

ABSTRACT
Antibody-drug conjugates (ADCs) are of great interest as targeted cancer therapeutics. Preparation of ADCs for early stage screening is constrained by purification and biochemical analysis techniques that necessitate burdensome quantities of antibody. Here we describe a method, developed for the maytansinoid class of ADCs, enabling parallel conjugation of antibodies in 96-well format. The method utilizes ∼100 µg of antibody per well and requires <5 µg of ADC for characterization. We demonstrate the capabilities of this system using model antibodies. We also provide multiple examples applying this method to early-stage screening of maytansinoid ADCs. The method can greatly increase the throughput with which candidate ADCs can be screened in cell-based assays, and may be more generally applicable to high-throughput preparation and screening of different types of protein conjugates.
KEYWORDS: Antibody, antibody-drug conjugate, conjugation, high-throughput, maytansinoid, protein chemistry
Abbreviations
- Dimethylacetamide
DMA
- size-exclusion high performance liquid chromatography
SE-HPLC
- size exclusion ultra performance liquid chromatography
SE-UPLC
- N2′-deacetyl- N2′-(3-mercapto-1-oxopropyl) maytansine
DM1
- N2′-deacetyl-N2′-(4-mercapto-4-methyl-1-oxopentyl) maytansine
DM4
- DM1 methyl thioether, DM1-Me N-hydroxysuccinimide
NHS
- 4-(2-pyridylthio)butanoic acid NHS ester
SPDB
- 4-(N-maleimidomethyl)cyclohexane carboxylic acid sulfo-NHS ester
sulfo-SMCC
- N-ethylmaleimide
NEM
- ultraviolet-visible
UV-vis
- epidermal growth factor receptor
EGFR.
Introduction
Monoclonal antibodies targeting antigens found on tumor cells have elicited much interest as cancer therapeutics, but have historically shown moderate single-agent activity. Antibody-drug conjugates (ADCs) are hybrid biotherapeutics that combine the specificity of monoclonal antibodies with potently cytotoxic small molecules.1,2 Approved ADCs have shown significant single agent activity in the clinic against several cancers, as well as favorable safety profiles in comparison with traditional chemotherapy.3-5 Properly designed, ADCs retain the binding activity of the parent antibody – and thus any inherent anti-tumor activity – and add to it the cell-killing activity of the conjugated small molecules. The chemical linkage between antibody and small molecule is intended to remain intact while the ADC is circulating, with release only after receptor-mediated endocytosis and catabolism of the antibody.
A widely used and clinically proven ADC format utilizes thiol-functionalized maytansinoids as the effector molecules.4,6,7 Maytansinoids DM1 and DM4 (Fig. 1A) embody the desired attributes of ADC effectors: high cytotoxic potency, facile conjugation to antibody, and favorable biophysical properties of antibody conjugates incorporating them. They are linked to antibodies via heterobifunctional crosslinkers that form thioether or disulfide linkages to the maytansinoid and amide linkages to solvent-exposed lysine residues on the antibody.8 (Fig. 1A)
Figure 1.
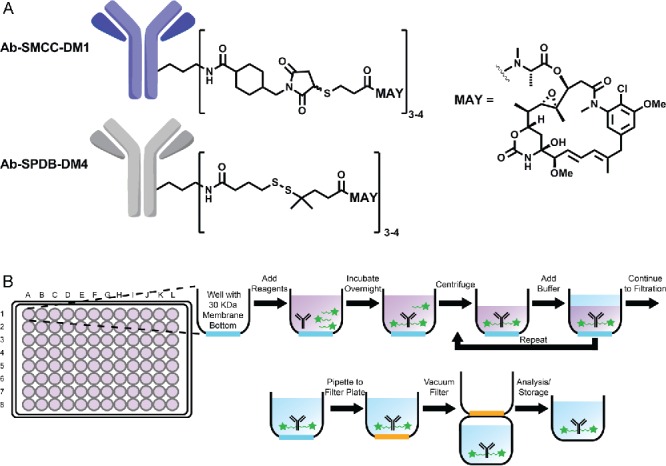
(A) Chemical structure of antibody-SMCC-DM1 and antibody-SPDB-DM4 conjugates. (B) Scheme of antibody conjugation described in this work.
Contemporary discovery of ADCs proceeds from identification of a molecular target to the generation of a panel of antibodies against it by suitable in vivo or in vitro methods.9 Such antibodies must then be screened for activity in the appropriate ADC format. Key attributes for the antibody portion of an ADC are less well-understood than for the effector, but they include specificity for target, high expression yield, and compatibility with the conjugation chemistry and effector molecule of choice. In addition, the final ADC must maintain favorable physicochemical properties (e.g., high affinity, low aggregate formation). Similarly, our understanding of the desired properties of ADCs is presently limited – in part due to the multiple internalization and processing steps involved in releasing the final cell-killing agent. As an example, specific high-affinity binding to target is necessary, but, even among multiple such clones, there may be significant differences in ADC activity both in vitro and in vivo.10 While the molecular bases for these differences remain poorly understood, it is reasonable to expect that differences in the precise binding mode (e.g., epitope, crosslinking) may lead to differences in biological fate (e.g., internalization into various compartments of the endocytic pathway). This has led to interest in combinations of unconjugated antibodies binding distinct epitopes of the same target,11-13 as well as identification of antibodies with improved internalization, either by screening14 or selection.15
Current antibody selection and purification techniques can provide large numbers of high affinity antibodies directed against a given target.16 Rapid selection of a lead ADC from among these candidates can be challenging because antibodies are time-consuming to produce and most existing conjugation techniques are low-throughput and material-intensive.
The two primary constraints driving conjugation throughput and antibody reagent consumption during ADC preparation are purification and characterization. For purification, many laboratories routinely use small gel filtration columns for early research scale conjugation of antibodies. While convenient, these columns suffer from fixed input volumes (typically ∼0.4 mL, corresponding to ∼1 mg or more of antibody), as well as high cost and very low throughput (maximum ∼20 in parallel, with the help of custom racks17). For characterization, ADCs are subject to a number of analyses (e.g., UV-vis, SEC, LC-MS) that require relatively large quantities of material – as much as 0.25 mg of ADC. Finally, replicate reactions are often carried out (for example, to titrate to a desired drug:antibody ratio (DAR)), which increases material requirements well above the 3 mg range. By contrast, the material needed to actually assay for cytotoxicity is typically well below 20 µg of ADC.
Use of isotype-specific secondary ADCs to “piggyback” the effector payload on an unmodified candidate immunoglobulin (for example, see ref 18) has served as a common shortcut. However, these secondary assays suffer inherent problems due to the dissociation of primary and secondary antibodies at low concentrations and the unknown effects of the secondary reagent on antibody internalization. They also provide no information on the properties of the eventual lead ADC, since no ADC is prepared.
To address this challenge, we describe here a platform for parallel preparation, purification and characterization of maytansinoid ADCs at the ∼100 µg scale in individual wells of a 96-well plate. The conjugates prepared can be assayed for various physicochemical properties, and then evaluated in cytotoxicity assays to rank antibody performance and to triage unstable or inactive candidate antibodies from the lead selection pool. Thus, this method both increases the throughput of evaluable antibodies and reduces the amount of reagent needed.
We established a single chromatographic assay to determine the essential physicochemical properties of each ADC (concentration, % monomer, DAR) using ∼5 ug of conjugate in <10 minutes. Our method allows antibody conjugation to be carried out at low concentrations (0.5-2 mg/mL) and small total quantities (50-200 µg). This enables screening of antibodies as direct conjugates following production at the ∼1 mg scale, increasing the breadth with which antibody space can be sampled for effective ADCs.
Using model antibodies, we show that this platform can generate well-characterized, specific, and potent maytansinoid ADCs. We also studied the effects of DAR on cytotoxic potency and show that, with properly designed cell assays, the DAR-potency relationship is linear over a wide range, reducing the need for DAR normalization (i.e., generating ADCs with a narrow final DAR range) to rank ADCs by potency. For cases where DAR normalization is desired, we also show that low pH conjugation can normalize the DAR, albeit with reduced ADC yields. These and other observations are presented in the context of case studies demonstrating the use of the platform for screening maytansinoid ADCs.
