

Abstract
One of the most promising classes of iron chelators are α-N-heterocyclic thiosemicarbazones with Triapine as the most prominent representative. In several clinical trials Triapine showed anticancer activity against hematological diseases, however, studies on solid tumors failed due to widely unknown reasons. Some years ago, it was recognized that “terminal dimethylation” of thiosemicarbazones can lead to a more than 100-fold increased activity, probably due to interactions with cellular copper depots. To better understand the structural requirements for the switch to nanomolar cytotoxicity, we systematically synthesized all eight possible N-methylated derivatives of Triapine and investigated their potential against Triapine-sensitive as well as -resistant cell lines. While only the “completely” methylated compound exerted nanomolar activity, the data revealed that all compounds with at least one N-dimethylation were not affected by acquired Triapine resistance. In addition, these compounds were highly synergistic with copper treatment accompanied by induction of reactive oxygen species and massive necrotic cell death.
Introduction
Due to the limited success of chemotherapeutic agents in the treatment of advanced cancer, novel anticancer drugs with different mechanisms of action need to be developed. One possibility is to target the deregulated iron metabolism of rapidly dividing cancer cells.1,2 Therefore, several iron chelators have been developed. The first candidate with potential anticancer activity was desferrioxamine (DFO),2 which entered clinical trials in the 1980s and showed remarkable results in leukemia3 as well as neuroblastoma patients.4 Nevertheless, subsequent studies demonstrated failure of DFO as a potent anticancer agent,5,6 which was at least in part connected with the very short plasma half-life time and low membrane permeability of this compound.7 Consequently, numerous other iron chelating drugs have been developed to overcome these limitations.2,8 One very promising class of iron chelators are α-N-heterocyclic thiosemicarbazones that harbor a N,N,S tridentate motif able to strongly coordinate to transition metal ions.9,10 The most prominent and best characterized member is 3-aminopyridine-2-carboxaldehyde thiosemicarbazone, also known as 3-AP or Triapine.7 Triapine is a highly efficient inhibitor of ribonucleotide reductase11 (via destruction of the iron-dependent tyrosyl radical12), a crucial enzyme for the synthesis of dNTPs. In addition, also other thiosemicarbazones like di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mt) and di-2-pyridylketone-4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC) are currently intensively investigated as drug candidates.13 With regard to the clinical situation, Triapine showed promising anticancer activity in several clinical phase I and II trials on patients with hematological diseases,14,15 while studies on solid tumors failed so far.16−18 The reason for this lack of efficiency against solid tumors is currently not fully understood, but one hypothesis is rapid development of resistance. Therefore, our group has recently generated a Triapine-resistant colon carcinoma cell line to investigate the mechanisms underlying acquired Triapine resistance in solid cancer cells. Interestingly, very rapid up-regulation of well-known multidrug resistance mechanisms, such as ABCB1 (P-glycoprotein) and protein kinase C, were found, although Triapine is only a weak substrate for ATP-binding cassette transporters.19
In order to improve thiosemicarbazone-based therapy, multiple novel derivatives have been developed in the last years resulting in the discovery of several compounds with a distinctly increased cytotoxicity. One of these compounds was the α-pyridyl thiosemicarbazone Dp44mT, which exhibited a more than 100-fold higher cytotoxicity than Triapine.20−22 This strongly increased activity was also shared by some other α-N-heterocyclic thiosemicarbazones published by our group and seems to be associated with the so-called “terminal dimethylation” (lack of hydrogen atoms at the terminal nitrogen atom).23,24 Interestingly, at least in the case of Dp44mT, the improved anticancer activity was so far explained by interaction with cellular copper ions, which results in oxidative stress and apoptosis induction.25−27 In order to fill the knowledge gap regarding the anticancer mechanisms of Triapine and the terminally dimethylated nanomolar cytotoxic derivatives, in this work, systematically all possible N-methylated derivatives of Triapine (Scheme 1) were synthesized and their physicochemical as well as anticancer properties were analyzed. In particular, we focused on the detailed structure–activity relationship, mode of action studies, and impact of acquired Triapine resistance on the activity of the novel derivatives.
Scheme 1. Library of the Investigated Compounds.

Reaction conditions: (i) (Boc)2O, sodium bis(trimethylsilyl)amide; (ii) NaH, MeI; (iii) n-BuLi, DMF; (iv) respective thiosemicarbazide (and conc. HCl).
Results
Synthesis and Characterization
The novel monomethylated 3-aminopyridine derivatives were synthesized by treatment of Boc-protected 3-amino-2-bromopyridine with methyl iodide in the presence of NaH. For dimethylation 3-amino-2-bromopyridine was directly treated with excess methyl iodide/NaH. Subsequently, in both cases, the bromo species were converted to the respective aldehydes using n-buthyllithium (n-BuLi) and dimethylformamide (DMF), which were finally reacted with the respective thiosemicarbazides (Scheme 1). For the monomethylated 3-aminopyridine derivatives, conc. HCl was added in the last step for deprotection, resulting in formation of the HCl salts (Scheme 1).
The 1H NMR spectra of the H2NNR2 series in dimethyl sulfoxide (DMSO)-d6 showed only one isomeric form with the N–NH signal around 11.1–11.4 ppm. This indicates the presence of the so-called E-isomer of 2-formylpyridine thiosemicarbazones.28 Also the HCl salts of the MeHNNR2 series resonated in this region (11.8–12.2). In contrast, for all three Me2NNR2 derivatives two sets of signals could be observed in DMSO-d6: the E-isomer again in the region 11.1–11.6 ppm and a second N–NH signal between 13.9–15.2 ppm attributed to the Z-isomer. This strong low field shift can be explained by the involvement of the N–NH moiety in an intramolecular hydrogen bond. This bond also strongly influences the shift of the HC=N proton (E-isomer, ∼8.5; Z-isomer, ∼7.5) and the corresponding carbon C=N signal (E-isomer, ∼142 ppm; Z-isomer, ∼132 ppm). Noteworthy, in the cases of Me2NNH2 and Me2NNHMe, the unpurified spectra showed both isomers, whereas after chromatographic purification only the pure E-isomer was present. In contrast, for Me2NNMe2 both isomers were observed in the NMR spectrum after column chromatography.
X-ray quality crystals of Me2NNH2 were obtained from a H2O/EtOH solution stored at 4 °C. Results of X-ray diffraction analysis are shown in Figure 1A, together with selected bond lengths and angles. Me2NNH2 crystallized in the monoclinic space group P21/c and adopts the Z-isomeric form, with a hydrogen bond between N1 and N4, in contrast to Triapine with E-configuration.23
Figure 1.

(A) X-ray crystal structure of Me2NNH2. The thermal ellipsoids are drawn at 50% probability levels. Selected bond lengths (Å) and angles (deg): C6–N3 1.2957(15), N3–N4 1.3649(14), N4–C7 1.3526(15), C7–S1 1.6850(12), C7–N5 1.3272(16) Å; θ(N2–C4–C5–C6) −5.88(17), θ(C5–C6–N3–N4) −0.84(18), θ(N3–N4–C7–S1) 177.52(8)°. (B) Three-dimensional fluorescence spectrum of MeHNNH2 in 1% DMSO/PBS (10 μM) at pH 7.4.
Due to the intrinsic fluorescence properties of Triapine,29 the series of new derivatives was also investigated by fluorescence spectroscopy (conditions: 10 μM in 1% DMSO/phosphate buffered saline (PBS), pH 7.4). Maximum of the excitation wavelength, emission wavelength, and intensity of emitted fluorescence light are listed in Table 1, and a 3D fluorescence plot of MeHNNH2 is shown in Figure 1B. Concerning the fluorescence intensity, no general trends could be observed, only the Me2NNR2 series showed distinctly lower molar intensity compared to the other derivatives. The maxima of the excitation wavelengths correlate as expected with the highest bands at the highest λmax in the UV/vis spectra. Noteworthy, the excitation maximum increases from the H2NNR2 series at ∼370 nm to the MeHNNR2 derivatives at ∼400 nm. However, unexpectedly it strongly decreases to ∼350 nm in the three Me2NNR2 compounds.
Table 1. Fluorescence Data of Triapine and Its Derivatives.
| compda | excitation maximum (nm) | emission maximum (nm) | counts per secondb |
|---|---|---|---|
| Triapine | 368 | 454 | 1,360,000 |
| H2NNHMe | 364 | 448 | 1,109,300 |
| H2NNMe2 | 368 | 422 | 1,530,600 |
| MeHNNH2 | 396 | 482 | 2,360,200 |
| MeHNNHMe | 392 | 482 | 1,524,900 |
| MeHNNMe2 | 396 | 492 | 378,600 |
| Me2NNH2 | 352 | 506 | 409,200 |
| Me2NNHMe | 352 | 500 | 731,300 |
| Me2NNMe2 | 352 | 482 | 670,700 |
DMSO stock solutions of all compounds were diluted with PBS (pH 7.4) to a final concentration of 10 μM (1% DMSO).
Measured at the maxima of the excitation and emission wavelengths.
The lipo-hydrophilic character of the compounds was studied at pH 7.4 via partitioning between n-octanol and water (Table 2). According to the pKa values of Triapine and H2NNMe2, both are neutral (100% HL) at pH 7.4,30,31 which can also be expected for the other derivatives. Thus, the logD7.4 values are considered to be equal to the logP values of the compounds.
