1 immunostainings (Figures 4H, I, N, and O) were often considerably dilated as compared to WT embryos (Figures 4G and M, arrow). The images show areas of these dilated blood vessels in which walls are discontinuous which would cause hemorrhage (arrows in figures 4H, I, N, and O). Severe vessel dilation, reduced vessel integrity, and hemorrhaging in Cre;myr-p110α embryos demonstrate that deregulated expression of class IA PI3K, p110α impacts vasculogenesis and angiogenesis.
Vascular endothelial growth factor-A (VEGF-A) mediates vasodilation leading to increased vascular permeability and stimulates vasculogenesis and angiogenesis [36]. To assess the relevance of the constitutively active PI3K signaling in the expression of VEGF-A, WT and Cre;myr-p110α embryos were analyzed for mRNA expression level of VEGF-A. Quantitative real-time reverse transcriptase-polymerase chain reaction (qRT-PCR) revealed significantly increased levels of VEGF-A mRNA (1.7-fold) in Cre;myr-p110α embryos with hemorrhage (Figure 5). This result connects hyperactive PI3K signaling to the regulation of VEGF expression and associated vascular embryonic development. The mRNA level of hypoxia-inducible factor α (HIF-1α), a heterodimeric transcription factor and a major regulator of VEGF-A, was also significantly increased in Cre;myr-p110α embryos (1.4-fold), whereas HIF-1β that is constitutively expressed in the cell was unchanged (Figure 5). Disorganized vascular structures caused by constitutively active PI3K signaling in Cre;myr-p110α embryos demonstrate that normal vascular development cannot tolerate overexpression of PI3K signaling during murine embryogenesis.
Figure 5. Cre;myr-p110α embryos increase VEGF-A and HIF-1α mRNA.
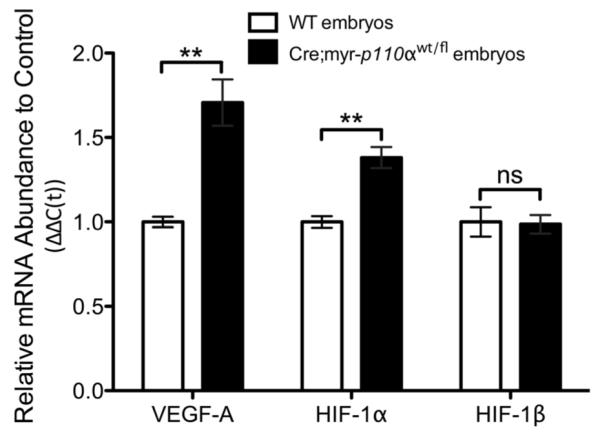
mRNA expression levels of VEGF-A, HIF-1α, and HIF-1β in WT and Cre;myr-p110α embryos analyzed by real-time qRT-PCR, the graphs of real-time qRT-PCR data quantified and normalized with respect to GAPDH. Mean fold increase compared with WT control embryos, was calculated after normalization. Error bars are presented as the mean ± SEM. Two-tailed unpaired t test was used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001. P > 0.05 was considered non-significant (ns). Data are representative of 2 independent experiments using 3~6 embryos per group.
3.5 Myristoylated-p110α activates PI3K signaling
The PI3K signaling pathway is complex with multiple regulatory controls. To determine the biochemical consequences of constitutive activation of p110α during development, we assayed the PI3K pathway activation status in Cre;myr-p110α embryos obtained between E9.5 and E11.5 by Western blotting with antibodies against PI3K signaling molecules. In Cre;myr-p110α embryos we observed significantly elevated levels of total p110α (4.7-fold) and phosphorylated AKT (p-AKT, 2.4-fold), which is the active form of AKT (Figures 6A and B), as compared to WT control embryos. In addition, total AKT level was slightly but significantly reduced (0.82-fold) and the p-AKT/AKT ratio was increased (2.9-fold) in Cre;myr-p110α embryos. Co-expression of eGFP, indicating expected expression of myr-p110α, was also confirmed in all transgenic embryos expressing myr-p110α (Figure 6A).
Figure 6. Cre;myr-p110α embryos activate AKT regardless of increased levels of PTEN and p85α.

