

Abstract
Objectives
Historically, mitochondrial disorders have been associated with predominantly multisystem or neurological symptoms. If present, hepatic complications were thought to be a late feature. Recently, mutations in at least 4 nuclear genes have been identified in infants presenting with rapidly progressive hepatic failure which may be precipitated by infection or drugs. We aimed to determine if hepatic mitochondrial DNA (mtDNA) depletion is associated with apparently isolated hepatic failure in individuals with acute liver failure (ALF) of known or unknown etiologies undergoing liver transplant (LT) In addition we wished to establish if there was an excess of mutations in gene known to cause hepatic mtDNA depletion.
Methods
Using previously established methods, we demonstrated that end stage liver disease from known causes did not lead to hepatic mtDNA depletion. Using thresholds derived from ROC analysis, 66% of cases with ALF had probable or definite mtDNA depletion, including 34% with definite mtDNA depletion.
Results
There was a small, but significant increase in the proportion of patients undergoing LT for ALF with heterozygous mutations known to lead to mtDNA depletion and hepatic failure compared with controls (P = 0.001).
Conclusions
Liver disease severe enough to require LT does not cause secondary mtDNA depletion. However, the majority of patients undergoing LT for ALF had reduced mtDNA content, which fell within the range seen in patients with classic mtDNA depletion. A subset of ALF patients have mutations in genes known to lead to mtDNA depletion and hepatic failure. Together, these results suggest defective mtDNA maintenance is associated with ALF.
Keywords: DGUOK, mitochondrial DNA depletion; next generation sequencing; POLG; Valproate
Introduction
Until recently, hepatic complications were thought to be a late feature of mitochondrial disorders, which are generally recognized as multisystem diseases presenting predominantly with neurological and muscular dysfunction (1). However, patients with mitochondrial disorders can present with isolated hepatic failure; indeed, patients may even present with apparent viral hepatitis (2-5).
Mitochondrial disorders may be due to defects in nuclear or mitochondrial genes. A subset of these defects leads to mitochondrial DNA (mtDNA) depletion (a reduction in cellular mtDNA content). These are usually caused by molecular defects in the nuclear genes responsible for mtDNA biogenesis, and the maintenance of mtDNA integrity and deoxynucleotide pools (6). Mutations in nine nuclear genes are known to cause mtDNA depletion (2, 3, 6-19).
Mutations in 4 of these nuclear genes have been identified in infants presenting with rapidly progressive hepatic failure: deoxyguanosine kinase (DGUOK), a gene coding for mitochondrial inner membrane protein, (MPV17), DNA polymerase γ (POLG), and TWINKLE, a DNA helicase, (C10orf2) (2-5, 17, 20-24). Patients with these mutations have presented with hepatic failure with (11, 12, 17, 20-22, 24-26) or without a neurological component (2-5, 24). Anecdotal evidence from published cases has suggested a link between primary mtDNA depletion and both drug- and viral-induced acute hepatic failure (3, 5, 27). In addition, 3 other genes have been shown to result in a reduced mtDNA content in the liver: ribonucleotide reductase M2 B (RRM2B), succinate-CoA ligase ADP-forming β subunit (SUCLA2), and thymidine phosphorylase (TP) without clear evidence of liver dysfunction in reported cases (28);
Currently, it is unclear how frequently mtDNA depletion is associated with isolated hepatic failure. The determination of mtDNA depletion requires the primarily affected organ to be assayed (3, 28); therefore, in the case of hepatic disorders, liver tissue is required. We have previously validated an assay utilizing real time quantitative polymerase chain reaction to evaluate mtDNA content relative to haploid nuclear DNA in muscle and liver specimens, thus establishing normal ranges for liver tissue (28). Since previous data has demonstrated mitochondrial dysfunction results from bile acid accumulation (29), it was first necessary to determine if hepatic disease, particularly of a cholestatic nature from known etiologies, severe enough to require liver transplant (LT), affects mtDNA content. If such liver dysfunction does not lead to secondary mtDNA depletion, then hepatic mtDNA content could be used as a tool to evaluate the association between clinically isolated hepatic failure in individuals with acute liver failure (ALF) and disordered mtDNA maintenance.
A recent systematic study demonstrated an association between heterozygous mutations in POLG and drug-induced hepatic injury following exposure to sodium valproate (30). We therefore also set out to establish the prevalence of mutations in nuclear encoded genes associated with hepatic mtDNA depletion, in patients with ALF.
Materials and Methods
Liver Specimens/ DNA Samples (Table 1)
Table 1.
