

ABSTRACT
Antibody-drug conjugates (ADCs) are complex therapeutic agents that use the specific targeting properties of antibodies and the highly potent cytotoxicity of small molecule drugs to selectively eliminate tumor cells while limiting the toxicity to normal healthy tissues. Two critical quality attributes of ADCs are the purity and stability of the active small molecule drug linked to the ADC, but these are difficult to assess once the drug is conjugated to the antibody. In this study, we report a enzyme deconjugation approach to cleave small molecule drugs from ADCs, which allows the drugs to be subsequently characterized by reversed-phase high performance liquid chromatography. The model ADC we used in this study utilizes a valine-citrulline linker that is designed to be sensitive to endoproteases after internalization by tumor cells. We screened several proteases to determine the most effective enzyme. Among the 3 cysteine proteases evaluated, papain had the best efficiency in cleaving the small molecule drug from the model ADC. The deconjugation conditions were further optimized to achieve complete cleavage of the small molecule drug. This papain deconjugation approach demonstrated excellent specificity and precision. The purity and stability of the active drug on an ADC drug product was evaluated and the major degradation products of the active drug were identified. The papain deconjugation method was also applied to several other ADCs, with the results suggesting it could be applied generally to ADCs containing a valine-citrulline linker. Our results indicate that the papain deconjugation method is a powerful tool for characterizing the active small molecule drug conjugated to an ADC, and may be useful in ensuring the product quality, efficacy and the safety of ADCs.
KEYWORDS: Antibody-Drug conjugate, enzyme deconjugation, HPLC, mass spectrometry, small molecule pharmaceutical analysis
Introduction
Antibody-drug conjugates (ADCs) are complex biotherapeutics that combine the specific targeting properties of monoclonal antibodies (mAbs) with the extreme potency of small molecule drugs.1-4 By utilizing the selectivity of mAbs, an efficacious dose of the cytotoxic drug can be targeted to the tumor site to kill the tumor cells while limiting the general toxicity of the highly potent cytotoxic agents to normal, healthy tissues.5-8 Three components are required to make an ADC: the antibody, a cytotoxic drug (i.e., the payload) and a linker that holds the antibody and small molecule drug together. The linker and drug are typically combined into a final small molecule intermediate termed the linker drug, which is then conjugated to the antibody. The linker portion should have sufficient stability to enable the ADCs to circulate in the bloodstream before reaching the tumor site without prematurely releasing the free drug and potentially damaging normal cells. Once the ADC is internalized within the target cell, the linker should be sufficiently labile to allow efficient release of the active free drug.9
Currently, the physicochemical characterization of ADCs is primarily focused on the assessment of the antibody. For example, size-exclusion chromatography can be used to determine the presence of size variants and charge variants can be observed via imaged capillary isoelectric focusing or ion exchange chromatography.10,11 As ADCs also carry small molecule drugs, specialized analytical assays must be used to determine attributes such as drug-to-antibody ratio (DAR),12,13 drug distribution,12,13 content of unconjugated free drug,14 and content of residual conjugation solvent.15 However, the small molecule drug (typical MW 500-1500 Da) accounts for only about 1% of the ADC molecule (average MW 150 kDa) by weight, and there are no assays currently available that can directly characterize the small molecule drug component of ADCs. The purity of the small molecule drug and linker components of ADCs are critical quality attributes. Low impurity levels must be maintained because impurities can potentially introduce new toxicities that affect patient safety or they can be sub-potent, leading to a decrease in efficacy.
It is also important to monitor the stability of the active drug linked to the ADC because it may degrade during storage, particularly in liquid formulations. In the past, however, it has been possible to detect only impurities that formed and were released from the antibody.14 Therefore, a method that can directly assess the purity, impurities and degradation products of the active drug linked to an ADC is highly desirable to ensure product quality, stability, and patient safety.