(A) Western blot analysis of p110α, p-AKT, AKT, and eGFP in WT and Cre;myr-p110α embryos during E9.5 to E11.5. (B) The graphs of Western blot signals of p110α, p-AKT, and AKT were quantified and normalized with respect to β-Actin levels. (C) Western blot analysis of p85α, p-PTEN, and PTEN in WT and Cre;myr-p110α embryos during E9.5 to E11.5. (D) The graphs of Western blot signals of p85α, p-PTEN, and PTEN were quantified and normalized with respect to β-Actin levels. Mean fold increase compared with WT control embryos, was calculated after normalization. Error bars are presented as the mean ± SEM. Two-tailed unpaired t test was used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001. P > 0.05 was considered non-significant (ns). Data are representative of 4 independent experiments using 5 embryos per group.
Further Western blots were done in order to investigate the possible feedback of this active p110α allele on PTEN and the p85α regulatory subunit that regulate this pathway. The embryos responded to overexpression of active p110α with increased protein levels of both PTEN and p85α. Total protein levels of the PI3K regulatory subunit p85α (1.7-fold) and PTEN (2-fold), and level of phosphorylated PTEN (p-PTEN, 1.9-fold), which is the less active form of PTEN, were also significantly increased in Cre;myr-p110α embryos (Figures 6C and D). Despite increased total PTEN and p-PTEN, the ratio of p-PTEN/PTEN was not changed as compared to WT control embryos. Our biochemical results indicate that in spite of increased PTEN level that should oppose PI3K activity, the myr-p110α is still overly active since levels of active p-AKT were 2.4-fold increased. The overall biochemical analysis shows that despite increased levels of regulatory proteins, embryonic lethality is due to increased activation of the PI3K/AKT pathway.
3.6 Myristoylated-p110α is ubiquitously expressed in mouse embryos although commonly reveals abnormal vascular phenotypes
Although the Rosa26 locus-driven myr-p110α is expected to be expressed in every cell type [34], the phenotype of Cre;myr-p110α embryos was largely associated with hemorrhages resulting from defective vascularization and vasodilation. To test the possibility that myr-p110α is silenced or expressed at low levels in non-endothelial tissues, whole-embryo imaging ex uterus for eGFP signal, a reporter co-inserted with myr-p110α into the Rosa26 locus, was performed to visualize myr-p110α expression in mouse embryos and confirm its global expression. Cre;myr-p110α embryos were bright green under a fluorescence stereomicroscope and expressed GFP in essentially all tissues with the exception of hemorrhaging regions due to the absorption of the light by extravasated red blood cells (Figure 7), although there was no enzyme-based amplification of GFP signal. This result demonstrates that myr-p110α is expressed ubiquitously under the control of Rosa26 promoter and causes embryonic death.
Figure 7. Myristoylated-p110α is ubiquitously expressed in all tissues.

(A) Cre;WT embryo at E15.5 in mixed genetic background with normal phenotype. (B) Cre;myr-p110α embryo at E15.5 in mixed genetic background displaying extensive hemorrhaging areas in the embryo. (C) Fluorescence image of WT embryo shown in (A), presenting no GFP signal. (D) Fluorescence image of Cre;myr-p110α embryo shown in (B), revealing significant levels of GFP signal. The fluorescent signals of GFP did not employ enzymatic amplification. Pictures are representative of total 14 embryos analyzed. Bar in panel (A) represents 0.5 cm and indicates scale for (A) to (D).
To further test the possibility that myr-p110α is preferentially expressed in vascular endothelial cells or silenced in other non-vascular cells, primary mouse embryonic fibroblasts (pMEFs) from WT and Cre;myr-p110α embryos at E12.5 were isolated and used for the detection of eGFP signal, as an indicative of myr-p110α expression. Fluorescence imaging observed GFP fluorescence in Cre;myr-p110α pMEFs, indicating the expression of myr-p110α (Figure 8A vs 8B). After confirming myr-p110α expression, the downstream effects of myr-p110α expression were investigated by performing Western blots of PI3K signaling proteins in Cre;myr-p110α or WT pMEFs (Figure 8E, F, G, and H). Cre;myr-p110α pMEFs showed significantly increased levels of total p110α (12.6-fold) and phosphorylated AKT (p-AKT, 8.6-fold), in comparison with WT control pMEFs (Figure 8E and F). In addition, total protein levels of both p85α (5.8-fold) and PTEN (1.7-fold), and level of phosphorylated PTEN (p-PTEN, 1.8-fold) were also significantly elevated in Cre;myr-p110α pMEFs (Figure 8G and H). Co-expression of eGFP was also confirmed by Western blot in Cre;myr-p110α embryos pMEFs (Figure 8G). These results indicate that myr-p110α is not only expressed in vascular endothelial cells but is also expressed in non-vascular fibroblast cells, and the PI3K/AKT signaling pathway is activated as an outcome of myr-p110α expression in Cre;myr-p110α pMEFs, as compared to WT pMEFs. In sum, these data enable us to unequivocally state that myr-p110α is expressed ubiquitously although the phenotype is predominantly associated with abnormal vascular development.