Results
Analytical methods
In order to rapidly characterize large numbers of maytansinoid ADCs, we established a size exclusion LC method using a narrow-bore, high-pressure UHPLC column (SE-UPLC). Chromatograms generated by SE-UPLC directly measure the level of soluble aggregate (or % monomer), as well as the DAR (by integration of the conjugate peak in the 252 and 280 nm channels).8,19 We found good correlation between % monomer and DAR as measured by SE-UPLC and standard ADC SE-HPLC methods (Fig. S1A, B; Table S1). Importantly, the SE-UPLC method can provide good data with as little as 3 µg of ADC and a run time of as little as 8 minutes, while the SE-HPLC methods require a minimum of ∼30 µg of material and a run time of 30 minutes. Furthermore, we found that the ADC concentration could be measured from the peak area in the 214 nm SE-UPLC chromatogram by fitting to a standard curve. Again, ADC concentration as well as DAR measured by SE-UPLC showed good correlation with measurements of identical samples made by UV-Vis spectrometry (Fig. S1C, Table S2).
Conjugation setup
An overview of the antibody conjugation platform is shown in Fig. 1B. Separate 200 µL conjugation reactions are set up in each well of a commercially available 96-well plate with a 30 kilodalton molecular weight cutoff filter at the bottom. Buffer and antibody are added to each well, followed by an in situ generated maytansinoid-linker-NHS ester (Fig. 1B), which reacts with primary amines of the antibody to give the final antibody-linker-maytansinoid conjugate. After the reaction is complete, unreacted small molecules are washed away by iterative rounds of centrifugation and buffer addition. The conjugated antibody product remains in the well throughout the ultrafiltration steps and is exchanged into the desired storage buffer. In preliminary experiments, the number of centrifuge/wash cycles was optimized by monitoring the average decrease in residual maytansinoid per wash cycle using an HPLC-based assay (Fig. S2) (see also the Cytotoxicity section below).
As an initial validation of the platform, we conjugated 3 well-characterized human IgG1 antibodies with 2 linker-payload formats (SMCC-DM1 and SPDB-DM4) at pH 8 in triplicate. The resulting ADCs were produced in good yields (60–80% for SMCC-DM1 and ∼50% for SPDB-DM4) and with >97% monomer (Fig. 2). The conjugations were also robust, in most cases showing little variation between replicates conjugated with the same quantity of in situ SMCC-DM1 or SPDB-DM4.
Figure 2.
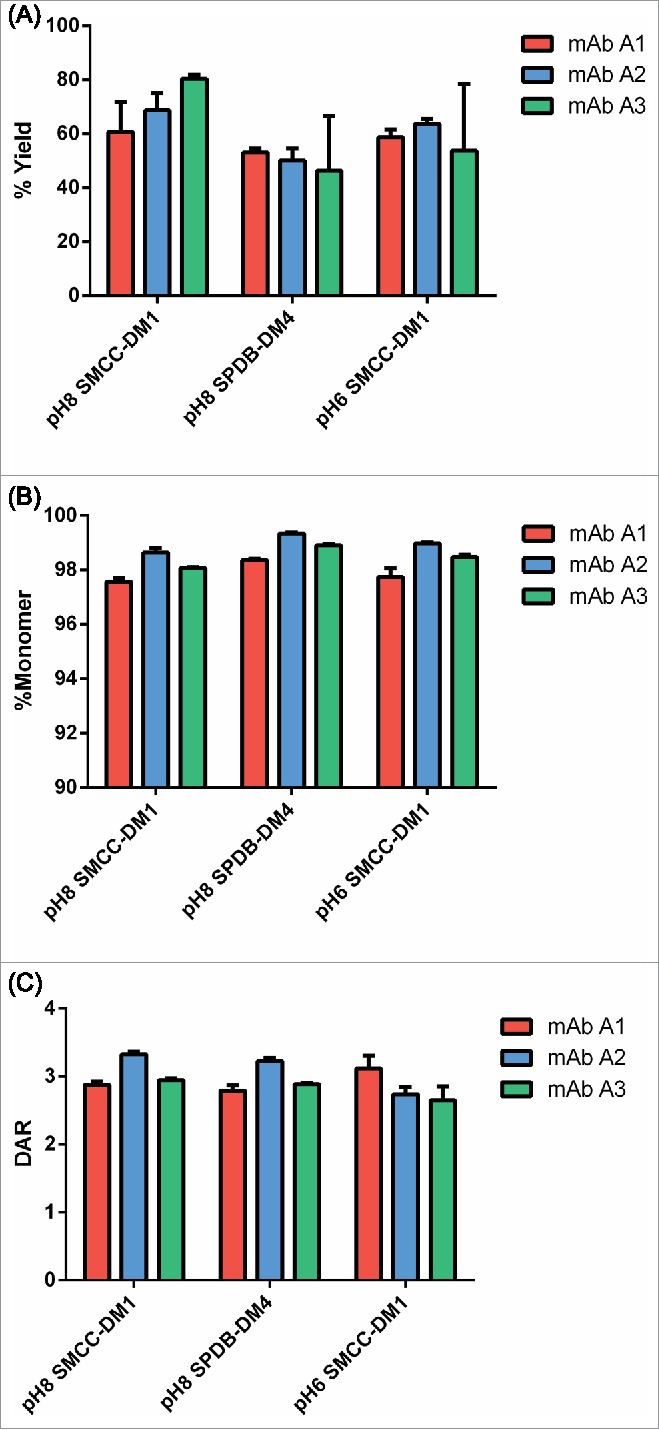
Initial qualification of conjugation using the microscale conjugation platform. Each of 3 antibodies was conjugated at microscale (3 replicates each, 100 µg per replicate) with SMCC-DM1 and SPDB-DM4 at pH 8, and with SMCC-DM1 at pH 6. (A) Yield as determined by volume measurement and concentration from SE-UPLC. (B) % Monomer and (C) DAR, each determined by SE-UPLC. Each bar is an average + SD of 3 replicates.
Titration curves relating antibody:SMCC-DM1 equivalency of the reaction to the DAR of the final ADC product were generated for each antibody conjugated (Fig. 3). These curves were also generated at 4 different antibody concentrations as part of the same simultaneous experiment. As expected, the curves were largely linear except at very high SMCC-DM1:antibody ratios (≥40 mole:mole). In every case, conjugates with DAR 3.0-4.0 could be generated for every antibody concentration tested.
Figure 3.
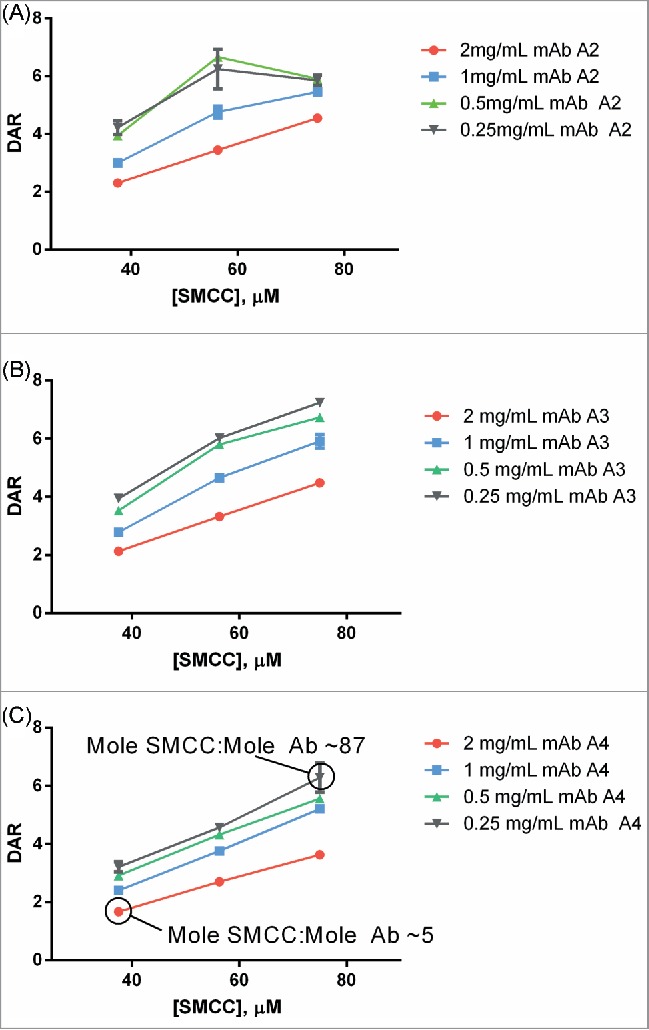
Example titrations of different human IgG1s with SMCC-DM1 using antibody stocks of different concentrations. Each point is the average of 2 replicate purified conjugation reactions. Each reaction contained 100 µL of antibody stock at the indicated concentration in a 200 µL reaction volume. Antibodies: (A) mAb A2; (B) mAb A3; (C) mAb A4.
