

ABSTRACT
The enormous diversity created by gene recombination and somatic hypermutation makes de novo protein sequencing of monoclonal antibodies a uniquely challenging problem. Modern mass spectrometry-based sequencing will rarely, if ever, provide a single unambiguous sequence for the variable domains. A more likely outcome is computation of an ensemble of highly similar sequences that can satisfy the experimental data. This outcome can result in the need for empirical testing of many candidate sequences, sometimes iteratively, to identity one which can replicate the activity of the parental antibody. Here we describe an improved approach to antibody protein sequencing by using phage display technology to generate a combinatorial library of sequences that satisfy the mass spectrometry data, and selecting for functional candidates that bind antigen. This approach was used to reverse engineer 2 commercially-obtained monoclonal antibodies against murine CD137. Proteomic data enabled us to assign the majority of the variable domain sequences, with the exception of 3–5% of the sequence located within or adjacent to complementarity-determining regions. To efficiently resolve the sequence in these regions, small phage-displayed libraries were generated and subjected to antigen binding selection. Following enrichment of antigen-binding clones, 2 clones were selected for each antibody and recombinantly expressed as antigen-binding fragments (Fabs). In both cases, the reverse-engineered Fabs exhibited identical antigen binding affinity, within error, as Fabs produced from the commercial IgGs. This combination of proteomic and protein engineering techniques provides a useful approach to simplifying the technically challenging process of reverse engineering monoclonal antibodies from protein material.
KEYWORDS: Antibody sequencing, mass spectrometry, phage display, proteomics, reverse engineering
Abbreviations
- mAb
monoclonal antibody
- V-domain
variable domain
- MS
mass spectrometry
- VH
variable heavy
- VL
variable light
- CDR
complementarity-determining region
- FR
framework
- Fab
antigen-binding fragment
- PBS
phosphate-buffered saline
- KD
equilibrium dissociation constant
- ELISA
enzyme-linked immunosorbent assay
- UPLC
ultra performance liquid chromatography
- LC/MS
liquid chromatography/ mass spectrometry
- LC/MS/MS
liquid chromatography/ tandem mass spectrometry
Introduction
In addition to their significance as human therapeutic and diagnostic agents, monoclonal antibodies (mAbs) are extensively used to support research activities in commercial and academic settings. The exquisite specificity of mAbs for different antigens is a property uniquely encoded by the amino acid sequence within the variable domains (V-domains) of each mAb. Regardless of the application, mAbs are typically produced by cell culture methods, and as such, a cell line or expression plasmid ordinarily provides a source of nucleic acid material from which the unique amino acid sequence can be easily derived by DNA sequencing methods. However, in cases where these materials are not available, for instance, if a hybridoma cell line has been irretrievably lost or an investigator only has access to protein material, then elucidation of the amino acid sequence would only be possible through direct sequencing of the mAb itself.
In recent years, mass spectrometry (MS) has become the tool of choice for sequencing of proteins. The most common method for determination of protein sequence and identity are bottom-up approaches where proteolytically derived peptide fragments are subjected to tandem MS, and those data are interpreted against a reference database of proteins. For most proteins, this approach enables identification of an unknown protein based on partial matching of the full protein sequence to the experimental tandem MS data set. In the case of mAbs, this approach can be used to determine the isotype and light chain type, but cannot be used to determine the complete sequences of V-domains as the enormous diversity that can result from V-J or V-D-J recombination and somatic hypermutation results in limited coverage of potential sequences within protein databases.
Given the limited database coverage, deducing the complete amino acid sequences of antibody V-domains by MS-based methods is challenging. In contrast to DNA sequencing, de novo MS sequencing will not typically lead to a single sequence. Isobaric (Leu/Ile) or near isobaric (Lys/Gln) amino acids can lead to ambiguous sequence assignments, as can isobaric residue pairs (e.g., Gly-Thr and Ala-Ser) or higher order fragments if these occur within proteolytic peptides that are resistant to MS fragmentation. In these cases, informed assignment may be guided by the sequence of a candidate germline gene segment, but when these sequence ambiguities map to hypervariable complementarity-determining regions (CDRs) of the antibody, this is not necessarily possible. A number of reports have described methods for partial de novo sequencing of antibody V-domains,1-6 but obtaining the complete sequences necessary to enable reverse engineering of a mAb from protein material remains a very challenging undertaking.
A number of commercial vendors now claim to offer de novo antibody sequencing services. However, only 2 reports in the literature described the complete determination of V-domain sequences for unknown antibodies and, importantly, demonstrated that the reverse-engineered mAb possessed the desired activity.2,6 Both of these reports utilized a bottom up proteomic approach that combined digestion with multiple proteases to generate overlapping peptide fragments and computational assignment of most of the sequence, but they differed in the approaches used to extend and/or correct the sequence information obtained by MS data interpretation. In the case of Castella et al.,2 a customized informatics approach termed ‘template proteogenomics’7 was used to computationally analyze the tandem MS data and rapidly assign most of the antibody sequence. However, this methodology was unable to determine the sequence of the CDR-H3 region, for which they had to resort to unspecified techniques, which may have included the use of non-MS based methods.5 In the case of Sousa et al.,6 chemical modification of Cys residues to thialysine, which can by cleaved by Lys-specific proteases, was used to help with identification of peptides containing regions of CDR1 and/or CDR3, and isotopic labeling of Lys and Arg residues in a tentative draft construct was used to identify regions of sequence errors by comparing digests of this material to the parental antibody by LC/MS/MS. As these studies showed, MS-based de novo sequencing can readily provide 90–98% of the V-domain sequences, but elucidating the residual sequence, which is typically within critical CDRs, is challenging and would benefit from complementary techniques that can quickly survey a large number of potential sequence solutions.
Herein we describe a combination of proteomic-based methods and phage display technology to determine the complete V-domain amino acid sequences for 2 commercially sourced mAbs against CD137. MS-based sequencing of proteolytic peptide fragments was used to derive the bulk of the sequence information. Phage display was then used to resolve ambiguous regions of sequence resulting from isobaric amino acids (Leu/Ile), or regions that were not fully fragmented in the mass spectrometer and could not be resolved by reference to antibody germline sequences. This approach enabled us to combinatorially explore a large repertoire of sequences that were compatible with the MS data, and functionally select for those that could bind the target antigen. In both cases, we were able to fully determine the V-domain sequences, and demonstrate that the reverse-engineered antigen-binding fragments (Fabs) exhibited equivalent CD137 binding affinities as Fabs generated from the commercial materials.
Results
General workflow
Our workflow (Fig. 1) consisted of a classical bottom up approach to determine much of the V-domain sequences. As we were only interested in determining the full sequence for the antibody V-domains, and content to limit constant region analysis to confirmation of the vendor-provided IgG subtype information, our process began with generation of Fabs to simplify the complexity of later MS analyses. Once generated, digestion of the Fabs with multiple proteases provided samples that were analyzed by LC/MS/MS, and these data were computationally analyzed against a translated database of functional V and J+C gene segments to identify candidate germline genes. Following the automated database searching, we evaluated the use of software to interpret MS/MS spectra and elucidate additional regions of sequence within the V-domains. However, the package that was tested (the Sherenga algorithm as part of the Agilent Spectrum Mill software) did not prove effective in this application and did not significantly shorten the time spent on manual interpretation. Accordingly, all subsequent interpretation of MS/MS spectra was performed manually, guided by the sequences of candidate germline genes. This analysis was carried out in an iterative manner until the maximum possible amount of V-domain sequence information had been extracted from the tandem MS data. Remaining regions of uncertain sequence were then addressed through the design of phage-displayed Fab libraries that contained limited regions of sequence diversity programmed according to mass constraints defined by the MS data. Functional binders were then selected from these libraries by panning against recombinant antigen. Individual clones were analyzed by DNA sequencing and phage ELISA prior to recombinant expression for a more accurate determination of their antigen binding affinity.