Figure 8. Myristoylated-p110α is not expressed only in vascular endothelial cells but also in non-vascular fibroblast cells.

(A) GFP image of WT pMEFs at passage 7, showing no GFP signal. (B) GFP image of Cre;myr-p110α pMEFs at passage 7, revealing significant levels of GFP signal. (C) WT pMEFs shown in (A) were stained with Hoechst 33342 to visualize nuclei. (D) Hoechst 33342 nucleic acid stains of Cre;myr-p110α pMEFs shown in (B). Pictures were taken with X100 magnification. Bar in panel (D) represents 100 μm and indicates scale for (A) to (D). (E) Western blot analysis of p110α, p-AKT, and AKT in WT and Cre;myr-p110α pMEFs. (F) The graphs of Western blot signals of p110α, p-AKT, and AKT. (G) Western blot analysis of p85α, p-PTEN, PTEN, and GFP in WT and Cre;myr-p110α pMEFs. (H) The graphs of Western blot signals of p85α, p-PTEN, and PTEN. For F and H, Western blot signals were quantified and normalized with respect to β-Actin and mean fold increase compared with signals from WT control pMEFs, was calculated after normalization. Error bars are presented as the mean ± SEM. Two-tailed unpaired t test was used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001. P > 0.05 was considered non-significant (ns). Data are representative of 4 independent experiments.
4 Discussion
4.1 Myristoylated-p110α activates PI3K/AKT signaling despite increased negative feedback regulatory proteins
Membrane targeting of p110α by myristoylation has been reported to enhance catalytic activity of PI3K similar to the active mutants E542K, E545K, and H1047R [21, 22]. In support of these findings, we show that embryos constitutively expressing myr-p110α overly activated the PI3K signaling pathway, with significantly increased levels of total p110α and phosphorylated AKT, a major signaling kinase downstream of PI3K. Our result shows that fetal embryonic lethality observed in Cre;myr-p110α embryos is associated with constitutive activation of the PI3K/AKT pathway.
The inhibition of p110α catalytic activity by the p85α regulatory subunit via tight binding is a good example of a widely used regulatory scheme, in which regulatory subunits of kinases are stabilized and maintain enzyme activity at a low level in the basal state, and subsequent activation of the enzyme occurs by a release of inhibitory interaction. The embryos expressing myr-p110α had increased levels of p85α. Elevated p85α level is likely attempted negative regulation of p110α activity following hyperactivation of PI3K signaling. However, inhibition of p110α activity potentially mediated by increased p85α is ineffective because myr-p110α is likely not removed from membrane association by p85α.
PTEN-mediated inhibition serves as one major mechanism that negatively regulates the PI3K signaling pathway [37]. PTEN tightly regulates the cellular level of PIP3 by dephosphorylation and functionally antagonizes PI3K activity. PTEN can be regulated via multiple mechanisms, including subcellular localization and posttranslational modifications, all of which potentially impact PTEN levels and/or activity [38]. Phosphorylation of the C-terminal tail stabilizes the PTEN protein but reduces its lipid phosphatase activity in relation to PIP3 by repelling it from the plasma membrane and by inhibiting interactions with PDZ domain–containing proteins [39, 40]. Conversely, dephosphorylation of the C-terminal tail of PTEN causes its recruitment to the membrane leading to increased phosphatase activity accompanied by rapid degradation, therefore keeping PTEN activity under tight control. The embryos expressing myr-p110α displayed both increased total protein level of PTEN and greater level of phosphorylated PTEN. This may be an outcome of sensing myr-p110α-driven PI3K signaling activation and negative regulation through a feedback inhibition. Since the ratio between total PTEN and p-PTEN is not changed, we expect overall PTEN activity to be increased due to the increased level of total PTEN. However, despite 2-fold increased PTEN levels that catalyses PIP3 into PIP2 and antagonizes PI3K signaling, negative feedback regulation by PTEN is not sufficient to block the downstream consequences of aberrantly hyperactive PI3K signaling induced by myr-p110α. The higher p-AKT level observed in Cre;myr-p110α embryos demonstrates the ineffective negative feedback loop and activated PI3K signaling.