Table 2. LogD7.4 Values (n-Octanol/Water) for the Triapine Derivatives [25 °C, pH = 7.40, 10 mM 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic Acid (HEPES) and I = 0.10 M (KCl)].
| λmax (nm) (water) | λmax (nm) (n-octanol) | logD7.4 | SD | logP predicted with Chemdraw | |
|---|---|---|---|---|---|
| Triapine | 360 | 376 | 0.85a | –0.02 | |
| H2NNHMe | 360 | 372 | 1.20 | 0.03 | 0.50 |
| H2NNMe2 | 362 | 374 | 1.30b | 0.88 | |
| MeHNNH2 | 386 | 396 | 1.40 | 0.10 | 0.28 |
| MeHNNHMe | 384 | 392 | 2.03 | 0.10 | 0.80 |
| MeHNNMe2 | 386 | 394 | 2.10 | 0.01 | 1.18 |
| Me2NNH2 | 348 | 364 | 1.21 | 0.02 | 1.07 |
| Me2NNHMe | 350 | 364 | 1.86 | 0.01 | 1.59 |
| Me2NNMe2 | 352 | 372 | 1.52 | 0.03 | 1.97 |
Stepwise methylation of Triapine up to MeHNNMe2 as expected increased the lipophilicity. Noteworthy, the methylation of NH2 → NHMe had a much stronger influence on the lipophilicity compared to the NHMe → NMe2 step. However, again trends of the Me2NNR2 derivatives did not correlate with all other compounds. Unexpectedly, the logD7.4 values were below that of the MeHNNR2 series, and , in addition, there was no stepwise increase from terminal NH2 to NMe2. In general, the obtained logD7.4/P values were much higher than the values predicted with ChemDraw software. This once again confirms that the calculation of logP values is often afflicted with errors (especially when isomerization or intermolecular bonding is not considered).
Synthesis and Investigation of Isomers
The fact that the physicochemical parameters (UV/vis and fluorescence maxima, lipophilicity) of the three derivatives of the Me2NNR2 series did not fit into the expected range prompted us to further investigate this set of compounds. As the NMR spectra of the Me2NNH2 series already indicated the presence of two isomers (in contrast to the other six compounds), we aimed to isolate both isomers as pure compounds. In the cases of Me2NNH2 and Me2NNHMe, the standard synthesis with chromatographic purification already yielded the pure E isomers. To obtain the respective Z isomers, the E isomers were stirred in acetonitrile for 24 h at 37 °C. This was necessary because solvents with a low donor number32 were previously reported to stabilize the Z isomer.28,33 Indeed, this approach resulted in partial conversion of the two compounds and allowed isolation of the pure Z isomers of Me2NNH2 and Me2NNHMe after chromatographic separation. For Me2NNHMe2, the isomers could not be separated due to a very fast interconversion.
As next step, the isomerization process of all nine compounds including the purified E and Z isomers of Me2NNH2 and Me2NNHMe were studied in PBS at pH 7.4 via high-performance liquid chromatography (HPLC) coupled to a mass spectrometry (MS) detector (Table 3).
Table 3. Isomerization Study in PBS at pH 7.4 via HPLC–MSa.
| 0 h |
24 h |
|||
|---|---|---|---|---|
| compd | % isomer 1 | % isomer 2 | % isomer 1 | % isomer 2 |
| Triapine | 100 (7.7)b | 100 | ||
| H2NNHMe | 100 (10.0) | 100 | ||
| H2NNMe2 | 100 (10.2) | 100 | ||
| MeHNNH2 | 100 (10.5) | 100 | ||
| MeHNNHMe | 99 (11.3) | 1 (9.8)b | 99 | 1 |
| MeHNNMe2 | 98 (11.3) | 2 (10.3) | 98 | 2 |
| (E)-Me2NNH2 | 100 (10.3) | 8 | 92 | |
| (Z)-Me2NNH2 | 94 (13.9) | 6 (10.3) | 6 | 94 |
| (E)-Me2NNHMe | 4 (16.3) | 96 (11.1) | 15 | 85 |
| (Z)-Me2NNHMe | 93 (16.3) | 7 (11.1) | 13 | 87 |
| Me2NNMe2 | c | c | c | c |
DMSO stock solutions of all compounds were diluted with PBS (pH 7.4) to a final concentration of 50 μM (1% DMSO) and immediately measured by HPLC–MS. All measured values are ±2%.
The numbers in brackets correspond to the HPLC–MS retention time (min) of the compounds.
In the case of Me2NNMe2, only one very broad peak was obtained, which could not be separated into the two isomers, presumably due to the fast interconversion on the column.
In aqueous solution, the N–NH protons, which are usually used for the assignment of E and Z isomer in organic solvents via NMR spectroscopy, are not detectable anymore. Thus, for the HPLC measurements, the respective retention times were used as assignment parameters (the different isomers were termed isomer 1 and 2). The measurements performed directly after dissolution in PBS showed a clear increase of the retention times with increasing number of methyl groups. Consequently, Triapine was found at 7.7 min, followed by MeHNNMe2 at 11.3 min, (Z)-Me2NNH2 at 13.9 min, and finally (Z)-Me2NNHMe at 16.3 min. For Me2NNMe2 only a very broad peak was observed, presumably due to fast interconversion of the isomers on the column. In contrast, the synthesized E-isomers are not in line with this trend, with (E)-Me2NNH2 at 10.3 min and (E)-Me2NNHMe at 11.1 min. After 24 h the isomeric pattern was identical for all derivatives, except for the purified E and Z isomers of Me2NNH2 and Me2NNHMe, which interconverted and reached an equilibrium with ∼90% isomer 2 and 10% isomer 1 (Figure 2). In contrast to all other derivatives, in the cases of Me2NNH2 and Me2NNHMe, isomer 2 was stabilized in PBS solution. This could explain why the measured UV/vis and fluorescence maxima as well as lipophilicity do not follow a clear trend within all nine compounds.
Figure 2.

HPLC-MS chromatograms of the interconversion of isomer 1 (black) and isomer 2 (red) of Me2NNH2 in PBS at pH 7.4.
Cytotoxicity
To assess the impact of the structural modifications in the Triapine backbone on the antitumor activity, Triapine and its eight derivatives were tested against the colon adenocarcinoma SW480, the ovarian carcinoma A2780, selected Triapine-resistant SW480/Tria cells,19 the cervix carcinoma KB-3-1 together with its ABCB1-overexpresing subline KBC-1, and finally the lung fibroblast line WI-38. To this end, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays were performed after 72 h of drug incubation.
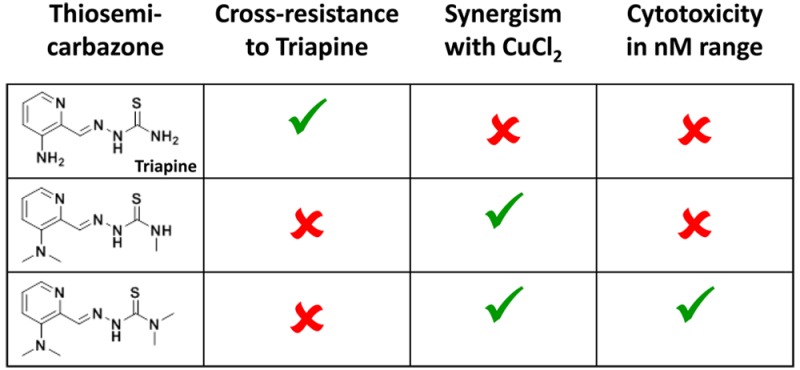
In the cancer cell models, all compounds revealed IC50 values in the low μM range with rather minor differences (Table 4). The only remarkable exception was Me2NNMe2, exerting extremely increased activity with IC50 values in the low nM range. With regard to the structural differences, in the chemosensitive A2780 and SW480, terminal monomethylation in the cases of H2NNHMe and MeHNNHMe resulted in decreased activity (e.g., in SW480 cells by 2.9- and 2.3-fold, respectively) compared to the terminal NH2 derivatives. In contrast, terminal dimethylation showed a tendency to increase the cytotoxicity in the cases of H2NNMe2 and MeHNNMe2. Also monomethylation of the pyridine NH2 slightly decreased the anticancer activity in both cells lines compared to the nonmethylated derivatives. Notably, dimethylation of the pyridine NH2 resulted in strong variances between the individual experiments, indicated by the rather high standard deviations of the respective IC50 values. Interestingly, these are exactly the derivatives where the presence of different isomers was observed (in the cases of Me2NNH2 and Me2NNMeH, both the isomeric mixture and the pure E and Z isomers were tested, however, without significant differences). Furthermore, clearly diminished activity of Me2NNH2 compared to Triapine as well as a dramatic increase from H2NNMe2 to Me2NNMe2 (43-fold in A2780 cells) was found, resulting in nanomolar activity for Me2NNMe2.
Table 4. IC50 Values (μM) of Triapine and Its Derivatives in Different Cancer Cell Lines after 72 h Drug Treatmenta.

***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05; n.s., not significantly different, calculated by student’s t test with Welch’s correction; n.t., not tested.
Interestingly, Triapine-resistant SW480/Tria cells responded differently as compared to the parental SW480 cells. In general, with increasing number of methylation sites also the cytotoxicity increased regardless whether methylation was at the pyridine NH2 or at the terminal NH2 position. Hence, the observed order of activity was Triapine < H2NNHMe < H2NNMe2 and Triapine < MeHNNH2 < Me2NNH2. Me2NNMe2 was again the most active thiosemicarbazone, with IC50 values in the nM range. Also the degree of resistance (resistance factors are shown in Table 4) markedly decreased with increased methylation of the pyridine NH2 and terminal NH2 groups. Thus, SW480/Tria cells were highly cross-resistant against Triapine derivatives with one methyl group (H2NNHMe and MeHNNH2). For MeHNNHMe, the activity was not diminished in the Triapine-resistant cell model. In contrast, for all thiosemicarbazones, with at least one NMe2 moiety, the activity was even higher in the Triapine-resistant cell line compared to the parental SW480 cells (in most cases the IC50 levels were ∼0.5-fold compared to the parental cells). However, due to the high standard deviation of the derivatives with dimethylation of the pyridine NH2, only MeHNNMe2 reached statistical significance. This clearly indicates that dimethylation of one of the two amino groups is able to efficiently overcome acquired Triapine resistance or even induce a tendency toward collateral sensitivity. To investigate also the contribution of ABCB1 expression on the activity, our panel was additionally tested on KB-3-1 cells in comparison to its highly ABCB1-overexpressing subline KBC-1. In agreement to our previous publication,19 KBC-1 displayed a weak (2.9-fold) resistance to Triapine and closely related derivatives like H2NNHMe, MeHNNH2, and MeHNNHMe, whereas drug resistance was reduced (not significant) by terminal dimethylation. However, dimethylation at the pyridine NH2 in the case of Me2NNH2 and Me2NNHMe induced significant collateral sensitivity of KBC-1 cells.