Figure 1.
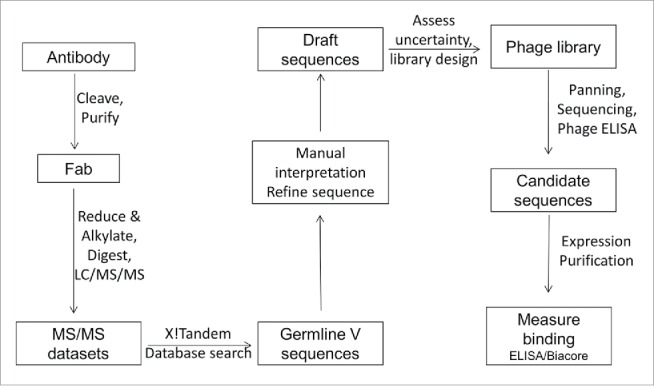
Schematic representation of the workflow used for determination of the light and heavy chain variable domain sequences.
Fab generation and proteomic analysis of mAbs LOB12.3 and 3H3
LOB12.3 and 3H3 are commercially available rat monoclonal antibodies that bind specifically to murine CD137 and can agonize the activity of this receptor.8,9 The heavy chain isotype of LOB12.3 has been described as IgG1, while that of 3H3 is IgG2a. Both IgGs were cleaved with papain to isolate Fabs and simplify the eventual LC/MS analysis by maximizing the amount of data that was collected on V-domain peptides compared to constant regions. It also improved the accuracy of intact mass data on these regions, which provided narrower mass constraints on any unidentified portions of the sequence during the determination process. For LOB12.3, cleavage with papain was not efficient and did not occur exclusively at a single site, rather, a short ladder of Fab species were generated that varied in length by 1–2 residues. The intact mass of the reduced LOB12.3 light chain was 23,316 Da, while the heavy masses were 24,717, 24,614 and 24,501 Da (Fig. 2A). This set of heavy chain masses suggested the C-terminus of the 2 smaller species differed by Cys (24,717 – 24,614Da) or Cys-Ile (24,717 – 24,501 Da) relative to the largest species. This allowed us to identify the location of papain cleavage within the IgG1 hinge region (Fig. 2A). A second round of LOB12.3 cleavage was performed with the alternate enzyme SpeB, which provided more efficient and specific cleavage at a similar site within the hinge region to that cleaved by papain. For 3H3, the experimental masses of the reduced light and heavy chains were 23,275 Da and 24,591 Da, respectively (Fig. 2B). A single tryptic digest of the Fc fragment was performed to specifically identify where papain cleavage had occurred in the 3H3 hinge sequence. From this analysis, together with intact mass data for the Fc region, we were able to assign the tryptic fragments to the sequence for rat IgG2a. We identified CTGSEVSSVFIFPPK as the N-terminal tryptic peptide derived from digestion of the Fc fragment. Accordingly, this enabled us to define the C-terminus of the papain-derived Fab heavy chain fragment (Fig. 2B), which allowed for a meaningful interpretation of the intact mass of the Fab heavy chain.
Figure 2.

Mass spectra and location of protease cleavage sites during LOB12.3 and 3H3 Fab generation. (A) Intact mass spectra of reduced Fab generated by papain digestion of LOB12.3 IgG. The rat IgG1 hinge region sequence is shown above the spectra and sites of cleavage by papain (open triangles) or SpeB (filled triangle) are shown. (B) Intact mass spectra of reduced Fab generated by papain digestion of 3H3 IgG. The rat IgG2a hinge region sequence is shown above the spectra and sites of cleavage by papain are shown.
Following isolation of the Fabs, several different proteases were used for digestion into overlapping peptide fragments. Trypsin was selected as it is the most widely used enzyme to digest proteins for MS analysis, based on its robustness and the size and charge properties of tryptic peptides often being highly favorable for determination of sequence by MS/MS. Chymotrypsin was also selected as it is also highly robust but orthogonal to trypsin in its specificity. GluC and AspN both cleave at sites distinct from trypsin and chymotrypsin, however, in our procedures, these enzymes were inefficient at cleaving the intact Fab. We therefore used a combination of either enzyme with trypsin, and this was very effective at cleaving larger tryptic peptides at the relevant internal acidic residues, leaving peptides where a larger portion of their sequence could be read from MS/MS data.
Our strategy for MS data interpretation began with an automated search of the tryptic and chymotryptic peptide data sets to identify candidate germline sequences encoding each V-domain. This analysis was performed with the software package X! Tandem10 which attempted matching of the acquired MS/MS spectra to model spectra based on peptide fragments derived from a database containing translated rat V-gene or J+C gene segments (see supplementary material for a summary of search outputs). For 3H3, the database search of the tryptic and chymotryptic data gave sufficient matches to V-domain framework peptides to identify a small number of closely related rat germline Vκ genes derived from the IGKV22 family, and VH genes derived from the IGHV5 family. For LOB12.3, this process also identified IGHV5 as the candidate germline gene family for the heavy chain V-region, and IGKV12 as the candidate germline gene family for the light chain. Assignment of the LOB12.3 light chain germline was tentative given the limited sequence coverage obtained from the automated search, but data from the GluC/trypsin digest was used to identify several additional peptides that were also consistent with IGKV12.
Having assigned the V-region germline genes for each antibody to specific families, manual interpretation of tandem MS data across the different digestion data sets was then guided by these germline sequences to progressively build out missing regions of the LOB12.3 and 3H3 sequences. For CDR-H3 peptides, where template sequence was much less apparent, additional strategies were employed, including identification of peptides using mass constraints from the intact protein mass, and analysis of the AspN/trypsin and/or chymotryptic digests to identify CDR-H3 peptides resulting from cleavage at conserved residues encoded within the J segment (i.e., VH-D101, VH-W103). During this process, ambiguities in the determined sequence were tracked (namely, Leu/Ile and Lys/Gln uncertainties or peptide subfragments lacking inter-residue MS/MS fragmentation data). Although most of the ambiguities could be resolved from overlapping peptides across the multiple digests, those that could not be fully resolved were either interpreted according to germline sequence homology, protease digestion specificity (e.g., to resolve Lys vs Gln) or carried forward to the library step.
Manual interpretation of the tryptic digest data for LOB12.3 enabled us to define the sequence of the C-terminal half of the light chain V-domain, including CDR-L2, CDR-L3 and the framework (FR) 4 region encoded by the J-segment. The GluC/trypsin and AspN/trypsin digests provided sufficient data to define much of this missing N-terminal region and part of CDR-L1, and the remainder of the light chain sequence was completed through manual analysis of the chymotryptic data set. Similarly, much of the LOB12.3 heavy chain V-domain sequence could be determined using the tryptic data alone, including CDR-H2. A peptide containing CDR-H3 and FR-H4 could be identified but not sequenced in the tryptic data, and CDR-H1 was not apparent in this data set. A peptide from the AspN/trypsin digest, resulting from cleavage at VH-D101 (present in 3 of 4 rat germline J-segments), was identifiable as covering the C-terminus of CDR-H3 through to the N-terminal region of CH1, and had a mass consistent with usage of the J segment IGHJ2. This in turn allowed identification of the remaining section of CDR-H3 as a single 875 Da (MH+) peptide in the same digest, and this could be sequenced. Examination of the chymotrypsin data enabled us to complete the sequence in the region containing CDR-H1, and provided additional information to resolve ambiguities across all segments. The combined analysis led to draft sequences for the LOB12.3 V-domains as shown in Fig. 3 (LOB12.3-Lv1 for the light chain; LOB12.3-Hv1 for the heavy chain). This draft sequence accounted for all positions in the V-domains, but the MS data could not distinguish between Leu/Ile residues (31 total, 8 of which were in CDRs) and a 114 Da motif in CDR-H3 that could either be a GlyGly or Asn residue. Representative MS/MS spectra covering some of the CDRs in LOB12.3 and their sequence interpretation are shown in Fig. S3. Importantly, the calculated masses for LOB12.3-Lv1 and LOB12.3-Hv1, with the appropriate constant regions appended, were within 4 Da of the experimental intact mass data obtained for the light and heavy chain components of LOB12.3 Fab (23,319 vs 23,316 Da for the light chain; 24,721 vs 24,717 Da for the heavy chain). The 3–4 Da deficit in calculated vs observed masses for the light chain and Fd fragments is routinely observed in our hands, and results from the limited reduction of intrachain disulfides under non-denaturing conditions.