4.2 Hyperactive PI3K signaling pathway disrupts embryonic vascular development
Constitutive expression of p110α resulted in early embryonic lethality in which embryos generally did not survive after E11.5 of embryonic development. This observation demonstrates that proper control of p110α is critical for normal embryonic development. By 9.5 days of embryonic development, endothelial cells, which line the interior surface of blood vessels, are fully differentiated [41]. The Cre;myr-p110α embryos began to die at E9.5, indicating that tight regulation of class 1A PI3K activity might be necessary for endothelial cell differentiation or the formation of the primitive vascular structure. Given the wide variety of cellular processes that PI3K signaling controls, a perplexing aspect of the phenotype of Cre;myr-p110α embryos is that most embryos survive to E9.5 since the Rosa26 locus-driven myr-p110α is expressed ubiquitously in every cell type (Figures 7 and 8). During post-implantation stages E5.5 to E9.5, successful embryo development and survival through implantation is mediated by the initial communication between the fetus and mother. Our results show that myr-p110α expressing embryos are able to establish the chorioallantoic placenta and maternally mediated fetal circulation in addition to establishing the basic body plan to maintain the embryo.
During E9.5 and E10.5, proliferation and differentiation markedly increase as organogenesis is rapidly accelerated in the embryo [42]. Organogenesis is accompanied by the recruitment of new blood vessels as the developing embryo needs nutrients to promote organ formation. At E11.5, pulsatile heartbeats circulate blood [43]. The mutant Cre;myr-p110α embryos often displayed enlargement of the pericardial cavity frequently filled with blood. Infrequency of Cre;myr-p110α embryo survival past E11.5, at which point adequate circulation becomes essential for the embryo, suggests that constitutively active p110α is associated with vascular insufficiency which often included pericardial bleeding, hemorrhaging in other regions, and overall indicated circulatory collapse.
Vasculogenesis/angiogenesis allows the vascular system to expand in order to deliver nutrients and oxygen, and to remove waste products [44]. It is interesting to note that the myr-p110α expressing embryos demonstrated broad areas of extravasated blood associated with extensive dilated blood vessels that were often discontinuous, implying that constitutively active PI3K signaling causes vascular rupture due to deregulated vasculogenesis/angiogenesis. Excessive vascular dilation can reduce blood pressure [45], which in association with hemorrhage may result in an inadequate blood supply to all organs including the heart and multiple organ failure. The defective vascularization impacted by myr-p110α activity may lead to loss of vessel integrity, increased vascular permeability, and hypotension with the result that the cardiovascular system fails to provide adequate oxygen and nutrients for cells during organogenesis, which leads to cell death and associated overall death of the embryo.
4.3 Loss and gain of function mutations of PI3K pathway reveal similar embryonic phenotypes
Other studies that manipulated PI3K pathways in the embryo caused disruption of vasculogenesis/angiogenesis and surprisingly the phenotypes were outwardly similar for both loss and gain of PI3K activity. Our study of increased PI3K activity during embryogenesis shows that vascular development is impaired. Consistent with our finding, homozygous PTEN deletion in the endothelium that increased PIP3 levels causes abnormal vascular remodeling and bleeding, and mouse embryonic loss occurs by E11.5 [26]; constitutively activated AKT by addition of a myristoylation sequence to AKT in the endothelium results in defective vessel patterning in addition to vessel congestion and breaching, and is embryonic lethal [29]. Loss of p110α activity reveals a similar phenotype in the embryo: p110α−/− mouse embryos die between E9.5 and E10.5 due to insufficient cell proliferation and defects in angiogenesis [46]; p85α−/−;p85β−/− embryos, which lack these two regulatory subunits of class 1A p110 catalytic subunits and have decreased PI3K activity resulting from impaired membrane localization of p110 catalytic subunit, also exhibit loss of vascular integrity and start to die at E12.5 [47]. Collectively, all data including our data supports the conclusion that fine-tuning of class 1A PI3Ks during embryo vasculogenesis/angiogenesis is vital for proper vessel development and integrity, and either too much or too little PI3K signaling activity disrupts vasculogenesis/angiogenesis during mouse embryo development.