Finally, the impact of our test panel on nonmalignant lung fibroblasts (WI38) was investigated. This cell line was in general less sensitive toward our compound panel than the cancer cell lines, especially A2780. However, it might be worth noting that the structure–activity relationship of WI-38 differed to some extent from the cancer cell models. Such, it displayed the highest sensitivity toward all compounds harboring a terminal dimethylation (H2NNMe2, MeHNNMe2, Me2NNMe2), with IC50 values ∼1 μM. This also implies that Me2NNMe2 showed no nanomolar activity in this cell model. Overall, these data indicate a cancer cell-specific activity window for several compounds of our panel including Me2NNMe2. This is also supported by preliminary toxicity tests of Me2NNMe2 in mice revealing no differences in the tolerability compared to, e.g., Triapine.
Despite their similarities in IC50 values (except Me2NNMe2), the morphological evaluations of the treated cells revealed distinct differences between the tested derivatives (exemplary images taken after 48 h incubation time are shown in Figure 3A). In general, the tested substance panel could be divided into two categories based on the induction of characteristic morphologic phenotypes: (1) induction of massive cell flattening resulting in distinctly increased cell size and surface area (especially pronounced for Triapine, H2NNHMe, and MeHNNH2), in contrast to (2) mild cell flattening accompanied by appearance of vesicular blebbing in the cytosol (Figure 3B; especially pronounced for MeHNNMe2, Me2NNHMe, and the nanomolar cytotoxic Me2NNMe2). Based on already available data for the nanomolar derivative Dp44mT,13,27,34 this could indicate a structure-dependent increasing impact of the compounds on lysosomal compartments.
Figure 3.

(A) Phase contrast images of SW480 cells treated with 2.5 μM of the indicated drugs for 48 h (200× magnification). (B) Vacuoles in SW480 cells induced by 24 h treatment of Me2NNMe2 shown at 400× magnification. At early time points, vacuolization was predominantly perinuclear, but eventually the vacuoles fill the entire cytoplasmic space. Scale bar: 10 μm.
Recently, Ishiguro et al. showed that Triapine differs from the terminally dimethylated nanomolar cytotoxic Dp44mT and P44mT (pyridine-2-carboxaldehyde 4,4-dimethyl-3-thiosemicarbazone) in its interaction with copper ions.25 Thus, we investigated the impact of preincubation with CuCl2 on the activity of our thiosemicarbazone panel (Figure 4). In agreement with the published data, CuCl2 (when given in excess or at least in a 1:1 ratio) had potent protective effects toward Triapine. In contrast, the nanomolar derivative Me2NNMe2 was highly synergistic with CuCl2 (with combination indices (CI values) down to 0.02, Supporting Information Figure S1), which is in line with the data on the nanomolar Dp44mT (Figure S2) and P44mT.25 In addition, strong synergism with CuCl2 was also observed for MeHNNMe2, Me2NNH2, and Me2NNHMe, and at higher concentrations with H2NNMe2 and MeHNNHMe (the strongest effects were observed for MeHNNMe2 where CI values below 0.0001 were calculated, Figure S1). 2′,7′-Dichlorofluorescein diacetate (DCF-DA) staining experiments suggested that this higher cytotoxic activity in the presence of CuCl2 is associated with induction of reactive oxygen species (ROS; Figure 5A). As higher drug concentrations of Me2NNHMe and Me2NNMe2 (5 μM drug with 10 μM CuCl2) decreased the DCF-DA signals to the level of untreated cells, tests for membrane integrity using the fluorescent dye propidium iodide (PI) were performed. These indicated that at the given conditions massive necrotic cell death (∼80% of the cells were found to be PI-positive) occurred (Figure 5B). Consequently, the lack of DCF-DA signal in these cells might be explained by enhanced membrane permeability (due to treatment-induced ROS), which could have reduced retention of DCF inside the cells.
Figure 4.

Impact of structural modifications of Triapine on the cytotoxicity in the presence of Cu(II) ions. Briefly, after 60 min preincubation with CuCl2 (10 and 30 μM), SW480 cells were treated for 72 h with the indicated concentrations of Triapine and its derivatives. Viability was determined using MTT assay. The values given are the mean ± the standard deviation of triplicates from one representative experiment out of three.
Figure 5.

(A) Intracellular ROS production in HL-60 cells after 30 min treatment of Triapine or its derivatives and 15 min prior to incubation with CuCl2. As positive control, H2O2 was used. DCF-DA fluorescence was measured using flow cytometry. (B) Percentage of dead cells in conditions according to the DCF-DA assay. Cells were stained with Hoechst/PI, and the percentage of PI-positive cells was determined microscopically. Ligand/copper(II) ion ratio, 1:1 and 1:2. The values given are the mean ± the standard deviation of three experiments. Significances were established using one-way ANOVA with Bonferroni’s multiple comparison test (***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05).
Discussion and Conclusion
Thiosemicarbazones are known for their promising biological activity, which resulted in their clinical development as anticancer drugs. The best investigated agents of this class of compounds are Triapine and Dp44mT.7,8,35 Noteworthy, Triapine has been tested in multiple clinical trials, showing promising activity in hematological diseases.13,14 However, there is so far no proof of anticancer activity of Triapine when given as monotreatment against solid tumors.36 The reasons for this are widely unexplored and make the development of novel thiosemicarbazone-based drugs interesting. Dp44mT has been well studied in multiple preclinical studies, indicating that this compound strongly differs from Triapine in several aspects. For example, Dp44mT was characterized by a distinctly higher (IC50 ≈ nM) anticancer activity and a very high affinity for copper(II) ions.13,26,27 Also some of the thiosemicarbazone derivatives developed in our group shared this shift of activity to the nM range.23 Our data indicated that this pronounced anticancer activity is associated with terminal nitrogen dimethylation but only in the absence of any NH2 functionality in the thiosemicarbazone backbone. Noteworthy, this increased activity was not accompanied by a similar increase in ribonucleotide reductase inhibition.23
In the here presented study, the structure–activity relationship of Triapine derivatives was investigated in detail using the complete panel of stepwise methylated Triapine analogues. Our new panel was synthesized with the aim to allow detailed structure–activity/mode of action studies as well as development of novel derivatives to overcome Triapine resistance. Overall, several (rather unexpected) conclusions can be drawn from our results: (1) in contrast to the expected correlation of increased methylation of Triapine with changes in physicochemical properties, the Me2NNR2 series showed unexpected behavior in terms of lipophilicity and characteristics of the UV/vis spectra. Subsequent studies revealed that this is based on isomerization and stabilization of a different isomer in buffered aqueous solution. Therefore, simple prediction of physicochemical properties does not seem to be possible for this class of compounds. (2) The shift of the cytotoxic activity to the nanomolar range was only observed for one derivative, namely, Me2NNMe2. This means that already the presence of only one hydrogen atom either at the terminal nitrogen or at the aminopyridine nitrogen distinctly reduced the anticancer potential of the drug by ∼100–1000-fold. This is in contrast to some reports on several Richardson-type dipyridyl compounds, which did not follow this structure–activity relationship when tested on SK-N-MC neuroepithelioma cells.22,37 In detail, in this cell line several of Richardson’s complexes with terminal NHR-moiety (R = methyl, ethyl, phenyl, or allyl) were of similar activity like the terminally dimethylated Dp44mT. However, a study of the same complexes in 28 different cell lines revealed an average IC50 value of 0.03 ± 0.01 μM for Dp44mT, whereas the four thiosemicarbazones with terminal NHR had average IC50 values of 0.20–1.70 μM.21 Thus, these average cytotoxicity data followed the trend of our study with distinctly increased activity of compounds without any NH group. Noteworthy, these data also suggest that the activity enhancement by complete dimethylation is cell context-dependent, which is in agreement with our data revealing much stronger differences in the case of A2780 compared to SW480 cells. (3) The impact of Triapine resistance on the activity of the novel derivatives was strongly associated with methylation of the pyridine and terminal NH2 group. SW480/Tria cells were cross-resistant or equally active to compounds with only monomethylation of one or both NH2-groups (H2NNHMe, MeHNNH2, and MeHNNHMe). In contrast, all compounds with at least one NH2-dimethylation were not affected by the Triapine-resistance of SW480/Tria. This clearly indicates that dimethylation of one of the two amino groups is a valuable tool to efficiently overcome acquired Triapine resistance and suggests a different mode of action. Furthermore, it shows that the nanomolar activity is not mandatory for circumventing Triapine resistance. (4) Preliminary studies using ABCB1-overexpressing cells indicate that the impact of this efflux pump differs within the Triapine derivatives. Such, in agreement to our previous publication,19 KBC-1 displayed a weak resistance to Triapine and closely related derivatives like H2NNHMe, MeHNNH2, and MeHNNHMe. In contrast, drug resistance was reduced (not significant) by terminal dimethylation. Dimethylation at the pyridine NH2 in the cases of Me2NNH2 and Me2NNHMe induced significant collateral sensitivity of KBC-1 cells. The nanomolar Me2NNMe2 showed no significant differences between KB-3-1 and KBC-1. This is in contrast to data on Dp44mT, where induction of collateral sensitivity was recently published in KBV-1 cells34,35 and might indicate some differences between Dp44mT and our nanomolar compound. (5) The observed hyperactivity on Triapine-resistant cancer cells positively correlated with the appearance of vesicular blebbing. This is a first hint on the molecular differences between classical thiosemicarbazones like Triapine and novel derivatives especially those with three and four methyl groups (MeHNNMe2 and Me2NNHMe as well as the nanomolar Me2NNMe2), which are able to circumvent Triapine resistance. (6) Also with respect to the interaction with copper(II) ions, distinct differences were observed in our study. Thus, compounds that are inefficient against SW480/Tria cells also showed reduced cytotoxicity in the presence of copper(II) ions. In contrast, hyperactive compounds (again especially MeHNNMe2, Me2NNHMe and the nanomolar Me2NNMe2) were highly synergistic with copper(II) treatment accompanied by induction of ROS and massive necrotic cell death. In general, ROS production by Me2NNMe2 is not surprising, as it is in good agreement with data on Dp44mT, reporting copper-dependent ROS production by this thiosemicarbazone with nanomolar cytotoxicity.25,27 However, our data indicate that the increased cytotoxicity in the presence of copper(II) ions is not the (only) reason for the nanomolar activity of some thiosemicarbazones, as also MeHNNMe2 and Me2NNHMe with a low micromolar cytotoxicity show this synergistic effect. Noteworthy, the reasons underlying this very specific interaction with copper ions are still not fully understood. Ishiguro et al., for example, suggested that copper(II) complexes of Triapine and, in general, thiosemicarbazones without terminal dimethylation are able to get desulfurized in aqueous basic media with formation of insoluble CuS.25 However, as already indicated by Ishiguro et al., our previous solution equilibrium studies on Triapine and several related derivatives did not show any indications for such an irreversible process. In contrast, Lovejoy et al. suggested that due to its “polyprotic nature” Dp44mT is specifically trapped in the acidic lysosomes after protonation of the first pyridyl moiety and subsequent copper binding by the second resulting in formation of a positively charged copper complex.27 Noteworthy, despite several similarities to Dp44mT, all our derivatives contain only one pyridyl group. Consequently, the mechanisms suggested for cytotoxic activity of Dp44mT cannot explain the differences observed with regard to the interaction with copper in our study. Solution equilibrium studies showed that terminal dimethylation of thiosemicarbazones significantly increased the copper(II) binding abilities compared to derivatives with a terminal NH2 moiety.38 However, already the stability of the copper(II) complex in the case of Triapine is outstandingly high with >99% [CuL]+ at 1 μM at pH 7.4. Thus, also the stability does not seem to be the crucial parameter, and probably other factors like the thermodynamics and kinetics of the reduction process in the cellular environment are important. Studies addressing these issues are currently in progress in our group. Nevertheless, the rapid necrotic cell death observed with some derivatives in the presence of copper should be also considered carefully with respect to a possible application in vivo, especially, as it is well-known that induction of necrosis can result in inflammatory reactions of the body.39 Moreover, it has to be considered that various cancer types are frequently associated high serum copper levels.40,41 Consequently, treatment of such patients with copper-synergizing thiosemicarbazones might be associated with increased occurrence of adverse effects due to ROS production.