The purpose of this study was to identify a specific enzyme that can cleave the small molecule drug from the linker and release the payload from an ADC such that RP-HPLC could be used to assess the quality of the active drug component of the ADC. There are 2 main classes of ADC linkers: cleavable and noncleavable. Cleavable linkers are designed to be sensitive to a specific enzyme, pH or reduction to trigger the release of free drug, while noncleavable linkers depend on complete degradation of the antibody after internalization of the ADC to release the free drug with the linker attached.16 The model ADC we used in this study utilized a cleavable valine-citrulline linker to connect the antibody and the small molecule drug. Fig. 1 shows the schematic structure of this model ADC; the actual ADC structure has been previously reported.17 The antibody component of this ADC was engineered to contain 2 unpaired cysteines that are covalently linked to 2 linker drugs via the valine-citrulline linker. The valine-citrulline linker is a lysosomal protease-sensitive linker that was incorporated into brentuximab vedotin (Adcetris®) and many other ADCs that are currently in clinical trials because it displays an optimal balance between plasma stability and intracellular protease cleavage.18 This linker is designed to be sensitive to cathepsin B, a cysteine endoprotease that recognizes and cleaves a dipeptide bond to free the drug from the ADC.
Figure 1.

Schematic diagram of the release of free drug from the model ADC by endoprotease.
Cathepsin B has been used to simulate the lysosomal medium that releases the active drug from ADCs from the valine-citrulline linker for in vitro efficacy assays.19 In these studies, cathepsin B was added to the ADC solutions and incubated at 37°C and the amount of the drug released was quantified by HPLC. This cathepsin B-mediated free drug release has also been used for the comparison and selection of linkers that can efficiently release the free drug from ADCs intracellularly, while demonstrating excellent stability in human plasma.20 In this work, we utilized this strategy to release the payloads from the model ADC and then characterized the purity and stability of the small molecule drug by HPLC. We screened several proteases, including cathepsin B, to identify the most effective enzyme that cleaves the free drug, and we found that papain, also a cysteine protease, cleaves the active drug from the ADC completely. This papain deconjugation approach allows the characterization of the purity and stability of the active drug portion of ADC molecules, which was not previously possible.
Results
Enzyme screening
In our model ADC, a valine-citrulline linker, which is sensitive to lysosomal protease, was used to conjugate the payload, a synthetically derivatized antibiotic, to a full-length IgG1 mAb. To find an enzyme that could cleave the linker and release the small molecule drug from the ADC, proteases from 3 protease families and different sources were screened. Because valine-citrulline is designed to be sensitive to cathepsin B, a type of cysteine protease that recognizes and cleaves a dipeptide bond to release the free drug from ADC, we tested cathepsin B and 3 other cysteine proteases with similar cleavage sites to determine if they have different activities and cleavage efficiencies. Papain is a protease commonly used to digest a mAb into 2 Fab fragments and an Fc fragment. Cathepsin L is a cysteine protease from the same cathepsin family. Because the valine-citrulline linker is not a natural dipeptide, proteases that can cleave it must do so via a non-specific mechanism of action. Therefore, in addition to cysteine proteases, we also chose 2 common aspartate proteases and 2 serine proteases with less specific cleavage site requirements to determine if they can also cleave the dipeptide bond on the linker. These enzymes are thought to function on different protein motifs, are generally less specific than the cysteine proteases and cleave the mAb into smaller pieces. The screening was performed by adding 50 ug of each enzyme into solutions containing 1 mg of ADC sample, and the solutions were then incubated in a 40°C chamber for 24 hours. Acetonitrile (ACN) was added to the sample solution at 2:1 ratio to precipitate the proteins. The precipitated samples were centrifuged at 14,000 rpm for 10 minutes, and then the supernatant was removed and analyzed by RP-HPLC to determine the amount of free drug released.
The amount of drug released from the model ADC by the different enzymes is shown in Fig. 2. The results indicated that the cysteine proteases cleave the payload from the valine-citrulline linker with different efficiencies. However, the 2 aspartate proteases and 2 serine proteases we tried showed little to no ability to release the small molecule drug. The valine-citrulline linker was designed to be sensitive to cathepsin B, and our study confirmed that cathepsin B does cleave the drug from the valine-citrulline linker and release the free drug, although the amount of the drug cleaved from the ADC was lower than expected. Additionally, we found that other cysteine proteases (papain, cathepsin L, and bromelain) were also able to cleave the payload from the linker at the same dipeptide position, and they worked more effectively than cathespin B, with papain yielding the highest amount of the free drug. Papain is a plant extract that costs 1/1000 that of cathepsin B and cathepsin L, which are extracted from tissues, and so papain is an affordable reagent for routine assays to evaluate the conjugated drug.
Figure 2.

Comparison of the release of the payload from the model ADC by different enzymes.