Low pH conjugation
Standard antibody-maytansinoid conjugation protocols use pH 7–8 for lysine/NHS chemistry.20 This provides for rapid reaction of the NHS ester, although hydrolysis of the NHS ester also competes with protein modification. This also leads to relatively steep, linear curves for drug incorporation. For the current platform, we sought conjugation conditions that would reduce the sensitivity of the reaction conditions to the antibody:NHS ratio, leading to similar final DARs in multiple reactions with varying antibody concentration. This would be especially useful for large numbers of target antibodies, or batches of antibodies that are not normalized to the same input concentration. We observed that conjugation of standard antibodies at pH 6 with a large excess of in situ SMCC-DM1 resulted in a flattened DAR titration curve at different input antibody stock concentrations compared with conjugation at pH 8 (Fig. 4A). The exact mechanism for this is not immediately obvious, and is likely to be a complex combination of both the slower reaction rate between antibody lysines and NHS ester, and the decreased hydrolysis rate of the NHS ester. Antibody conjugations at pH 6 had not reached completion at ∼24 hrs, so reaction times were extended to this duration for conjugation under these conditions (Fig. 4B).
Figure 4.
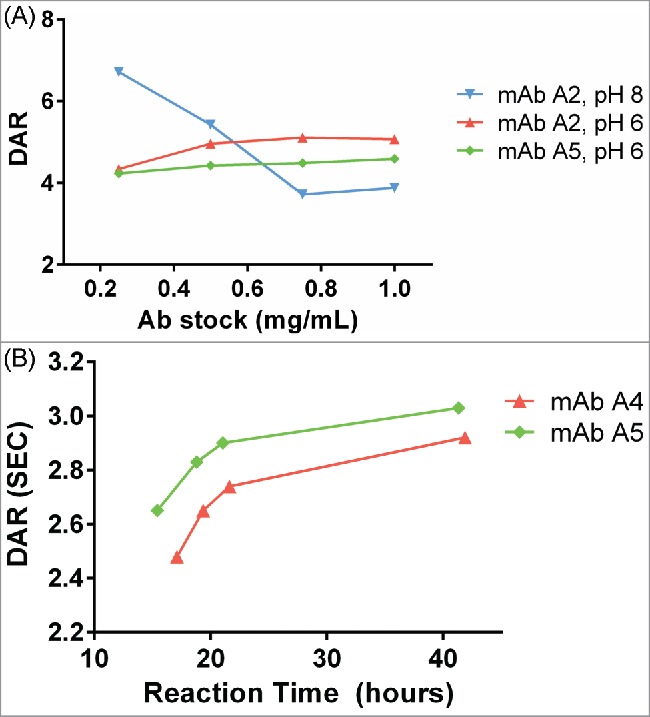
(A) Dependence of DAR on antibody concentration at pH 8 and pH 6. At pH 8, as antibody concentration increases, the DAR steeply decreases. At pH 6, the DAR is insensitive to antibody concentration changes. For pH 8, the concentration of SMCC-DM1 is 38 µM for all points; for pH 6, the concentration of SMCC-DM1 is 150 µM for all points. Each point is the average of 2 microscale reactions. (B) Reaction progress at pH 6 was measured by setting up a reaction in an autosampler vial and measuring the DAR of the crude reaction product by SE-UPLC at the indicated times.
Cytotoxicity
Antibody A2 recognizes the myeloid cell surface marker CD33, a widely used target for ADC therapy. Several test conjugates of Antibody A2 with SMCC-DM1 were produced at both pH 8 and pH 6 and purified in the 96-well format described. These conjugates were active in a cell-killing assay (IC50 ∼100-300 pM) against the CD33-positive MOLM-13 leukemia cell line (Fig. 5A), and the activity was similar to a conjugate produced at larger scale using conventional methods. The lack of non-specific cytotoxicity (as judged by cell killing in the presence of an excess of unconjugated antibody) also suggests that free maytansinoid species are effectively removed by the iterative ultrafiltration purification strategy. In order to examine this final point more closely, conjugates were prepared and purified to different extents (by removing from the plate after 3, 6, 9 and 12 washes). These partially purified conjugates were then assayed for activity on the CD33-negative JeKo-1 lymphoma cell line to measure the amount of non-specific cytotoxicity (Fig. 5B). After only 3 washes, some non-specific potency was observed; however, additional washes beyond 6 did not decrease non-specific cell killing.
Figure 5.
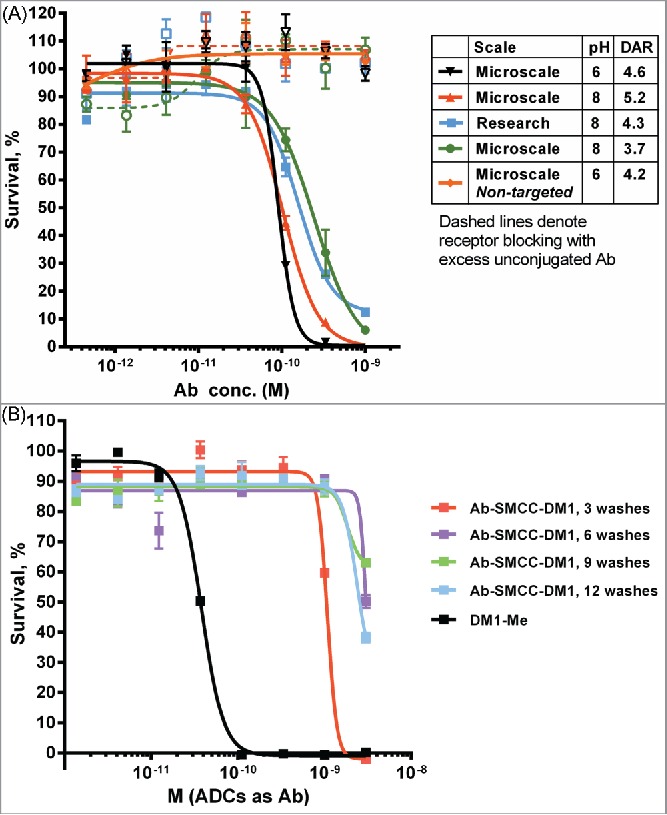
Cytotoxic potency of maytansinoid ADCs produced by microscale methods. (A) Microscale anti-CD33 conjugates produced at both pH 8 and pH 6 were compared with conjugate produced using standard methods were assayed for cytotoxic potency against MOLM-13 cells, both alone (solid curves) and in the presence of saturating unconjugated antibody (1 μM) to block specific uptake (dashed curves). (B) Cytotoxicity assay of identical mAb A2-SMCC-DM1 ADCs which were purified by the indicated number of wash steps. The cell line used (JeKo-1) does not express the antigen recognized by mAb 2, hence the cytotoxicity observed is due only to nonspecific uptake or cytotoxic impurities. Each wash consisted of the addition of fresh buffer and ultrafiltration. (C) mAb A6-SMCC-DM1 conjugates were generated with a range of DAR using microscale conjugation at pH 8. These conjugates were then assayed for cytotoxic potency on a high antigen (EGFR) density cell line (MDA-MB-483). The kill curves are plotted with antibody concentration as the X-axis. (D) The same data as in part C, but plotted with DM1 concentration as the X-axis.
Figure 5.