Figure 3.

LOB12.3 variable domain sequences described in this work. LOB12.3-Lv1/Hv1 represent the initial assembly of proteomic data into light and heavy V-domain sequences, where X represents ambiguous regions of sequence (gray highlighting = 113 Da; green = 114 Da). Alignment of this draft to closest V, D and J translated germline gene segments is shown below. LOB12.3-Lv2/Hv2 were refined draft sequences, as defined in Table 1, and ambiguous regions in this light/heavy pair were programmed into a Fab phage display library. Further refinement after phage display led to LOB12.3-Lv3/Hv3, and finally 2 unique light/heavy pairings (LOB12.3-Lv4/Hv4 and LOB12.3-Lv4/Hv5) that were part of recombinant Fab fragments. Sequence numbering is according to Kabat et al.18 CDRs are shown in bold and were defined according to the sequence definition of Kabat et al.,18 except for CDR-H1, which is the combined sequence and structural definition.19
Using both tryptic and chymotryptic digests, the majority of the 3H3 light and heavy chain V-domains were straightforward to define, with the major exceptions being the CDR-H2 region, and the CDR3/FR4 regions for both chains. We assigned VL39 as a Gln residue instead of the germline Lys, as we did not observe any tryptic peptides resulting from cleavage at that site, despite efficient cleavage throughout the rest of the protein. Tryptic peptides that covered both CDR3 regions were readily identified, but at 4.9 and 5.9 kDa for the light and heavy chains, respectively, were far too large to yield useful sequence information after fragmentation. Sequences for the CDR-H2 and CDR-L3/FR-L4 regions were determined using data from the combination digest of GluC and trypsin, which allowed partial sequence determination in this region, and provided mass information to allow placement of previously unidentified chymotryptic peptides as part of these regions. These peptides enabled completion of the light chain sequence and CDR-H2, except for isobaric ambiguities with Leu/Ile and 2 dipeptide motifs within CDR-H2.
Sequencing of the 3H3 CDR-H3 region required alternative strategies. Since the sequence immediately N-terminal to CDR-H3 had been assigned, the chymotryptic digest data was searched for peptides with a marker ion at 324 m/z, which can be generated as the b2 ion from a peptide with an N-terminal sequence of YC (i.e., corresponding to VH91–92). This identified one previously unassigned peptide, and although the MS/MS data for this peptide was weak, an alternatively cleaved chymotryptic peptide lacking the N-terminal tyrosine was also identified. The MS/MS data from this peptide (Fig. 4) allowed determination of its sequence to be CT(I/L)DGY, thereby defining the N-terminal portion of CDR-H3. A similar method was used to search for chymotryptic peptides cleaved at the conserved VH-W103, which might be expected to contain the C-terminal portion of CDR-H3. This was done by searching for unassigned chymotryptic peptides that generated a fragment ion of 205 m/z, a diagnostic y1 ion for peptides containing a C-terminal tryptophan. This led to identification of the peptide fragment YFDFW, which matches part of the IGHJ1 germline sequence. This provided much narrower mass constraints on the remaining sections of CDR-H3.
Figure 4.
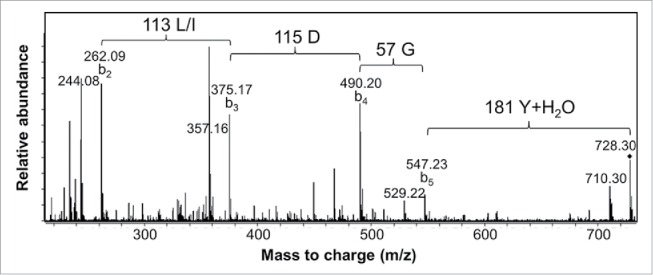
MS/MS spectrum of a 3H3 chymotryptic peptide containing part of CDR-H3. Interpretation of the fragmentation observed from the 728.3 Da parent ion led to assignment of the sequence as CT(L/I)DGY where C is modified with a carbamidomethyl group. Additional peaks arise primarily by water loss from the labeled b ion peaks.
Given the preceding identification of 2 aspartate residues in this section (C-T-I/L-D-G-Y……Y-F-D-F-W), we were then able to identify a single peptide in the AspN/trypsin data set (MH+ = 1495.6 Da) that contained the remaining unidentified sections of CDR-H3. The fragmentation data for this peptide was insufficient for complete sequencing, but it indicated the presence of an additional tyrosine residue in CDR-H3. An additional chymotryptic peptide was then identified, resulting from cleavage after this tyrosine and VH-W100e, and this was partially sequenced as XXSHW (where XX = SerGly or GlySer). This enabled completion of draft sequences for the 3H3 V-domains as shown in Fig. 5 (3H3-Lv1 and 3H3-Hv1). The draft accounted for all positions in the sequence, but the MS data could not distinguish between Leu/Ile residues (27 total, 4 of which were in CDRs), the aforementioned SerGly/GlySer and a 114 Da motif in CDR-H3 that could either be GlyGly or Asn, and 2 isobaric dipeptide motifs in CDR-H2. Representative MS/MS spectra covering additional CDRs in 3H3 and their sequence interpretation are shown in Fig. S4. The calculated masses for all possible sequences of 3H3-Lv1 and 3H3-Hv1, with the appropriate constant regions appended, were within 4 Da of the experimental intact mass data obtained for the light and heavy chain components of 3H3 Fab (23,278 vs 23,275 Da for the light chain; 24,594 vs 24,591 Da for the heavy chain).
Figure 5.