Summarizing, we systematically synthesized a panel of N-methylated Triapine derivatives and investigated their biological modes of action. Our data gave new insights into the coherences between methylation, cross-resistance to Triapine, nanomolar cytotoxicity, and synergism with copper(II) ions. The data, on the one hand, reveal that stepwise methylation of Triapine results in a change of the mode of action, which might be associated with the interaction of the intracellular copper balance. However, on the other hand, these effects do not seem to be responsible for the increased cytotoxic activity of some derivatives into the nanomolar range.
Experimental Section
tert-Butyl-(2-bromopyridin-3-yl)carbamate (1), tert-butyl (2-formylpyridin-3-yl)carbamate (2), Triapine, and H2NNMe2 were synthesized as previously reported.23 All other solvents and chemicals were purchased from commercial suppliers and used without further purification. Elemental analyses were performed by the Microanalytical Laboratory of the University of Vienna and are within ±0.4%, confirming >95% purity. All chromatographic separations were performed on silica gel. UV/vis spectra were recorded on an Agilent 8453 spectrophotometer from 200 to 1000 nm in PBS buffer pH 7.4 (<0.5% DMSO). ESI–MS spectrometry was carried out with a Bruker Esquire3000 ion trap spectrometer (Bruker Daltonics, Bremen, Germany). Expected and experimental isotope distributions were compared. 1H and 13C one- and two-dimensional NMR spectra were recorded in DMSO-d6, with a Bruker Avance III 500 MHz FT-NMR spectrometer at 500.10 (1H) and 125.75 (13C) MHz at 298 K. The residual 1H and 13C present in DMSO-d6 were used as internal references. Abbreviations for NMR data: py = pyridine, q,py = quaternary carbon of pyridine. The fast relaxation of the HCl salts MeHNNH2 and MeHNNHMe prevented the exact assignment of all protons and the detection of all carbon signals. Fluorescence measurements were performed on a Horiba FluoroMax-4 spectrofluorometer and processed using the FluorEssence v3.5 software package. All tested solutions had a concentration of 1 × 10–5 M. Scans were run at room temperature in 1% DMSO/PBS pH 7.4 with excitation and emission slit widths of 4 nm.
Synthesis
3-Aminopyridine-2-carbaldehyde N-Methylthiosemicarbazone (H2NNHMe)
tert-Butyl (2-formylpyridin-3-yl)carbamate (2) (147 mg, 0.66 mmol) was dissolved in EtOH (3 mL)/H2O (1 mL), and 4-methylthiosemicarbazide (70 mg, 0.59 mmol) as well as conc. HCl (0.3 mL) were added. The reaction mixture was refluxed for 3 h and cooled to RT. The precipitate was filtered, washed with EtOH, and dried in vacuo. H2NNHMe·HCl was dissolved in water at 70 °C, and 10% aq. NaHCO3 (0.8 mL) was added. After 1 h the precipitate was filtered off, washed with cold EtOH and Et2O and dried in vacuo. Yield: 110 mg (89%). Anal. Calcd for C8H11N5S (Mr 209.27 g/mol): C, 45.91; H, 5.30; N, 33.47; S, 15.32. Found: C, 45.57; H, 5.47; N, 33.27; S, 15.12. UV/vis (PBS), λmax, nm (ε, M–1 cm–1): 259 (17800), 284 (17470), 359 (23750). ESI-MS in MeOH (negative): m/z 210 [M + H]+. 1H NMR (DMSO-d6): δ 11.35 (s, 1H, N–NH), 8.35–8.29 (m, 2H, HC=N and NHCH3), 7.84 (dd, 3J = 4 Hz, 4J = 1 Hz, 1H, Hpy), 7.16 (dd, 3J = 8 Hz, 4J = 1 Hz, 1H, Hpy), 7.08 (dd, 3J = 8 Hz, 3J = 4 Hz, 1H, Hpy), 6.46 (s, 2H, NH2), 3.03 (d, 3J = 4 Hz, 3H, NHCH3) ppm. 13C NMR (DMSO-d6): δ = 177.6 (C=S), 149.3 (C=N), 144.3 (Cq, py), 137.7 (Cpy), 133.5 (Cq, py), 124.8 (Cpy), 122.7 (Cpy), 31.7 (CH3) ppm.
tert-Butyl(2-bromopyridin-3-yl)(methyl)carbamate (3)
tert-Butyl-(2-bromopyridin-3-yl)carbamate (1) (1.0 g; 3.66 mmol) was dissolved in dry DMF (20 mL) at 0 °C and 60% NaH in mineral oil (183 mg, 4.58 mmol) was slowly added. The solution was stirred for 20 min, and MeI (0.26 mL, 4.18 mmol) was added dropwise. After 30 min at 0 °C the solution was additionally stirred for 1 h at RT. The reaction was quenched with water (20 mL), and the solvents were evaporated under reduced pressure. The residue was dissolved in water and extracted with Et2O. The organic layers were washed with H2O, 0.1 M HCl, sat. NaHCO3 solution, and brine, dried over Na2SO4, evaporated under reduced pressure, and dried in vacuo. n-Hexane was added to precipitate the product. The product was used without further purification. Yield: 0.84 g (85%). 1H NMR (DMSO-d6): δ 8.37–8.30 (m, 1H, Hpy), 7.90 (dd, 3J = 8 Hz, 4J = 2 Hz, 1H, Hpy), 7.51 (dd, 3J = 5 Hz, 3J = 8 Hz, 1H, Hpy), 3.11 and 3.08 (s, 3H, NCH3), 1.48 and 1.30 (s, 9H, (CH3)3) ppm.
tert-Butyl(2-formylpyridin-3-yl)(methyl)carbamate (4)
Compound 3 (1.0 g; 3.48 mmol) was dissolved in dry THF (15 mL) at −78 °C and n-BuLi (2.6 mL, 4.18 mmol) was slowly added. After 1 h, DMF (0.31 mL, 4.18 mmol) was added, and the reaction mixture was allowed to slowly warm up to RT. Subsequently, after addition of 1.5 M HCl (1.5 mL) the pH was adjusted to about 7 with Na2CO3. The solution was extracted with EtOAc (3×), H2O (2×), and brine (1×), dried over Na2SO4, evaporated under reduced pressure and dried in vacuo. Yield: 0.69 g (82%). 1H NMR (DMSO-d6): δ 10.01 (br. s, 1H, CHO), 8.70 (dd, 3J = 5 Hz, 4J = 1 Hz, 1H, Hpy), 7.94 (dd, 3J = 8 Hz, 4J = 1 Hz, 1H, Hpy), 7.73 (dd, 3J = 8 Hz, 3J = 5 Hz, 1H, Hpy), 3.15 (br. s, 3H, NCH3), 1.46 and 1.23 (s, 9H, (CH3)3) ppm.
3-(Methylamino)pyridine-2-carbaldehyde Thiosemicarbazone Hydrochloride (MeHNNH2)
The synthesis of MeHNNH2 was already reported in literature, but using a different synthetic strategy.42 Compound 4 (100 mg, 0.42 mmol) was dissolved in EtOH (3.5 mL)/H2O (0.65 mL) and thiosemicarbazide (42.4 mg, 0.47 mmol) as well as conc. HCl (176 μL) were added. The red solution was refluxed for 4 h and cooled to RT. The orange precipitate was isolated by filtration, washed with cold EtOH, and dried in vacuo. Yield: 81 mg (78%). Anal. Calcd for C8H11N5S·HCl (Mr = 245.73 g/mol): C, 39.10; H, 4.92; N, 28.50. Found: C, 39.10; H, 5.00; N, 28.23. ESI-MS in MeOH (negative): m/z 208 [M–HCl–H]−. UV/vis (PBS), λmax, nm (ε, M–1 cm–1): 263 (10690), 291 (11620), 383 (11370). 1H NMR (DMSO-d6): δ 11.84 (s, 1H, NH), 8.51 (s, 1H), 8.41 (s, 2H), 8.02 (d, 3J = 8 Hz, 1H), 7.58 (br. s, 2H), 7.27 (v. br. s, 1H), 2.94 (s, 3H, NCH3) ppm. 13C NMR (DMSO-d6): δ = 178.6 (C=S), 145.8 (Cq, py), 129.0 (Cq,py), 126.7 (Cpy), 124.5 (Cpy), 30.3 (NHCH3) ppm (due to the fast relaxation of the HCl salt not all of the 13C signals could be observed).