Despite involvement in many central aspects of cellular processes, it appears that increased or reduced PI3K signaling is perhaps surprisingly compatible with relatively normal early embryonic development, since Cre;myr-p110α embryos implant and undergo germ layer differentiation, gastrulation, and subsequent establishment of the body plan in a manner that appears normal. It is not until the embryonic stage when organs are established and grow rapidly that deregulation of PI3K signaling pathway manifests pathologically and then it most often leads to problems in vasculogenesis/angiogenesis. Either this early stage of embryo development does not require precise regulation of PIP3 levels or other systems enable proper regulation of PIP3 levels at this early stage. Other pathways that generate PIP3 could make p110α regulation redundant in the early embryo when there is a loss of p110α activity. Increased p110α catalytic activity as studied here could potentially be more effectively ameliorated by increased PTEN at this early embryonic stage. Many aspects of the role of p110α in the very early stages of embryonic development still remain to be elucidated.
Supplementary Material
Supplemental Figure 1. CMV-Cre is X-linked and express Cre recombinase in early stages of mouse embryogenesis but X-chromosome inactivation is not cause of phenotypic variability shown in Cre;myr-p110α embryos. PCR products were generated from purified genomic DNA of the yolk sacs dissected from embryos. Cre;myr-p110α (lanes 1 to 6) or Cre;WT (lanes 7 to 12) embryos harvested from CMV-Cre homozygous mother and myr-p110αwt/fl father crosses were either female or male, whereas Cre;myr-p110α embryos harvested from myr-p110αwt/fl mother and hemizygous CMV-Cre father crosses were all females (lanes 13 to 18). From myr-p110αwt/fl mother and hemizygous CMV-Cre father crosses, all Cre harboring embryos were females (lanes 13 to 18 and 25 to 27) and embryos not harboring Cre gene were all males (lanes 19 to 24 and 28 to 30). Embryos were randomly chosen at the embryonic days of 10.5 to E12.5. The PCR products were visualized in a 1.5% agarose gel electrophoresis. Representative results of genomic PCR detecting 441 bp of male-specific gene SRY and 245 bp of the autosomal gene myogenin (Myog) found in both males and females.
Acknowledgements
We would like to acknowledge Dr. Radu Stan for the assistance with the LEICA MZ16 stereomicroscope and QCapture-pro software, Dartmouth Transgenic and Genetic Construct Shared Resource for animal maintenance, and Immunology COBRE core C.
Funding for this work was provided by NIH grants U54 CA151662-01, 5P30GM103415-03, P30 CA023108-27, and R01CA157664.
Abbreviations
- Cre
Cre recombinase gene
- DMEM
Dulbecco’s modified Eagle medium
- ES
embryonic stem
- H&E
hematoxylin and eosin
- HIF-1α
hypoxia-inducible factor α
- Myog
myogenin
- myr-p110α
myristoylated p110α
- pMEFs
primary mouse embryonic fibroblasts
- PBS
phosphate-buffered saline
- PI3K
phosphatidylinositol 3-kinase
- PIP3
phosphatidylinositol-3,4,5-trisphosphate
- PIP2
phosphatidylinositol-4,5-bisphosphate
- PECAM-1
platelet endothelial cell adhesion molecule
- RTKs
receptor tyrosine kinases
- SRY
sex determination region on the Y chromosome
- SEM
standard error of the mean
- VEGF
vascular endothelial growth factor
Footnotes
Conflict of interest: Authors declare nothing to disclose.
Contributor Information
Mee Rie Sheen, Department of Microbiology and Immunology, Geisel School of Medicine at Dartmouth, Hanover, NH 03755, United States.
Sandra L. Warner, Department of Microbiology and Immunology, Geisel School of Medicine at Dartmouth, Hanover, NH 03755, United States; Norris Cotton Cancer Center, Lebanon, NH 03756, United States
Jennifer L. Fields, Department of Microbiology and Immunology, Geisel School of Medicine at Dartmouth, Hanover, NH 03755, United States; Norris Cotton Cancer Center, Lebanon, NH 03756, United States
Jose R. Conejo-Garcia, Tumor Microenvironment and Metastasis Program, the Wistar Institute, Philadelphia, PA 19104, United States
Steven Fiering, Department of Microbiology and Immunology, Geisel School of Medicine at Dartmouth, Hanover, NH 03755, United States; Department of Genetics, Geisel School of Medicine at Dartmouth, Hanover, NH 03755, United States.
