

Researchers review how random changes and our environment (for example, diet) determines our life span.
Keywords: Epigenetics, aging, histones, chromatin
Abstract
Over the past decade, a growing number of studies have revealed that progressive changes to epigenetic information accompany aging in both dividing and nondividing cells. Functional studies in model organisms and humans indicate that epigenetic changes have a huge influence on the aging process. These epigenetic changes occur at various levels, including reduced bulk levels of the core histones, altered patterns of histone posttranslational modifications and DNA methylation, replacement of canonical histones with histone variants, and altered noncoding RNA expression, during both organismal aging and replicative senescence. The end result of epigenetic changes during aging is altered local accessibility to the genetic material, leading to aberrant gene expression, reactivation of transposable elements, and genomic instability. Strikingly, certain types of epigenetic information can function in a transgenerational manner to influence the life span of the offspring. Several important conclusions emerge from these studies: rather than being genetically predetermined, our life span is largely epigenetically determined; diet and other environmental influences can influence our life span by changing the epigenetic information; and inhibitors of epigenetic enzymes can influence life span of model organisms. These new findings provide better understanding of the mechanisms involved in aging. Given the reversible nature of epigenetic information, these studies highlight exciting avenues for therapeutic intervention in aging and age-associated diseases, including cancer.
INTRODUCTION
Aging is a complex multifactorial biological process shared by all living organisms. It is manifested by a gradual decline of normal physiological functions in a time-dependent manner. Organismal aging holds significant importance for human health because it increases susceptibility to many diseases, including cancer, metabolic disorders, such as diabetes, cardiovascular disorders, and neurodegenerative diseases (1–4). On the other hand, cellular senescence, also called replicative senescence, is a specialized process, considered to be a potential endogenous anticancer mechanism, during which there is irreversible growth arrest in response to potentially oncogenic stimuli (5). It is also considered a potential trigger to cause tissue remodeling during embryonic development and following tissue damage, which also requires a proliferation arrest (6). Cellular senescence bears many similarities to the aging process but also shows distinct features. Although the causes of aging are poorly understood, there are continued efforts to delineate longevity pathways conserved among all eukaryotes. In recent years, great strides have been made by numerous studies that efficiently categorized the cellular and molecular hallmarks of aging (7). Among these hallmarks, epigenetic alterations represent one crucial mechanism behind the deteriorated cellular functions observed during aging and in age-related disorders. By definition, epigenetics represents the reversible heritable mechanisms that occur without any alteration of the underlying DNA sequence. Although the chromosomes in our genome carry the genetic information, the epigenome is responsible for the functional use and stability of that valuable information; that is, it connects the genotype with the phenotype (8–11). These epigenetic changes can either be spontaneous or driven by external or internal influences. Epigenetics potentially serves as the missing link to explain why the pattern of aging is different between two genetically identical individuals, such as identical twins, or, in the animal kingdom, between animals with identical genetic makeup, such as queen bees and worker bees (1, 12, 13). Although longevity studies on the human population have shown that genetic factors could explain a fraction (20 to 30%) of the differences observed in life spans of monozygotic twins, the majority of the remainder of variation is thought to have arisen through epigenetic drift during their lifetime (9, 10, 12, 14, 15). Similarly, different environmental stimuli, including diet, cause differential alterations of stored epigenetic information to create a striking contrast in physical appearance, reproductive behavior, and life span of queen and worker honeybees, despite their identical DNA content (16). In turn, the resulting variability in the pattern of epigenetic information within individual cells in the population during aging leads to transcriptional drift and genomic instability (Fig. 1). Being established by enzymes, epigenetic information is reversible. Hence, epigenetics holds great prospects for targeting by therapeutic interventions, as opposed to genetic changes, which are currently technically irreversible in humans. Accordingly, delineating and understanding the epigenetic changes that happen during aging is a major ongoing area of study, which may potentially lead the way to the development of novel therapeutic approaches to delay aging and age-related diseases.
Fig. 1. Overview of epigenetic changes during aging.
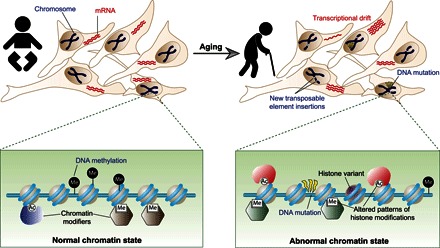
In young individuals, the cells within each cell type have a similar pattern of gene expression, determined in large part by each cell having similar epigenetic information. During aging, the epigenetic information changes sporadically in response to exogenous and endogenous factors. The resulting abnormal chromatin state is characterized by different histone variants being incorporated, altered DNA methylation patterns, and altered histone modification patterns, resulting in the recruitment of different chromatin modifiers. The abnormal chromatin state in old cells includes altered transcription patterns and transcriptional drift within the population. The abnormal chromatin state in old cells also leads to new transposable elements being inserted into the genome and genomic instability, including DNA mutations.
There are different types of epigenetic information encoded within our epigenome, including but not limited to the presence or absence of histones on any particular DNA sequence, DNA methylation, chromatin remodeling, posttranslational modifications of the histone proteins, structural and functional variants of histones, and transcription of noncoding RNAs (ncRNAs) (1, 2, 10). Together, these different types of epigenetic information comprise our epigenome and are important determining factors behind the function and fate of all cells and tissues, in both unicellular and multicellular organisms. Invariably, each of these different types of epigenetic information is functionally significant for the process of aging (Fig. 1). Growing evidence in recent years also clearly implicates chromatin structure, which carries much of the epigenetic information, as a major player during the aging process. The basic unit of chromatin structure is the nucleosome, which consists of 147 base pairs of DNA wrapped around a histone octamer that comprises two copies of each core histone protein, H2A, H2B, H3, and H4 (17, 18). The addition of linker histones, such as histone H1, and other nonhistone proteins, such as heterochromatin protein 1 (HP1), facilitates the formation of higher-order repressive chromatin structures, such as heterochromatin (19). Packaging of the genomic DNA into the highly organized chromatin structure regulates all genomic processes in the nucleus, including DNA replication, transcription, recombination, and DNA repair, by controlling access to the DNA (2).
Studies on the aging of mammals are rather limited by the long life span of the commonly used model organisms. Thus, both nonvertebrate and invertebrate organisms, with their shorter life span and ease of genetic and environmental manipulations, gained popularity among researchers in the aging field as experimental models for aging studies. Among them, budding yeast or Saccharomyces cerevisiae is a highly informative organismal model for aging studies with its genetic tools, short life span, and fully sequenced genome (20, 21). Despite being unicellular, yeast has been an excellent model to identify and characterize conserved basic biological processes, including aging. Yeast has been extensively used to identify genes and interventions responsible for life span extension and to gain insights into the aging processes of all eukaryotic organisms. In parallel, over the years, studies on invertebrate organisms, such as Drosophila melanogaster (flies) and Caenorhabditis elegans (worms), and certain vertebrate models, such as mice, zebrafish, naked mole rats, and, most recently, African turquoise killifish, have also provided invaluable information to help us understand the complexity of the process of aging and the influence of overlapping pathways on the outcome (22, 23).
Hutchinson-Gilford progeria syndrome (HGPS) and Werner syndrome are rare human genetic disorders characterized by premature aging phenotypes with a shortened life span. This group of diseases resembles physiological aging to a certain extent, serving as excellent models to gain insight into the biology of aging in humans (24, 25). These diseases are due to either a mutation in genes encoding the DNA repair machinery or the A-type lamin, leading to disorganized chromatin structures. The causative mutations behind these progeria syndromes indicate that genomic instability and chromatin deterioration are causes of human aging. Furthermore, the knowledge we gain from understanding the molecular pathology of these human premature aging diseases provides us with useful information to understand the complex aging process. Individuals with HGPS do not recapitulate all aging phenotypes because they usually show segmental progeria affecting multiple tissues. By recapitulating some molecular and cellular changes that are characteristics of the natural aging process, these models provide us with a unique opportunity to understand the aging process in a human model (24, 25).
Studies in humans and these powerful models of aging reveal that, similar to all other biological structures inside the cells, the epigenome suffers from a progressive loss in its configuration during aging. This results in a profound change in the chromosomal architecture, genomic integrity, and gene expression patterns (1, 10). Where examined, these effects are mostly conserved all the way from single-celled organisms, such as budding yeast, to complex multicellular eukaryotes. These conserved mechanisms help us gain a clearer picture of the aging process. Here, we will discuss the epigenetic changes that have emerged in recent years to significantly influence the aging process, from studies on single-celled model organisms to human models of aging. We will synthesize the recent developments in this area and highlight potential future directions.