3-(Methylamino)pyridine-2-carbaldehyde N-Methylthiosemicarbazone Hydrochloride (MeHNNHMe)
Compound 4 (98.7 mg, 0.418 mmol) was dissolved in EtOH (1.15 mL)/H2O (0.55 mL) and 4-methylthiosemicarbazide (34.8 mg, 0.46 mmol) as well as conc. HCl (0.15 mL) were added. The solution was refluxed for 3 h and cooled to RT. The orange precipitate was separated by filtration, washed with cold EtOH, and dried in vacuo. Yield: 87 mg (80%). Anal. Calcd for C9H13N5S·HCl (Mr = 223.30 g/mol): C, 41.61; H, 5.43; N, 26.96. Found: C, 41.59; H, 5.43; N, 26.89. ESI-MS in MeOH (negative): m/z 222 [M–HCl–H]−. UV/vis (PBS), λmax, nm (ε, M–1 cm–1): 267 (15323), 286 (15334), 383 (14583). 1H NMR (DMSO-d6): δ 12.01 (s, 1H, N-NH), 9.01 (s, 1H, NH), 8.38 (s, 1H, HC=N), 8.03 (dd, 3J = 5 Hz, 4J = 2 Hz, 1H, Hpy), 7.66–7.57 (m, 2H, Hpy), 7.39 (v. br. s., 1H, NH), 3.06 (d, 3H, 3J = 5 Hz, S=CNHCH3), 2.95 (s, 3H, CqpyNHCH3) ppm. 13C NMR (DMSO-d6): δ = 178.3 (C=S), 145.7 (Cq, py), 129.4 (Cpy), 126.5 (Cpy), 122.9 (Cq, py), 31.6 (S=CNHCH3), 30.3 (CpyNHCH3) ppm (due to the fast relaxation of the HCl salt not all of the 13C signals could be observed).
3-(Methylamino)pyridine-2-carbaldehyde N,N-Dimethylthiosemicarbazone Hydrochloride (MeHNNMe2)
Compound 4 (100 mg, 0.42 mmol) was dissolved in EtOH (1.15 mL)/H2O (0.55 mL) and 4,4-dimethyl-3-thiosemicarbazide (50 mg, 0.42 mmol) as well as conc. HCl (133 μL) were added. The solution was refluxed for 4 h and stored at 4 °C overnight. The brown precipitate was separated by filtration, washed with cold isopropanol, and dried in vacuo. Yield: 62 mg (54%). Anal. Calcd for C10H15N5S·HCl (Mr = 273.73 g/mol): C, 43.87; H, 5.89; N, 25.58; S, 11.71. Found: C, 43.85; H, 5.92; N, 25.36; S, 11.75. ESI-MS in MeOH (negative): m/z 236 [M–HCl–H]−. UV/vis (PBS), λmax, nm (ε, M–1 cm–1): 264 (22980), 288 (13530), 384 (14860). 1H NMR (DMSO-d6): δ 12.15 (s, 1H, N-NH), 9.55 (br. s, 1H, NHCH3), 8.96 (s, 1H, HC=N), 8.02 (dd, 3J = 4 Hz, 4J = 2 Hz, 1H, Hpy), 7.73–7.65 (m, 2H, Hpy), 3.37 (s, 6H, N(CH3)2), 3.04 (s, 3H, NHCH3) ppm. 13C NMR (DMSO-d6): δ = 180.2 (C=S), 145.3 (Cq, py), 138.8 (C=N), 128.5 (Cq, py), 128.2 (Cpy), 126.0 (Cpy), 124.2 (Cpy), 41.7 (N(CH3)2), 30.3 (NCH3) ppm.
2-Bromo-N,N-dimethylpyridin-3-amine (5)
3-Amino-2-bromopyridine (1.5 g, 8.67 mmol) was dissolved in dry DMF (30 mL) at 0 °C, and 60% NaH (956 mg, 23.9 mmol) was slowly added. The solution was stirred for 20 min, and MeI (1.24 mL, 19.94 mmol) was added dropwise. After 30 min at 0 °C and 1 h at RT the reaction was quenched with water (3 mL). The solvents were evaporated under reduced pressure; the residue dissolved in water and extracted with Et2O. The organic layer was washed with H2O, 0.1 M HCl, sat. NaHCO3 solution and brine, dried over Na2SO4, evaporated under reduced pressure and dried in vacuo. The mineral oil was removed by column chromatography (pure n-hexane and subsequently pure ethyl acetate to elute the product). Yield: 1.24 g (71%). 1H NMR (DMSO-d6): δ 8.01 (dd, 3J = 5 Hz, 4J = 2 Hz, 1H, Hpy), 7.56 (dd, 3J = 8 Hz, 4J = 2 Hz, 1H, Hpy), 7.38 (dd, 3J = 8 Hz, 3J = 5 Hz, 1H, Hpy), 2.76 (s, 6H, NCH3) ppm.
3-(Dimethylamino)picolinaldehyde (6)
Compound 5 (600 mg; 2.98 mmol) was dissolved in dry THF (12 mL), cooled to −78 °C, and n-BuLi (3.8 mL, 5.96 mmol) was slowly added. After 1 h, DMF (0.3 mL, 3.87 mmol) was added, and the reaction mixture was allowed to slowly warm up to RT. Subsequently, 1 M HCl (2 mL) was added and the pH adjusted to about 7 with Na2CO3. The product was extracted with EtOAc (3×) and washed with H2O (2×) and brine (1×), dried over Na2SO4, evaporated under reduced pressure, and dried in vacuo. Purification was performed via column chromatography (EtOAc/hexane, 6:1) Yield: 0.24 g (54%). 1H NMR (DMSO-d6): δ 9.93 (d, J = 1 Hz, 1H, CHO), 8.20 (dd, 3J = 4 Hz, 4J = 1 Hz, 1H, Hpy), 7.53–7.50 (m, 1H, Hpy), 7.46 (dd, 3J = 9 Hz, 3J = 4 Hz, 1H, Hpy), 2.91 (s, 6H, NCH3) ppm.
3-(Dimethylamino)pyridine-2-carbaldehyde Thiosemicarbazone (Me2NNH2)
Compound 6 (150 mg, 1.00 mmol) was dissolved in EtOH (2 mL)/H2O (1 mL), and thiosemicarbazide (91 mg, 1.00 mmol) was added. The reaction mixture was refluxed for 3 h, cooled to RT, and the solvent was removed under reduced pressure. Purification was performed via column chromatography. Pure ACN was used until the first impurity had been eluted and afterward ACN/MeOH (9:1). Yield: 94 mg (42%). Anal. Calcd for C9H13N5S (Mr = 223.30 g/mol): C, 48.41; H, 5.87; N, 31.36; S, 14.36. Found: C, 48.32; H, 5.89; N, 31.16; S, 14.25. ESI-MS in MeOH (positive): m/z 246 [M + Na]+. UV/vis (PBS), λmax, nm (ε, M–1 cm–1): 292 (14190), 348 (9530). 1H NMR (DMSO-d6): E-isomer δ 11.59 (s, 1H, NH), 8.41 (s, 1H, HC=N), 8.26 (s, 1H, NH2), 8.23 (dd, 3J = 5 Hz, 4J = 1 Hz, 1H, Hpy), 7.52 (dd, 3J = 8 Hz, 4J = 1 Hz, 1H, Hpy), 7.50 (s, 1H, NH2), 7.33 (dd, 3J = 8 Hz, 3J = 5 Hz, 1H, Hpy), 2.74 (s, 6H, N(CH3)2). 13C NMR (DMSO-d6): E-isomer δ = 178.8 (C=S), 149.9 (Cq, py), 144.4 (Cq, py), 142.8 (Cpy), 141.9 (C=N), 126.3 (Cpy), 124.8 (Cpy), 44.7 (N(CH3)2) ppm.
Synthesis of the pure Z isomer: 20 mg of the pure E-isomer was dissolved in 20 mL of acetonitrile and stirred at 37 °C for 24 h. After removal of the solvent, pure Z-isomer was isolated after column chromatography with 100% acetonitrile as eluent.
1H NMR (DMSO-d6): Z-isomer δ 13.92 (s, 1H, NH), 8.53 (s, 1H, NH2), 8.35 (dd, 3J = 5 Hz, 4J = 1 Hz, 1H, Hpy), 8.18 (s, 1H, NH2), 7.71 (dd, 3J = 8 Hz, 4J = 1 Hz, 1H, Hpy), 7.49 (s, 1H, HC=N), 7.47 (dd, 3J = 8 Hz, 3J = 5 Hz, 1H, Hpy), 2.82 (s, 6H, N(CH3)2) ppm. 13C NMR (DMSO-d6): Z-isomer δ = 179.3 (C=S), 150.4 (Cq, py), 144.2 (Cq, py), 141.4 (Cpy), 131.2 (C=N), 128.0 (Cpy), 125.6 (Cpy), 45.0 (N(CH3)2) ppm.
3-(Dimethylamino)pyridine-2-carbaldehyde N-Methylthiosemicarbazone (Me2NNHMe)
Compound 6 (70 mg, 0.47 mmol) was dissolved in EtOH (2 mL), and 4-methylthiosemicarbazide (49.5 mg, 0.47 mmol) was added. The solution was stirred for 1 h at RT and 4 h at 50 °C. Subsequently, the solvent was evaporated under reduced pressure. Purification was performed via column chromatography using ACN/CHCl3 (10:1) and ACN/MeOH (95:5) for final elution of the product. Yield: 93 mg (83%). Anal. Calcd for C10H15N5S (Mr 237.32 g/mol): C, 50.61; H, 6.37; N, 29.51; S, 13.51. Found: C, 50.57; H, 6.39; N, 29.23; S, 13.51. ESI-MS in MeOH (negative): m/z 236 [M–H]−. UV/vis (PBS), λmax, nm (ε, M–1 cm–1): 290 (26410), 348 (19380). 1H NMR (DMSO-d6): E-isomer δ 11.60 (s, 1H, N-NH), 8.42 (s, 1H, HC=N), 8.25 (dd, 3J = 4 Hz, 4J = 1 Hz, 1H, Hpy), 8.20–8.16 (m, 1H, NHCH3), 7.52 (dd, 3J = 8 Hz, 4J = 1 Hz, 1H, Hpy), 7.33 (dd, 3J = 8 Hz, 3J = 4 Hz, 1H, Hpy), 3.04 (d, 3J = 5 Hz, 3H, NHCH3), 2.74 (s, 6H, N(CH3)2). 13C NMR (DMSO-d6): E-isomer δ = 178.5 (C=S), 150.0 (Cq, py), 144.8 (Cq. py), 142.9 (Cpy), 141.1 (C=N), 126.2 (Cpy), 124.7 (Cpy), 44.8 (N(CH3)2), 31.4 (CH3) ppm.