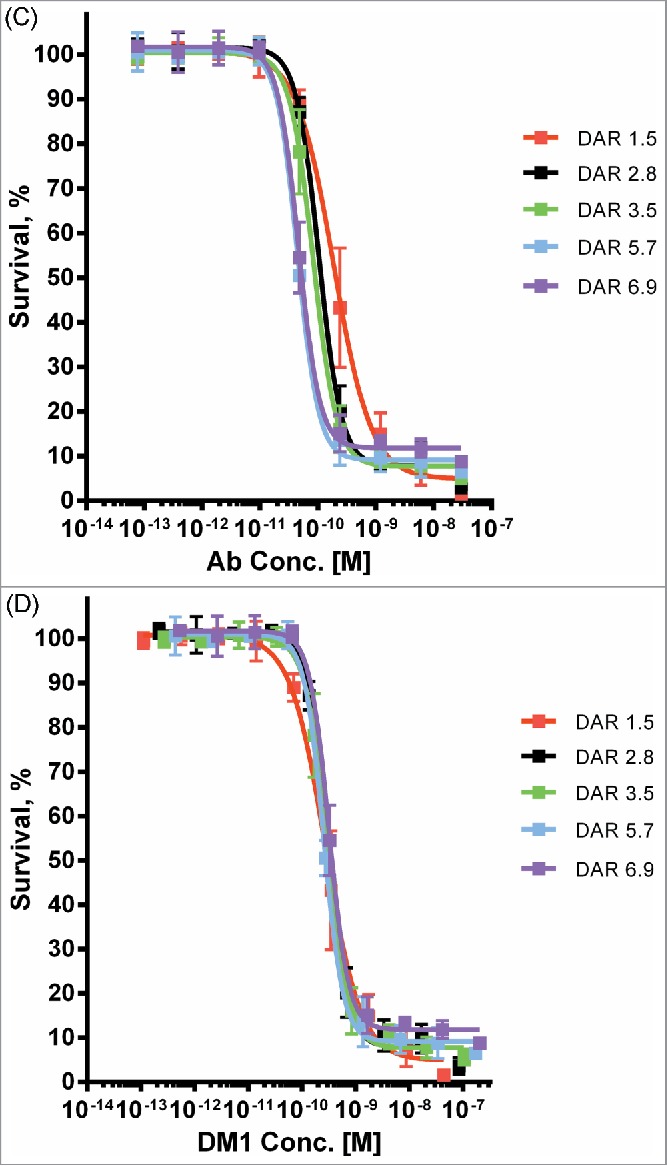
Continued.
Finally, to investigate the relationship between DAR and cytotoxic potency, we used microscale conjugation to generate a set of 5 SMCC-DM1 conjugates of ML66, an anti-EGFR antibody that lacks intrinsic pro-apoptotic activity, with DAR spanning 1.5 to 6.9. When assayed on a high-expressing EGFR+ cell line (MDA-MB-483), the cytotoxic potency of these ADCs showed a clear increase with increasing DAR (Fig. 5C), with the fitted IC50 values spanning about a 4-fold range (Fig. S3). Indeed, when cell killing is plotted as a function of the DM1 concentration (rather than the antibody concentration), the curves largely overlap (Fig. 5D).
Antibody lead triage
Proof-of-concept
As proof-of-concept, the microscale antibody-maytansinoid conjugation platform was used to examine a panel of 19 anti-Antigen B human IgG1 antibodies that were prescreened for favorable biophysical properties and produced at the 15-80 mg scale. The entire panel of 19 antibodies was conjugated with SPDB-DM4 at pH 8 using duplicate reactions each with 100 µL of antibody stock (100 µL corresponded to ∼150 µg). All 19 antibodies gave sufficient quantity of ADC for cytotoxicity assay, with the average DAR 3.1 ± 0.5 (see Table S3 for full conjugation results).
To compare ADCs produced at microscale with material from larger scale, a subset of 10 of the Antigen B targeting antibodies was conjugated with SPDB-DM4 using conventional research scale techniques (3-8 mg of input Ab). Simultaneous 6-day continuous exposure cytotoxicity assays were run comparing the potency of the microscale and research scale ADCs, using both antigen-positive and antigen-negative cell lines. Comparison of the targeted IC50s showed good agreement and Bland-Altman analysis21,22 measured the average bias between methods as 2.6-fold, within the practical range for cell-based screening assays (Fig. 6A, Fig. S4). Importantly, comparison of ranking of ADCs using these methods illustrates that research scale and microscale conjugates identify essentially the same set of lead ADCs (Fig. 6B).
Figure 6.
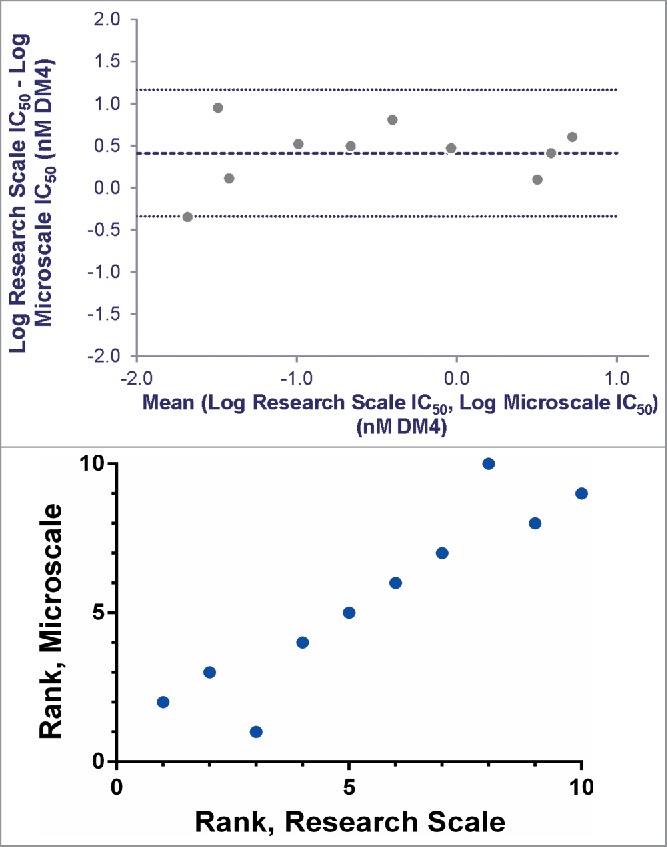
Agreement between IC50 values determined for research and microscale SPDB-DM4 ADCs targeting Antigen B. For each antibody, conjugates made by both methods were simultaneously assayed for cytotoxicity on cells expressing Antigen B. (A) Agreement between research and microscale ADCs depicted in a Bland-Altman plot comparing IC50 values in log space. The mean of the differences between assays = 0.41, so the bias is 100.41, corresponding to a 2.6-fold mean difference between microscale and research scale, with microscale appearing more potent. The standard deviation of the differences between assays = 0.38, so the 95% confidence interval of a pair of microscale and research scale IC50s = 100.76 = 5.6-fold above and below the mean. (B) ADCs targeting Antigen B were ranked based on cytotoxic IC50 using research scale and microscale conjugates, and the rank order is compared.
Use test with human antibodies
As a use test, 85 antibodies against Antigen C, which were not prescreened for any biophysical or biological properties, were expressed on the 1-2 mg scale. The entire library was conjugated with SMCC-DM1 using microscale conjugation.
As a broad test of the effects of conjugation at pH 6, the entire panel of 85 antibodies was conjugated with SMCC-DM1 at pH 6 with duplicate 100 µL reactions (antibody stocks ranged in concentration from 0.37-1.26 mg/mL). Analysis of the reaction products showed 24 conjugates were produced with >20 µg and monomer >90%. Of the 61 antibodies that did not meet these criteria, most (56/61) conjugated with inadequate yield. These 61 antibodies were re-conjugated at pH 8 using constant volumes of antibody stock (100 µL per reaction) and SMCC-DM1 (final concentration = 38 µM). This round of conjugation yielded 25 additional ADCs at >20 µg and DAR 2-6. Finally, the remaining ADC products from the first 2 rounds of conjugation were pooled and re-analyzed by SE-UPLC, giving an additional 15 ADCs meeting the yield, DAR and monomer criteria, for a combined success rate of 75% (64 out of 85 antibodies) (Table 1 and Tables S4A-C). The average DAR for the entire set was 3.4 ± 1.1.
Table 1.
Summary of conjugation reactions for 85 antibodies against Antigen C.
| Input antibodies | Conjugates* | Success rate | |
|---|---|---|---|
| First round (pH 6) | 85 | 24 | 28% |
| Second round(pH 8) | 61 | 25 | 41% |
| Two rounds pooled | 36 | 15 | |
| TOTAL | 85 | 64 | 75% |
54-252 ug of antibody used per round.
Success defined as DAR 2-6, monomer > 90%.
These 64 ADCs targeting Antigen C were assayed using a 5-day continuous exposure cytotoxicity assay in 384-well format. The cell line used was a high antigen density cell line against which unconjugated antibodies targeting Antigen C show no cytotoxic activity. As expected, the IC50 values spanned a broad range from ∼50 pM to ∼10 nM (based on DM1 concentration, (Fig. 7)). The lower value of 50 pM is similar to the IC50 for unconjugated cell-permeable maytansinoid species, and is therefore the expected maximal maytansinoid ADC potency for this cell line.
Figure 7.