Source of liver specimens/DNA samples
| n | |
|---|---|
| Previously healthy liver transplant (LT) donors (healthy controls) | 65 |
| Individuals with known mtDNA depletion (mtDNA depletion controls) | 18 |
| Patients with liver disease of known etiology requiring LT (LT controls) | 28 |
| Extra hepatic biliary atresia | 12 |
| PFIC1 | 6 |
| Wilson Disease | 5 |
| Tyrosinemia Type 1 | 3 |
| Alagille Syndrome | 2 |
| Healthy individuals* (population controls) | 205 |
| Patients with fulminant hepatic failure (FHF) undergoing LT (FHF test group) | 45 |
| Viral-induced FHF | 10 |
| Drug-induced FHF | 17” |
| FHF due to unknown cause | 18 |
DNA was obtained from liver samples except in this group of individuals in whom DNA was obtained from blood samples.
one sample was too degraded to extract DNA, and in a further sample the DNA was insufficient quality to perform sequencing.
DNA from unrelated healthy subjects was obtained from excess tissue from 65 LT donors, provided by the National Institute of Health-sponsored Liver Tissue Cell Distribution System for Minneapolis, Minnesota and Pittsburgh, Pennsylvania (healthy controls). Although delay in freezing, tissue mishandling or length of time in storage has been demonstrated to not affect copy number (Ref Dimmcok, Tang), all liver tissue used from the LTCDS was flash frozen within 30 minutes of harvesting. An additional 205 DNA samples from unrelated healthy subjects were obtained through a separate institutional review board-approved protocol, and were used to evaluate the population frequency of detected mutations.
DNA from liver specimens from 18 patients with molecularly proven mtDNA depletion syndromes (mtDNA depletion homozygous or compound heterozygous for deleterious mutations in DGUOK, MPV17, POLG, or TWINKLE) were selected from those submitted to the Mitochondrial Diagnostic Laboratory at Baylor College of Medicine or the Genetics Center, Children's Hospital of Wisconsin (mtDNA depletion controls).
Individuals with end stage liver disease, requiring LT, as a result of clearly defined, non-mitochondrial etiology according to the Liver Tissue Cell Distribution System registry, were identified to evaluate if end stage liver disease leads to secondary hepatic mtDNA depletion. These LT cases (LT controls; n=28) comprised 12 patients with extra hepatic biliary atresia; six with progressive familial intrahepatic cholestasis type 1(PFIC-1); five with Wilson disease; three with tyrosinemia type 1; and two with Alagille syndrome.
DNA samples were also obtained from the liver tissue of 45 LT patients following ALF due to ‘unknown’, ’drug-induced’, or ‘viral’ etiology (ALF test), via the Liver Tissue Cell Distribution System.
Where appropriate, studies were performed with the consent of the patient or their legal guardian. All studies were performed according to protocols approved by the Institutional Review Board of Children's Hospital of Wisconsin and in accordance with the Declaration of Helsinki.
Hepatic mtDNA Content Analysis
DNA was extracted from control and ALF test specimens and mtDNA content was determined as described previously (28, 31). To allow for appropriate statistical comparison, the normally distributed cycle threshold difference (ΔCt) was used. mtDNA content in healthy controls (n=65) and LT controls (n=28) were compared with the mtDNA depletion controls to determine if they could be distinguished from each other based on mtDNA content alone. Thresholds for defining probable and definite depletion were determined using a receiver operated curve analysis (supplemental figure, http://links.lww.com/MPG/A236 [Receiver operator curve comparing cycle threshold in patients with proven mitochondrial {mtDNA} depletion {mtDNA depletion controls} vs healthy controls and patients with hepatic failure {LT controls}. Based on these data the following thresholds were defined: probable mtDNA depletion {100% sensitivity, 96% specificity at 8.19 cycle difference} and definite mtDNA depletion {100% specificity, 84% sensitivity at 7.47 cycle difference.}]). These thresholds were then applied to the ALF test group.
Molecular Genetic Analysis
ALF test samples underwent the following molecular genetic analyses to evaluate if they had pathologic variants in 7 genes that have been associated with reduced hepatic mtDNA content (28): bi-directional dideoxy (Sanger) sequencing of coding exons for POLG, MPV17, ribonucleotide reductase M2 B(RRM2B), succinate-CoA ligase ADP-forming β subunit (SUCLA2), DGUOK, thymidine phosphorylase (TP) and TWINKLE, (28); in addition they were subject to long-range PCR with 454 sequencing utilizing the titanium upgrade (Roche, California), as described previously (4). In addition, 270 controls (65 healthy controls and 205 population controls) were also subject to Sanger dideoxy sequence-based evaluation of all exons in which a variant was found in the ALF test samples. The 65 healthy control samples were also subject to screening by long-range PCR and subsequent 454 sequencing. A pathogenic mutation was defined according to ACMG criteria. For POLG it was defined as pathogenic; for the other genes we defined a mutation as a variant published as pathogenic in HGMD Biobase with a population frequency of less than 5% that after review of the primary literature, the authors agreed would meet criteria for clinical reporting.