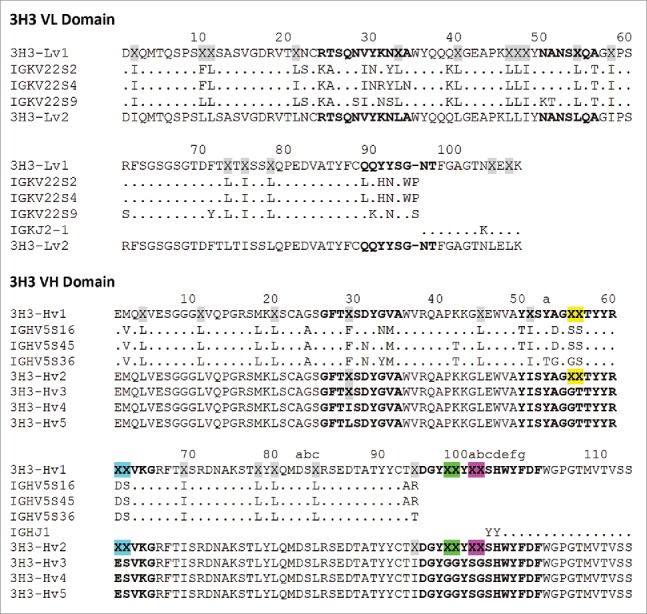
3H3 variable domain sequences described in this work. 3H3-Lv1/Hv1 represent the initial assembly of proteomic data into light and heavy V-domain sequences, where X represents ambiguous regions of sequence (gray highlighting = 113 Da; yellow = 158 Da; blue = 216 Da; green = 114 Da; magenta = 144 Da). An alignment of this draft to closest V and J translated germline gene segments is show below (a germline D-segment could not be assigned based on the initial draft sequence). 3H3-Lv2/Hv2 were refined draft sequences, as described in Table 1, and ambiguous regions in this light/heavy pair were programmed into 2 Fab phage display libraries. Further refinement after phage display led to 3H3-Lv2/Hv3, and finally 2 unique light/heavy pairings (3H3-Lv2/Hv4 and 3H3-Lv2/Hv5) that were part of recombinant Fabs. Sequence numbering is according to Kabat et al.18 CDRs are shown in bold and were defined according to the sequence definition of Kabat et al.,18 except for CDR-H1, which is the combined sequence and structural definition.19
Resolution of draft sequence uncertainties by phage display
Having extracted as much sequence information as possible for LOB12.3 and 3H3 Fabs through analysis of the MS data and reconciling this with the putative germline sequences, we turned our attention to the undefined elements within the draft sequences. Although the masses of these elements were known and could be used to restrict the possible sequence solutions, the sheer number of these (particularly Leu/Ile uncertainties) and their dispersion across the light and heavy chain V-domains was such that construction of a combinatorial library to include all possible solutions would be technically challenging, if not impossible. To reduce the complexity of such a library, we fixed the sequence in many of these regions based on considerations that included homology to the putative germline sequences, whether the region was within or outside of a CDR, and overall complexity of the final library design. Most of the Leu/Ile uncertainties were assigned based on germline homology, though we also considered the chymotryptic data set as this protease shows partial cleavage at Leu residues, but rarely cleaves after Ile.11 We also assigned a 114Da motif in CDR-H3 of LOB12.3 as GlyGly based on high homology in this CDR to the germline D-segment IGHD1–6. These details are summarized in Table 1. Updated draft sequences, LOB12.3-Lv2/Hv2 and 3H3-Lv2/Hv2 (Fig. 3 and 5), reflected these changes and the remaining sequence uncertainties were then programmed into phage display libraries.
Table 1.
Ambiguous regions of sequence and strategy for resolving these.
| Sequence region | Mass (Da) | †Possible Sequence(s) | Fix or randomize sequence? | Rationale |
|---|---|---|---|---|
| 3H3-VL: 2, 10, 33, 40, 46, 48, 58, 75, 78 3H3-VH: 4, 45, 51, 69, 80 LOB12.3-VL: 2, 15, 18, 21, 46, 48, 54, 73, 75, 83, 85, 106 LOB12.3-VH: 4, 20, 45, 78, 80, 82c |
113 |
Leu or Ile |
Fix |
Fix to Leu or Ile according to germline homology |
| 3H3-VL: 11, 21, 47, 54, 73, 104, 106 3H3-VH: 11, 20, 78, 82c LOB12.3-VL: 11, 47, 78, 104 LOB12.3-VH: 11, 18 |
113 |
Leu or Ile |
Fix |
Fix as Leu based on germline homology & chymotryptic cleavage at these positions |
| LOB12.3-VH: 98-99 |
114 |
GlyGly or Asn |
Fix |
Fix as GlyGly as “YGGYSE” motif is exact match to +1 translation of IGHD1-6 D-segment germline |
| LOB12.3-VL: 29, 33, 89 LOB12.3-VH: 51 |
113 |
Leu or Ile |
Randomize |
Although germline homology could have been used to fix as Leu or Ile, these site were randomized given the CDR location and low overall complexity of LOB12.3 library |
| 3H3-VH: 29, 94 LOB12.3-VH: 28, 56, 100c |
113 |
Leu or Ile |
Randomize |
Localized in or near CDR. Germline homology non-guiding, or not applicable. Randomize to Leu/Ile in library (in case of 3H3-VH29, Leu and Ile were encoded in two separate libraries) |
| 3H3-VH: 55-56 | 158 | AlaSer, SerAla, GlyThr, ThrGly | Randomize | Localized in CDRs. Germline homology non-guiding, therefore randomize in library |
| 3H3-VH: 61-62 | 216 | GluSer, AspThr | Randomize | |
| 3H3-VH: 98-99 | 114 | GlyGly or Asn | Randomize | |
| 3H3-VH: 100a-100b | 144 | SerGly or GlySer | Randomize |
Based on mass constraints and germline homology considerations.
Phage-displayed Fab libraries were generated encoding variants of the LOB12.3v2 and 3H3v2 sequences diversified according to the information shown in Table 1. The displayed Fabs were chimeric in nature, containing human kappa and IgG1 constant regions fused to the rat V-domains. A single LOB12.3v2 library was generated containing Leu/Ile diversity at 3 sites in the light chain, and another 4 in the heavy chain. Two 3H3v2 libraries were generated containing the same diversity at 5 distinct sites in the heavy chain, but differing from each other in their sequence at VH29 (Leu or Ile). Libraries were panned against recombinant murine CD137-Fc fusion protein for 3 rounds, and all 3 libraries showed marked enrichment based on phage titers after each round of selection. Individual clones were sequenced after the 2nd and 3rd rounds, leading to identification of clearly defined sequence preferences at many of the randomized positions (Table 2). A lack of consensus was observed at some of the randomized positions, suggesting that antigen binding was negligibly affected by the alternative sequences encoded at those positions within the libraries.
Table 2.
Distribution of sequences following 2 (R2) or 3 (R3) rounds of phage display selection.
| Sequence region | †Possible sequence(s) | Sequences at R2 | Sequences at R3 |
|---|---|---|---|
| *3H3-VH: 94 |
Leu or Ile |
15% Leu, 85% Ile |
4% Leu, 96% Ile |
| *3H3-VH:55-56 |
GlyThr, AlaSer, SerAla, ThrGly |
74% GlyThr, 16% AlaSer, 5% SerAla, 5% ThrGly ---------------------------- 28% GlyThr, 14% GlyAla, 14% GlySer, 11% ThrSer, 7% ThrThr, 6% AlaSer, 5% AlaAla, 5% SerThr, 2% AlaThr, 2% ThrAla, 2% SerSer, 2% SerAla, 2% ThrGly |
49% GlyThr, 31% AlaSer, 20% SerAla ---------------------------- 19% GlyThr, 19% ThrSer, 15% GlyAla, 15% ThrThr, 12% AlaSer, 8% SerAla, 8% GlySer, 4% ThrAla |
| *3H3-VH: 61-62 |
GluSer, AspThr |
94% GluSer, 6% AspThr ---------------------------- 46% GluSer, 45% AspSer, 6% GluThr, 3% AspThr |
95% GluSer, 5% AspThr ---------------------------- 58% AspSer, 40% GluSer, 2% AspThr |
| *3H3-VH: 98-99 |
GlyGly or Asn |
100% GlyGly |
100% GlyGly |
| *3H3-VH: 100a-100b |
SerGly or GlySer |
95% SerGly, 5% GlySer ---------------------------- 39% SerGly, 31% SerSer, 29% GlyGly, 2% GlySer |
100% SerGly ---------------------------- 54% SerSer, 27% SerGly 19% GlyGly |
| LOB12.3-VL: 29 |
Leu or Ile |
14% Leu, 86% Ile |
0% Leu, 100% Ile |
| LOB12.3-VL: 33 |
Leu or Ile |
39% Leu, 61% Ile |
36% Leu, 64% Ile |
| LOB12.3-VL: 89 |
Leu or Ile |
90% Leu, 10% Ile |
100% Leu, 0% Ile |
| LOB12.3-VH: 28 |
Leu or Ile |
28% Leu, 72% Ile |
18% Leu, 82% Ile |
| LOB12.3-VH: 51 |
Leu or Ile |
31% Leu, 69% Ile |
0% Leu, 100% Ile |
| LOB12.3-VH: 56 |
Leu or Ile |
22% Leu, 78% Ile |
18% Leu, 82% Ile |
| LOB12.3-VH: 100c | Leu or Ile | 57% Leu, 43% Ile | 64% Leu, 36% Ile |
Frequency of sequences satisfying MS-data and germline homology constraints are shown. Consensus sequences are shown in bold, and represent positions where >70% of clones had an identical sequence within these constraints. For completeness, where the library encoded additional sequences outside of these constraints, the full set of sequence distributions is shown in italic text.