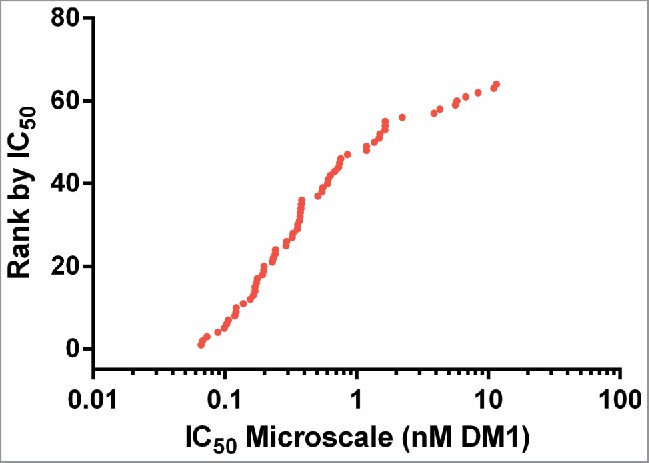
Cytotoxic potency of 64 anti-Antigen C antibodies as SMCC-DM1 conjugates.
Use test with murine antibodies
As an additional use test, the microscale conjugation method was used to test 17 murine antibodies (IgG1 and IgG2a isotypes) against Antigen D. These hybridoma-derived antibodies were not prescreened in any other fashion. In our experience, compared with human antibodies, murine antibodies exhibit lower yields and higher propensity to aggregate when conjugated with maytansinoids (unpublished results). This may be due in part to differences in isoelectric point or thermal stability. Initial attempts to conjugate murine antibodies at microscale with SMCC-DM1 at pH 6 were not encouraging due to low yields. Conjugation at pH 8 showed better yields, but lower success rates compared with human antibodies. Interestingly, the yields from successful murine conjugations were not drastically lower than for human antibodies, suggesting that the mechanism for higher failure rates might be related to some stochastic aggregation event during purification. Test antibody conjugations suggested a success rate of 1 in 5 reactions might be expected. Therefore, we attempted conjugation of these 17 antibodies at pH 8 using up to 6 replicate conjugations/antibody (97 total reactions). Gratifyingly, we were able to produce 15 out of 17 conjugates (88% success rate) with DAR between 3.0-4.0 (Fig. 8A and Table S5). Furthermore, the success rate was higher than expected, with most antibodies (65%) showing a success rate of 1 in 3 or higher. Analysis of these conjugates for binding and cytotoxicity showed that none lost the ability to bind antigen upon conjugation (Fig. S5) and that a range of cytotoxic IC50 values were again observed (Fig. 8B). As expected, no direct correlation between cytotoxicity and either Kd or antibodies bound per cell was observed for this set of antibodies, further highlighting the need for functional screens as part of ADC discovery campaigns.
Figure 8.
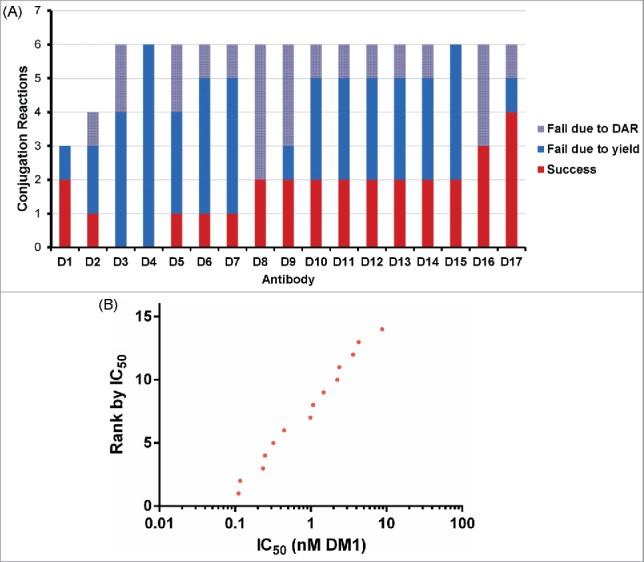
Conjugation and screening of murine antibodies targeting Antigen D. (A) Summary of 97 reactions to conjugate murine antibodies with SMCC-DM1 showing success/failure rates for conjugation of each antibody. (B) Cytotoxic potency of 14 anti-Antigen D antibodies as SMCC-DM1 conjugates.
Discussion
Maytansinoid ADCs have shown intriguing efficacy and safety in the clinic,4,23 leading to increased interest in applying this technology across a range of tumor antigens to produce novel cancer therapeutics. It is not readily obvious which antibodies will make potent ADCs, so improved techniques for conjugating antibodies are valuable for accelerating early lead screening. We therefore set out to develop a platform for direct antibody-maytansinoid conjugation with convenient reaction, purification, and analysis using a 96-well format.
We aimed to generate quantities of ADC sufficient for biochemical characterization and for multiple rounds of cell assays. After optimization of our SE-UPLC analysis method (requiring ∼5 µg of ADC) and factoring in material needed for cytotoxicity analysis (2-5 µg per cell line), the minimum goal was set at the production of 20 µg of ADC per reaction. Based on this, the working scale for conjugation was set at 50-150 µg of antibody per reaction. In order to accommodate these quantities and volumes of reagents, as well as the more dilute antibody stocks typically obtained from early stage expression and purification, we re-evaluated our conjugation strategy and established conjugation reaction conditions needed to give ADCs within a desired DAR range.
We experimented with various ADC purification strategies to find a method that could be readily implemented in a 96-well format. In our hands, high-throughput gel filtration chromatography did not adequately remove small molecule maytansinoid impurities. This criterion is critical, as such contaminants can be potently cytotoxic, and thus result in false positives when the resulting ADCs are screened in cell assays. By comparison, centrifugal ultrafiltration showed more variability in yields, but stringently removed small molecule impurities. We were able to optimize purification by ultrafiltration to give minimal non-specific cytotoxicity (Fig. 5B). In addition, our ADC panels are routinely assayed for cytotoxicity against antigen-negative cells sensitive to the maytansinoid payload as an additional control.
We have alluded to the importance of DAR as an ADC quality attribute and, in particular, for interpreting the potency ranking of candidate ADCs. For antibodies that lack intrinsic cytotoxic activity, we expect that cytotoxic potency will scale linearly with DAR because the amount of maytansinoid delivered will be equivalent to the number of internalization events factored by the DAR. Indeed, we find that this is the case for target antigens that lack intrinsic cytotoxic activity and cell lines with moderate to high antigen density (Fig. 5C).
When implementing a new method that replaces an existing one, a key question is whether the new method generates comparable data, leading to similar decisions. Our comparison between cytotoxic IC50 values of microscale and research scale ADCs targeting Antigen B (Fig. 6A) demonstrates the utility of this method for ranking candidate ADCs at the research stage. The forced rank comparison (Fig. 6B) further emphasizes that the same lead antibody pool is identified by both screens. Analysis of this limited data set (Fig. 6A, Fig. S4) does reveal a small bias (2.6-fold) toward higher potency in the microscale conjugates. Sources for this effect are unclear, but could include differences in purification, handling, or concentration measurements. Accounting for the observed bias, the data fall within a 95% confidence interval of 5.6-fold above and below the mean. This degree of variation would not lead to very different decisions on the ADCs to pursue, particularly at the early research stage, as demonstrated by the comparison of rank ordering by each method. Future efforts to refine the biochemical methods, as well as analyze larger data sets, may better characterize the differences between methods (both in statistical magnitude and in biochemical mechanism). Having demonstrated the practical utility of microscale conjugation, we propose that it can adequately substitute for research scale conjugation using the linker and effector combinations tested thus far.
Using the 85 antibody library against Antigen C, we tested the use of microscale conjugation to screen ADCs from non-prescreened antibody libraries. Although our initial titration screens indicated slightly lower but comparable yields at pH 6 vs pH 8 for test antibodies (Fig. 2), conjugation of this early stage library at pH 6 gave a lower success rate than at pH 8. It is unclear why this pH dependence is observed, but we suspect that antibody aggregation plays a role in the total yields observed for microscale conjugation, and the lower pH condition, combined with the presence of organic solvents and other reaction components, may subject antibodies to greater stress than pH 8. Given the variable tendencies of antibodies to aggregate,24 it is difficult to predict how general this effect would be. Ultimately, for the naïve library against Antigen C we achieved a conjugation success rate of 76%, enabling screening of a large number of unique ADCs against this target using <600 µg of each antibody as input.
Despite the need to carry out downstream humanization, murine antibodies continue to be a convenient tool for early stage antibody and ADC development. Therefore, we applied our current microscale platform to a panel of murine antibodies to test its performance. Again, we observe a wide range of cytotoxic potencies in the resulting panel of ADCs, and notably no loss of antigen binding following lysine conjugation. We suspect that conjugation of murine antibodies will generally require slightly higher input quantities of antibody compared with fully human antibodies. However, our example screen enabled 88% success rate for ADC screening using input of only 500-600 µg of murine antibody. The ease with which this new capability was added underscores the adaptability of the microscale conjugation method to quickly provide libraries of ADCs using small quantities of input antibody.