Results
Hepatic mtDNA Content
Data from 63 of 65 healthy controls and 15 of18 mtDNA depletion controls have been previously published (28). These control samples showed a stable mtDNA content across age, gender and stated ethnicity (28).
The mtDNA content measured by cycle threshold difference (ΔCt) in samples from patients undergoing LT due to end stage liver disease of non-mitochondrial etiology (LT controls) were not statistically different from the normal range (i.e. the range defined by the healthy control group; Figure 1). However, the mtDNA content for several samples from patients with Tyrosinemia type 1, PFIC-1, Wilson's disease or Alagille syndrome was lower than that in the healthy control sample with the lowest mtDNA content encroaching the range seen in patients with mutations in DGUOK. As expected, the mtDNA content for liver samples from patients with confirmed mtDNA depletion syndromes designated as the mtDNA depletion group had marked mtDNA depletion, with an mtDNA content <25% of the mean for healthy controls.
Figure 1.
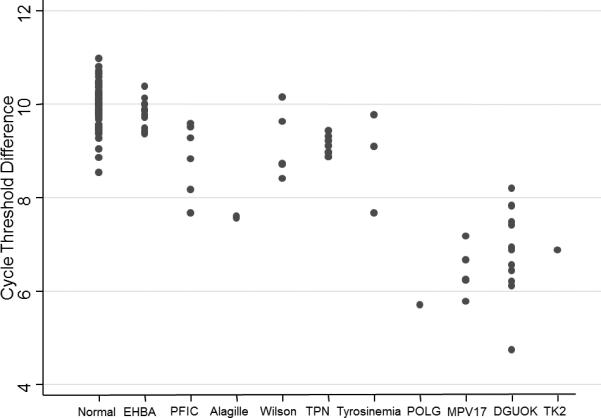
Mitochondrial DNA content in hepatic failure. mtDNA depletion was not seen in patients with hepatic failure of non-mitochondrial etiology, although a small reduction in mtDNA was seen in patients with progressive familial intrahepatic cholestasis type 1 (PFIC1), Alagille syndrome (Alagille), Wilson disease (Wilson), and tyrosinemia type 1 (Tyrosinemia). Conversely, liver tissue from patients with two mutations in POLG, MPV17, DGUOK, and TK2 show marked mtDNA depletion, with mtDNA content less than 25% of the mean value for normal controls.
Utilizing a receiver operated curve analysis we compared mtDNA content in the mtDNA depletion controls with healthy controls and LT controls; the area under the curve was 0.9932. Using these data we were able to establish the threshold for probable mtDNA depletion (100% sensitivity, 96% specificity) at 8.19 cycles different; and definite mtDNA depletion (100% specificity, 84% sensitivity) at 7.47 cycles different (supplemental figure, http://links.lww.com/MPG/A236). Individual samples were reviewed in light of the thresholds, demonstrating that two patients with PFIC-1, two with Alagille syndrome, and one with Tyrosinemia type 1 were classified as having probable mtDNA depletion.
Hepatic mtDNA Depletion in Patients with ALF
DNA was obtained from 44 of the 45 samples in the ALF test group; one sample from a patient with drug-induced ALF was too degraded to extract DNA. Hepatic mtDNA content was significantly lower in patients undergoing LT for ALF (ALF test) compared with healthy controls plus LT controls (mean = 7.86, P = 3.94 × 10^−25). Furthermore, mtDNA content remained significantly lower in the ALF test group than controls when the ALF test group was split into subgroups defined by the cause of ALF: ‘unknown’: mean = 7.51; t-test P = 4.79 × 10^−24; ‘drug-induced’: mean = 8.61; t-test P = 4.79 × 10^−7; ‘viral’: mean = 7.32; t-test P = 1.58 × 10^−15;
Applying the probable and definite thresholds for mtDNA depletion derived from the receiver operated curve analysis to the ALF test group, 29 of 44 (66%) patients had probable or definite mtDNA depletion, with 15 of 44 (34%) of these falling into the definite mtDNA depletion category (Figure 2, Table 2). Interestingly, a lower proportion of patients with drug-induced liver failure exhibited mtDNA depletion than patients with ALF due to unknown or viral etiologies. Regardless of the age cut-off used, there was no difference in the proportion of samples from pediatric patients and adult patients with mtDNA depletion.
Figure 2.