References
- [1].Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- [2].Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat. Rev. Cancer. 2005;5:921–929. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- [3].Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- [4].Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–320. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
- [5].Domin J, Waterfield MD. Using structure to define the function of phosphoinositide 3-kinase family members. FEBS Lett. 1997;410:91–95. doi: 10.1016/s0014-5793(97)00617-0. [DOI] [PubMed] [Google Scholar]
- [6].Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu. Rev. Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- [7].Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu. Rev. Cell. Dev. Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- [8].Yu J, Wjasow C, Backer JM. Regulation of the p85/p110alpha phosphatidylinositol 3′-kinase. Distinct roles for the n-terminal and c-terminal SH2 domains. J. Biol. Chem. 1998;273:30199–30203. doi: 10.1074/jbc.273.46.30199. [DOI] [PubMed] [Google Scholar]
- [9].Matsuda M, Mayer BJ, Hanafusa H. Identification of domains of the v-crk oncogene product sufficient for association with phosphotyrosine-containing proteins. Mol. Cell. Biol. 1991;11:1607–1613. doi: 10.1128/mcb.11.3.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mayer BJ, Jackson PK, Baltimore D. The noncatalytic src homology region 2 segment of abl tyrosine kinase binds to tyrosine-phosphorylated cellular proteins with high affinity. Proc. Natl. Acad. Sci. USA. 1991;88:627–631. doi: 10.1073/pnas.88.2.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Moran MF, Koch CA, Anderson D, Ellis C, England L, Martin GS, et al. Src homology region 2 domains direct protein-protein interactions in signal transduction. Proc. Natl. Acad. Sci. USA. 1990;87:8622–8626. doi: 10.1073/pnas.87.21.8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, et al. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr. Biol. 1997;7:776–789. doi: 10.1016/s0960-9822(06)00336-8. [DOI] [PubMed] [Google Scholar]
- [13].Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- [14].Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol. Cell. Biol. 1998;18:1379–1387. doi: 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fruman DA, Mauvais-Jarvis F, Pollard DA, Yballe CM, Brazil D, Bronson RT, et al. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85 alpha. Nat. Genet. 2000;26:379–382. doi: 10.1038/81715. [DOI] [PubMed] [Google Scholar]
- [16].Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- [17].Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH, et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc. Natl. Acad. Sci. USA. 2009;106:16996–17001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Klippel A, Reinhard C, Kavanaugh WM, Apell G, Escobedo MA, Williams LT. Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol. Cell. Biol. 1996;16:4117–4127. doi: 10.1128/mcb.16.8.4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc. Natl. Acad. Sci. USA. 2005;102:18443–18448. doi: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hayashi N, Titani K. N-myristoylated proteins, key components in intracellular signal transduction systems enabling rapid and flexible cell responses. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010;86:494–508. doi: 10.2183/pjab.86.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang H, Liu G, Dziubinski M, Yang Z, Ethier SP, Wu G. Comprehensive analysis of oncogenic effects of PIK3CA mutations in human mammary epithelial cells. Breast Cancer Res. Treat. 2008;112:217–227. doi: 10.1007/s10549-007-9847-6. [DOI] [PubMed] [Google Scholar]
- [22].Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc. Natl. Acad. Sci. USA. 2005;102:802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu Z, Roberts TM. Human tumor mutants in the p110alpha subunit of PI3K. Cell Cycle. 2006;5:675–677. doi: 10.4161/cc.5.7.2605. [DOI] [PubMed] [Google Scholar]
- [24].Bader AG, Kang S, Vogt PK. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc. Natl. Acad. Sci. USA. 2006;103:1475–1479. doi: 10.1073/pnas.0510857103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Renner O, Blanco-Aparicio C, Grassow M, Canamero M, Leal JF, Carnero A. Activation of phosphatidylinositol 3-kinase by membrane localization of p110alpha predisposes mammary glands to neoplastic transformation. Cancer Res. 2008;68:9643–9653. doi: 10.1158/0008-5472.CAN-08-1539. [DOI] [PubMed] [Google Scholar]