Based on MS-data constraints and germline homology considerations.
As both 3H3 libraries (which differed only at VH29) gave similar sequencing outputs after CD137 selection, the combined data are represented in the table.
Binding characteristics of reverse-engineered LOB12.3 and 3H3 Fab variants
Based on the selection outputs from phage display libraries, we further refined the draft sequences of LOB12.3 and 3H3 by incorporating the consensus residue(s) identified after panning (Fig. 3: LOB12.3-Lv3/LOB12.3-Hv3; Fig. 5: 3H3-Lv2/3H3-Hv3). For this purpose, we defined consensus to mean those randomized positions where >70% of the mass compatible clones had the same sequence after 2 or 3 rounds of panning (Table 2). In the revised draft sequences, 2 ambiguous Leu/Ile positions remained within the sequence of LOB12.3 at VL33 and VH100c. For 3H3, sequences at each of the randomized sites had consensed after antigen binding selections, and only VH29 was considered ambiguous as the separate libraries encoding Leu and Ile at this position had both shown strong enrichment upon CD137 selection. We looked within the pool of sequenced clones to identify whether we had a set of Fab variants that could be quickly analyzed by phage ELISA to confirm antigen binding and also compare the effect of Leu vs Ile at these remaining ambiguous positions (i.e., Leu and Ile-containing Fab variants at VL33 and VH100c in LOB12.3; VH29 in 3H3, and otherwise matching the sequences of LOB12.3-Lv3/Hv3 or 3H3-Lv2/Hv3). For LOB12.3, we identified clones coding for each of the 4 desired sequences. For 3H3, we identified a clone containing Ile at position VH29 (and otherwise matched to sequences 3H3-Lv2/Hv3), but not a corresponding clone encoding Leu at this position. However, we found a closely related pair of clones encoding either Leu or Ile at VH29, Asp at VH61 (instead of Glu), and otherwise matching the 3H3-Lv2/Hv3 sequences in the V-domains. We reasoned that comparison of these 3 clones would allow us to examine the effect of Leu vs Ile at position VH29, albeit in a VH-D61 background, and assess any difference in binding caused by Asp vs Glu at this position (though the similar frequency of GluSer and AspSer-containing sequences at VH61-62 in the library outputs suggested any difference would be minimal).
Using a competitive phage ELISA, we measured binding of the different LOB12.3 and 3H3 chimeric Fab variants to murine CD137 (Fig. 6A). In these assays, the set of 4 LOB12.3 variants exhibited similar binding affinities that were all within 2-fold of each other (IC50 values 2.0 – 3.9 nM). Similarly, the three 3H3 Fab variants also had binding affinities that were within 2-fold of each other (IC50 values 3.1 – 6.2 nM). These data indicated that the reverse-engineered sequences selected by phage display bound CD137 with high affinity, and that leucine vs isoleucine differences at positions VL33/VH100c in LOB12.3 and VH29 in 3H3 have negligible effect on the antigen binding affinity.
Figure 6.
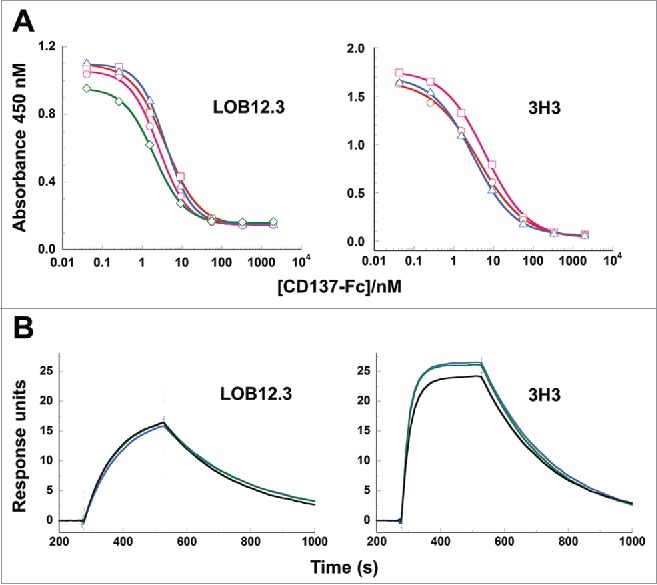
Binding of reverse-engineered LOB12.3 and 3H3 Fab variants to murine CD137. A) Competition phage ELISA measuring relative CD137 binding affinities to phage displayed LOB12.3 or 3H3 Fab variants. LOB12.3 Fab variants contained LOB12.3-Lv3/Hv3 light/heavy V-region pairs, where the sequence at VL33/VH100c was Leu/Leu (green), Leu/Ile (blue), Ile/Leu (magenta) or Ile/Ile (red). IC50 values for displacement of 50% of bound Fab-phage was between 2.0 – 3.9 nM for all 4 variants. 3H3 Fab variants contained 3H3-Lv2/Hv3 V-region pairs, where the sequence at VH29 was Ile (blue), or contained a VH-E61D substitution and Ile (red) or Leu (magenta) at VH29. IC50 values for displacement of 50% of bound Fab-phage was between 3.1 – 6.2 nM for all 3 variants. B) Sensorgram traces showing near identical binding kinetics for 11.1 nM injections of Fab samples over an immobilized CD137 surface. Reverse-engineered Fab variants (blue = Fab1; green = Fab2) are compared to the corresponding reference Fabs generated by partial proteolysis of the commercially-sourced parental IgG (black = Fabp). Binding kinetics calculated from the full set of surface plasmon resonance data are shown in Table 3.
Reverse-engineered 3H3 and LOB12.3 Fabs are functionally indistinguishable from parental fabs
As a final test of the functional equivalence of the reverse-engineered antibody sequences, we recombinantly expressed synthetic Fabs for comparison to reference Fab samples generated by proteolytic cleavage of the commercially-sourced IgGs. Two LOB12.3 Fab variants were expressed, differing only in having Ile or Leu at position VH100c. The paired V-domain sequences within these Fabs corresponded to LOB12.3-Lv4/Hv4 (LOB12.3-Fab1) and LOB12.3-Lv4/Hv5 (LOB12.3-Fab2); the full Fab sequences and MS data are shown in Fig. S1. Position VL33 was fixed to Leu in these 2 Fabs as this is the germline residue and the phage ELISA data (Fig. 6A) indicated equivalent binding affinity for the Ile-containing variants. For 3H3, 2 Fab variants were expressed that differed only in having Leu or Ile at position VH29. The paired V-domain sequences within these Fabs corresponded to 3H3-Lv2/Hv4 (3H3-Fab1) and 3H3-Lv2/Hv5 (3H3-Fab2), and the full Fab sequences and MS data are shown in Fig. S2. Recombinant Fabs contained the appropriate rat constant regions (Igκ/IgG1 for LOB12.3; Igκ/IgG2a for 3H3) and a C-terminal His tag on the heavy chain to facilitate purification. All recombinant samples, and the parental Fabs derived by proteolytic cleavage of IgG, underwent a final size-exclusion chromatographic purification step to ensure that the material used in binding studies was monomeric.