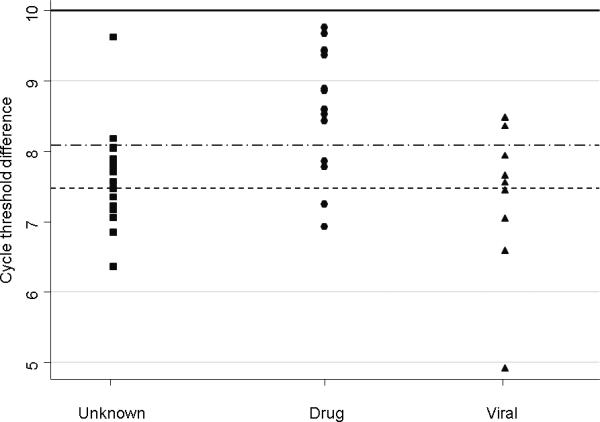
Mitochondrial DNA content in test patients with acute liver failure (ALF) due to unknown, drug-induced, or viral etiology. Thresholds for probable mtDNA depletion (100% sensitivity, 96% specificity at 8.19 cycle difference: upper dashed line) and definite mtDNA depletion (100% specificity, 84% sensitivity at 7.47 cycle difference: lower dashed line) were defined using receiver operated curve analysis. Mean cycle difference for normal liver tissue was 10 (based on healthy control data, solid line), and 8.6 is two standard deviations below the mean.
Table 2.
Demographics, Mitochondrial copy number in Liver obtained from individuals with acute liver failure is detailed with mutations detected in DGUOK and POLG.
| Category | Agent | Race | Age (Years) | Gender | dCT | POLG | DGUOK |
|---|---|---|---|---|---|---|---|
| Unknown | C | 12 | M | 6.35 | c.3199T>A (p.S1067T) |
c.494A>T (p.E165V) |
|
| Unknown | C | 3.5 | M | 6.84 | |||
| Unknown | H | 5 | M | 7.05 | |||
| Unknown | C | 7 | M | 7.16 | |||
| Unknown | U | 5 | 7.20 | ||||
| Unknown | U | 13 | F | 7.21 | |||
| Unknown | U | 2.2 | 7.34 | ||||
| Unknown | A | 38 | M | 7.46 | |||
| Unknown | U | 16 | F | 7.56 | c.3428A>AG (p.E1143G) |
||
| Unknown | C | 7 | M | 7.70 | c.1760C>T (p.P587PL) c.752C>T (p.T251I) |
||
| Unknown | A | 42 | F | 7.74 | |||
| Unknown | U | 2.2 | M | 7.74 | c.509A>G (p.Q170R) |
||
| Unknown | C | 42 | F | 7.82 | |||
| Unknown | U | 55 | F | 7.85 | |||
| Unknown | C | 24 | F | 7.88 | |||
| Unknown | H | 17 | M | 8.03 | |||
| Unknown | C | 62 | M | 8.04 | |||
| Unknown | N | 5.5 | M | 8.17 | |||
| Unknown | U | 60 | 9.61 | c.509A>G (p.Q170R) |
|||
| Drug | Acetaminophen | C | 5.6 | F | * | ||
| Drug | Isoniazid | U | 9.5 | M | 6.92 | ||
| Drug | Valproic Acid | C | 2.9 | F | 7.24 | c.1399G>A (p.A467T) |
|
| Drug | Herbal Infusion | A | 38 | F | 7.77 | ||
| Drug | Acetaminophen | C | 48 | F | 7.85 | ||
| Drug | Acetaminophen | C | 34 | M | 8.42 | ||
| Drug | Acetaminophen | U | 35 | F | 8.52 | ||
| Drug | Carbon Terachloride | C | 31 | F | 8.58 | ||
| Drug | Acetaminophen | C | 32 | F | 8.59 | ||
| Drug | Isoniazid | C | 53 | M | 8.85 | c.509A>G (p.Q170R) |
|
| Drug | Acetaminophen | N | 39 | F | 8.88 | ||
| Drug | Valproic Acid | C | 16 | F | 9.36 | ||
| Drug | Acetaminophen | U | 15 | M | 9.41 | ||
| Drug | Acetaminophen | C | 29 | F | 9.43 | ||
| Drug | Acetaminophen | C | 60 | F | 9.66 | * | |
| Drug | Acetaminophen | C | 41 | F | 9.75 | ||
| Viral | ND | B | 26 | F | 4.91 | ||
| Viral | Hepatitis A | N | 14 | M | 6.58 | ||
| Viral | ND | C | 39 | M | 7.04 | ||
| Viral | Hepatitis A | U | 26 | F | 7.44 | ||
| Viral | ND | C | 29 | F | 7.55 | ||
| Viral | Hepatitis A | C | 1.8 | M | 7.65 | ||
| Viral | Influenza | C | 1.9 | M | 7.93 | ||
| Viral | Hepatitis A | C | 24 | F | 8.35 | ||
| Viral | ND | A | 39 | M | 8.47 | ||
| Viral | ND | A | 39 | M | 8.47 |
Key:
Race: A = African American, B= Asian/Pacific Islander C= White Non-Hispanic, N= Native American H= White Hispanic, U= Unknown
Agent: ND= Not Documented
Insufficient quantity or quality of DNA
Genetic Analysis in Patients with ALF
Genetic analysis was possible for 43 of 45 samples in the ALF test group; one sample was too degraded to extract DNA and DNA was of insufficient quality to analyze in another sample. We detected 7 (16%) pathogenic mutations in hepatic mtDNA depletion genes in the 43 ALF test samples and 7 (3%) in the 270 controls (healthy controls + population controls; Fisher's exact test P = 0.001), when using a conservative definition of pathogenic mutations (i.e. only those mutations currently predicted to be pathogenic).