Optimization of deconjugation conditions
As papain showed the best activity in cleaving the small molecule drug from the model ADC, we further optimized the deconjugation conditions for papain and compared the results with those for cathepsin B cleavage. We first evaluated the rate of drug release to determine the incubation time for each enzyme. For this experiment, ADC samples were incubated with papain or cathepsin B for various times up to 24 hours at 40°C. RP-HPLC was used to measure the rate of free drug release at different time points, and the result is shown in Fig. 3. Papain can cleave the drug from ADC at a faster rate than cathepsin B; the drug concentration in the papain sample reached a plateau after 8 hours, indicating complete free drug release after 8 hours at 40°C. In the cathepsin B-treated ADC sample, the amount of free drug released gradually increases over the 24 hour window, and the drug release is only about 50% of the papain treated sample after 24 hours. The cathepsin B sample does not reach a plateau, although the rate of release decreases significantly after 8 hours.
Figure 3.
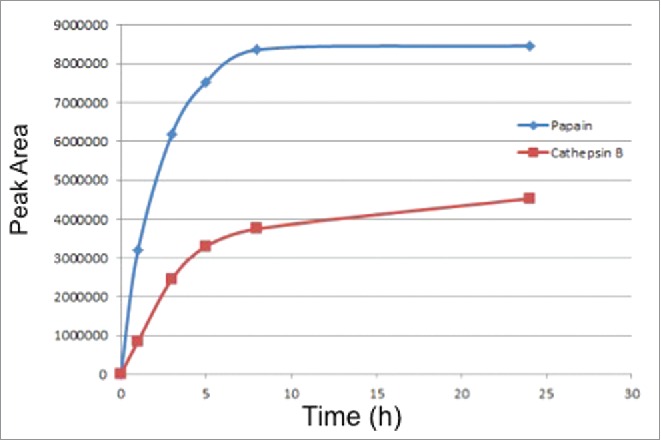
Comparison of release rates of the small molecule free drug from the ADC by papain and cathepsin B.
Enzyme activity can be affected by temperature and pH. We thus examined the deconjugation reaction at 30°C, 40°C, and 50°C and pH 4-7 to optimize the release conditions for papain and cathepsin B. The enzymatic cleavage efficiency was evaluated based on the amount of the free drug released. The results show that both the temperature and pH affect the activity of cathepsin B (Fig. 4). A temperature of 40°C and pH 5, which is the pH of lysosomes where cathepsin B functions in vivo, were found to be the optimal conditions for cathepsin B activity. On the other hand, the amount of free drug released from the ADC by papain is quite consistent through the temperature range of 30°C-50°C and pH 4-7. As the activity of papain is less susceptible to temperature and pH, it is a more robust enzyme to use for the analyzing the small molecule drug conjugated to the ADCs.
Figure 4.

Effect of temperature and pH on the drug release from ADC by papain and cathepsin B.
Another factor that needs to be considered when choosing the appropriate deconjugation temperature is the stability of the small molecule drug during the incubation. We evaluated the stability of the small molecule drug standard in the presence of the 2 enzymes in solutions that were incubated at 30°C, 40°C and 50°C for 24 hours. The small molecule drug was found to be very stable at 40°C or lower; however a small amount of degradation was observed when the sample was incubated at 50°C for a day. Therefore, 40°C was selected as the deconjugation temperature to ensure the integrity of the small molecule drug during the enzyme digestion.
Using the optimized conditions, ADC samples were treated with cathepsin B and papain, and the results were compared to a control ADC sample that was not treated with any enzyme (Fig. 5). All three samples were prepared in 20 mM histidine acetate buffer (pH 6) and incubated at 40°C for 8 hours. ACN was added to the samples after deconjugation to precipitate the protein, and the samples were then centrifuged to isolate the precipitated protein. The small molecule drug that was conjugated to the model ADC has a very strong UV absorption at 606 nm and exhibits a dark blue color even after conjugation to the antibody. While precipitated naked antibody is white, the model ADC is blue because the blue drug is incorporated. In the control sample, the supernatant is colorless and the precipitate is dark blue, indicating that all of the payload is conjugated to the ADC and there is no free drug released into the solution. In the cathepsin B-treated sample, both the supernatant and the precipitated protein are blue, suggesting that a portion of the free drug was released from the ADC while some remained conjugated to the antibody. In the papain-treated sample, the precipitate is white and the supernatant is darker blue because all of the drug has been released from the ADC, resulting in the precipitated protein returning to its original white color. This visual evidence suggests that the small molecule drug was completely released from the ADC by papain digestion. We also confirmed the complete cleavage by determining the amount of the small molecule drug released by quantitating with a standard of the released drug analyzed with HPLC and comparing it to the theoretical amount of the drug present on the mAb. The mAb used in the model ADC was engineered to have 2 unpaired thiol groups available to react with the linker; thus, the theoretical DAR of the ADC is 2.0. The actual DAR of this model ADC was determined to be 2.1 by hydrophobic interaction chromatography. The amount of drug cleaved from the ADC by papain is consistent with the amount of small molecule drug on the ADC calculated based on the actual DAR of the ADC and its concentration.