Synthesis of the Z-isomer: 20 mg of the pure E isomer was dissolved in 8 mL of acetonitrile and stirred at 37 °C for 24 h. After removal of the solvent, pure Z-isomer was isolated after column chromatography using 100% acetonitrile and for final elution acetonitrile/MeOH (95:5). 1H NMR (DMSO-d6): Z-isomer δ 13.97 (s, 1H, N-NH), 8.86 (m, 1H, NHCH3), 8.34 (dd, 3J = 5 Hz, 4J = 1 Hz, 1H, Hpy), 7.71 (dd, 3J = 4 Hz, 4J = 1 Hz, 1H, Hpy), 7.47 (s, 1H, HC=N), 7.46 (dd, 3J = 8 Hz, 4J = 5 Hz, 1H, Hpy), 3.02 (d, 3J = 5 Hz, 3H, NHCH3), 2.81 (s, 6H, N(CH3)2) ppm. 13C NMR (DMSO-d6): Z-isomer δ = 178.7 (C=S), 150.3 (Cq, py), 144.3 (Cq. py), 141.3 (Cpy), 130.8 (C=N), 127.8 (Cpy), 125.5 (Cpy), 45.0 (N(CH3)2), 31.5 (CH3) ppm.
3-(Dimethylamino)pyridine-2-carbaldehyde N,N-Dimethylthiosemicarbazone (Me2NNMe2)
Compound 6 (107 mg, 0.71 mmol) was dissolved in EtOH (3 mL), and 4,4-dimethyl-3-thiosemicarbazide (85 mg, 0.71 mmol) was added. The solution was stirred at RT for 1 h and at 50 °C for 4 h, and subsequently the solvent was evaporated under reduced pressure. Purification was performed via column chromatography using EtOAc/Hexane (9:1) and EtOAc/MeOH (95:5) for final elution of the product. Finally, the product was stirred in diethyl ether (2 mL) at 35 °C for 3 h, the solid was filtered off and dried in vacuo. Yield: 60 mg (34%). Anal. Calcd for C11H17N5S (Mr 251.35 g/mol): C, 52.56; H, 6.82; N, 27.86; S, 12.76. Found: C, 50.58; H, 6.77; N, 27.68; S, 12.54. ESI-MS in MeOH (positive): m/z 252 [M + H]+. UV/vis (PBS), λmax, nm (ε, M–1 cm–1): 280 (19380), 352 (11750). 1H NMR (DMSO-d6): E-isomer 11.05 (s, 1H, NH), 8.49 (s, 1H, HC=N), 8.25–8.22 (m, 1H, Hpy), 7.51 (dd, 3J = 8 Hz, 4J = 1 Hz, 1H, Hpy), 7.30 (dd, 3J = 8 Hz, 3J = 4 Hz, 1H, Hpy), 3.31 (s, 6H, S=CN(CH3)2), 2.75 (s, 6H, CpyN(CH3)2). Z-isomer δ 15.23 (s, 1H, NH), 8.35 (dd, 3J = 5 Hz, 4J = 1 Hz, 1H, Hpy), 7.72 (dd, 3J = 8 Hz, 4J = 1 Hz, 1H, Hpy), 7.71 (s, 1H, HC=N), 7.46 (dd, 3J = 8 Hz, 3J = 5 Hz, 1H, Hpy), 3.36 (s, 6H, S=CN(CH3)2), 2.83 (s, 6H, CpyN(CH3)2) ppm. 13C NMR (DMSO-d6): E-isomer δ 181.2 (C=S), 149.5 (Cq, py), 145.2 (Cq, py), 143.0 (Cpy), 142.8 (C=N), 126.2 (Cpy), 124.4 (Cpy), 44.8 (CpyN(CH3)2), 42.7 (S=CN(CH3)2). Z-isomer δ 180.6 (C=S), 149.9 (Cq, py), 144.3 (Cq, py), 140.1 (Cpy), 133.6 (C=N), 127.9 (Cpy), 125.2 (Cpy), 45.0 (CpyN(CH3)2), 41.3 (S=CN(CH3)2) ppm.
Crystallographic Structure Determination
X-ray diffraction measurements were performed on a Bruker D8 VENTURE system equipped with a multilayer monochromator and a Mo K/a INCOATEC microfocus sealed tube (λ = 0.71073 Å). A single crystal of approximate dimensions 0.30 mm × 0.20 mm × 0.10 mm was coated with Paratone-N oil, mounted at room temperature on a Hampton Research 0.3–0.4 mm CryoLoop, and cooled to 100 K under a stream of N2 maintained by a KRYOFLEXI low-temperature apparatus. The crystal was positioned at 35 mm from the detector and 775 frames were collected, each for 10 s over 1° scan width. The data were processed using SAINT software.43 The structures were solved by direct methods and refined by full-matrix least-squares techniques. Data were corrected for absorption effects using the multiscan method (SADABS44). Non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were placed at calculated positions and refined as riding atoms in the subsequent least-squares model refinements. The isotropic thermal parameters were estimated to be 1.2 respectively 1.5 times the values of the equivalent isotropic thermal parameters of the atoms to which hydrogens were bound. The following computer programs were used: structure solution SHELXS-97;45 refinement SHELXL-Version 2013/3,45 OLEX2;46 molecular diagrams ORTEP;47 Processor: Intel Xeon CPU E3-1270 V2 @ 3.50 GHz; scattering factors.48
Crystal data for Me2NNH2: C9H13N5S, Mr = 223.30, monoclinic, P21/c, a [Å] = 11.8688(4), b [Å] = 7.6283(3), c [Å] = 13.1597(5), β = 115.668(2), V = 1073.89(7) Å3, Z = 4, ρcalcd = 1.381 g/cm3, μ = 0.276 mm–1, λ(Mo–Kα) = 0.71073 Å, T = 100 K, 2θmax = 60.27°, 17994 reflections measured, 3162 unique (Rint = 0.0413), R1 = 0.0354, wR2 = 0.0931, GOF = 1.046. Crystallographic data have been deposited at the Cambridge Crystallographic Data Center with number CCDC1449031.
Interconversion Studies of the Isomers by HPLC–MS
Sample preparation: DMSO stock solutions of the compounds were diluted with PBS (pH 7.4) to a final concentration of 50 μM (1% DMSO), followed by immediate LC–MS measurements. LC–MS system: The chromatographic separation was performed with an Atlantis T3 C18 reversed-phase column (150 mm × 2.1 mm, 3 μm particle size) from Waters (Milford, USA). As a mobile phase a gradient prepared from water containing 1% (v/v) acetonitrile and 0.1% (v/v) formic acid (eluent A) and acetonitrile containing 1% (v/v) water and 0.1% (v/v) formic acid (eluent B) was used. The mobile phase was kept constant at 10% B for 1 min. Then, B was increased to 50% within 5 min and kept for 2 min. Subsequently, B was increased to 90% within 0.1 min and kept for 0.9 min to flush the column, followed by reconstitution of the starting conditions within 0.1 min and re-equilibration with 10% B for 8.9 min (total analysis time = 18 min). Due to the high affinity of thiosemicarbazones for metal ions even within a HPLC system, the measurements were carried out on an inert HPLC system (1260 Infinity Bioinert Quaternary LC System, Agilent Technologies), controlled by an Agilent OpenLAB CDS ChemStation Edition Rev. C.01.06[61] software, coupled to an Amazon L electrospray ionization ion trap mass spectrometry system (Bruker Daltonics). The LC–MS runs were performed in positive ionization mode with the following optimized parameters: flow rate 0.2 mL/min, injection volume 5 μL, column temperature 25 °C, and autosampler temperature 5 °C, drying gas 10 L/min (350 °C), nebulizer pressure 35 psi, and capillary voltage 4000 V. The HyStar 3.2 and Data Analysis 4.0 software package (Bruker Daltonics) were used for instrument control and data processing.
Determination of the Distribution Coefficients (D7.4)
D7.4 values of all compounds were determined by the traditional shake-flask method in n-octanol/buffered aqueous solution at pH 7.4 at 25 ± 0.2 °C as described previously.30 Two parallel experiments were performed for each sample. The compounds were dissolved at 30–45 μM in the n-octanol presaturated aqueous solution of the buffer (10 mM HEPES) at constant ionic strength (0.10 M KCl). The aqueous solutions and n-octanol with 1:1 phase ratio were gently mixed with 360° vertical rotation for 3 h to avoid emulsion formation, and the mixtures were centrifuged at 5000 rpm for 5 min by a temperature controlled centrifuge (Sanyo) at 25 °C. Estimated logP values of the neutral forms of the compounds were calculated using ChemDraw Ultra program (Version 10.0, 1986–2005 Cambridge Soft.).
Cell Lines and Culture Conditions
The following human cancer cell lines were used in this study: the colon carcinoma-derived cell line SW480 and lung fibroblasts WI-38 (obtained from the American Tissue Culture Collection), the ovarian carcinoma-derived cell line A2780 (obtained from Sigma-Aldrich), the acute promyelocytic leukemia-derived cell line HL-60 (obtained from Dr. M. Center, Kansas State University), the cervix carcinoma-derived cell line KB-3-1 and the colchicine resistant subline KBC-1 (obtained from Dr. D. W. Shen, Bethesda, Maryland). SW480 and WI-38 cells were grown in MEM with 10% FCS and A2780, KB-3-1, KBC-1, and HL-60 cells were cultured in RPMI 1640 supplemented with 10% FCS. SW480/Tria cells were generated by continuous exposure of SW480 cells to increasing concentrations of Triapine (starting point, 0.05 μM; end point, 20 μM) over a period of one year.19 Triapine was administered to the cells once every other week at the day after passage, when cells had attached to the culture flasks.