When considering DGUOK, a higher mutation rate was found in the ALF test samples than controls (Fisher's exact test P = 0.014). In the ALF test group 3 (7%) samples had the p.Q170R mutation (20) and 1 (2%) had the p.E165V mutation (3), while 4 (1%) of the controls had mutations, all p.Q170R.
A higher rate of POLG mutations was also found in the ALF test samples than in the controls (Fisher's exact test P = 0.036). In the ALF test group, 3 (7%) samples had mutations, 1 had the p.E1143G variant, 1 had the p.A467T variant, and 1 had both the p.T251I and p.P587L variants (which are frequently reported on the same chromosome and are considered one mutation; (26)), and in the control group 3 (1%) had mutations, all having both the p.T251I and p.P587L variants.
No pathogenic mutations were detected in TP, RRM2B, or MPV17 or SUCLA2 in either group, and no mutations in TWINKLE that are currently considered pathogenic were detected.
Discussion
The diagnosis of mtDNA depletion as a cause of liver failure is clinically challenging, with patients often presenting with no other features to suggest a mitochondrial etiology (2, 3, 20, 32-35). Hepatic mtDNA depletion may go unrecognized in many cases, or may only come to light when a subsequent affected child is born (3).
The results of this study demonstrate that the presence of hepatic disease severe enough to require LT does not, by itself, lead to secondary mtDNA depletion (LT control group; Figure 1), including hepatic diseases known to reduce mitochondrial function such as bile accumulation as a result of cholestasis caused by biliary atresia (29). The reduced mtDNA content in the two patients with Alagille syndrome in the LT control group was a surprising finding. The underlying mechanism behind this reduction in mtDNA content may be related to the pivotal role of NOTCH/MYC signaling in mitochondrial biogenesis (36). Thus one may postulate that these cases with JAG1 mutations may have reduced Notch signaling thus reduced mtDNA replication, suggesting an alternate genetic etiology leading to a reduction in hepatic mtDNA content. Further evaluation in a larger cohort of patients with Alagille syndrome is required before firm conclusions can be drawn.
Definite mtDNA depletion was demonstrated in 34% (15/44) of ALF test patients, and 66% (29/44) were identified as having probable or definite mtDNA depletion, based on mtDNA content. Definite/probable mtDNA depletion was identified in patients irrespective of the underlying cause of ALF. This demonstrates that there is a strong association between hepatic mtDNA depletion and ALF in cases in which there appears to have been an environmental trigger, i.e. drug-induced and viral ALF. However this study was not designed to establish causality. Importantly, mtDNA depletion was found in both adults and children with ALF, and there was no significant association between mtDNA depletion and age.
Heterozygous mutation ‘carriers’ were found at a significantly higher rate in patients undergoing LT for ALF than in the control group (P = 0.001); most notably DGUOK. The status of the p.Q170R variant remains controversial, as it is more frequent in the general population than other variants in DGUOK (3), and is more frequent than would be expected given the published frequency of the severe hepatocerebral phenotype. However, this mutation has been demonstrated to cause infantile onset hepatocerebral mtDNA depletion in one infant, in trans with a null mutation (20) and more recently been shown to cause later onset myopathy in a cohort of patients (37). This glutamine is found within the highly constrained alpha helix domain of the protein (GERP 5.67, Phastcons 1.0 at the variant nucleotide). Furthermore, the p.Q170R variant in DGUOK is at a highly conserved position with all homologous proteins having a glutamine and none having an arginine. In silico prediction, by POLYPHEN 2 (38), predicts that this change is possibly damaging. However, the effects of the p.Q170R variant in DGUOK in isolation have not been determined by enzymatic testing and, to date, no individuals homozygous for this variant have been published. For the healthy controls we have no data on ethnicity.
We also saw a higher rate of carriers of pathogenic POLG mutations in patients undergoing LT for ALF. This is consistent with recent data describing a higher rate of heterozygous POLG mutations in patients with sodium valproate-induced liver toxicity than in ethnically matched controls (odds ratio 23.6, 95% confidence interval 8.4–65.8) (30). Our data suggest that POLG mutations may be a general risk factor for liver disease and not specific to drug-induced liver injury.