THE HETEROCHROMATIN LOSS MODEL OF AGING
One of the earlier proposed models of aging was the “heterochromatin loss model of aging” (26, 27). This model suggests that the loss of heterochromatin that accompanies aging leads to changes in global nuclear architecture and the expression of genes residing in those regions, directly or indirectly causing aging and cellular senescence. As with any other model of aging, the heterochromatin loss model is supported by experimental data, but there are also confounding observations. Loss of transcriptional silencing due to decay of the heterochromatin occurs during aging in all eukaryotes examined from yeast to humans (26–30), and there is evidence that accelerating or reversing this process can either shorten or lengthen life span, respectively (Fig. 2). Gene silencing requires the absence of histone acetylation within heterochromatin regions. Accordingly, treatment with histone deacetylase (HDAC) inhibitors or deletion of genes encoding HDACs, such as yeast SIR2 or its sirtuin counterparts in metazoan species, shortens life span, whereas chemical activation or overexpression of SIR2 or sirtuins extends life span (31, 32). Yeast Sir2 was first recognized as an H4 K16Ac deacetylase, whereas mammalian SIRT1 is an H3 K9Ac or H4 K16Ac deacetylase (32). However, it is now appreciated that sirtuins deacetylate not only histones but also many other transcriptional regulators, and indeed, their roles in aging extend far beyond heterochromatin, including genome maintenance (33). It has been previously observed in budding yeast that redistribution of Sir proteins from the silent mating–type loci to sites of increased genomic instability during aging causes loss of silencing from heterochromatic mating–type loci to cause sterility and promote aging (34). Similarly, the mammalian homolog of Sir2, SIRT1, has been shown to repress repetitive elements and other regions across the mouse genome. However, in response to DNA damage, SIRT1 is redistributed from these loci to DNA breaks, resulting in alterations in gene expression that is comparable to those in the aging mice brain (33). Therefore, repression of heterochromatic repeat elements seems to be evolutionarily conserved and extremely critical in life span maintenance (33, 35). Another example where it is clear that loss of heterochromatin promotes aging is at the ribosomal DNA (rDNA) locus, where loss of silencing of the rDNA promotes genomic instability and aging in budding yeast (36, 37). Further evidence supporting the heterochromatin loss theory comes from the analysis of chromatin structures from HGPS patients and, most recently, from a Werner syndrome stem cell model of premature aging (10, 38–41). These disorders exemplify how deregulation of heterochromatin, illustrated by global loss of heterochromatin marks (discussed more below) and altered heterochromatin structure, accelerates aging. Although some of the defects observed in premature aging patients have also been observed in aged individuals (39), it is still debatable whether the findings are equally applicable to physiological aging.
Fig. 2. Alterations in chromatin structure during aging leads to activation of retrotransposons.
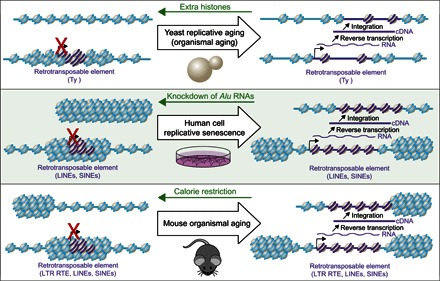
The schematic at the top depicts the reduction in the level of bulk histone proteins during aging (for example, as seen in budding yeast), leading to a more open chromatin structure and subsequent transcriptional activation of the normally silenced Ty elements. The resulting transcripts from the retrotransposable elements are reverse-transcribed into cDNAs that reinsert elsewhere into the genome of old cells. Overexpression of histones reverses the loss of histones, reduces Ty retrotransposition, and extends replicative life span. The schematic in the middle depicts the heterochromatin reorganization that occurs during aging, as seen, for example, in tissue culture cells during replicative senescence. The normally heterochromatinized retrotransposable elements become euchromatinized, leading to their transcription and transposition elsewhere into the genome. Knockdown of the Alu element transcripts in human adult stem cells leads to escape from senescence and reduced genomic instability. The schematic at the bottom depicts the heterochromatin reorganization that occurs in mouse tissues during aging and is accompanied by activation of retrotransposable elements (RTEs). Calorie restriction reduces the level of retrotransposition in the aged mice genome.
A rather important unresolved paradox for the heterochromatin loss model of aging was the observation of loss of heterochromatin during aging at the same time as the formation of senescence-associated heterochromatin foci (SAHF), which are domains of heterochromatin over proliferation-promoting genes in senescent cells (42). For a long time, SAHF have been perceived as another difference between the aging process and replicative senescence (43). However, very recently, global reorganization of genome architecture has been reported in senescent cells using Hi-C (43) and FAIRE (44) analyses, leading to a proposed two-step mechanism for the formation of SAHF. This two-step mechanism consists of heterochromatin decondensation in senescent cells, as seen during organismal aging, followed by the heterochromatinization of formerly euchromatin regions (Fig. 2) that spatially cluster to form the compact SAHF structures. This process has been referred to as heterochromatin redistribution (45) and provides a unifying theme to resolve the paradox.
GLOBAL HISTONE PROTEIN REDUCTION DURING AGING
Recently, the aging research field has experienced a leap from the earlier paradigm of the heterochromatin loss model. Not only is the heterochromatin reorganized during aging, but a global loss of core histone proteins from the genome during aging has also been observed in multiple scenarios, and this has been shown to be a cause of aging in yeast.
In budding yeast, replicative aging is accompanied by loss of approximately half of the core histone proteins (46, 47). The extensive nucleosome loss from the entire yeast genome during replicative aging (Fig. 2) results in global increased transcription in aged cells (48). These global changes in transcription and chromatin structure were only detectable because of the development of novel normalization approaches for genomic data sets (49). Notably, during yeast replicative aging, not only transcriptionally silent regions showed marked derepression but also all genomic regions showed transcriptional up-regulation, presumably because of increased access of the transcription machinery to the DNA sequences (48).
The drastic decline of the core histone proteins in S. cerevisiae is due to the reduced protein synthesis of the histones (Fig. 2) (47). Clearly, the fact that the levels of histone transcripts, like all other transcripts, actually increase during replicative aging in yeast (46, 47, 50) has no functional consequence because of the reduced histone protein synthesis in old yeast. Other cells may indeed use transcriptional regulation to reduce histone levels during aging because quiescent muscle stem cell aging is accompanied by reduced histone transcript levels (51). Supply of extra H3 and H4 histone proteins, either from an inducible promoter, by deleting the genes encoding the Hir repressor of histone gene transcription, or by deleting the gene encoding the protein Tom1 that is involved in the degradation of excess histone proteins, results in life span extension of yeast, identifying loss of histone proteins as a cause of aging (47, 52). The pathway of life span extension used by supplying extra histones appears to be independent from the pathway of calorie restriction (CR) in budding yeast (47), which is a widely accepted way to extend life span of seemingly all species (53). The life span extension that results from lithium exposure in worms is accompanied by increased histone gene expression (54), although it is not known whether the increased histone gene expression played a causative role in life span extension. The reduced bulk core histone protein levels observed during replicative aging is not restricted to budding yeast and has also been observed during aging in worms (55), during replicative aging of human diploid primary fibroblasts (56), and in senescent human cells (57). Notably, global histone loss has not yet been reported in mitotically dividing mammalian tissues in vivo. In human primary fibroblasts, the reduced synthesis of new histones during replicative senescence was a consequence of the shortened telomeres that activate the DNA damage response (56), potentially explaining the mechanism by which telomere shortening limits the number of cell divisions. Hence, loss of core histones may be a more generalized phenomenon observed with aging in many organisms.
Decreased levels of core histone proteins in aged cells would likely cause a marked effect on the chromatin landscape by providing inappropriate access to the genetic material. The consequences of having limiting amounts of histone proteins in the DNA during age progression have been elaborately examined in a recent global analysis of chromatin from aged cells in budding yeast, in comparison to the young rejuvenated population (48). Most of the nucleosomal positions are maintained in aged cells, whereas nucleosome occupancy is reduced to half the normal proportion and nucleosome locations on particular DNA sequences become less stringent or fuzzier. Additionally, partial overexpression of histone proteins H3 and H4 partially reversed the transcriptional defects observed during aging. It will be intriguing to investigate in the future whether the longevity effect conferred by supplying extra histones is equally pertinent in multicellular organisms.