Our continued screening efforts using human IgG1 antibodies have focused on pH 8 conjugation with 2–4 replicate reactions per antibody (i.e., 200-400 µg of antibody) to generate 10-25 member panels of ADCs. In practice, the choice of experimental design (i.e., replicate numbers, reaction pH, scale) for a particular antibody panel and target is determined by multiple considerations, including the type of antibodies (e.g., murine vs. human), total size of the input antibody library, the quantity available of each library member, and the range of DAR tolerated in the final ADC panel. As noted above, murine antibodies require a greater number of replicates and thus more input material, but microscale conjugation still offers significant advantage over research scale techniques in terms of material use and throughput. The DAR range required largely depends on reagent availability and biology of the target antigen. If, as in the 3 cases presented, the cell line(s) used for screening are high in antigen density and do not demonstrate antibody activity, then a narrow DAR range may not be necessary due to the approximately linear relationship between DAR and cytotoxic IC50 (Fig. 5D). In cases where there is significant antibody activity, or a broader range of input antibody concentrations, then the DAR can be normalized either using low pH conjugation or by setting up multiple pH 8 reactions per clone (effectively titrating as shown in Fig. 3). As illustrated by the examples here, we have observed success rates of 70-100% for conjugation of antibody panels using less than 1 mg of input antibody (as low as ∼200 µg in some cases). We estimate an 70-90% success rate for research scale (5 mg input) conjugation of naïve hybridoma-derived antibody libraries, suggesting that the microscale ADC conjugation platform offers similar overall conjugation success rate with dramatically higher throughput and lower reagent demands. For example, the conjugation campaign carried out for Antigen C, consisting of 146 antibody reactions and 182 ADCs analyzed, was completed by one individual over the course of 12 normal working days.
Through the course of multiple screenings, we have observed a general agreement between yield and conjugate quality for micro- and research scale conjugations: some antibodies conjugated well at both scales, while the few available antibodies that conjugated poorly at research scale also conjugated poorly at microscale (data not shown). Future studies will more rigorously examine the ability of microscale conjugation to predict the compatibility of antibodies with conjugation reactions. Additional data on the effects of antibody prescreening on conjugation outcomes may also enable novel screening workflows that augment (or even replace) prescreening with microscale conjugation to enrich antibody libraries in members suitable for therapeutic development as ADCs.
Our methods constitute a substantial advance in screening and characterization of ADCs, as few descriptions of protein conjugation at this scale and throughput have appeared in the literature. Notably, Lyon et al. described a highly parallel ADC screening technique in a patent application, using conjugation of antibodies through reduced interchain disulfides with a mixture of maleimido-caproyl-monomethyl auristatin F (mcMMAF), Alexa 647-maleimide, and NEM as capping reagent.25,26 This platform uses hybridoma supernatants as inputs, so no quantitation of input antibody quantity or quality is made. Depending on the culture volume the resulting fluorescent ADC was used for a binding assay or for a 2-point cytotoxicity assay. By contrast, the method here is intended for use at a later stage of antibody screening, and so uses purified antibody as input and provides material sufficient for both a multi-point cytotoxicity assay and an SEC assay to assess conjugate aggregation.
The use of ultrafiltration for purification allows for stringent removal of small molecules independent of any tags or handles incorporated in the protein, allowing for direct screening of putative candidate macromolecules agnostic of their structural or sequence features. Zimmerman et al. also recently described an elegant screening approach that could be used to test mutant antibodies incorporating an azide amino acid for their reactivity in a strain-promoted azide-alkyne cycloaddition reaction, and for the cytotoxicity of the resulting MMAF conjugate.27 In principle, this platform provides relative uniformity in DAR due to the site-specific conjugation chemistry employed, and appears capable of impressive throughput. However, it is reliant on specialized robotic equipment, and the ADCs produced are both aglycosylated and His6-tagged, features which may decrease the generality of this approach.
Advances in cell handling and analysis techniques have allowed massively parallel screening of small molecules for a desired phenotype.28 Current techniques for antibody conjugation generally lack the throughput to fully leverage these capabilities. The microscale conjugation techniques described here begin to address this opportunity. Moreover, the past decade has seen increased interest in protein conjugation, with new and intriguing chemistry being developed29,30 as well as ambitious synthetic targets being pursued and produced.31,32 Some of these chemical approaches have been applied to ADCs by conjugating known small molecule effectors to IgG scaffolds,27,33,34 but given the diversity of chemistries, protein scaffolds35,36 and potential payloads,37 it seems likely that interest in generating and screening diverse types of conjugates (e.g., ref. 38) for therapeutic and diagnostic applications will increase in the coming years. Additional screening approaches could also be enabled by microscale conjugation of different payloads, such as fluorophores,39 to better characterize uptake of different binding agents or across different targets. Finally, we demonstrated the applicability of microscale conjugation to ADC lead selection, but it is reasonable to expect this may be applied to proof-of-concept studies for new targets or antibody/targeting protein formats, screening of conjugation reaction conditions, and, with integration of the proper biophysical instrumentation,40 for screening of formulation or developability41 of candidate ADCs at early stages.
In summary, we described the development and implementation of a 96-well plate based platform for conjugating antibody libraries in parallel at greatly reduced scale, and demonstrated its utility for screening the resulting maytansinoid ADCs. The platform can produce small quantities of ADC with quality matching that of larger-scale preparation methods, while enabling much higher throughput than conventional benchtop conjugation with comparable time and effort. Finally, the process is relatively simple to implement and requires little investment in specialized instrumentation.
Materials and Methods
Reagents and antibodies
For the proof-of-concept studies and for screening ADCs targeting Antigens B and C, all antibodies used were humanized IgG1 (ImmunoGen, Inc.; Novartis). For Antigen D, antibodies were murine IgG1 or IgG2a (ImmunoGen, Inc.).
Preparation of linker-drug adduct
Drug and linker were reacted prior to use in antibody conjugation. The final reaction conditions for sulfo-SMCC-DM1 adduct were 60% dimethylacetamide (DMA) (SAFC: W2750), 40% 50 mM sodium succinate pH 5.0 (Sigma: S2378), 1.5 mM sulfo-SMCC in DMA (Pierce: 22322), 1.8 mM DM1 in DMA. The DMA and 50 mM succinate pH 5.0 were combined first before adding the sulfo-SMCC and DM1 (ImmunoGen42). After 10 minutes, excess DM1 was capped by spiking 50 mM NEM in ethanol (Sigma: E1271; Pharmco-AAPER: 111000200) at 1% by volume.
The final reaction conditions for SPDB-DM4 conjugation were 60% DMA (SAFC: W2750), 40% 50 mM sodium succinate pH 5.0 (Sigma: S2378), 1.0 mM SPDB in DMA (ImmunoGen internal), 1.2 mM DM4 in DMA (ImmunoGen7). The DMA and 50 mM sodium succinate pH 5.0 were combined first before adding the SPDB and DM4.
Conjugation conditions
For pH 6 reactions using sulfo-SMCC-DM1, 100 μL of each antibody (0.25–1.0 mg/mL), 50 μL 0.2 M sodium phosphate pH 6 (BDH: 67447; Mallinckrodt Chemicals: G02672), 20 μL PBS (Gibco: 10010), 20 μL sulfo-SMCC-DM1 adduct, and 10 μL DMA were combined in a microtiter plate with a 30 kDa molecular weight cut-off (MWCO) membrane bottom (Pall: 5035).
For pH 8 reactions using sulfo-SMCC-DM1, 100 μL of each antibody (0.25–1.0 mg/mL), 50 μL 0.2 M sodium phosphate pH 8 (BDH: 67447; Mallinckrodt Chemicals: G02672), 25 μL phosphate-buffered saline (PBS) (Gibco: 10010), 5 μL sulfo-SMCC-DM1 adduct, and 20 μL DMA were combined in a microtiter plate with a 30 kDa MWCO membrane bottom (Pall: 5035 or 8035).
For reactions using SPDB-DM4, 100 μL of each antibody (0.25–1.0 mg/mL), 50 μL 0.2 M sodium phosphate pH 8 (BDH: 67447; Mallickrodt Chemicals: G02672), 25 μL PBS (Gibco: 10010), 5 μL SPDB-DM4 adduct, and 20 μL DMA were combined in a microtiter plate with a 30 kDa MWCO membrane bottom (Pall: 5035).