Figure 5.

Picture of the ADC samples treated with cathepsin B and papain compared to the control ADC sample without enzyme.
Method assessment
Cathepsin B released the small molecule drug from the ADC, but not completely, so it can only be used to qualitatively evaluate how well the linker holds the small molecule drug and antibody together. Because papain can cleave the small molecule drug from the ADC completely, digestion with papain can be used to quantitatively measure the amount of drug, as well as different impurities and degradants, on the ADC. These impurities include process-related impurities, free linker, and other species with an intact maleimide group that can conjugate to the mAb. Once conjugated to the mAb, these impurities will typically remain in the drug substance even after purification of the ADC. In order to evaluate the suitability of the enzyme deconjugation method for quantification, several parameters were assessed, including specificity, precision, and accuracy. To demonstrate the specificity of the papain-mediated drug release, we compared the papain-treated ADC with 2 control samples: 1) the ADC without papain; and 2) the naked antibody (i.e., without conjugated drug) digested by papain. All samples were prepared in 20 mM histidine acetate buffer (pH 6) and incubated at 40°C for 8 hours. The chromatograms of the supernatant after protein precipitation are shown in Fig. 6. The free drug peak from the papain-treated ADC sample matches the small molecule drug standard, confirming the release of free drug from the ADC. Both control samples are very clean, with no peaks observed in the chromatogram, indicating the cleavage is highly specific and no artifacts are introduced in the sample from papain digestion. Therefore, the papain deconjugation can be used for assessing the purity of the small molecule drug conjugated to the ADC.
Figure 6.

Chromatograms of free drug released from ADC by papain compared with the free drug standard with 2 controls: ADC without papain and naked monoclonal antibody treated with papain.
Next, we assessed the performance of this papain deconjugation method using validation criteria that are typically used to evaluate the suitability of a method for an early development quality control assay; the data from this method assessment is summarized in Table 1. Both the system precision and method precision were evaluated. The former was demonstrated by 5 replicate injections of a 100 μg/mL small molecule drug standard solution, while the latter was demonstrated by 5 replicate sample preparations of ADC samples digested by papain to evaluate the consistency of the papain digestion procedure. The method precision was determined to be 2.2%, based on the %RSD of the free drug peak area from HPLC, indicating good precision of the method. Linearity was assessed in the concentration range 0.5 µg/mL to 500 µg/mL; the correlation coefficient is 0.9999. Spike recovery, another key parameter to evaluate the accuracy of the method, was determined by spiking 100 µg/mL of the small molecule drug standard solution into the naked antibody sample, which was then digested by papain in the same way used for the ADC sample. The mean recovery was 103.5%, indicating no sample loss during the digestion and sample preparation procedure. The method performance results demonstrated that the papain digestion of the ADC is robust and accurate, and therefore suitable for quantitative usage.
Table 1.
Summary of method performance.
| Parameter | Results |
|---|---|
| System precision (n = 5) | %RSD = 0.2% |
| Method precision (n = 5) | %RSD = 2.2% |
| LOQ in ADC sample | 1µg/mL |
| Calibration curve | y = 73937x–48032 (r2 = 0.9999)range: 0.5–500µg/mL |
| Spike recovery (n = 3) | 103.5% |
To determine if the papain deconjugation has general application to other ADCs with the same enzyme-cleavable linker, 3 ADCs that have the same valine-citrulline linker, but different drug and antibody components, were analyzed. The samples were all treated with papain using the optimized preparation conditions, and the released drug was analyzed by RP-HPLC. Fig. 7 shows the chromatograms of the 3 different free drugs cleaved from the 3 ADCs by papain. These results demonstrated that papain can cleave the dipeptide bond on the valine-citrulline linker independent of the small molecule drug and antibody that comprise the ADC. We therefore conclude that this deconjugation approach can be applied to other ADCs with valine-citrulline linkers.