GENOMIC INSTABILITY RESULTING FROM CHROMATIN RELAXATION DURING AGING AND THE “AGING BY TRANSPOSITION” MODEL
A more relaxed chromatin structure, due to heterochromatin loss or histone loss, is predictive of not only transcriptional deregulation (as discussed above) but also genome instability. Indeed, the reduced nucleosome occupancy in old yeast led to increased genomic perturbations, including higher levels of DNA breaks, damaged foci formation, inter- and intrachromosomal translocations, insertion of mitochondrial DNA into the nuclear genome, and high levels of retrotransposition (48). Notably, partial overexpression of histone proteins H3 and H4 partially reversed the transcriptional defects observed during aging and reduced retrotransposition, indicating that the increased retrotransposition in old yeast was a consequence of histone loss during aging (48). Increased levels of DNA breaks or unrepaired DNA damage, as illustrated by the formation of γ-H2AX foci (a hallmark of DNA damage), have been observed in aged cells from multiple species, including aged mice, senescent human cells, and cells derived from patients with premature aging disorders (58–61). This suggests that DNA damage may be a driving force behind organismal aging, although the full extent of the role of DNA damage during age progression still needs to be explored.
It is a long-standing belief that accumulation of DNA mutations over a period of time affects organismal aging because mutation accumulation happens as a function of age in different organisms, such as flies or mice and, in some cases, also correlates with life span (62, 63). However, a recent study indicated that accumulation of somatic DNA mutations, at least in budding yeast, does not have a causal role in aging (64). Therefore, it is yet to be conclusively determined whether the persistent DNA damage observed in aging cells is the result of the increased susceptibility of the accessible genome to accrue DNA damage with time or the inability of aged cells to efficiently repair the damage, or a combination of both. Notably, another recent analysis of the DNA repair transcriptome of liver in species with significant life span differences revealed that the longer-lived species express certain DNA repair genes at higher levels compared to species with shorter life span, suggesting a superior genome maintenance mechanism in the longer-lived species, which could potentially be responsible for their longer life span (65).
Another form of genomic instability observed during aging that results from heterochromatin decay is increased mobility of endogenous genetic elements, called retrotransposable elements (Fig. 2). Retrotransposable elements have been implicated in the aging process by several lines of evidence across organisms, including budding yeast, flies, worms, and mice, and even during cellular senescence in adult human stem cells (Fig. 2) (44, 48, 66–70). Retrotransposable elements are tightly silenced by heterochromatin in young cells or organisms. However, the loss of heterochromatin with aging leads to increased expression of otherwise silent retrotransposons, essentially causing transposition of these elements, because the transcripts are reverse-transcribed to make a genomic cDNA copy that gets integrated elsewhere into the genome (Figs. 1 and 2). The end result is increased mobility of retrotransposable elements within genomes, resulting in disruption of cellular homeostasis during aging (71). Molecular insight into this process was recently provided in studies of SIRT6 in mouse cells. While SIRT6 is best known for being a deacetylase for H3 K9Ac and H3 K56Ac (32), it also exhibits adenosine 5′-diphosphate (ADP)–ribosylase activity. Accordingly, SIRT6-mediated mono–ADP-ribosylation of KAP1 was shown to promote its interaction with HP1 and the packaging of L1 LINE elements into repressive heterochromatin (72). During aging, SIRT6 is sequestered away from the L1 elements to, presumably, DNA breaks elsewhere in the genome, resulting in the activation of the retrotransposons (72). Proof that the loss of chromatin-mediated silencing of retrotransposable elements during the course of aging directly leads to their increased transposition during aging came from the ability to repress Ty transcription and retrotransposition in old yeast by overexpressing histones (Fig. 2) (48). Life span–extending interventions, such as CR, have also been shown to counteract the increased expression of retrotransposons in aged mice (Fig. 2) (66).
The increased mobility of retrotransposons, observed in the genomes of aged cells and tissues from multiples species, provides strong evidence in favor of a recently hypothesized model of aging: aging by transposition (73). According to this model, these mobile elements and their transposases are the driving force to cause structural dysregulation of the genome to manifest aging phenotypes. Indeed, in an adult stem cell model of ex vivo aging, entry into senescence was accompanied by increased transcription from the SINE/Alu retrotransposable elements and persistent DNA damage foci (70). Experimental suppression of the transcripts from Alu elements reversed the arrested phenotype and eradicated the DNA damage foci, directly indicating that retrotransposon transcription was driving the entry into senescence (Fig. 2). Given the many recent links between activation of retrotransposable elements and aging, it is interesting to note that transposition also becomes more frequent during cancer development (74, 75), wherein aging is the highest risk factor for most cancers. Neurodegeneration, another disease of aging, is also characterized by increased retrotransposition (76, 77). Future studies will hopefully illuminate the full influence of transposition on the process of aging and the contribution of the age-dependent increase in transposition to these other human diseases.
Notably, the processes described in this section are unlikely to function independently from each other, and one event may lead to another during the aging process. For example, heterochromatin decay and histone loss lead to retrotransposition and changes in gene expression. But what causes the heterochromatin decay and block in histone protein synthesis during aging in the first place? We are currently limited in our knowledge of the sequence of the causal events during aging in healthy individuals. Attempts should be made in the near future to define the cascade of events during age progression to attain a comprehensive view of the aging process and to identify the initial causative events.
HISTONE VARIANT CHANGES WITH AGING
In addition to the canonical histone proteins H2A, H2B, H3, and H4, there are nonallelic variants of most of the histone proteins that are sometimes distinctly different in their primary sequence and are crucial for regulating chromatin dynamics by performing specialized functions (78, 79). Unlike core histone proteins, the age-associated changes observed for histone variants are more diversified and somewhat disparate, depending on which model organisms are studied. Histone H3.3 is a histone H3 variant that is incorporated into the genome in a replication-independent manner, in contrast to the replication-dependent histone variants H3.1 and H3.2 (80). H3.3 is expressed constitutively in metazoan cells, unlike the canonical H3.1 that is mainly expressed in the S phase. Therefore, it seemed rather predictable that H3.3 would play a key role in chromatin maintenance during senescence, when cells are no longer dividing. Indeed, H3.3 is the major form of histone H3 in the chromatin of senescent human cells, along with an N-terminally cleaved form of the H3.3 variant, which is termed H3.3cs1 (81). It appears that excess incorporation of H3.3 can drive senescence because ectopic expression of either H3.3 or H3.3cs1, but not H3.1, was able to induce senescence in the absence of any oncogenic stimuli (81). The induction of senescence was most robust for the cleaved variant H3.3cs1, revealing the importance of proteolytic processing of H3.3 during senescence. The deposition of H3.3cs1 onto DNA was mainly attributed to the histone chaperones ASF1a and UBN1 and putatively to HIRA in senescent cells (81). Notably, deletion of the yeast counterpart of HIRA, the Hir complex, extended replicative life span of yeast, but this is more likely due to the resulting increase in histone gene expression that counters the loss of core histones during aging (47). A similar correlation of increased H3.3 incorporation during aging has also been observed in the aged mouse brain, where H3.1 levels gradually decline with age and H3.3 gradually accumulates (82). The incorporation of H3.3 into actively transcribed DNA regions in neuronal cells and its efficient turnover in neuronal cells are crucial to maintain cell type–specific gene expression as well as neuronal plasticity and cognition (82). It appears that the histone variant H3.3 and its cleavage should be considered as important new players in maintaining functional chromatin structure during senescence.
There are multiple different variants of histone H2A, and one in particular has been implicated in aging. The H2A variant macroH2A is a prominent feature of the SAHF (83, 84). MacroH2A’s function is generally assumed to be transcriptional repression (85, 86). Although macroH2A has not been presumed to be important for the formation of SAHFs, it was suggested that macroH2A is crucial to maintain the transcriptional silencing at those sites (83, 84). The level of macroH2A increases in an age-dependent manner during replicative senescence in cultured human fibroblast cells and also in several tissues of aged mice and primates (87). More recently, enrichment of macroH2A1 at sites encoding the senescence-associated secretory phenotype (SASP) genes has been reported in response to oncogenic stimuli, promoting SASP transcriptional activation and propagating senescence through a positive feedback loop in a paracrine manner (88). The SASP leads to endoplasmic reticulum stress that triggers the production of reactive oxygen species. The subsequent DNA damage and activation of the ATM signaling pathway are important for the removal of macroH2A1 from the SASP genes, which results in their transcriptional repression. Hence, this study demonstrates a role for macroH2A1 in transcriptional activation. Basically, a massive redistribution of macroH2A1 occurs during senescence, with the macroH2A1 leaving the SASP genes and potentially relocalizing to SAHFs, along with other heterochromatin marks (88, 89). Hence, macroH2A1 appears to be a critical regulator of chromatin structure and chromatin dynamics during senescence.