Measurement of Fab binding affinities was performed by surface plasmon resonance on a Biacore instrument, by injecting each Fab sample at a range of concentrations over a chip surface containing immobilized CD137-Fc. In this format, antigen binding was monovalent and free of avidity effects that could otherwise mask subtle differences in binding affinity. Each of the reverse-engineered Fabs had CD137 binding affinities that were nearly identical to the corresponding reference Fabs (Fig. 6B and Table 3), and can be considered equivalent within the margin of variability for KD determinations. Thus, the reverse-engineered Fabs for LOB12.3 and 3H3 faithfully reproduced the activity of Fabs generated from the commercial antibody samples. Consistent with the phage ELISA data, each pair of reverse-engineered Fabs for LOB12.3 and 3H3 had near indistinguishable affinities, which indicates that Leu and Ile perform equivalently at positions VH100c in LOB12.3 and VH29 in 3H3.
Table 3.
Binding affinities of reverse-engineered 3H3 and LOB12.3 Fab variants to CD137.
| Fab sample | ka (x 105M−1s−1) | kd (x 10−3s−1) | KD (nM) |
|---|---|---|---|
| LOB12.3-Fab1 (VH-I100c) | 4.29 | 3.68 | 8.60 |
| LOB12.3-Fab2 (VH-L100c) | 5.29 | 3.75 | 7.06 |
| LOB12.3-Fabp | 4.50 | 3.74 | 8.30 |
| 3H3-Fab1 (VH-I29) | 25.4 | 4.63 | 1.82 |
| 3H3-Fab2 (VH-L29) | 28.8 | 4.83 | 1.68 |
| 3H3-Fabp | 22.1 | 4.51 | 2.05 |
LOB12.3-Fab1, -Fab2 and 3H3-Fab1, -Fab2 were reverse-engineered Fab variants, while LOB12.3-Fabp and 3H3-Fabp were Fabs derived by partial proteolysis of the parental (i.e., commercially obtained) IgGs. Standard errors for fitting of data to a 1:1 binding model were less than one unit in the third significant digit for all computed ka, kd and KD values.
Discussion
In this work, we have demonstrated the value of combining a library display technology with MS-based protein sequencing methods to enable determination of the complete amino acid sequence of antibody V-domains. While MS-based sequencing is a powerful technique that can determine the majority of this sequence, there will always be uncertainties or gaps in the derived sequence due to limitations in this technology and the extensive natural diversity of antibody CDRs that cannot be captured by protein databases. For instance, distinguishing the isobaric isomers Leu and Ile is not possible in MS/MS data, except in some cases with specially designed fragmentation protocols that require MS instruments different to the one used in this work.12,13 Although these 2 residues are structurally similar, antigen binding may be affected if an incorrect assignment is made, particularly within a region that makes antigen contacts or can affect the folded antibody conformation. In our work, we found that 6 of 9 library positions encoded as Leu/Ile showed a marked preference for one or the other residue following antigen binding selection. Although we did not formally demonstrate this, such data is certainly suggestive of differences in binding affinity for Leu vs Ile at each of these positions. Similarly, incomplete fragmentation in MS/MS data can lead to short segments of unresolved CDR sequence, where multiple solutions can satisfy the available mass and (if applicable) germline homology constraints. Thus, phage or an alternative display technology is well suited to efficiently identifying the most appropriate candidate(s) for evaluation from this large pool of potential sequences.
While phage display technology enables generation and screening of up to 109 unique sequences, we generated small libraries that only randomized a limited number of unresolved sites within or adjacent to CDRs. For all other unresolved regions, we inferred the sequence based on mass constraints and homology to rat antibody germline genes. Striking an appropriate balance between programming unresolved regions into a library versus assigning these based on homology and mass constraints is an important, but subjective decision. Larger libraries may seem appealing, as it reduces the risk of making an incorrect assignment that affects binding, but the technical difficulty of constructing libraries escalates as the number and dispersion of randomized sites increases. It is also likely to increase the number of independent sequences that appear functionally equivalent. On the other hand, one can attempt to infer the sequence for most or all unresolved regions, such that one or a small number of antibody variants can be expressed without need for library generation. However, this approach increases the risk of making an incorrect assignment that results in a reverse-engineered product failing to accurately reproduce the activity of the parental mAb. This was the outcome experienced by Sousa et al.,6 notable as one of only 2 previous reports describing the reverse engineering of a mAb of unknown sequence, where the final product exhibited weaker binding activity than the parental antibody, despite several iterations of draft sequence correction and gene synthesis.
Ultimately, the complexity of a mAb protein sequencing project will be very dependent on the specific sequences under study. As found in this work and the work of others, regions encompassing CDRs are the most problematic, in particular CDR-H3 given the more extreme divergence from germline sequences due to junctional editing in combination with somatic hypermutation. We believe that the set of 4 digestions used in this work (trypsin, chymotrypsin, AspN + trypsin, GluC + trypsin) can produce a set of overlapping peptide fragments that suffices for sequencing of most V-domains, but recognize that some antibodies may present unique challenges. For example, the ability to generate small to medium sized proteolytic fragments for sequencing of CDR-H3 was greatly aided by the presence of 3 frequently conserved residues at or near the boundary of this CDR, namely Arg/Lys at VH94, Asp at VH101, and Trp at VH103 (trypsin, AspN and chymotrypsin cleavage sites, respectively). While Trp is >99% conserved at VH103, Arg/Lys at VH94 and Asp at VH101 are conserved less strongly (∼80% and 75%, respectively).14 Sequencing of CDR-H3 was more straightforward for a heavy chain containing all 3 of these conserved residues (as in LOB12.3), but the absence of one (as in 3H3) or both of these can substantially increase the difficulty. While phage or an alternative display technology could cope with resolving greater gaps in MS-derived sequence information than those encountered in this work, gaps that are greater than 3–5 contiguous residues could easily push this beyond the capability of these technologies, particularly if combined with additional sites of uncertainty.
In conclusion, we have developed an efficient method for reverse engineering of mAbs by combining phage display with de novo protein sequencing. We successfully applied this approach to derive the V-domain sequences for 2 commercially available mAbs of unknown sequence, and believe this strategy will be useful to others with a need to sequence antibodies in the absence of DNA material.
Materials and methods
Reagents
Rat anti-murine CD137 monoclonal antibodies 3H3 and LOB12.3 were purchased from Bio X Cell. Proteases (trypsin, chymotrypsin, GluC and AspN) were purchased from Promega, unless otherwise noted. Murine CD137-Fc fusion protein was obtained from R&D Systems, and all other biochemicals were from Sigma-Aldrich unless otherwise noted.
Fab cleavage and purification
Antibodies were cleaved into Fab/Fc regions using immobilized papain (Pierce) and SpeB (Genovis) following the manufacturers' protocols. Following cleavage, the reaction mixture was reduced with DTT to convert (Fab')2 to Fab, and this was purified by anion exchange chromatography on a QHP column (GE Healthcare), using a gradient between 10 mM Tris, pH 8.0 buffer and phosphate-buffered saline (PBS). Purified Fab was dialyzed first against PBS containing reduced and oxidized glutathione (1 mM each), and then oxidized glutathione (1 mM) to cap unpaired cysteines in the upper hinge region and prevent unwanted formation of (Fab')2 dimers. Cleaved Fabs were characterized by size-exclusion chromatography combined with multi-angle light scattering, and intact protein MS after reduction with 25 mM DTT in PBS for 30 min at 37 °C.