- [26].Hamada K, Sasaki T, Koni PA, Natsui M, Kishimoto H, Sasaki J, et al. The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 2005;19:2054–2065. doi: 10.1101/gad.1308805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- [28].Jiang BH, Liu LZ. PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv. Cancer Res. 2009;102:19–65. doi: 10.1016/S0065-230X(09)02002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sun JF, Phung T, Shiojima I, Felske T, Upalakalin JN, Feng D, et al. Microvascular patterning is controlled by fine-tuning the Akt signal. Proc. Natl. Acad. Sci. USA. 2005;102:128–133. doi: 10.1073/pnas.0403198102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jiang BH, Zheng JZ, Aoki M, Vogt PK. Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc. Natl. Acad. Sci. USA. 2000;97:1749–1753. doi: 10.1073/pnas.040560897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature. 2008;453:662–666. doi: 10.1038/nature06892. [DOI] [PubMed] [Google Scholar]
- [32].Takahashi K, Mitsui K, Yamanaka S. Role of ERas in promoting tumour-like properties in mouse embryonic stem cells. Nature. 2003;423:541–545. doi: 10.1038/nature01646. [DOI] [PubMed] [Google Scholar]
- [33].Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc. Natl. Acad. Sci. USA. 2003;100:14920–14925. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zambrowicz BP, Imamoto A, Fiering S, Herzenberg LA, Kerr WG, Soriano P. Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proc. Natl. Acad. Sci. USA. 1997;94:3789–3794. doi: 10.1073/pnas.94.8.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bates DO, Harper SJ. Regulation of vascular permeability by vascular endothelial growth factors. Vascul. Pharmacol. 2002;39:225–237. doi: 10.1016/s1537-1891(03)00011-9. [DOI] [PubMed] [Google Scholar]
- [37].Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- [39].Birle D, Bottini N, Williams S, Huynh H, deBelle I, Adamson E, et al. Negative feedback regulation of the tumor suppressor PTEN by phosphoinositide-induced serine phosphorylation. J. Immunol. 2002;169:286–291. doi: 10.4049/jimmunol.169.1.286. [DOI] [PubMed] [Google Scholar]
- [40].Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, et al. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. USA. 1999;96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Papaioannou VE, Behringer RR. Early embryonic lethality in genetically engineered mice: diagnosis and phenotypic analysis. Vet. Pathol. 2012;49:64–70. doi: 10.1177/0300985810395725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rugh R. The Mouse: Its Reproduction and Development. Oxford University Press; 1990. [Google Scholar]
- [43].Jones EA, Baron MH, Fraser SE, Dickinson ME. Measuring hemodynamic changes during mammalian development. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H1561–1569. doi: 10.1152/ajpheart.00081.2004. [DOI] [PubMed] [Google Scholar]
- [44].Persson AB, Buschmann IR. Vascular growth in health and disease. Front. Mol. Neurosci. 2011;4:14. doi: 10.3389/fnmol.2011.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Duarte JD, Cooper-DeHoff RM. Mechanisms for blood pressure lowering and metabolic effects of thiazide and thiazide-like diuretics. Expert Rev. Cardiovasc. Ther. 2010;8:793–802. doi: 10.1586/erc.10.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J. Biol. Chem. 1999;274:10963–10968. doi: 10.1074/jbc.274.16.10963. [DOI] [PubMed] [Google Scholar]
- [47].Brachmann SM, Yballe CM, Innocenti M, Deane JA, Fruman DA, Thomas SM, et al. Role of phosphoinositide 3-kinase regulatory isoforms in development and actin rearrangement. Mol. Cell. Biol. 2005;25:2593–2606. doi: 10.1128/MCB.25.7.2593-2606.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. CMV-Cre is X-linked and express Cre recombinase in early stages of mouse embryogenesis but X-chromosome inactivation is not cause of phenotypic variability shown in Cre;myr-p110α embryos. PCR products were generated from purified genomic DNA of the yolk sacs dissected from embryos. Cre;myr-p110α (lanes 1 to 6) or Cre;WT (lanes 7 to 12) embryos harvested from CMV-Cre homozygous mother and myr-p110αwt/fl father crosses were either female or male, whereas Cre;myr-p110α embryos harvested from myr-p110αwt/fl mother and hemizygous CMV-Cre father crosses were all females (lanes 13 to 18). From myr-p110αwt/fl mother and hemizygous CMV-Cre father crosses, all Cre harboring embryos were females (lanes 13 to 18 and 25 to 27) and embryos not harboring Cre gene were all males (lanes 19 to 24 and 28 to 30). Embryos were randomly chosen at the embryonic days of 10.5 to E12.5. The PCR products were visualized in a 1.5% agarose gel electrophoresis. Representative results of genomic PCR detecting 441 bp of male-specific gene SRY and 245 bp of the autosomal gene myogenin (Myog) found in both males and females.