HISTONE MODIFICATION CHANGES DURING AGING
The histone components of chromatin are subject to a wide variety of posttranslational modifications. Each modification, either alone or in combination with others, enables regulation of the use of the underlying DNA sequence. Functionally, histone modifications either disrupt the chromatin organization or provide new binding surfaces for recruitment of other proteins to specific regions of the chromatin. The type of posttranslational modification, the amino acid that is modified, and the other histone posttranslational modifications nearby determine their exact functional outcome. The functional complexity of the steadily rising number (currently well over 1000) of these histone modifications is yet to be fully understood (90–92). The presence of this vast array of modifications on histones orchestrates the functional responses, which are highly diversified. These range from regulation of gene transcription, DNA repair, DNA replication, effective condensation of chromatin, and many others, which affect almost all biological processes in eukaryotes, including aging. The histone posttranslational modifications are added and removed by enzymes, usually in a tightly regulated manner. However, during aging, the abundance, activity, and function of some of these enzymes change, leading to alterations in the epigenome.
Among the histone modifications that are known to affect the longevity process, the most prominent ones are acetylation and methylation of lysine residues. In budding yeast, acetylation present on the globular domain of histone H3 on lysine 56 (H3 K56Ac) and acetylation at the N-terminal tail of histone H4 on lysine 16 (H4 K16Ac) both influence replicative aging, although through distinct mechanisms. It seems that either the dynamic nature or a balanced level of each acetylation mark is crucial for the cell because either inability to acetylate these residues or the presence of mutations that mimic permanent acetylation reduces replicative life span of budding yeast (46, 47). Levels of H3 K56Ac decrease during yeast aging (46, 47). Meanwhile, deletion of the genes encoding the relevant HDACs that remove H3 K56Ac, Hst3, and Hst4 leads to a shortened yeast life span (46, 93) and genomic instability (46), including a high frequency of loss of heterozygosity (94). The notion that there is a precise level of H3 K56Ac for optimal life span is borne out by the fact that overexpression of Hst3 shortens life span, whereas addition of one extra gene copy of HST3 or HST4 extends life span (95). Similarly, deletion of the yeast histone acetyltransferase (HAT) that acetylates H3 K56Ac, Rtt109, or the histone chaperone Asf1, which assists in the acetylation, causes a shortened life span (47). Exactly how a slightly elevated level of H3 K56Ac promotes longevity, whereas too much or too little H3 K56Ac shortens life span, is not clear. However, H3 K56Ac has been implicated in driving chromatin assembly, transcriptional regulation of genes including the histones themselves, genomic stability, and DNA replication (96–98); therefore, any of these functions may be optimized with slightly more H3 K56Ac to promote longevity.
In contrast to the reduced levels of H3 K56Ac during aging, the levels of H4 K16Ac increase during yeast replicative aging (46). Similar changes in the levels of these two histone acetylation marks also occur during replicative aging in cycling human fibroblasts, where there is a substantial decrease in H3 K56Ac and an increase in H4 K16Ac in late-passage cells (56). The increased level of H4 K16Ac in old yeast cells is attributed to the progressive decline in levels of the relevant HDAC Sir2 during aging (46). Overexpression of Sir2 extends life span (99). In agreement, deletion of SAS2, which encodes the HAT that acetylates H4 K16, extends life span (46). Hence, the increase in H4 K16Ac that occurs during aging is driving aging, or pro-aging, whereas manipulations that reduce H4 K16Ac levels extend life span. The pro-aging influence of H4 K16Ac accumulation has been suggested to manifest through effects on the chromatin structure at the telomeres (46), which is reminiscent of the heterochromatin loss model of aging. Notably, this work (46) provided direct evidence for aging regulation through modification of H4 K16Ac by Sir2, the founding member of the sirtuin protein family, in addition to the already existing aging theories for this protein. The other modes by which Sir2 or sirtuins have been proposed to influence aging (responding to nutrient availability through inflammatory response, apoptosis, DNA repair, cell cycle, mitochondrial functions, etc.) will not be discussed here further because they are not directly related to epigenetics and are covered in other recent reviews (100, 101).
Other HATs and HDACs have also been implicated in aging. Deletion of a component of the NuA4 HAT extends yeast replicative life span (52). NuA4 acetylates histone H4 but also has several nonhistone substrates that may be relevant to its role in limiting longevity, such as Pck1p, which encodes a phosphoenolpyruvate carboxykinase enzyme, Tap42p, a putative downstream member in the mammalian target of rapamycin (mTOR) pathway, and/or Nnt1p, a putative nicotinamide N-methyltransferase (102). Deletion of genes expressing components of the histone H3 deacetylase complex Rpd3 extends replicative life span of yeast (103) and organismal life span of flies (104). Rpd3-mediated deacetylation appears to limit longevity through multiple pathways (105), highlighting the pleiotropic effects of altering levels of histone acetylation. Further studies are required to fully understand the molecular mechanisms used by these HATs and HDACs to influence life span.
Histone acetylation also appears to play an important role in the aging brain. Global histone hypoacetylation occurs in the repeated DNA elements in aged mice brain, which suggests a loss of chromatin integrity with aging (106). Aged mice brains also show memory impairment that has been proposed to be due to failure to up-regulate acetylation at histone H4 K12, which is a histone mark promoting transcription elongation, and due to the resulting altered gene expression pattern. Restoration of H4 K12 acetylation recovered both gene expression and learning behavior in aged mice, signifying the critical role of this histone acetylation in aged mice brains (107).
The trends in changed levels of histone methylation during aging have also been extensively examined recently. Significant changes during aging have been seen for trimethylation marks on histone H3 lysines 4, 9, 27, and 36 (H3 K4me3, H3 K9me3, H3 K27me3, and H3 K36me3, respectively), mostly as changes in the methylation marks that indicate loss of heterochromatic structure with aging. In parallel, reduction in HP1 levels was evident during aging in whole flies, with a decreased level of H3 K9me2 and altered levels of H3 K4me3 (29). However, when the profiles of the repressive histone marks were analyzed in aging fly heads, an increase in the level of H3 K9me3 was observed, signifying the importance of tissue-specific expression pattern changes during aging in multicellular animals (108). Histone methylation pattern analysis in aged SAMP8 (senescence-accelerated mouse prone 8) brain tissues revealed decreased H4 K20me1 and H3 K36me3 and an increase in H3 K27me3 (109). Fibroblast cells collected from individuals with HGPS showed decreased H3 K9me3 and H3 K27me3 with the expected reduction of HP1α (40), given that HP1 binds to H3 K9me3. There was also an accompanying increase in transcript levels from the pericentric satellite DNA (40). The same study also revealed an increase in the level of H4 K20me3, which serves as a signature epigenetic mark of constitutive heterochromatin, demonstrating a shift in the epigenetic landscape in HGPS patients (40). Although the global pattern of histone methylation differs in different organisms, presumably due to differential involvement of longevity pathways during the aging process, in general, the trend mostly reveals the increase in appearance of activating histone methylation marks and disappearance of repressive histone methylation marks during aging, signifying the loss of compact chromatin architecture in aging cells, tissues, or organisms (110). In recent years, some efforts have been made in either certain stem cell populations or tissue types to delineate the differences between young populations and the aged counterparts or to determine the regulatory mechanisms from high-throughput sequencing approaches, including chromatin immunoprecipitation–sequencing (ChIP-seq) and micrococcal nuclease–sequencing (MNase-seq) (51, 111–114). However, examination of bulk modification levels from cell populations or tissues misses important additional information on gene-specific effects that would hopefully be available soon from profiling histone modifications by ChIP-seq, eventually in individual cells, during aging.
Evidence of histone lysine methylation playing a causal role during aging comes from studies modulating the levels of histone methyltransferases and demethylases. RNA interference (RNAi) screens in C. elegans identified several histone methyltransferases, including ASH-2, SET-2, and SET-9, and several demethylases, including UTX-1, as potential factors influencing the outcome of aging (55, 115). Reduction in the level of H3 K4me3 methylation, which serves as a signature of active chromatin, by knockdown of the H3 K4 methyltransferase SET-2 or the components of the regulatory complex of H3 K4me3, ASH-2 and WDR-5, caused life span extension in fertile worms. In contrast, knockdown of the H3 K4me3 demethylase protein RBR-2 shortened life span of worms. On the other hand, overexpression of RBR-2 has also been shown to be sufficient for extending life span of worms (116). It is unclear how reduced levels of H3 K4me3 increase longevity, but it is evident that a limiting amount of this modification is favorable for optimal C. elegans life span (117). Consistent with the findings in C. elegans, flies deficient in Lid, the RBR-2 ortholog in D. melanogaster, have shortened life span (117, 118). However, this is only evident in male flies, implying that the effect of this demethylase may be sex-specific in some organisms. Alteration in the level of H3 K27me3 also influences life span of both worms and flies. Contrary to H3 K4me3, H3 K27me3 is a prominent mark of repressed chromatin. Attenuation of the H3 K27me3 demethylase UTX-1 in C. elegans extends life span through an insulin-dependent pathway (119, 120). The opposing effects of H3 K4me3 and H3 K27me3 on longevity are not surprising, given their opposing effects on transcription where they function as a toggle switch to determine developmental patterns of gene expression (121). However, high H3 K27me3 does not always extend life span. In D. melanogaster, evidence suggests that heterozygous mutations in components of Polycomb repressive complex 2 (PRC2), namely E(z) and Esc, decrease the global level of H3 K27me3 and extend life span in male flies (117, 122). These results reveal species- and sex-specific discrepancies and indicate that excess chromatin-mediated repression may sometimes be detrimental to life span.