Synergism is expressed by the combination index (CI) according to Chou and Talalay49 using CalcuSyn software (Biosoft, Ferguson, MO, USA). CI < 0.9, CI = 0.9–1.2, or CI > 1.2 represent synergism, additive effects, and antagonism, respectively.
Cytotoxicity Tests in Cancer Cell Lines
To determine cell viability, either 2 × 104 cells/mL of SW480, SW480/Tria, WI-38, KB-3-1 and KBC-1 or 3 × 104 cells/mL of A2780 cells were plated on 96-well plates (100 μL/well) and allowed to recover for 24 h. Then, cells were exposed to the test drugs with the indicated concentrations for 72 h. In combination experiments the cells were preincubated for 1 h with CuCl2 (10 or 30 μM). Anticancer activity was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-based vitality assay (EZ4U; Biomedica, Vienna, Austria) following the manufacturer’s recommendations. Cytotoxicity was calculated using the Graph Pad Prism software (using a point-to-point function) and was expressed as IC50 values calculated from full dose–response curves (drug concentrations inducing a 50% reduction of cell number in comparison to untreated control cells cultured in parallel).
Microscopy
Phase contrast images (Figure 3A) were taken at a 200× magnification at the Nikon eclipse Ti-e fluorescence microscope after incubation with Triapine and its derivatives (2.5 μM) for 48 h. The magnified cells (Figure 3B) were photographed after 24 h of treatment at 400× magnification at the Nikon eclipse Ti-e fluorescence microscope with a sCMOS pco.edge camera.
Measurement of Intracellular ROS
2′,7′-Dichlorofluorescein diacetate (DCF-DA) was used to detect the production of ROS.50 DCF-DA stock solutions (33.4 mM) in DMSO were stored at −20 °C. HL-60 cells (5 × 105 cells per sample in 500 μL of phenol-free Hanks balanced salt solution) were incubated with DCF-DA for 30 min. Subsequently, Triapine and its derivatives were added in the indicated concentrations for further 30 min. CuCl2 was added as indicated (1, 2, 5, or 10 μM) to the samples 15 min prior to addition of the thiosemicarbazones. After incubation, the mean fluorescence intensity was measured by flow cytometry using a FACSCalibur instrument (Becton Dickinson, Palo Alto, CA, USA). A concentration of 200 μM H2O2 was used as the control.
Hoechst 33258/PI Staining
A staining of Hoechst 33258 combined with propidium iodide (PI) was used to measure the percentage of dead cells. Therefore, HL-60 cells (5 × 105 cells per sample in 500 μL of phenol-free Hanks balanced salt solution) were incubated with Hoechst/PI (1 μg/mL/2.5 μg/mL) for 30 min. Subsequently, Triapine and its derivatives were added in the indicated concentrations for further 30 min. CuCl2 was added as indicated (2, 5, or 10 μM) to the samples 15 min prior to addition of the thiosemicarbazones. Pictures were taken at 200× magnification at the Nikon eclipse Ti-e fluorescence microscope and PI-positive cells were counted as percentage of Hoechst-positive cells. At least 300 cells were counted per sample.
Acknowledgments
We thank Alexander Roller for X-ray diffraction measurement and refinement. This work was supported by the Austrian Science Fund (FWF) grant P22072-B11 (to W.B.), the Hungarian Research Foundation OTKA (PD103905), and the J. Bolyai Research Scholarship of the Hungarian Academy of Sciences. S.H. was financed by the Initiative Krebsforschung (given to P.H.) and S.K. by the Mahlke geb. Obermann-Stiftung (given to B.K.).
Glossary
Abbreviations
- ABCB1
P-glycoprotein
- DCF-DA
2′,7′-dichlorofluorescein diacetate
- DFO
desferrioxamine
- dNTP
deoxynucleotide triphosphate
- Dp44mT
di-2-pyridilketone 4,4-dimethyl-3-thiosemicarbazone
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- P44mT
pyridine-2-carboxaldehyde 4,4-dimethyl-3-thiosemicarbazone
- PI
propidium iodide
- ROS
reactive oxygen species
- Triapine
3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP)
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00342.
Author Contributions
∥ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Torti S. V.; Torti F. M. Ironing out cancer. Cancer Res. 2011, 71, 1511–1514. 10.1158/0008-5472.CAN-10-3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Wong J.; Lovejoy D. B.; Kalinowski D. S.; Richardson D. R. Chelators at the cancer coalface: desferrioxamine to Triapine and beyond. Clin. Cancer Res. 2006, 12, 6876–6883. 10.1158/1078-0432.CCR-06-1954. [DOI] [PubMed] [Google Scholar]
- Estrov Z.; Tawa A.; Wang X. H.; Dube I. D.; Sulh H.; Cohen A.; Gelfand E. W.; Freedman M. H. In vitro and in vivo effects of deferoxamine in neonatal acute leukemia. Blood 1987, 69, 757–761. [PubMed] [Google Scholar]
- Donfrancesco A.; Deb G.; Dominici C.; Pileggi D.; Castello M. A.; Helson L. Effects of a single course of deferoxamine in neuroblastoma patients. Cancer Res. 1990, 50, 4929–4930. [PubMed] [Google Scholar]
- Selig R. A.; White L.; Gramacho C.; Sterling-Levis K.; Fraser I. W.; Naidoo D. Failure of iron chelators to reduce tumor growth in human neuroblastoma xenografts. Cancer Res. 1998, 58, 473–478. [PubMed] [Google Scholar]
- Blatt J. Deferoxamine in children with recurrent neuroblastoma. Anticancer Res. 1994, 14, 2109–2112. [PubMed] [Google Scholar]
- Kalinowski D. S.; Richardson D. R. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol. Rev. 2005, 57, 547–583. 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Gutierrez E.; Kovacevic Z.; Saletta F.; Obeidy P.; Suryo Rahmanto Y.; Richardson D. R. Iron chelators for the treatment of cancer. Curr. Med. Chem. 2012, 19, 2689–2702. 10.2174/092986712800609706. [DOI] [PubMed] [Google Scholar]
- Antholine W. E.; Knight J. M.; Petering D. H. Inhibition of tumor cell transplantability by iron and copper complexes of 5-substituted 2-formylpyridine thiosemicarbazones. J. Med. Chem. 1976, 19, 339–341. 10.1021/jm00224a030. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Kalinowski D. S.; Kovacevic Z.; Siafakas A. R.; Jansson P. J.; Stefani C.; Lovejoy D. B.; Sharpe P. C.; Bernhardt P. V.; Richardson D. R. Thiosemicarbazones from the old to new: iron chelators that are more than just ribonucleotide reductase inhibitors. J. Med. Chem. 2009, 52, 5271–5294. 10.1021/jm900552r. [DOI] [PubMed] [Google Scholar]
- Finch R. A.; Liu M.; Grill S. P.; Rose W. C.; Loomis R.; Vasquez K. M.; Cheng Y.; Sartorelli A. C. Triapine (3-aminopyridine-2-carboxaldehyde- thiosemicarbazone): A potent inhibitor of ribonucleotide reductase activity with broad spectrum antitumor activity. Biochem. Pharmacol. 2000, 59, 983–991. 10.1016/S0006-2952(99)00419-0. [DOI] [PubMed] [Google Scholar]
- Aye Y.; Long M. J.; Stubbe J. Mechanistic Studies of Semicarbazone Triapine targeting human ribonucleotide reductase in vitro and in mammalian cells tyrosyl radical yuenching not involving reactive oxygen species. J. Biol. Chem. 2012, 287, 35768–35778. 10.1074/jbc.M112.396911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebacher N. A.; Lane D. J.; Jansson P. J.; Richardson D. R. Glucose modulation induces lysosome formation and increases lysosomotropic drug sequestration via the P-glycoprotein drug transporter. J. Biol. Chem. 2016, 291, 3796–3820. 10.1074/jbc.M115.682450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles F. J.; Fracasso P. M.; Kantarjian H. M.; Cortes J. E.; Brown R. A.; Verstovsek S.; Alvarado Y.; Thomas D. A.; Faderl S.; Garcia-Manero G.; Wright L. P.; Samson T.; Cahill A.; Lambert P.; Plunkett W.; Sznol M.; DiPersio J. F.; Gandhi V. Phase I and pharmacodynamic study of Triapine, a novel ribonucleotide reductase inhibitor, in patients with advanced leukemia. Leuk. Res. 2003, 27, 1077–1083. 10.1016/S0145-2126(03)00118-8. [DOI] [PubMed] [Google Scholar]
- Karp J. E.; Giles F. J.; Gojo I.; Morris L.; Greer J.; Johnson B.; Thein M.; Sznol M.; Low J. A phase I study of the novel ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) in combination with the nucleoside analog fludarabine for patients with refractory acute leukemias and aggressive myeloproliferative disorders. Leuk. Res. 2008, 32, 71–77. 10.1016/j.leukres.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attia S.; Kolesar J.; Mahoney M. R.; Pitot H. C.; Laheru D.; Heun J.; Huang W.; Eickhoff J.; Erlichman C.; Holen K. D. A phase 2 consortium (P2C) trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) for advanced adenocarcinoma of the pancreas. Invest. New Drugs 2008, 26, 369–379. 10.1007/s10637-008-9123-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox J. J.; Hotte S. J.; Kollmannsberger C.; Winquist E.; Fisher B.; Eisenhauer E. A. Phase II study of Triapine in patients with metastatic renal cell carcinoma: a trial of the National Cancer Institute of Canada Clinical Trials Group (NCIC IND.161). Invest. New Drugs 2007, 25, 471–477. 10.1007/s10637-007-9044-9. [DOI] [PubMed] [Google Scholar]