For reactions run under non-standard conditions, amounts of DMA and PBS were adjusted to maintain 10% total DMA and 200 μL total reaction volume.
After all of the reagents were combined, the plate was sealed (Whatman: 7704-0001) and vortexed to mix. Reactions were allowed to proceed for at least 18 hours at room temperature, or 24 hours in the case of pH 6 reaction.
Conjugate purification
To remove excess linker-drug from the reaction mixture at the end of the reaction time, the plate was placed on top of a deep well microtiter plate (Axygen Scientific: PDW20C) and spun in a centrifuge (1500 × g for 10 minutes) then 200 μL buffer was added to each well. These steps were repeated 5 more times increasing the spin time to 15 minutes.
After iterative centrifugations and buffer additions, the purified conjugates were moved to a microtiter plate with 0.2 μm membrane bottom (Pall: 5042). A vacuum manifold (Pall: 5017) was then used to filter the conjugates into a round bottom microtiter plate (Greiner Bio-One: 650261).
SEC analysis
Five microliters of each sample was injected onto an HPLC system (Agilent 1200 UPLC) equipped with an SEC column (BEH 200 Å, 4.6 × 150 mm column; Waters: 186005225) with a 0.3 or 0.35 mL/min flow rate using a 170.8 mM potassium phosphate (Sigma: P0662, Sigma: P3786), 212 mM potassium chloride (Sigma: P9541), 15% isopropanol (Fisher: BP26324), pH 7.0 mobile phase. Absorbances were measured at 214 nm, 252 nm, and 280 nm. The area under the curve (AUC) is used to calculate DARs as previously described.8 Concentration was extrapolated from the AUC of 214 nm using a standard curve (data not shown).
UV-Vis analysis
All spectrophotometric measurements were taken with an Agilent 8453 with absorbance at 600 nm subtracted as a baseline. DARs and concentrations were calculated as previously described.8
In vitro cytotoxicity assay
Cytotoxicity assays shown in Fig. 5A-C were carried out as previously described.43 For screening of ADCs against Antigen C, human cancer cells expressing Antigen C were cultured in EMEM medium (ATCC, Manassas, VA) supplemented with 10% fetal bovine serum, 10 µg/mL insulin (Sigma, St. Louis, MO), penicillin and streptomycin (Mediatech, Herndon, VA). Cells were plated on 384-well plates (Grenier Bio-One #781091, Monroe, NC), at a dilution of 400 cells in 22.5 µL growth medium. The cells were incubated for 16-24 h at 37°C, 5% CO2. ADCs were diluted 1:100 in growth medium, then diluted serially 1:4 and 2.5 µL added to cells in triplicate with a Beckman FX automated liquid handler. Plates were sealed with gas permeable plate seals (Thermo Fisher Scientific #241205, Waltham, MA) and cultured for 5 d at 37°C, 5% CO2. Cell viability was measured by addition of 12.5 µL CellTiter-Glo reagent (Promega #7573, Madison, WI) followed by shaking in the dark for 30 min. The luminescence intensity of the plates read on an Enspire 2300 multimode plate reader (Perkin Elmer, Shelton CT).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to acknowledge Erin K. Maloney, Laura Bartle, Olga Ab, Ling Dong, and Yulius Setiady for expert assistance with cell assays; and Thomas A. Keating for helpful discussions on the manuscript.
References
- 1.Chari RV, Miller ML, Widdison WC. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl 2014; 53:3796-827; PMID:24677743; http://dx.doi.org/ 10.1002/anie.201307628 [DOI] [PubMed] [Google Scholar]
- 2.Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med 2013; 64:15-29; PMID:23043493; http://dx.doi.org/ 10.1146/annurev-med-050311-201823 [DOI] [PubMed] [Google Scholar]
- 3.Teicher BA, Doroshow JH. The promise of antibody-drug conjugates. N Engl J Med 2012; 367:1847-8; PMID:23134386; http://dx.doi.org/ 10.1056/NEJMe1211736 [DOI] [PubMed] [Google Scholar]
- 4.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Dieras V, et al.. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 2012; 367:1783-91; PMID:23020162; http://dx.doi.org/ 10.1056/NEJMoa1209124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Younes A, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, Ramchandren R, Bartlett NL, Cheson BD, et al.. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin's lymphoma. J Clin Oncol 2012; 30:2183-9; PMID:22454421; http://dx.doi.org/ 10.1200/JCO.2011.38.0410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chari RV. Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc Chem Res 2008; 41:98-107; PMID:17705444; http://dx.doi.org/ 10.1021/ar700108g [DOI] [PubMed] [Google Scholar]
- 7.Widdison WC, Wilhelm SD, Cavanagh EE, Whiteman KR, Leece BA, Kovtun Y, Goldmacher VS, Xie H, Steeves RM, et al.. Semisynthetic maytansine analogues for the targeted treatment of cancer. J Med Chem 2006; 49:4392-408; PMID:16821799; http://dx.doi.org/ 10.1021/jm060319f [DOI] [PubMed] [Google Scholar]
- 8.Singh R, Erickson HK. Antibody-cytotoxic agent conjugates: preparation and characterization. Methods Mol Biol 2009; 525:445-67, xiv; PMID:19252846; http://dx.doi.org/ 10.1007/978-1-59745-554-1_23 [DOI] [PubMed] [Google Scholar]
- 9.Shih HH. Discovery Process for Antibody-Based Therapeutics In: Tabrizi MA, Bornstein GG, Klakamp SL, editors. Development of Antibody-Based Therapeutics. New York: Springer: 2012. pp. Nine-32. [Google Scholar]
- 10.Polson AG, Williams M, Gray AM, Fuji RN, Poon KA, McBride J, Raab H, Januario T, Go M, et al.. Anti-CD22-MCC-DM1: an antibody-drug conjugate with a stable linker for the treatment of non-Hodgkin's lymphoma. Leukemia 2010; 24:1566-73; PMID:20596033; http://dx.doi.org/ 10.1038/leu.2010.141 [DOI] [PubMed] [Google Scholar]
- 11.Baselga J, Gelmon KA, Verma S, Wardley A, Conte P, Miles D, Bianchi G, Cortes J, McNally VA, et al.. Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol 2010; 28:1138-44; PMID:20124182; http://dx.doi.org/ 10.1200/JCO.2009.24.2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Kasus T, Schechter B, Lavi S, Yarden Y, Sela M. Persistent elimination of ErbB-2/HER2-overexpressing tumors using combinations of monoclonal antibodies: relevance of receptor endocytosis. Proc Natl Acad Sci U S A 2009; 106:3294-9; PMID:19218427; http://dx.doi.org/ 10.1073/pnas.0812059106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koefoed K, Steinaa L, Soderberg JN, Kjaer I, Jacobsen HJ, Meijer PJ, Haurum JS, Jensen A, Kragh M, et al.. Rational identification of an optimal antibody mixture for targeting the epidermal growth factor receptor. MAbs 2011; 3:584-95; PMID:22123060; http://dx.doi.org/ 10.4161/mabs.3.6.17955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Owen SC, Patel N, Logie J, Pan G, Persson H, Moffat J, Sidhu SS, Shoichet MS. Targeting HER2+ breast cancer cells: Lysosomal accumulation of anti-HER2 antibodies is influenced by antibody binding site and conjugation to polymeric nanoparticles. J Control Release 2013; 172(2):395-404; PMID:23880472; http://dx.doi.org/ 10.1016/j.jconrel.2013.07.011 [DOI] [PubMed] [Google Scholar]
- 15.Zhou Y, Marks JD. Discovery of internalizing antibodies to tumor antigens from phage libraries. Methods Enzymol 2012; 502:43-66; PMID:22208981; http://dx.doi.org/ 10.1016/B978-0-12-416039-2.00003-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krebs B, Rauchenberger R, Reiffert S, Rothe C, Tesar M, Thomassen E, Cao M, Dreier T, Fischer D, et al.. High-throughput generation and engineering of recombinant human antibodies. J Immunol Methods 2001; 254:67-84; PMID:11406154; http://dx.doi.org/ 10.1016/S0022-1759(01)00398-2 [DOI] [PubMed] [Google Scholar]