Mechanistic insights into the role of histone modifications during aging have come from studies of H3 K9me3. Cells and tissues from a progeria mouse model had increased levels of H3 K9me3 and the methyltransferase SUV39H1, along with defective DNA repair within heterochromatin (123). Following DNA damage, compact chromatin structures undergo rapid chromatin remodeling to provide access to DNA repair factors. However, increased H3 K9me3 levels may possibly hinder the heterochromatin remodeling, leading to sustained DNA damage, early senescence, and a shortened life span (123). Indeed, depletion of the major H3 K9me3 methyltransferase SUV39H1 rescued the defect in DNA repair and premature aging, leading to an extended life span (123).
Most recently, H3 K36me3 has been found to promote longevity. A high-throughput histone mutation screen in S. cerevisiae for identifying mutants with altered life span pinpointed this modification (124). Whereas the methyltransferase mutant for H3 K36 (set2Δ) had a shortened replicative life span, the H3 K36 demethylase mutant (rph1Δ) had an increased life span (124). Reduction of H3 K36me3 during replicative aging in yeast leads to a more open chromatin conformation that exposes cryptic promoters, and the resulting cryptic transcripts may limit life span. Deletion of the relevant demethylase generates a more compact chromatin conformation, resulting in life span extension (124). Although this study also identified increased cryptic transcripts during C. elegans aging, it remains to be validated whether a similar mechanism also applies in mammals. An interesting correlation exists between the gene expression change during aging and the level of H3 K36me3 in gene bodies in C. elegans and D. melanogaster (125). The gene bodies of genes with a robust expression change during aging are marked with almost undetectable levels of H3 K36me3 in both organisms, suggesting a conserved mechanism for this modification in promoting longevity by preventing age-dependent changes in mRNA expression. Inactivation or depletion of the methyltransferase met-1 resulted in a global reduction of H3 K36me3 level, increased changes in gene expression during aging, and shortening of life span in C. elegans (125). Together, these studies suggest that loss of histone H3 K36 methylation during aging leads to aberrant gene expression, reminiscent of a transcriptional drift-like effect, which presumably limits life span. In this light, it is of interest that a recent report showed that pharmaceutical suppression of transcriptional drift extends C. elegans life span by delaying the onset of mortality (126). It is tempting to speculate that deterioration of the epigenome during aging is responsible for the transcriptional drift that causes the onset of mortality (Fig. 1).
Histone ubiquitination is another modification that has been implicated in yeast replicative life span. A genome-wide screen for long-lived strains in yeast identified components of the histone deubiquinase module (DUBm) of the SAGA (Spt-Ada-Gcn5-acetyltransferase) complex (127), including SCF73, the yeast ortholog of human Ataxin-7. This protein serves as an adapter protein in the complex, which links the core SAGA complex with DUBm. Although the complex contains both histone acetyltransferase and DUB activities, life span analysis of different mutants suggests that a specific reduction in DUB function may regulate life span in yeast because strains that lack components of DUBm showed marked life span extension. Life span extension was not evident in strains lacking the acetylation-specific SAGA components but was seen with yeast lacking Spt8 (52), which recruits the SAGA complex to promoters. Further analysis revealed that the life span extension observed in DUBm mutant strains is due to their involvement in multiple longevity pathways, including Sir2-dependent pathways (127). Histone ubiquitination regulates transcription and DNA damage response (128), and it will be interesting to determine which of these functions of histone ubiquitination is relevant to longevity. In the future, it will be important to determine whether the role of histone ubiquitination in aging observed in yeast also holds true for multicellular organisms.
NUCLEOSOME REMODELING AND AGING
Although the nucleosome structure itself is rather rigid, the cell has evolved complex machineries to enable specific nucleosomes at precise genomic locations to become very dynamic, as required, to facilitate different genomic processes. This regulated alteration of nucleosome architecture during different biological events is mediated mainly by adenosine 5′-triphosphate (ATP)–dependent nucleosome remodeling complexes (129). Using the energy of ATP hydrolysis, ATP-dependent nucleosome remodelers work closely with other activities, including histone-modifying enzymes and histone chaperones, to package, evict, or slide nucleosomes in a highly regulated manner (130). Mutations and ablation of ATP-dependent nucleosome remodeler function have been linked to several disease syndromes and cancers (130, 131). Recently, some members of the ATP-dependent nucleosome remodeler families have also been linked to the aging process.
Both physiological aging and premature aging syndromes reveal signs of extensive alteration of chromatin structure, followed by increased susceptibility to persistent DNA damage (132). Although several studies suggest that altered chromatin conformations occur with age (43, 44), almost no direct connections established the involvement of ATP-dependent nucleosome remodelers until recently. The C. elegans homolog of the catalytic subunit of the NURD chromatin remodeling complex, LET-418/Mi2, has recently been identified as a longevity-determining factor because let-418 deficiency leads to prolonged life span and increased environmental stress resistance. This phenotype is partially dependent on the transcription factor DAF-16/FOXO. Genetic interaction analysis further suggests that let-418 may serve as a life span determinant by acting through the germ cell loss pathway. This protein is highly evolutionarily conserved, and indeed, it has been found that these increased longevity and enhanced stress resistance functions are also conserved in fruit flies and plants (133). One study that aims to identify the molecular basis for the age-related defect in chromatin structure used cells from HGPS patients and noted that components of the NURD ATP-dependent nucleosome remodeler complex, including RBBP4 and RBBP7, were substantially reduced in HGPS cells (132). Conversely, experimental depletion of RBBP4 and RBBP7 in cell culture caused an increase in endogenous DNA damage similar to what has been observed in HGPS cells and cells from aged individuals. Furthermore, it appeared likely that the defect in chromatin structure precedes the increased endogenous DNA damage observed under the experimental conditions, suggesting that the altered chromatin structure due to loss of NURD function leads to DNA damage or a defect in DNA repair. Loss of NURD subunits was evident not only in HGPS cells but also in cells collected from aged individuals. In summary, loss of NURD subunits seems to be an early event during both physiological and premature aging, which contributes to alterations in the chromatin structure, making the genome more susceptible to DNA damage (132). Although the exact mechanism behind the declining level of NURD components is yet to be determined, this study implicates the functional decline of an ATP-dependent nucleosome remodeling complex in aging.
Compelling evidence for involvement of an ATP-dependent nucleosome remodeling complex in limiting longevity comes from a large-scale study that measured the effect of single gene deletions on replicative life span of budding yeast (134). The absence of the Isw2 ATP-dependent nucleosome remodeler allowed yeast cells to live longer (124). Further experimental evidence suggests that the extended life span was through the same pathway as CR but was somewhat distinct from the well-studied suppression of the TOR signaling pathway during CR. Rather, in this case, a novel pathway is used, which involves up-regulation of several stress response genes that are also up-regulated as a response to genotoxic stresses during CR. In the absence of Isw2, a subset of these genes, including Rad51, is activated. Rad51 is a crucial factor required for homologous recombination (HR)–mediated DNA repair pathways (135). Transcriptional activation of Rad51 may provide the cells with the potential advantage to efficiently repair DNA using HR, empowering the cells to live longer. In agreement with this prediction, yeast life span was extended by addition of a single extra gene copy of RAD51 (134). An earlier study that investigated the mechanism of life span regulation by the FOXO transcription factor DAF-16 in worms identified the ATP-dependent nucleosome remodeler SWI/SNF as a crucial cofactor in transcriptional activation of DAF-16 target genes. The well-known prolongevity effect of DAF-16/FOXO is dependent on SWI/SNF because RNAi inactivation of core subunits of the SWI/SNF complex abolished DAF-16/FOXO–mediated life span extension (136). Deletion of the catalytic subunit of the ATP-dependent nucleosome remodeler Chd1 has also been shown to extend yeast replicative life span in a comprehensive analysis of replication life span in almost 4700 deletion mutants (52). Given the conserved roles of the ATP-dependent nucleosome remodelers from yeast to humans, it will be intriguing to explore the potential roles of these remodelers to modulate life span in mammals.