Although population stratification could account for the differences, the majority of controls were from Wisconsin and would be expected to be mostly Caucasian. Since the common pathogenic mutations in DGUOK and POLG show increased frequency in those of North European ancestry, if there is stratification it would be expected to favor the null hypothesis.
Given that a causal gene can only be identified in 20% of patients with multisystemic mtDNA depletion (39), the failure to detect pathogenic variants in the majority of our cohort with reduced mtDNA content is not surprising. Rather, it suggests that currently unrecognized genes may lead to reduction in hepatic mtDNA content, predisposing to liver dysfunction. Consequently, further work is required to identify such mutations and elucidate the mechanisms leading to reduced mtDNA content. Given the costs associated with the Sanger methods employed in this study, there is a potential need for larger-scale, genome-wide sequencing to evaluate the pathophysiology and predisposition towards mtDNA depletion and ALF.
Our results, in combination with recent data (27, 30), further suggest that carriers of mutations in genes associated with mtDNA depletion, specifically POLG and DGUOK, are at increased risk of liver failure. Therefore, the prescription of hepatotoxic drugs in particular isoniazid and sodium valproate should be considered carefully in individuals carrying, or who are suspected of carrying mutations in mtDNA depletion genes. These data suggest that being a carrier for mutations, even without depletion may be a potential mechanism underlying ‘idiosyncratic’ drug reactions in a subset of cases. Furthermore, in conjunction with previously published data (5, 40), our data suggests a more aggressive role for vaccines against hepatotropic viruses in those with hepatic mtDNA depletion. Given the relatively low population incidence of mtDNA depletion, and the expense of testing, there is currently insufficient evidence to recommend sequence-based testing for these genetic disorders before initiating potentially hepatotoxic therapy in patients. However, these recommendations may change as the cost of sequencing falls.
In summary, end stage liver disease severe enough to require a LT does not cause secondary mtDNA depletion. The majority of individuals in this study undergoing LT for ALF had reduced hepatic mtDNA content, which fell within the range seen in patients with genetically caused mtDNA depletion. A subset of ALF cases have single variants in genes known to lead to mtDNA depletion and hepatic failure. Together, these results suggest an important role for mtDNA replication and maintenance in ALF.
Supplementary Material
Acknowledgements
The authors would like to acknowledge Lin-Ya Tang who performed PCR analysis on liver tissue samples from many of the early healthy controls.
Source of Funding:
Normal and Pathologic human liver were obtained through the Liver Tissue Cell Distribution System, Minneapolis, Minnesota; Pittsburgh, Pennsylvania & Richmond, Virginia, which was funded by NIH Contract #N01-DK-7-0004 / HHSN267200700004C.
Abbreviations
- mtDNA
mitochondrial DNA
- DGUOK
deoxyguanosine kinase
- MPV17
mitochondrial inner membrane protein
- POLG
DNA polymerase γ
- TWINKLE
a DNA helicase located in chromosome 10, open reading frame 2 (also known as C10orf2)
- LT
liver transplant
- ALF
acute liver failure
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text, and links to the digital files are provided in the HTML text of this article on the journal's Web site (www.jpgn.org).
Conflicts of Interest: The authors have no conflict of interest.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Daniel Helbling, Division of Genetics, Department of Pediatrics, Medical College of Wisconsin, Milwaukee, WI, USA. dhelbling@mcw.edu.
Adam Buchaklian, Division of Genetics, Department of Pediatrics, Medical College of Wisconsin, Milwaukee, WI, USA. abuchaklian@mcw.edu.
Jing Wang, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA. JWang7@bcm.edu.
Lee-Jun Wong, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA. LJWong@bcm.edu.
David Dimmock, Division of Genetics, Department of Pediatrics, Medical College of Wisconsin, Milwaukee, WI, USA..