- 17.Stefano JE, Busch M, Hou L, Park A, Gianolio DA. Micro- and mid-scale maleimide-based conjugation of cytotoxic drugs to antibody hinge region thiols for tumor targeting. Methods Mol Biol 2013; 1045:145-71; PMID:23913146; http://dx.doi.org/ 10.1007/978-1-62703-541-5_9 [DOI] [PubMed] [Google Scholar]
- 18.Kellner C, Bleeker WK, Lammerts van Bueren JJ, Staudinger M, Klausz K, Derer S, Glorius P, Muskulus A, de Goeij BE, et al.. Human kappa light chain targeted Pseudomonas exotoxin A–identifying human antibodies and Fab fragments with favorable characteristics for antibody-drug conjugate development. J Immunol Methods 2011; 371:122-33; PMID:21756911; http://dx.doi.org/ 10.1016/j.jim.2011.06.023 [DOI] [PubMed] [Google Scholar]
- 19.Wakankar A, Chen Y, Gokarn Y, Jacobson FS. Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs 2011; 3:161-72; PMID:21441786; http://dx.doi.org/ 10.4161/mabs.3.2.14960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hermanson GT. Bioconjugate Techniques, 2nd edition. London: Academic Press; 2008. [Google Scholar]
- 21.Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet 1986; 1:307-10; PMID:2868172; http://dx.doi.org/ 10.1016/S0140-6736(86)90837-8 [DOI] [PubMed] [Google Scholar]
- 22.Sun D, Whitty A, Papadatos J, Newman M, Donnelly J, Bowes S, Josiah S. Adopting a practical statistical approach for evaluating assay agreement in drug discovery. J Biomol Screen 2005; 10:508-16; PMID:16093560; http://dx.doi.org/ 10.1177/1087057105275725 [DOI] [PubMed] [Google Scholar]
- 23.Trneny M, Verhoef G, Dyer MJS, Yehuda DB, Patti C, Canales M, Lopez A, Awan F, Montgomery P, et al.. Starlyte Phase II study of Coltuximab Ravtansine (CoR, SAR3419) Single Agent: Clinical Activity and Safety in Patients with Relapsed/Refractory (R/R) Diffuse Large B-cell Lymphoma (DLBCL; NCT01472887). American Society for Clinical Oncology presentation. 2014. abstr. #8506. [Google Scholar]
- 24.Lowe D, Dudgeon K, Rouet R, Schofield P, Jermutus L, Christ D. Aggregation, stability, and formulation of human antibody therapeutics. Adv Protein Chem Struct Biol 2011; 84:41-61; PMID:21846562; http://dx.doi.org/ 10.1016/B978-0-12-386483-3.00004-5 [DOI] [PubMed] [Google Scholar]
- 25.Lyon RP, Benjamin DR, Ryan MC. Methods for Screening Antibodies. WO2011109308 A1. WIPO Patent Application. 2011. pp. PCT/US2011/026534. [Google Scholar]
- 26.Lyon RP, Ryan MC, Kostner H, Meyer EB, Sutherland MS, Yu C, Gordon KA, Benjamin DR. Development of Parallel Conjugation and Assay Methodologies to Screen for Antibodies with Optimal Properties for use as Antibody-Drug Conjugates; 2010; Washington, DC: abstr. #4394. [Google Scholar]
- 27.Zimmerman ES, Heibeck TH, Gill A, Li X, Murray CJ, Madlansacay MR, Tran C, Uter NT, Yin G, et al.. Production of site-specific antibody-drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconjug Chem 2014; 25:351-61; PMID:24437342; http://dx.doi.org/ 10.1021/bc400490z [DOI] [PubMed] [Google Scholar]
- 28.Michael S, Auld D, Klumpp C, Jadhav A, Zheng W, Thorne N, Austin CP, Inglese J, Simeonov A. A robotic platform for quantitative high-throughput screening. Assay Drug Dev Technol 2008; 6:637-57; PMID:19035846; http://dx.doi.org/ 10.1089/adt.2008.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stephanopoulos N, Francis MB. Choosing an effective protein bioconjugation strategy. Nat Chem Biol 2011; 7:876-84; PMID:22086289; http://dx.doi.org/ 10.1038/nchembio.720 [DOI] [PubMed] [Google Scholar]
- 30.Spicer CD, Davis BG. Selective chemical protein modification. Nat Commun 2014; 5:4740; PMID:25190082; http://dx.doi.org/ 10.1038/ncomms5740 [DOI] [PubMed] [Google Scholar]
- 31.McGinty RK, Kim J, Chatterjee C, Roeder RG, Muir TW. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature 2008; 453:812-6; PMID:18449190; http://dx.doi.org/ 10.1038/nature06906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Popp MW, Dougan SK, Chuang TY, Spooner E, Ploegh HL. Sortase-catalyzed transformations that improve the properties of cytokines. Proc Natl Acad Sci U S A 2011; 108:3169-74; PMID:21297034; http://dx.doi.org/ 10.1073/pnas.1016863108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Fang T, Boons GJ. Preparation of well-defined antibody-drug conjugates through glycan remodeling and strain-promoted azide-alkyne cycloadditions. Angew Chem Int Ed Engl 2014; 53:7179-82; PMID:24862406; http://dx.doi.org/ 10.1002/anie.201402606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drake PM, Albers AE, Baker J, Banas S, Barfield RM, Bhat AS, de Hart GW, Garofalo AW, Holder P, et al.. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug Chem 2014; 25:1331-41; PMID:24924618; http://dx.doi.org/ 10.1021/bc500189z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jost C, Pluckthun A. Engineered proteins with desired specificity: DARPins, other alternative scaffolds and bispecific IgGs. Curr Opin Struct Biol 2014; 27C:102-12; PMID:25033247; http://dx.doi.org/ 10.1016/j.sbi.2014.05.011 [DOI] [PubMed] [Google Scholar]
- 36.Byrne H, Conroy PJ, Whisstock JC, O'Kennedy RJ. A tale of two specificities: bispecific antibodies for therapeutic and diagnostic applications. Trends Biotechnol 2013; 31:621-32; PMID:24094861; http://dx.doi.org/ 10.1016/j.tibtech.2013.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderl J, Faulstich H, Hechler T, Kulke M. Antibody-drug conjugate payloads. Methods Mol Biol 2013; 1045:51-70; PMID:23913141; http://dx.doi.org/ 10.1007/978-1-62703-541-5_4. [DOI] [PubMed] [Google Scholar]
- 38.Dirksen A, Madsen M, Dello Iacono G, Matin MJ, Bacica M, Stankovic N, Callans S, Bhat A. Parallel synthesis and screening of peptide conjugates. Bioconjug Chem 2014; 25:1052-60; PMID:24824568; http://dx.doi.org/ 10.1021/bc500129w [DOI] [PubMed] [Google Scholar]
- 39.Yang J, Chen H, Vlahov IR, Cheng JX, Low PS. Evaluation of disulfide reduction during receptor-mediated endocytosis by using FRET imaging. Proc Natl Acad Sci U S A 2006; 103:13872-7; PMID:16950881; http://dx.doi.org/ 10.1073/pnas.0601455103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao H, Graf O, Milovic N, Luan X, Bluemel M, Smolny M, Forrer K. Formulation development of antibodies using robotic system and high-throughput laboratory (HTL). J Pharm Sci 2010; 99:2279-94; PMID:20014026; http://dx.doi.org/ 10.1002/jps.22008 [DOI] [PubMed] [Google Scholar]
- 41.Samra HS, He F. Advancements in high throughput biophysical technologies: applications for characterization and screening during early formulation development of monoclonal antibodies. Mol Pharm 2012; 9:696-707; PMID:22263524; http://dx.doi.org/ 10.1021/mp200404c [DOI] [PubMed] [Google Scholar]
- 42.Liu C, Tadayoni BM, Bourret LA, Mattocks KM, Derr SM, Widdison WC, Kedersha NL, Ariniello PD, Goldmacher VS, et al.. Eradication of large colon tumor xenografts by targeted delivery of maytansinoids. Proc Natl Acad Sci U S A 1996; 93:8618-23; PMID:8710920;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whiteman KR, Johnson HA, Mayo MF, Audette CA, Carrigan CN, LaBelle A, Zukerberg L, Lambert JM, Lutz RJ. Lorvotuzumab mertansine, a CD56-targeting antibody-drug conjugate with potent antitumor activity against small cell lung cancer in human xenograft models. MAbs 2014; 6:556-66; PMID:24492307; http://dx.doi.org/ 10.4161/mabs.27756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fleming MS, Zhang W, Lambert JM, Amphlett G. A reversed-phase high-performance liquid chromatography method for analysis of monoclonal antibody-maytansinoid immunoconjugates. Anal Biochem 2005; 340:272-8; PMID:15840500; http://dx.doi.org/ 10.1016/j.ab.2005.02.010 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.