DNA METHYLATION CHANGES DURING AGING
DNA methylation is one of the most extensively studied and best-characterized epigenetic modifications during aging (Fig. 3) (137). In young cells, the majority of CpGs within the genome have cytosine methylation. CpG methylation within promoters leads to transcriptional repression, through the formation of compact chromatin structures, such as heterochromatin. Conversely, promoters of genes that are highly expressed are devoid of DNA methylation, hence their name—CpG islands. DNA methylation is particularly important during development, when it is used to silence genes in tissues in which their expression is never going to be needed, or at developmental stages in which they no longer need to be expressed. As identical twins age, the pattern of DNA methylation becomes more and more divergent because of epigenetic drift caused by environmental factors or spontaneous stochastic errors in the process of transmission of DNA methylation (12). Epigenetic drift leads to unpredictable differences in the methylome among aging individuals (Fig. 3). However, some of the methylation changes that occur with age are directional and involve specific regions of the genome. This fact indicates that at least part of the DNA methylation changes during aging are not stochastic but could be associated with biological mechanisms involved in the aging process. DNA methylation, as well as life span, also differs between organisms of identical genetic makeup, such as queen and worker honeybees, when subjected to different environmental stimuli, such as diet, which results in an altered gene expression pattern. Strikingly, similar methylome profiles were experimentally found following RNAi-mediated silencing of a DNA methyltransferase enzyme, Dnmt3, in laboratory-bred bees (16). Although it is yet to be determined whether the diet-induced DNA methylation changes are indeed responsible for causing the transcriptional shift observed in these cases, it provides great promise for unraveling a key concept of epigenetic reprogramming.
Fig. 3. Summary of DNA methylation changes during aging.
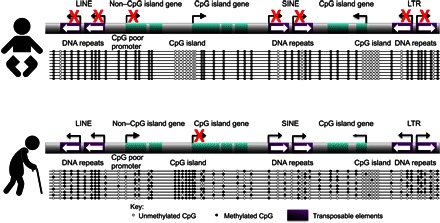
Young mammalian cells are characterized by DNA hypermethylation over the genome, with the exception of CpG islands within the promoters of expressed genes. In particular, DNA repeats, such as LINE, SINE, and long terminal repeat (LTR) transposable elements, are heavily DNA-methylated, helping to maintain them in a constitutive heterochromatin state. During aging, there is general DNA hypomethylation over the genome, which mostly occurs in a stochastic manner within the cell population. Loss of DNA methylation leads to activation of normally silenced DNA sequences like the transposable elements. However, DNA methylation also increases in a nonstochastic manner over the CpG islands of certain genes, correlating with their heterochromatinization and silencing.
With a few exceptions, mammalian aging is more commonly associated with CpG hypomethylation, especially at repetitive DNA sequences (Fig. 3) (138–144). This is likely to be at least partly responsible for the loss of heterochromatin during aging. Loss of CpG methylation at repetitive sequences will heighten the risk of retrotransposition events and, hence, genomic instability during aging, given that the retrotransposons comprise much of the repetitive DNA. The global decrease in DNA methylation upon aging may be attributed to the progressive decline in levels of the DNA methyltransferase DNMT1 (137). The contribution of DNA methylation to aging is further suggested by a Dnmt1+/− mouse model that showed reduced age-dependent impaired learning and memory function (145). In addition to the general DNA hypomethylation that occurs during aging, progressive age-dependent loss of DNA methylation also occurs at specific gene promoters, including ITGAL and IL17RC, leading to their transcriptional induction and subsequent autoimmune responses (146, 147). Simultaneous with the general and localized DNA hypomethylation during aging, hypermethylation occurs at specific CpG sites of the genome, presumably to repress expression of specific genes (Fig. 3) (137, 142, 148, 149). A recent analysis also observed loss of DNA methylation from cis regulatory elements, such as enhancer regions in pancreatic β cells, and suggested the possibility that transcription factor binding may hinder the ability of DNA methyltransferases to eventually recognize those sites, resulting in aberrant gene expression in aged cells (150). The hypothesis seems promising but is yet to be addressed during aging in different cells, tissues, or organisms. However, our current knowledge about the limited tissue types tested so far did not show a striking global correlation between DNA methylation and alterations in gene expression profile during hematopoietic stem cell aging or in whole blood during aging (151, 152).
The development of novel next-generation sequencing technologies for genome-wide assessment of DNA methylation levels has permitted the confirmation and extension of earlier studies. A comparison between the methylome of CD4+ T cells from newborn and centenarian individuals further solidifies the notion of global decreases in DNA methylation with aging, accompanied by heterogeneous DNA methylation in the centenarian genome (153). Generally, age-related DNA methylation changes are more prominent in CpG islands (Fig. 3), but tissue-specific biases are observed more frequently outside those sites (45, 149). DNA methylation changes occur at several specific sites during aging in a highly reproducible way, but the complete functional relevance of these changes is yet to be explored and understood. However, the reproducibility of DNA methylation changes during aging at some sites is such that age prediction can be made fairly accurately by analyzing DNA methylation patterns in only three specific CpG sites, at least for blood DNA (154).
Not surprisingly, given their functional interdependence, DNA methylation and histone modifications are intertwined to exert the changes observed during aging. Numerous studies have reported age-induced DNA hypermethylation of specific loci that contain known Polycomb group protein (PcG) target genes in both mice and humans (142, 149, 154–157). PcGs repress gene expression by H3 K27 trimethylation in a dynamic manner that is reversible by H3 K4 methylation, as needed, during development. Whereas hypermethylation of DNA upon aging is enriched at the genomic regions carrying bivalent histone marks (that is, both H3 K4me3 and H3 K27me3) at poised promoters, DNA hypomethylation co-occurs with the histone modification marks H3 K9Ac, H3 K27Ac, H3 K4me1, H3 K4me2, and H3 K4me3 that are found mostly at enhancer regions (158, 159). During replicative senescence, there are alterations in DNA methylation patterns globally and at specific sites, as observed during organismal aging. Notably, cell passage numbers and the population doublings can be accurately predicted from the methylation pattern at specific CpG sites (160). This altered DNA methylation pattern during replicative senescence correlated with the change of expression of SIRT1 (161). This might result from the effect of SIRT1 on DNA methylation of PcG target genes, although the mechanistic details of this phenomenon are unclear (162). One of the remaining challenges in the analyses of DNA methylation during aging is to identify the causal pathways that contribute to the functional decline of the DNA methylome during aging.
The age-dependent changes in the DNA methylome are reminiscent of those occurring in cancer. Global hypomethylation at the repetitive regions and site-specific hypermethylation at certain promoters have also been reported during cancer, suggesting a potential connection between age-dependent DNA methylation changes and increased cancer risk observed in the elderly population (163, 164). Notably, local hypermethylation events during aging sometimes occur at the promoters of tumor-suppressor genes, potentially preceding cell transformation events. The analogy between cancer and aging also extends to the PcG target genes because both aging and cancer are accompanied by DNA hypermethylation of these genes. Hence, a better understanding of the reasons behind the changes in the DNA methylome during aging may help us to also understand the causes of cancer.
ncRNAs AND AGING
ncRNAs are the most recent players in the epigenetics field, influencing seemingly all biological processes in virtually all organisms. The advent and widespread use of deep sequencing has provided unbiased insights into the eukaryotic genome, including the existence and functional roles of ncRNAs. In contrast to earlier beliefs, it is now widely accepted that approximately 60 to 90% of the human genome is transcribed, with some variability observed in other organisms (165, 166), giving rise to an enormous array of ncRNAs (165–169). Until recently, most of the studies focused on the short ncRNAs, but the functional importance of long ncRNAs (lncRNAs) is now becoming apparent. Although it is likely that the majority of these functions are epigenetic, with the ncRNAs having significant influence on modulating gene expression and chromatin packaging, the complete array of biological functions of ncRNAs is yet to be understood (170, 171). Disruption of ncRNA function has been implicated in numerous disease conditions, such as cancer, neurodegenerative disorders, cardiovascular disorders, and aging (172, 173).