Figure 7.

Chromatograms showing the release of free drugs from 3 ADCs that have different small molecule drugs conjugated to mAb by the same valine-citrulline (vc) linker.
Stability of the conjugated drug on the ADC
We also assessed the stability of the small molecule drug conjugated to the model ADC by cleaving the drug with papain. The model ADC contains 50 mg/mL ADC in 20 mM histidine acetate buffer (pH 6.0). The samples were tested at T(0) to examine the purity and impurities present in the original ADC sample. The sample was stored at 5°C and at elevated temperatures (30°C and 40°C) to allow formation of potential degradation products at more stressed conditions. The chromatogram of the cleaved free drug tested at different temperatures after one month is shown in Fig. 8. The chromatogram at T(0) represents the impurity profile of the small molecule drug conjugated to the ADC in the original ADC sample. Since we demonstrated that the small molecule drug remains stable during the enzyme digestion at 40°C for 24 hours, all the impurities observed at T(0) are actually impurities of the active drug conjugated to the ADC. The overall purity of the small molecule drug linked to the ADC is 98.1%, as determined by the peak area% on the HPLC. The most abundant impurity (0.8%) has retention time 22.4 min. The m/z of the [M+H]+ of this impurity is 925, 2 units lower than that of the [M+H]+ of 927 for the payload. It was identified as a molecule resulting from the conversion of one of the hydroxyl groups to ketone by loss of 2 hydrogens. Other impurities were also observed at 10.5 min ([M+H]+=1050), 15.1 min ([M+H]+=1032), and 19.9 min ([M+H]+=1032). All these impurities were generated in the synthesis of the small molecule drug and were observed in the payload prior to conjugation. The chromatograms at one month showed changes in the impurity profiles at different temperatures. The results indicate that the active drug on the ADC is stable at 5°C for at least one month, while degradation was observed at elevated temperatures (30°C and 40°C). The impurity peak at 14.1 min (degradant 1) grew significantly (from 0.2% at T(0) to 0.8%) after storage of the ADC at 40°C for one month. One new degradation product at retention time 24.5 min (degradant 2) was also observed. Degradant 1 was identified as the isomer of the active drug mostly likely formed by the rotation of an α, β-unsaturated ketone. Degradant 2 was formed by the loss of a methoxy group on the drug.
Figure 8.

Stability of active drug conjugated on ADC stored at different temperatures for one month.
Discussion
Analysis of the attributes of small molecule active drug conjugated to ADCs is critically important for drug product quality control, but also very challenging. Impurities can reduce potency of the molecule and lead to increased toxicities that can affect patient safety. An enzymatic deconjugation approach was developed for the characterization of the small molecule drug linked to ADCs. Papain, a cysteine protease, was found to cleave the drug from the valine-citrulline linker completely during incubation with our model ADC. The papain digestion approach demonstrated excellent specificity, robustness, and accuracy, and therefore can be used for quantitation purposes. Our study also showed that papain can cleave the valine-citrulline linker on ADCs composed of antibodies and drugs that are different from those in the model ADC, demonstrating the general application of this deconjugation approach to ADCs that incorporate the same linker. This method was also used to study the purity and stability of the small molecule drug conjugated to the model ADC, and the results provided valuable information about the impurity profile and degradation pathway of the small molecule drug conjugated to the ADC. This enzymatic deconjugation approach is thus a new tool for characterizing the payload on ADC molecules, and, as the method demonstrated excellent specificity, precision and accuracy, it may be valuable for the assessment of ADC product quality.