References
- 1.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 2.Wong LJ, Brunetti-Pierri N, Zhang Q, Yazigi N, Bove KE, Dahms BB, Puchowicz MA, et al. Mutations in the MPV17 gene are responsible for rapidly progressive liver failure in infancy. Hepatology. 2007;46:1218–1227. doi: 10.1002/hep.21799. [DOI] [PubMed] [Google Scholar]
- 3.Dimmock DP, Zhang Q, Dionisi-Vici C, Carrozzo R, Shieh J, Tang LY, Truong C, et al. Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase. Hum Mutat. 2008;29:330–331. doi: 10.1002/humu.9519. [DOI] [PubMed] [Google Scholar]
- 4.Goh V, Helbling D, Biank V, Jarzembowski J, Dimmock D. Next-generation sequencing facilitates the diagnosis in a child with twinkle mutations causing cholestatic liver failure. J Pediatr Gastroenterol Nutr. 2012;54:291–294. doi: 10.1097/MPG.0b013e318227e53c. [DOI] [PubMed] [Google Scholar]
- 5.Lutz RE, Dimmock D, Schmitt E, Zhang Q, Tang L- Y, Reyes C, Truemper E, et al. De Novo mutations in POLG presenting with acute liver failure or encephalopathy. Journal of Pediatric Gastroenterology and Nutrition. 2009 doi: 10.1097/MPG.0b013e31817d9cad. [DOI] [PubMed] [Google Scholar]
- 6.Spinazzola A, Zeviani M. Disorders of nuclear-mitochondrial intergenomic signaling. Gene. 2005;354:162–168. doi: 10.1016/j.gene.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 7.Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chretien D, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–780. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 8.Carrozzo R, Dionisi-Vici C, Steuerwald U, Lucioli S, Deodato F, Di Giandomenico S, Bertini E, et al. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain. 2007;130:862–874. doi: 10.1093/brain/awl389. [DOI] [PubMed] [Google Scholar]
- 9.Elpeleg O, Miller C, Hershkovitz E, Bitner-Glindzicz M, Bondi-Rubinstein G, Rahman S, Pagnamenta A, et al. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet. 2005;76:1081–1086. doi: 10.1086/430843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galbiati S, Bordoni A, Papadimitriou D, Toscano A, Rodolico C, Katsarou E, Sciacco M, et al. New mutations in TK2 gene associated with mitochondrial DNA depletion. Pediatr Neurol. 2006;34:177–185. doi: 10.1016/j.pediatrneurol.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 11.Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen A, Lonnqvist T. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain. 2007;130:3032–3040. doi: 10.1093/brain/awm242. [DOI] [PubMed] [Google Scholar]
- 12.Horvath R, Hudson G, Ferrari G, Futterer N, Ahola S, Lamantea E, Prokisch H, et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain. 2006;129:1674–1684. doi: 10.1093/brain/awl088. [DOI] [PubMed] [Google Scholar]
- 13.Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- 14.Ostergaard E, Christensen E, Kristensen E, Mogensen B, Duno M, Shoubridge EA, Wibrand F. Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion. Am J Hum Genet. 2007;81:383–387. doi: 10.1086/519222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ostergaard E, Hansen FJ, Sorensen N, Duno M, Vissing J, Larsen PL, Faeroe O, et al. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain. 2007;130:853–861. doi: 10.1093/brain/awl383. [DOI] [PubMed] [Google Scholar]
- 16.Sarzi E, Goffart S, Serre V, Chretien D, Slama A, Munnich A, Spelbrink JN, et al. Twinkle helicase (PEO1) gene mutation causes mitochondrial DNA depletion. Ann Neurol. 2007;62:579–587. doi: 10.1002/ana.21207. [DOI] [PubMed] [Google Scholar]
- 17.Spinazzola A, Viscomi C, Fernandez-Vizarra E, Carrara F, D'Adamo P, Calvo S, Marsano RM, et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38:570–575. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- 18.Wong LJ, Naviaux RK, Brunetti-Pierri N, Zhang Q, Schmitt ES, Truong C, Milone M, et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum Mutat. 2008;29:E150–E172. doi: 10.1002/humu.20824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaibani A, Shchelochkov OA, Zhang S, Katsonis P, Lichtarge O, Wong LJ, Shinawi M. Mitochondrial neurogastrointestinal encephalopathy due to mutations in RRM2B. Arch Neurol. 2009;66:1028–1032. doi: 10.1001/archneurol.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freisinger P, Futterer N, Lankes E, Gempel K, Berger TM, Spalinger J, Hoerbe A, et al. Hepatocerebral mitochondrial DNA depletion syndrome caused by deoxyguanosine kinase (DGUOK) mutations. Arch Neurol. 2006;63:1129–1134. doi: 10.1001/archneur.63.8.1129. [DOI] [PubMed] [Google Scholar]