Protein digestion
Proteins were denatured and reduced by combining the protein in PBS with 2 volumes of 8 M guanidinium hydrochloride, 130 mM Tris pH 7.5, 25 mM DTT, and incubating for 30 minutes at 37 °C, followed by alkylation with 40 mM iodoacetamide for 30 minutes at room temperature. Proteins were then buffer exchanged to the digestion buffer by multiple rounds of concentration and addition using Amicon Ultra 0.5 mL 10 kDa concentrators (EMD Millipore). Digestions were performed under the following conditions: trypsin, 1:20 w/w trypsin:protein in 100 mM Tris pH 7.6, 2 M urea; chymotrypsin, 1:20 w/w chymotrypsin:protein in 100 mM Tris pH 7.6; GluC/trypsin 2:5:100 w/w/w GluC:trypsin:protein in 100 mM Tris pH 7.6 containing 0.8 M urea; AspN/trypsin 2:5:100 w/w/w AspN:trypsin:protein in 100 mM Tris pH 7.6 containing 2 M urea. Coverage of the V-domain sequences by peptide fragments generated in each of the 4 different digests is summarized in Fig. S5.
LC/MS and spectral interpretation
LC/MS/MS analyses were performed with an Acquity UPLC system equipped with a 1.0 × 100 mm CSH-C18 column (Waters Corporation), coupled to an Agilent 6520 Q-TOF mass spectrometer (Agilent Technologies). Digested Fab samples were separated using a 2–50% gradient over 27 minutes at a flow rate of 0.1 mL/min, where mobile phase A = aqueous 0.08% v/v formic acid, 0.02% v/v trifluoroacetic acid and mobile phase B = 0.08% v/v formic acid, 0.02% v/v trifluoroacetic acid in acetonitrile. MS spectra were acquired using a gas flow of 10 L/min at 300 °C, with the capillary set at 3500 V. MS/MS spectra were acquired using data-dependent acquisition, with isolation width set at medium, and the collision energy determined in a m/z and z-dependent method by the Agilent ramped formula with a slope of 2.93 and offset of 6.75 for +1 and +2 ions, and a slope of 3.6 and offset of −4.8 for ions of +3 and higher charge. Data were analyzed using vendor-supplied (Agilent v.B.04.00) MassHunter Qualitative Analysis software.
Database searching of spectral data was done using the X! Tandem algorithm,10 with user-defined databases containing either translated rat germline V-region genes, or a set of joined J-C gene products (see supplemental material). The V-region database contained translated sequences for all rat Vκ, Vλ, and VH genes defined as “functional” or “open reading frame” in the IMGT15 repertoire, which can be accessed online at http://www.imgt.org (136 Vκ, 8 Vλ and 138 VH sequences at the time of this work; database file provided in supplementary material). The J-C database was created by appending amino acid sequences of the germline-encoded rat J-segments (7 kappa, 4 lambda and 4 heavy chain J-segments) to the N-terminal end of all possible rat constant regions of the same type (i.e., 2 kappa constant regions, 4 lambda constant regions and 4 heavy chain IgG constant regions containing CH1-hinge-CH2-CH3). This led to 46 protein sequences in the J-C database (7×2 = 14 kappa J-C sequences; 4×4 = 16 lambda J-C sequences and 4×4 = 16 heavy chain J-C sequences; database file provided in supplementary material). Searches against the 2 databases were run separately, and only for the tryptic and chymotryptic datasets as this was sufficient to identify the light and heavy chain germline gene families for both mAbs, and help with confirmation of the suspected IgG subclass as described by the vendor. A summary of the output of these searches is provided in the supplementary material, and MS/MS spectra for all relevant peptide matches in the automated search were manually examined to assess accuracy.
Construction of phage display libraries and fab expression constructs
Synthetic gene sequences and oligonucleotides were ordered from GeneArt (Thermo Fisher Scientific), or Integrated DNA Technologies. To generate phage display constructs, synthetic genes encoding the draft VL and VH regions of LOB12.3 and 3H3 (LOB12.3-Lv2/LOB12.3-Hv2 and 3H3-Lv2/3H3-Hv2) were cloned into an in-house phage display vector to give plasmids which encoded chimeric rat V-domain/human constant region Fabs fused at the 3' end of the heavy chain gene to codons 249–406 of M13 gene III. Two such templates were generated for the 3H3 Fab, differing only in the codon for VH29 (Leu or Ile). Codons to be randomized within each Fab contained TAA or TGA stop triplets to ensure that only mutagenized sequences would encode full length open reading frames. Degenerate oligonucleotides were then used to randomly mutate target codons by site-directed mutagenesis according to the method of Kunkel et al.16 Oligonucleotides used in generating each Fab library were as follows (degenerate codons underlined):
LOB12-LC1: 5′-GTCAGGCAAGCCAGGATMTCGGTAA-TTGGMTCGCATGGTATCATCAGAAACC-3′
LOB12-LC2: 5′GATATCGGCATCTATTATTGTMTCCA-GGCAT-ATGGTGCACC-3′
LOB12-HC1: 5′-GTGCAGCAAGCGGCTTCMTCTTCAG-CTACTTTGATATGG-3′
LOB12-HC2: 5′-CTGGAATGGGTTGCAAGCMTCAGTC-CGGATGGTAGCMTCCCGTATTATCGTGATAGC-3′
LOB12-HC3: 5′-CTATGGTGGTTATAGCGAAMTCGATTATTGGGGTCAGG-3′
3H3-1: 5′-CATTAGTTATGCGGGCRSTRSTACCTATTA-CCGGGASWCTGTGAAGGGCCGTTTC-3′
3H3-2a: 5′-GCAACGTATTATTGTACCMTCGATGGTT-ATGGCGGTTACRGTRGTTCGCATTGGTATTTTG-ATTTC-3′
3H3-2b: 5′-GCAACGTATTATTGTACCMTCGATGGTT-ATAACTACRGTRGTTCGCATTGGTATTTTGATT-TC-3′
Libraries were electroporated into Escherichia coli XL-1 Blue cells (Agilent Technologies), and ∼100 clones were sequenced from each starting library to ensure adequate representation of the desired sequences at each randomized position. Methods for propagation of Fab-displaying phagemid particles, and antigen binding selections using murine CD137-Fc immobilized onto microtiter plates were as previously described.17
Binding and activity assays
The relative affinities of selected Fab variants for binding to CD137 were initially measured as gene III fusion proteins on the surface of phagemid particles using a competitive phage ELISA assay. Briefly, 96-well half area polystyrene high bind microplate plates (Corning) were coated overnight at 4 °C with 100 μL of 5 μg/mL murine CD137-Fc. Wells were blocked and washed, then serial dilutions of soluble CD137-Fc competitor and a subsaturating concentration of Fab phage were added in 25 μL of 1% w/v skim milk in PBS. After 2 h the plates were washed, and bound phagemid were labeled with anti-M13 monoclonal antibody-horseradish peroxidase conjugate (GE Healthcare), and assayed. Binding affinities (IC50) were calculated as the concentration of competing CD137-Fc required to reduce maximal phagemid binding by 50%.
To calculate equilibrium dissociation constants (KD) for binding of soluble Fab proteins to CD137, association and dissociation rate constants were measured by surface plasmon resonance on a Biacore T200 instrument (GE Healthcare). Mouse anti-human IgG mAb (Fcγ fragment specific, GE Healthcare) was covalently immobilized onto a CM4 biosensor chip via primary amino groups. Following capture of murine CD137-Fc onto the anti-human IgG surface, binding of Fab variants was measured by injecting samples diluted over the concentration range 0.137–100 nM in instrument running buffer (HBS-EP+ buffer, GE Healthcare) at a flow rate of 75 μL/min. Following each binding measurement, residual Fab and CD137-Fc were desorbed from the chip surface by injection of 50 μL of 3 M MgCl2 at 50 μL/min. Each binding measurement was performed in triplicate and binding profiles were analyzed by nonlinear regression using a simple monovalent binding model (Biacore T200 Evaluation Software version 2.0, GE Healthcare).