- Traynor A. M.; Lee J. W.; Bayer G. K.; Tate J. M.; Thomas S. P.; Mazurczak M.; Graham D. L.; Kolesar J. M.; Schiller J. H. A phase II trial of triapine (NSC# 663249) and gemcitabine as second line treatment of advanced non-small cell lung cancer: Eastern Cooperative Oncology Group Study 1503. Invest. New Drugs 2010, 28, 91–97. 10.1007/s10637-009-9230-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklos W.; Pelivan K.; Kowol C. R.; Pirker C.; Dornetshuber-Fleiss R.; Spitzwieser M.; Englinger B.; van Schoonhoven S.; Cichna-Markl M.; Koellensperger G.; Keppler B. K.; Berger W.; Heffeter P. Triapine-mediated ABCB1 induction via PKC induces widespread therapy unresponsiveness but is not underlying acquired triapine resistance. Cancer Lett. 2015, 361, 112–120. 10.1016/j.canlet.2015.02.049. [DOI] [PubMed] [Google Scholar]
- Yuan J.; Lovejoy D. B.; Richardson D. R. Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: in vitro and in vivo assessment. Blood 2004, 104, 1450–1458. 10.1182/blood-2004-03-0868. [DOI] [PubMed] [Google Scholar]
- Whitnall M.; Howard J.; Ponka P.; Richardson D. R. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 14901–14906. 10.1073/pnas.0604979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson D. R.; Sharpe P. C.; Lovejoy D. B.; Senaratne D.; Kalinowski D. S.; Islam M.; Bernhardt P. V. Dipyridyl thiosemicarbazone chelators with potent and selective antitumor activity form iron complexes with redox activity. J. Med. Chem. 2006, 49, 6510–6521. 10.1021/jm0606342. [DOI] [PubMed] [Google Scholar]
- Kowol C. R.; Trondl R.; Heffeter P.; Arion V. B.; Jakupec M. A.; Roller A.; Galanski M.; Berger W.; Keppler B. K. Impact of metal coordination on cytotoxicity of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (Triapine) and novel insights into terminal dimethylation. J. Med. Chem. 2009, 52, 5032–5043. 10.1021/jm900528d. [DOI] [PubMed] [Google Scholar]
- Heffeter P.; Pirker C.; Kowol C. R.; Herrman G.; Dornetshuber R.; Miklos W.; Jungwirth U.; Koellensperger G.; Keppler B. K.; Berger W. Impact of terminal dimethylation on the resistance profile of alpha-N-heterocyclic thiosemicarbazones. Biochem. Pharmacol. 2012, 83, 1623–1633. 10.1016/j.bcp.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro K.; Lin Z. P.; Penketh P. G.; Shyam K.; Zhu R.; Baumann R. P.; Zhu Y. L.; Sartorelli A. C.; Rutherford T. J.; Ratner E. S. Distinct mechanisms of cell-kill by triapine and its terminally dimethylated derivative Dp44mT due to a loss or gain of activity of their copper(II) complexes. Biochem. Pharmacol. 2014, 91, 312–322. 10.1016/j.bcp.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson P. J.; Sharpe P. C.; Bernhardt P. V.; Richardson D. R. Novel thiosemicarbazones of the ApT and DpT series and their copper complexes: identification of pronounced redox activity and characterization of their antitumor activity. J. Med. Chem. 2010, 53, 5759–5769. 10.1021/jm100561b. [DOI] [PubMed] [Google Scholar]
- Lovejoy D. B.; Jansson P. J.; Brunk U. T.; Wong J.; Ponka P.; Richardson D. R. Antitumor activity of metal-chelating compound Dp44mT is mediated by formation of a redox-active copper complex that accumulates in lysosomes. Cancer Res. 2011, 71, 5871–5880. 10.1158/0008-5472.CAN-11-1218. [DOI] [PubMed] [Google Scholar]
- Pessôa M.; Andrade G. F.; Paoli Monteiro V. R.; Temperini M. L. 2-Formylpyridinethiosemicarbazone and methyl derivatives: spectroscopic studies. Polyhedron 2001, 20, 3133–3141. 10.1016/S0277-5387(01)00928-7. [DOI] [Google Scholar]
- Kowol C. R.; Trondl R.; Arion V. B.; Jakupec M. A.; Lichtscheidl I.; Keppler B. K. Fluorescence properties and cellular distribution of the investigational anticancer drug triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone) and its zinc(II) complex. Dalton Trans. 2010, 39, 704–706. 10.1039/B919119B. [DOI] [PubMed] [Google Scholar]
- Enyedy É. A.; Zsigó É.; Nagy N. V.; Kowol C. R.; Roller A.; Keppler B. K.; Kiss T. Complex-formation ability of salicylaldehyde thiosemicarbazone towards ZnII, CuII, FeII, FeIII and GaIII ions. Eur. J. Inorg. Chem. 2012, 25, 4036–4047. 10.1002/ejic.201200360. [DOI] [Google Scholar]
- Kowol C. R.; Nagy N. V.; Jakusch T.; Roller A.; Heffeter P.; Keppler B. K.; Enyedy É. A. Vanadium (IV/V) complexes of Triapine and related thiosemicarbazones: synthesis, solution equilibrium and bioactivity. J. Inorg. Biochem. 2015, 152, 62–73. 10.1016/j.jinorgbio.2015.08.023. [DOI] [PubMed] [Google Scholar]
- Gutmann V. Empirical parameters for donor and acceptor properties of solvents. Electrochim. Acta 1976, 21, 661–670. 10.1016/0013-4686(76)85034-7. [DOI] [Google Scholar]
- Pessôa M. M.; Andrade G. F.; Monteiro V. R. P.; Temperini M. L. 2-Formylpyridinethiosemicarbazone and methyl derivatives: spectroscopic studies. Polyhedron 2001, 20, 3133–3141. 10.1016/S0277-5387(01)00928-7. [DOI] [Google Scholar]
- Jansson P. J.; Yamagishi T.; Arvind A.; Seebacher N.; Gutierrez E.; Stacy A.; Maleki S.; Sharp D.; Sahni S.; Richardson D. R. Di-2-pyridylketone 4, 4-dimethyl-3-thiosemicarbazone (Dp44mT) overcomes multidrug resistance by a novel mechanism involving the hijacking of lysosomal P-glycoprotein (Pgp). J. Biol. Chem. 2015, 290, 9588–9603. 10.1074/jbc.M114.631283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson P. J.; Kalinowski D. S.; Lane D. J.; Kovacevic Z.; Seebacher N. A.; Fouani L.; Sahni S.; Merlot A. M.; Richardson D. R. The renaissance of polypharmacology in the development of anti-cancer therapeutics: Inhibition of the “Triad of Death” in cancer by Di-2-pyridylketone thiosemicarbazones. Pharmacol. Res. 2015, 100, 255–260. 10.1016/j.phrs.2015.08.013. [DOI] [PubMed] [Google Scholar]
- Miah A.; Harrington K.; Nutting C. Triapine in clinical practice. Eur. J. Clin. Med. Oncol. 2010, 2, 1–6. [Google Scholar]
- Lovejoy D. B.; Sharp D. M.; Seebacher N.; Obeidy P.; Prichard T.; Stefani C.; Basha M. T.; Sharpe P. C.; Jansson P. J.; Kalinowski D. S. Novel second-generation di-2-pyridylketone thiosemicarbazones show synergism with standard chemotherapeutics and demonstrate potent activity against lung cancer xenografts after oral and intravenous administration in vivo. J. Med. Chem. 2012, 55, 7230–7244. 10.1021/jm300768u. [DOI] [PubMed] [Google Scholar]
- Enyedy E. A.; Nagy N. V.; Zsigo E.; Kowol C. R.; Arion V. B.; Keppler B. K.; Kiss T. Comparative solution equilibrium study of the interactions of copper(II), iron(II) and zinc(II) with Triapine (3-aminopyridine-2-carbaldehyde thiosemicarbazone) and related ligands. Eur. J. Inorg. Chem. 2010, 11, 1717–1728. 10.1002/ejic.200901174. [DOI] [Google Scholar]
- Moriwaki K.; Chan F. K. Necrosis-dependent and independent signaling of the RIP kinases in inflammation. Cytokine Growth Factor Rev. 2014, 25, 167–174. 10.1016/j.cytogfr.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denoyer D.; Masaldan S.; La Fontaine S.; Cater M. A. Targeting copper in cancer therapy:‘Copper That Cancer’. Metallomics 2015, 7, 1459–1476. 10.1039/C5MT00149H. [DOI] [PubMed] [Google Scholar]
- Jungwirth U.; Kowol C. R.; Hartinger C.; Keppler B. K.; Berger W.; Heffeter P. Anticancer activity of metal complexes: involvement of redox processes. Antioxid. Redox Signaling 2011, 15, 1085–1127. 10.1089/ars.2010.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.-C.; Lin T.-S.; Cory J. G.; Cory A. H.; Sartorelli A. C. Synthesis and biological activity of 3-and 5-amino derivatives of pyridine-2-carboxaldehyde thiosemicarbazone. J. Med. Chem. 1996, 39, 2586–2593. 10.1021/jm9600454. [DOI] [PubMed] [Google Scholar]
- SAINT-Plus, version 8.32b; Bruker AXS Inc.: Madison, WI, 2013. [Google Scholar]
- SADABS-2012/1 Bruker AXS 2013 area detector scaling and absorption correction.
- Sheldrick G. M.SHELXS-97; Program for Crystal Structure Solution; University Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Johnson G. K.Report ORNL-5138; Oak Ridge National Laboratory; Oak Ridge, TN, 1976. [Google Scholar]
- International Tables for X-ray Crystallography; Kluwer Academic Press: Dodrecht, The Netherlands, 1992; Vol. C, Tables 4.2.6.8 and 6.1.1.4. [Google Scholar]
- Heffeter P.; Atil B.; Kryeziu K.; Groza D.; Koellensperger G.; Korner W.; Jungwirth U.; Mohr T.; Keppler B. K.; Berger W. The ruthenium compound KP1339 potentiates the anticancer activity of sorafenib in vitro and in vivo. Eur. J. Cancer 2013, 49, 3366–3375. 10.1016/j.ejca.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes A.; Fernandes E.; Lima J. L. Fluorescence probes used for detection of reactive oxygen species. J. Biochem. Biophys. Methods 2005, 65, 45–80. 10.1016/j.jbbm.2005.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.