Material and methods
Reagents and materials
Acetonitrile (ACN, HPLC grade) was purchased from J. T. Baker (Phillipsburg, NJ, USA). Trifluoroacetic acid (TFA) and histidine acetate were purchased from Sigma-Aldrich (St. Louis, MO, USA). Deionized water was from an in-house Milli-Q system (Millpore, Billerica, CA, USA). Cathepsin B (human liver; catalog number 219362) and papain (catalog number 5125) were purchased from EMD Millipore (Billerica, MA, USA). Cathepsin L (human liver; catalog number C6854) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Pepsin from porcine gastric mucosa (catalog number P7000) was purchased from Sigma (St. Louis, MO, USA). Bromelain (catalog number AC40283) was purchased from Fisher Scientific (Pittsburgh, PA, USA). Trypsin (MS grade; catalog number 90057) was purchased from Thermo Scientific (Waltham, MA, USA). Cathespin D (human liver; catalog number J66506) and α-chymotrypsin (bovine pancreases: catalog number J65266) were purchased from Alfa Aesar (Ward Hill, MA, USA). All four ADC samples and the active small molecule drug standards used in this study were provided by Genentech (South San Francisco, CA, USA). The four antibodies were full-length IgG1 antibodies produced in Chinese hamster ovary cells. The four drugs conjugated to the ADCs were a synthetically derivatized natural product, monomethyl auristatin A, and 2 different DNA-damaging toxin dimers. In particular, the model ADC was an anti-S. aureus antibody conjugated to a highly efficacious antibiotic via the valine-citrulline linker.17
Sample prepare procedure
The model ADC sample was dissolved in 20 mM histidine acetate buffer (pH 6) at 50 mg/mL. Other ADC samples were prepared in 20 mM histidine acetate buffer (pH 6) at 20 mg/mL. The ADC samples were digested by aliquoting 20 µL of the ADC solution and 50 µL of papain (0.5 mg/mL in water) into a vial and then incubating for 8 hours at 40°C. Following incubation, the proteins in the samples were precipitated by adding 200 µL ACN. The precipitated samples were then centrifuged at 14,000 rpm for 10 minutes and the supernatant was transferred to a vial for HPLC analysis. The standard solution of the small molecule drug was prepared in ACN:water (1:1) diluent at concentration of 0.1 mg/mL.
LC-MS conditions
The cleaved small molecule drug was analyzed by RP-HPLC using a Poroshell SB-Aq column (150 × 3.0 mm, 2.7 µm, Agilent, Sunnyvale, CA, USA). Initial conditions were set at 80% solvent A (0.05% TFA in water) and 20% solvent B (ACN). Solvent B was increased to 50% in 20 minutes using a linear gradient and then to 90% in 10 minutes. Solvent B was held at 90% for 5 min followed by a re-equilibration step at 20% B for 5 min. The flow rate was maintained at 0.5 mL/min, the column temperature was set at 25°C, and detection wavelength was 225 nm, and the injection volume was 10 µL.
The HPLC system was coupled with an LCQ Fleet MS detector purchased from Thermo Scientific (Waltham, MA, USA). The instrument was equipped with an ESI source and was operated in a positive mode with capillary voltage at 3.5kV, capillary temperature at 350°C, sheath flow rate at 45 mL/min and Auxiliary flow rate at 10 mL/min. Full scan spectra were collected over the m/z range of 200–2000. Thermo Xcalibur software was used to control the instrument and for data processing.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Isabella De-Jong from Genentech's protein formulation department for providing the model ADC drug product and Jack Sadowsky from Genentech's ADC conjugation group for providing the other ADC samples. We are also grateful to Stefan Koenig and Sigrid Hubbell from Genentech for manuscript review and helpful discussions.
References
- 1.Larson RA, Sievers EL, Stadtmauer EA, Löwenberg B, Estey EH, Dombret H, Theobald M, Voliotis D, Bennett JM, Richie M, et al.. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005; 104:1442-1452; PMID:16116598; http://dx.doi.org/ 10.1002/cncr.21326 [DOI] [PubMed] [Google Scholar]
- 2.Alley SC, Okeley NM, Senter PD, Antibody-drug conjugates: targeted drug delivery for cancer. Curr Opin Chem Biol 2010; 14:529-537; PMID:20643572; http://dx.doi.org/ 10.1016/j.cbpa.2010.06.170 [DOI] [PubMed] [Google Scholar]