The occurrence of ncRNAs has been highly conserved through evolution; even budding yeast is now appreciated to have many ncRNAs. ncRNA transcription from the rDNA locus, which is silent under normal conditions, serves as a life span determinant of this unicellular organism through its function to regulate rDNA stability (174). Mutations that prevent expression of the ncRNAs from the rDNA locus lead to life span extension in budding yeast (174). As discussed above, expression of the ncRNAs from Alu elements during adult human stem cell aging ex vivo promotes entry into senescence, and knockdown of the Alu transcripts reverses senescence (70). During aging of both worms and mice, a decline in either mRNA or protein level has been observed for Dicer (175), suggesting likely defective small ncRNA (sncRNA) processing in these organisms during aging. Indeed, the majority of microRNAs (miRNAs), a class of sncRNAs, have been shown to be down-regulated with age (175–177). Notably, this phenotype can be reversed following CR in mice (175). In the absence of Dicer in mice, there are signs of early senescence, signifying its role in longevity (175). A decrease in Dicer levels has also been observed in adipocytes collected from elderly humans, suggesting the possibility of a conserved mechanism of sncRNA dysregulation during aging in mammals (175).
miRNAs are sncRNAs that negatively control their target gene expression posttranscriptionally and have been implicated in aging. Although miRNAs do not alter the chromatin structure, they are considered mediators of epigenetics because they lead to heritable changes in gene expression that do not involve a change in DNA sequence. The best-characterized examples of roles of miRNAs during aging come from studies in C. elegans. Several miRNAs have been shown to be involved in modulating life span and in controlling tissue aging (178, 179). One of the most prominent examples includes the regulation of aging by the miRNA lin-4 and its pro-aging target miRNA lin-14 (178). Loss of function of lin-4 shortens life span, whereas overexpression of lin-4 extends life span. In contrast, knocking down lin-14, even in adult animals, extends life span (178). The key question is what are the target genes of lin-14 that are regulating aging? Of all the miRNAs reported in C. elegans, more than a quarter are differentially expressed during aging, including some of the miRNA families conserved with humans (176, 177, 179). Whereas some miRNAs have an antiaging effect by promoting longevity in C. elegans, others show a pro-aging effect by antagonizing longevity (177, 179, 180). Even the expression pattern of some miRNAs serves as a predictor of longevity. However, the evolutionarily conserved nature of some of these miRNAs indicates that their role in life span regulation likely extends beyond this organism to larger eukaryotes, including humans. Differential expression of miRNAs is also evident in mice and humans during aging, although the differences are not always present in all organs (181–184).
One of the most notable features of human aging is neurodegeneration and reduced brain function. Premature death of neurons is considered to be a major feature of neurodegenerative diseases. Studies of neurodegenerative processes implicate ncRNAs. Whereas some of the miRNAs correlate with neuroprotection, others clearly contribute toward neurodegenerative diseases and/or aging (173). For example, the mir-34 family appears to be an important determinant for brain aging in flies and maybe also in worms (185). There are higher levels of mir-34 in Alzheimer’s disease mouse model brains and samples collected from Alzheimer’s disease patients (186, 187). The prosurvival factor BCL2 and the antiaging deacetylase SIRT1 both serve as targets of mir-34, and the expression of the latter correlates inversely with mir-34 expression (181, 188), revealing a potential mechanism for mir-34 function in the aged brain. Similarly, another miRNA, mir-144, seems to be enriched in aged brains and may also contribute to age-associated neurodegeneration through down-regulation of key protective factors (173). Genetic studies have demonstrated that the longevity-modulating miRNAs lin-4 and lin-14 in C. elegans function in the same pathway as DAF-2 and DAF-16 (178), modulating life span through the insulin/insulin-like growth factor 1 (IGF-1) signaling pathway. There are reports of multiple miRNAs involved in the mTOR pathway, although experimental evidence of these miRNAs regulating life span is still limited (181). Clearly, changes in miRNA levels during aging are a means to regulate target gene expression, in addition to chromatin changes.
lncRNAs themselves also serve as important regulators of transcription through their interaction with chromatin or chromatin-associated factors, modulating aging and senescence directly or indirectly. One such example includes a specific lncRNA, Gas5, which is highly expressed in aged mice brain and has been associated with impaired learning (189). Another bona fide example is H19 lncRNA, a differentially spliced product from the H19 gene located at the IGF2/H19 imprinted locus, which interacts with methyl-CpG–binding domain protein 1 to form a complex to repress expression of an imprinted gene network in mice (190). Loss of imprinting from the IGF2/H19 locus has been shown during aging in both mice and human prostate associated with reexpression of certain inactive genes and loss of binding of chromatin-associated protein CTCF (191). This may serve as a potential reason for increased prostate cancer occurrence in aging men, further supporting the connection between aging and cancer development. Other lncRNAs that are implicated in major senescence-associated pathways, such as p53/p21 pathways, are differentially expressed in proliferating early passage compared to senescent late-passage fibroblast cells (192, 193). Similarly, differential expression patterns of various lncRNAs have been implicated in the pathophysiology of another age-onset neurological disorder, Huntington’s disease, wherein some of these lncRNAs have been postulated to modulate chromatin architecture and/or transcription (194, 195). Other ncRNAs, mostly products of the RNAi pathway, are involved in heterochromatin assembly in repetitive DNA elements in diverse organisms (196). Here, they function to target histone-modifying activities in DNA repeats through reader proteins. Other ncRNAs act to organize the three-dimensional organization of chromosomes or as boundaries or insulators to prevent the spreading of heterochromatin (197). Whether the changes in chromatin structure that occur during aging are due to changes in these particular ncRNAs is yet to be examined.
TRANSGENERATIONAL EPIGENETIC CHANGES THAT AFFECT AGING
According to biological dogma, genetics governs all the inherited traits across generations, and epigenetic modifications are reset upon passage through the germ line. However, over the years, this notion was challenged when evidence of epigenetic inheritance through meiosis became acknowledged in certain processes, such as flower symmetry and color in plants, or coat color and size in mice (198, 199). Recently, longevity mediated by histone methylation was shown to be epigenetically inherited for several generations (198), implicating transgenerational epigenetic inheritance for the first time in the regulation of life span. Deficiencies in either of the three components of H3 K4me3 methylase complex (ASH-2, WDR-5, or SET-2), in only the parental generation, resulted in life span extension in C. elegans in the three subsequent generations, in the absence of methylase deficiency in these offsprings. However, only the parents with the deficiencies in the H3 K4me3 regulatory complex, and not their wild-type long-lived offspring, had reduced global H3 K4me3 levels. Hence, altered histone methylation per se was not transgenerationally inherited. Instead, microarray analysis revealed that there were persistent changes in gene expression throughout the generations upon manipulation of the H3 K4me3 regulatory complex in the parents (198), which could potentially be responsible for the transgenerational inheritance of long life span. Further experimentation is needed to identify the pathways responsible for the transgenerational inheritance of longevity and to explore whether this epigenetic memory is generalizable to other species. A useful approach to study the inheritance of aging phenotypes would be to follow the lead of a recent study examining epigenetic germ line inheritance of diet-induced obesity and insulin resistance in mice (200). This study used in vitro fertilization to ensure exclusive inheritance through the gametes and showed that the parental high-fat diet renders the offspring more susceptible to developing obesity and diabetes. It is tempting to speculate that this novel mode of inheritance may illustrate how epigenetics could have contributed to evolution, whereby the ancestors’ environmental exposure determined the fate of the descendants. Given the intriguing nature of the subject, more studies will undoubtedly further explore this exciting direction in the near future.
LIFE SPAN–EXTENDING REGIMENS THAT FUNCTION, AT LEAST IN PART, THROUGH EPIGENETIC CHANGES
There is considerable interest in extending health span as well as life span. Accordingly, improvements in our knowledge of the basic mechanisms that cause aging have led to the field that identifies different ways to promote longevity. Experimental manipulations involving genetic, epigenetic, environmental, or even pharmacological interventions in different model organisms have been extremely informative for demonstrating the potential of manipulating the processes under study, to delay aging. Of all the hallmarks of aging, epigenetic alterations have attracted attention as a promising avenue for life span extension because, unlike most of other age-promoting processes, epigenetic alterations can be modulated by inhibition or stimulation of the relevant enzymes. Thus, interventions targeting epigenetic information hold immense potential to extend life span and to counteract age-associated diseases, including cancer and neurodegenerative disorders. In this section, we will discuss the relationship of well-known life span–extending interventions with epigenetics.