Expression and purification of recombinant rat fabs
Synthetic genes encoding light and heavy chain sequences (protein sequences shown in Fig. S1, S2) were cloned into separate mammalian expression constructs, both containing an upstream signal sequence and cytomegalovirus promoter element. LOB12.3 and 3H3 Fab variants encoded a rat kappa constant region in the light chain. Heavy chain constant regions (IgG1 for LOB12.3; rat IgG2a for 3H3 variants) were fused at the C-terminus to a His-tag motif to enable affinity purification of the assembled Fab product from culture medium. HEK293F cells were transiently co-transfected with the light and heavy chain-containing plasmids using 293fectin™ in serum-free Freestyle™ medium according to the supplier's recommended procedures (Invitrogen). Cell culture supernatants were harvested 6–7 days after transfection, filtered through a 0.22 μm sterile filter, and the His-tagged Fabs were purified in batch mode using Superflow Ni-NTA resin (Qiagen) according to the manufacturers' protocol. Eluted Fab proteins were analyzed by SDS-PAGE to estimate quantity and purity, and were then buffer exchanged into PBS by passing them over a Hiprep 26/10 desalting column (GE Healthcare) and characterized by intact protein MS (reduced and non-reduced) and size exclusion chromatography combined with multi-angle light scattering.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Qingwu Meng for assistance in performing some of the enzyme digests in this study.
References
- 1.Bandeira N, Pham V, Pevzner P, Arnott D, Lill JR. Automated de novo protein sequencing of monoclonal antibodies. Nat Biotechnol 2008; 26:1336-8; PMID:19060866; http://dx.doi.org/ 10.1038/nbt1208-1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castellana NE, McCutcheon K, Pham VC, Harden K, Nguyen A, Young J, Adams C, Schroeder K, Arnott D, Bafna V, et al.. Resurrection of a clinical antibody: Template proteogenomic de novo proteomic sequencing and reverse engineering of an anti-lymphotoxin-alpha antibody. Proteomics 2011; 11:395-405; PMID:21268269; http://dx.doi.org/ 10.1002/pmic.201000487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dekker L, Wu S, Vanduijn M, Tolic N, Stingl C, Zhao R, Luider T, Pasa-Tolic L. An integrated top-down and bottom-up proteomic approach to characterize the antigen-binding fragment of antibodies. Proteomics 2014; 14:1239-48; PMID:24634104; http://dx.doi.org/ 10.1002/pmic.201300366 [DOI] [PubMed] [Google Scholar]
- 4.Perdivara I, Deterding L, Moise A, Tomer KB, Przybylski M. Determination of primary structure and microheterogeneity of a beta-amyloid plaque-specific antibody using high-performance LC-tandem mass spectrometry. Anal Bioanal Chem 2008; 391:325-36; PMID:18369607; http://dx.doi.org/ 10.1007/s00216-008-1941-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pham V, Henzel WJ, Arnott D, Hymowitz S, Sandoval WN, Truong BT, Lowman H, Lill JR. De novo proteomic sequencing of a monoclonal antibody raised against OX40 ligand. Anal Biochem 2006; 352:77-86; PMID:16545334; http://dx.doi.org/ 10.1016/j.ab.2006.02.001 [DOI] [PubMed] [Google Scholar]
- 6.Sousa E, Olland S, Shih HH, Marquette K, Martone R, Lu Z, Paulsen J, Gill D, He T. Primary sequence determination of a monoclonal antibody against alpha-synuclein using a novel mass spectrometry-based approach. Int J Mass Spectrom 2012; 312:61-9; http://dx.doi.org/ 10.1016/j.ijms.2011.05.005 [DOI] [Google Scholar]
- 7.Castellana NE, Pham V, Arnott D, Lill JR, Bafna V. Template proteogenomics: Sequencing whole proteins using an imperfect database. Mol Cell Proteomics 2010; 9:1260-70; PMID:20164058; http://dx.doi.org/ 10.1074/mcp.M900504-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, Brown TJ, Emswiler J, Raecho H, Larsen CP, et al.. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med 1997; 186:47-55; PMID:9206996; http://dx.doi.org/ 10.1084/jem.186.1.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taraban VY, Rowley TF, O'Brien L, Chan HT, Haswell LE, Green MH, Tutt AL, Glennie MJ, Al-Shamkhani A. Expression and costimulatory effects of the TNF receptor superfamily members CD134 (OX40) and CD137 (4-1BB), and their role in the generation of anti-tumor immune responses. Eur J Immunol 2002; 32:3617-27; PMID:12516549; http://dx.doi.org/ [DOI] [PubMed] [Google Scholar]
- 10.Craig R, Beavis RC. TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 2004; 20:1466-7; PMID:14976030; http://dx.doi.org/ 10.1093/bioinformatics/bth092 [DOI] [PubMed] [Google Scholar]
- 11.Poston CN, Higgs RE, You J, Gelfanova V, Hale JE, Knierman MD, Siegel R, Gutierrez JA. A quantitative tool to distinguish isobaric leucine and isoleucine residues for mass spectrometry-based de novo monoclonal antibody sequencing. J Am Soc Mass Spectrom 2014; 25:1228-36; PMID:24845350; http://dx.doi.org/ 10.1007/s13361-014-0892-1 [DOI] [PubMed] [Google Scholar]
- 12.Armirotti A, Millo E, Damonte G. How to discriminate between leucine and isoleucine by low energy ESI-TRAP MSn. J Am Soc Mass Spectrom 2007; 18:57-63; PMID:17010643; http://dx.doi.org/ 10.1016/j.jasms.2006.08.011 [DOI] [PubMed] [Google Scholar]
- 13.Lebedev AT, Damoc E, Makarov AA, Samgina TY. Discrimination of leucine and isoleucine in peptides sequencing with orbitrap fusion mass spectrometer. Anal Chem 2014; 86:7017-22; PMID:24940639; http://dx.doi.org/ 10.1021/ac501200h [DOI] [PubMed] [Google Scholar]
- 14. http://www.bioinf.org.uk/abysis/searches/distributions/distributions.html abYsis, version 2.3.3. [Google Scholar]
- 15.Giudicelli V, Duroux P, Ginestoux C, Folch G, Jabado-Michaloud J, Chaume D, Lefranc MP. IMGT/LIGM-DB, the IMGT comprehensive database of immunoglobulin and T cell receptor nucleotide sequences. Nucleic Acids Res 2006; 34:D781-4; PMID:16381979; http://dx.doi.org/ 10.1093/nar/gkj088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kunkel TA, Bebenek K, McClary J. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol 1991; 204:125-39; PMID:1943776; http://dx.doi.org/ 10.1016/0076-6879(91)04008-C [DOI] [PubMed] [Google Scholar]
- 17.Baca M, Presta LG, O'Connor SJ, Wells JA. Antibody humanization using monovalent phage display. J Biol Chem 1997; 272:10678-84; PMID:9099717; http://dx.doi.org/ 10.1074/jbc.272.16.10678 [DOI] [PubMed] [Google Scholar]
- 18.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of Proteins of Immunological Interest. 5th ed Bethesda, MD: Public Health Service, National Institutes of Health, 1991 [Google Scholar]
- 19.Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol 1987; 196:901-17; PMID:3681981; http://dx.doi.org/ 10.1016/0022-2836(87)90412-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.