- 3.LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastuzumab emtansine: a unique antibody-drug conjugate in development of human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res 2011; 17:6437-6447; PMID:22003071; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0762 [DOI] [PubMed] [Google Scholar]
- 4.Chari RVJ. Targeted cancer therapy: Conferring specificity to cytotoxic drugs, Acc. Chem Res 2008; 41:98-107; PMID:17705444; http://dx.doi.org/16151407 10.1021/ar700108g [DOI] [PubMed] [Google Scholar]
- 5.Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol 2005; 23:1137-46; PMID:16151407; http://dx.doi.org/ 10.1038/nbt1141 [DOI] [PubMed] [Google Scholar]
- 6.Chari RV. Targeted delivary of chemotherapeutics tumor-activated prodrug therapy. Adv Drug Delivery Rev 1998; 31:89-104; PMID:10837619; http://dx.doi.org/16087399 10.1016/S0169-409X(97)00095-1 [DOI] [PubMed] [Google Scholar]
- 7.Lambert JM. Drug-conjugated monoclonal antibodies for the treatment of cancer. Curr Opin Pharmacol 2005; 5:543-549; PMID:16087399; http://dx.doi.org/ 10.1016/j.coph.2005.04.017 [DOI] [PubMed] [Google Scholar]
- 8.Payne G. Progress in immunoconjugate cancer therapeutics. Cancer Cell 2003; 3:207-212; PMID:12676579; http://dx.doi.org/ 10.1016/S1535-6108(03)00057-6 [DOI] [PubMed] [Google Scholar]
- 9.Perez HL, Cardarelli PM, Deshpande S, Gangwar S, Schroedar GM, Vite GD, Borzilleri RM. Antibody-drug conjuages: current status and future directions. Drug Discovery Today 2014; 19:869-881; PMID:24239727; http://dx.doi.org/ 10.1016/j.drudis.2013.11.004 [DOI] [PubMed] [Google Scholar]
- 10.Wakankar A, Chen Y, Gokarn Y, Jacobson FS. Analytical methods for physiochemical characterization of antibody drug conjugates. mAbs 2011; 3(2): 161-172; PMID:21441786; http://dx.doi.org/ 10.4161/mabs.3.2.14960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alley SC, Anderson KE, Analytical and bioanalytical technologies for characterizing antibody-drug conjugates. Curr Opin Chem Biol 2013; 17:406-411; PMID:23570980; http://dx.doi.org/ 10.1016/j.cbpa.2013.03.022 [DOI] [PubMed] [Google Scholar]
- 12.Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al.. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res 2004; 10:7063-7070; PMID:15501986; http://dx.doi.org/ 10.1158/1078-0432.CCR-04-0789 [DOI] [PubMed] [Google Scholar]
- 13.Sanderson RJ, Hering MA, James SF, Sun MM, Doronina SO, Siadak AW, Senter PD, Wahl AF. In vivo drug-linker stability of an anti-CD30 dipeptide-linked auristatin immunoconjugate, Clin. Cancer Res 2005; 11:843-852. [PubMed] [Google Scholar]
- 14.Li Y, Gu C, Gruenhagen J, Zhang K, Yehl P, Chetwyn NP, Medley CD. A size exclusion -reversed phase two dimensional-liquid chromatography methodology for stability and small molecule related species in antibody drug conjugates. J Chromatogr A 2015; 1393:81-88; PMID:2581855819769391 [DOI] [PubMed] [Google Scholar]
- 15.Medley CD, Kay J, Li Y, Gruenhagen J, Yehl P, Chetwyn NP. Quantification of residual solvents in antibody drug conjugates using gas chromatography. Anal Chim Acta 2014; 850:92-96; PMID:2544116519769391 [DOI] [PubMed] [Google Scholar]
- 16.Ducry L, Stump B, Antibody-drug conjugate: lining cytotoxic payload to monoclonal antibodies. Bioconjugate Chem 2010; 21:5-13; PMID:19769391 [DOI] [PubMed] [Google Scholar]
- 17.Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, Depalatis L, Raab H, Hazenbos WL, Morisaki JH, et al.. Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 2015; 527:323-328; PMID:26536114; http://dx.doi.org/ 10.1038/nature16057 [DOI] [PubMed] [Google Scholar]
- 18.Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, et al.. Development of potent monoclonal antibody auristatin conjugates for cancer therapy, Nat. Biotechnol 2003; 21:778-784; PMID:12778055.9873731 [DOI] [PubMed] [Google Scholar]
- 19.Dubowchik GM1, Firestone RA, Padilla L, Willner D, Hofstead SJ, Mosure K, Knipe JO, Lasch SJ, Trail PA. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjugate Chem 2002; 13:855-869; PMID:12121142; http://dx.doi.org/9873731 10.1021/bc025536j [DOI] [PubMed] [Google Scholar]
- 20.Dubowchik GM, Firestone RA. Cathepsin B-sensitive depepetide prodrugs, 1. A model study of structural requirements for efficient release of doxorubicin. Bioorg Med Chem Lett 1998; 8:3341-3346; PMID:9873731; http://dx.doi.org/ 10.1016/S0960-894X(98)00609-X [DOI] [PubMed] [Google Scholar]