- 21.Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, Anbinder Y, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337–341. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- 22.Naviaux RK, Nguyen KV. POLG mutations associated with Alpers' syndrome and mitochondrial DNA depletion. Ann Neurol. 2004;55:706–712. doi: 10.1002/ana.20079. [DOI] [PubMed] [Google Scholar]
- 23.Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- 24.Dimmock DP, Dunn JK, Feigenbaum A, Rupar A, Horvath R, Freisinger P, Mousson de Camaret B, et al. Abnormal neurological features predict poor survival and should preclude liver transplantation in patients with deoxyguanosine kinase deficiency. Liver Transpl. 2008;14:1480–1485. doi: 10.1002/lt.21556. [DOI] [PubMed] [Google Scholar]
- 25.Van Goethem G, Luoma P, Rantamaki M, Al Memar A, Kaakkola S, Hackman P, Krahe R, et al. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology. 2004;63:1251–1257. doi: 10.1212/01.wnl.0000140494.58732.83. [DOI] [PubMed] [Google Scholar]
- 26.Tang S, Wang J, Lee NC, Milone M, Halberg MC, Schmitt ES, Craigen WJ, et al. Mitochondrial DNA polymerase gamma mutations: an ever expanding molecular and clinical spectrum. J Med Genet. 2011;48:669–681. doi: 10.1136/jmedgenet-2011-100222. [DOI] [PubMed] [Google Scholar]
- 27.Saneto RP, Lee IC, Koenig MK, Bao X, Weng SW, Naviaux RK, Wong LJ. POLG DNA testing as an emerging standard of care before instituting valproic acid therapy for pediatric seizure disorders. Seizure. 2010;19:140–146. doi: 10.1016/j.seizure.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dimmock D, Tang LY, Schmitt ES, Wong LJ. Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin Chem. 2010;56:1119–1127. doi: 10.1373/clinchem.2009.141549. [DOI] [PubMed] [Google Scholar]
- 29.Krahenbuhl L, Schafer M, Krahenbuhl S. Reversibility of hepatic mitochondrial damage in rats with long-term cholestasis. J Hepatol. 1998;28:1000–1007. doi: 10.1016/s0168-8278(98)80349-8. [DOI] [PubMed] [Google Scholar]
- 30.Stewart JD, Horvath R, Baruffini E, Ferrero I, Bulst S, Watkins PB, Fontana RJ, et al. Polymerase gamma gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology. 2010;52:1791–1796. doi: 10.1002/hep.23891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Venegas V, Wang J, Dimmock D, Wong LJ. Real-time quantitative PCR analysis of mitochondrial DNA content. Curr Protoc Hum Genet. 2011 doi: 10.1002/0471142905.hg1907s68. Chapter 19:Unit 19 17. [DOI] [PubMed] [Google Scholar]
- 32.Filosto M, Mancuso M, Tomelleri G, Rizzuto N, Dalla Bernardina B, DiMauro S, Simonati A. Hepato-cerebral syndrome: genetic and pathological studies in an infant with a dGK mutation. Acta Neuropathol (Berl) 2004;108:168–171. doi: 10.1007/s00401-004-0872-9. [DOI] [PubMed] [Google Scholar]
- 33.Mousson de Camaret B, Taanman JW, Padet S, Chassagne M, Mayencon M, Clerc-Renaud P, Mandon G, et al. Kinetic properties of mutant deoxyguanosine kinase in a case of reversible hepatic mtDNA depletion. Biochem J. 2007;402:377–385. doi: 10.1042/BJ20060705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salviati L, Sacconi S, Mancuso M, Otaegui D, Camano P, Marina A, Rabinowitz S, et al. Mitochondrial DNA depletion and dGK gene mutations. Ann Neurol. 2002;52:311–317. doi: 10.1002/ana.10284. [DOI] [PubMed] [Google Scholar]
- 35.Tadiboyina VT, Rupar A, Atkison P, Feigenbaum A, Kronick J, Wang J, Hegele RA. Novel mutation in DGUOK in hepatocerebral mitochondrial DNA depletion syndrome associated with cystathioninuria. Am J Med Genet A. 2005;135:289–291. doi: 10.1002/ajmg.a.30748. [DOI] [PubMed] [Google Scholar]
- 36.Tammam J, Ware C, Efferson C, O'Neil J, Rao S, Qu X, Gorenstein J, et al. Down-regulation of the Notch pathway mediated by a gamma-secretase inhibitor induces anti-tumour effects in mouse models of T-cell leukaemia. Br J Pharmacol. 2009;158:1183–1195. doi: 10.1111/j.1476-5381.2009.00389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ronchi D, Garone C, Bordoni A, Gutierrez Rios P, Calvo SE, Ripolone M, Ranieri M, et al. Next-generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions. Brain. 2012;135:3404–3415. doi: 10.1093/brain/aws258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarzi E, Bourdon A, Chretien D, Zarhrate M, Corcos J, Slama A, Cormier-Daire V, et al. Mitochondrial DNA depletion is a prevalent cause of multiple respiratory chain deficiency in childhood. J Pediatr. 2007;150:531–534. 534, e531–536. doi: 10.1016/j.jpeds.2007.01.044. [DOI] [PubMed] [Google Scholar]
- 40.Shieh JT, Berquist WE, Zhang Q, Chou PC, Wong LJ, Enns GM. Novel deoxyguanosine kinase gene mutations and viral infection predispose apparently healthy children to fulminant liver failure. J Pediatr Gastroenterol Nutr. 2009;49:130–132. doi: 10.1097/MPG.0b013e31819de7a6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.