Some of the most effective life span extension regimens function at least in part through epigenetic pathways (Fig. 4). Of all the experimental manipulations known to date, by far the most accessible means toward life span extension is dietary restriction (DR) or CR, which has profound effects in multiple organisms, including yeast, flies, worms, mice, and even primates (53). Although CR has been shown to act through multiple different pathways, the precise unified mechanism by which CR exerts its life span–extending effects still needs to be defined (201). However, evidence suggests that epigenetic alterations, including DNA methylation and histone modifications, may play crucial roles in life span extension by CR (201, 202). For example, CR is thought to be important for regulating DNA methylation patterns at individual loci in the genome, but not the global DNA methylation pattern, and this may occur with CR increasing the expression level and/or activities of certain DNMT enzymes, such as DNMT1 and DNMT3b (9, 201). In agreement, some DNMT inhibitors have displayed preventive potential against age-associated diseases, such as cancer, implying the importance of studying these compounds to promote a healthy life span. Similarly, the regulation of histone modifications, such as histone acetylation and deacetylation, may play roles in regulating life span during CR. This is solidified by numerous studies performed on class III HDAC enzymes (Sir2 in yeast and its mammalian sirtuin orthologs) in multiple model organisms, wherein activation of this enzyme class has frequently been associated with CR and life span extension (Fig. 4), suggesting that there may be a protective mechanism conferred by deacetylation during aging. Comparative analysis of the transcriptome of the brains of rats maintained on a normal diet or CR regime during aging reveals that CR largely prevents the differential gene expression that is observed during aging (203). Furthermore, CR induces specific gene expression patterns in the cerebral cortex that offer neuroprotection, potentially by inducing mir-98-3p, which itself alters the activity of HDACs and HATs (203). In relation to this, the ability of HDAC inhibitors to reactivate key tumor suppressor genes has been extensively analyzed for their potential application as anticancer agents to treat age-related increases in carcinogenesis (201). Clearly, some of the antiaging effects of CR are through the down-regulation of the nutrient-sensing mTOR pathway (Fig. 4), and this has been discussed elsewhere (204).
Fig. 4. Schematic showing how some external interventions trigger longevity, often at least partly through stimulating autophagy.
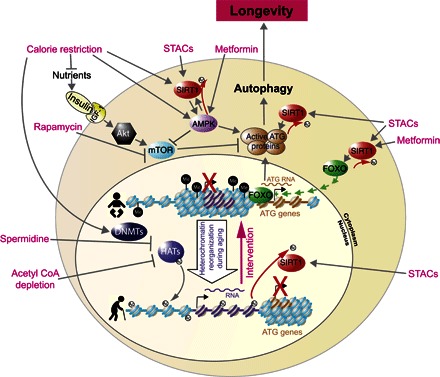
The pink writing refers to dietary, chemical, or therapeutic interventions that can extend life span, in at least some organisms (described in the text). Arrows indicate stimulating effects, and blocked lines indicate inhibitory effects. This schematic is not meant to be exhaustive but highlights the pathways that alter the epigenetic information and autophagy.
Because of the challenges for humans in adherence to a CR regimen, significant research emphasis has been placed on finding effective “CR mimetics.” These are compounds or drugs that can essentially mimic the prolongevity effects of CR in humans by using the same pathways as CR, without actual restriction of calorie intake. In this context, several new, as well as already existing marketed, drugs are being assessed (205, 206). Given that the sirtuins promote longevity in diverse species, they have attracted considerable interest as drug targets. The first sirtuin-activating compounds (STACs) to be identified included plant-derived metabolites, such as flavones, stilbenes, anthocyanidins, and chalcones (207). The best known of these is resveratrol. Resveratrol and synthetic STACs have been shown to induce physiological changes and gene expression changes that are similar to those that are induced by CR and to extend life span in many model organisms (207). The ability of resveratrol to extend life span in mice depends on them being fed a high-fat diet because there was no extended life span when mice were given resveratrol on a standard diet (208). Although resveratrol stimulates Sir2/SIRT1 activity in vitro (209), its stimulatory interaction with SIRT1 depended on binding to the 7-amino-4-methylcoumarin (AMC) fluorophore within the histone peptide substrate used to measure deacetylase activity (210). The fluorophore used in the in vitro HDAC assay mimics bulky hydrophobic amino acids, and resveratrol only stimulates SIRT1-mediated deacetylation of natural substrates that have large hydrophobic residues C-terminal to the acetyl lysine (211–214). The recent structure of the complex of SIRT1, resveratrol, and an AMC-peptide (210) is likely to guide the computational search and/or chemical design of better compounds to stimulate SIRT1 deacetylase activity against its native substrates. Promisingly, the synthetic STACs SRT1720 and SRT2104 extend the life span of obese mice and protect against age-related changes in multiple tissues (215).
The antidiabetic drug metformin also induces effects similar to CR (216). Diabetes is considered an age-associated disease, and disturbances in insulin signaling and carbohydrate homeostasis may essentially lead to other age-related complications, including cancer, if untreated. Along with its antidiabetic properties, metformin supplementation has been shown to increase life span and health span in C. elegans (217, 218) and male mice (219), together with some of the benefits seen with CR. A recent study of more than 180,000 people showed that diabetes patients being treated with metformin not only lived longer than other diabetic patients but also lived longer than the healthy control sample (220). Until recently, metformin was thought to primarily function by regulating the stress response and metabolic pathways (216). However, a very recent report provided evidence for epigenetic regulation potentially contributing to the mechanism of action of metformin. Increased levels of SIRT1 were observed in human subjects treated with metformin (221). Further exploration is needed to fully understand how metformin mechanistically extends life span in humans. However, given that metformin is approved by the U.S. Food and Drug Administration for use in humans, commercially available, and relatively cheap, it has the potential to be revolutionary for increasing human health span and life span.
Several compounds that reduce global histone acetylation also extend life span. Spermidine, a naturally occurring polyamine, directly inhibits HATs, maintaining histone H3 in a hypoacetylated state (222). Polyamines result in resistance to stress and reduced rates of cell necrosis during chronological aging in yeast and human cells and extend the life span of flies, worms, and human cells. In agreement, administration of spermidine through diet in aging mice also reduced age-related oxidative stress (222). Another potential way to reduce histone acetylation globally is to feed a diet that depletes acetyl–coenzyme A (CoA), the donor for acetylation reactions. But how does reducing acetylation extend life span? Depletion of acetyl-CoA by starvation of mammalian cells in culture and fasting in mice has been recently shown to induce autophagy (223). In agreement, spermidine also induced autophagy, extending chronological life span in yeast (223, 224). Autophagy may be regarded as a mechanism of cellular “cleansing,” and its up-regulation has clear cellular benefits that would promote longevity (225). Accordingly, note that stimulating autophagy is a common theme among multiple interventions that extend life span (Fig. 4) (226). Recently, histone acetylation has also been implicated in the mechanism whereby autophagy is activated in metazoans. Fly life span is extended and autophagy is induced when acetyl-CoA levels were reduced in fly brains (224). The mechanism wherein high acetyl-CoA levels block autophagy is not totally clear, but it likely involves histone acetylation. In one study, the HAT p300, whose substrates include histone H3, was required for high levels of acetyl-CoA to block autophagy (223). In another study, high levels of H4 K16Ac (the substrate of Sir2/SIRT1) down-regulated autophagy, and low levels of H4 K16Ac induced autophagy (227). The natural increase in H4 K16Ac that occurs during aging, because of Sir2 loss, could lead to the decline of autophagy, contributing to the aging process and diseases of aging. Hence, dietary and pharmacological manipulations that decrease acetyl-CoA levels might extend life span by stimulating autophagy. Notably, SIRT1 also stimulates autophagy, wherein SIRT1 induces FOXO-mediated expression of autophagy pathway components and deacetylates major players of the autophagy induction pathway (Fig. 4) (228). Indeed, the longevity-inducing role of CR appears to be through the SIRT1-dependent induction of autophagy (229). Hence, it is tempting to speculate that multiple different manipulations to extend life span, such as CR, acetyl-CoA depletion, polyamines, and sirtuin activation, all function through a common pathway to stimulate autophagy, potentially through an effect on histone acetylation (Fig. 4).
An exciting new way to improve health span and, subsequently, life span in mice is to specifically kill off senescent cells with senolytic agents (230). Given the epigenetic changes that are required for the maintenance of senescence, another potential avenue to extend life span may be to develop senolytic drugs that target specific epigenetic enzymes.
CONCLUSION AND FUTURE DIRECTIONS (RESETTING THE AGING CLOCK)
The aging process is unquestionably complex. During an organism’s lifetime, aging cells undergo a plethora of changes and accumulate macromolecular damage. The aging phenotypes are manifested by the summation of alterations of many different signaling cascades. Here, we attempted to provide an overview of some of the major changes in aging cells, tissues, and organisms that have come to light in recent years through the lens of chromatin and epigenetic regulation. Genetic and environmental manipulations are unequivocally important to decipher the effect of any specific factor over the longevity process. It is becoming apparent mechanistically that many of those factors that do affect longevity act primarily through the modification of the epigenome. Undoubtedly, epigenetic influences over the aging process need to be incorporated into our current understanding of aging. Almost all classes of epigenetic alterations reported so far influence longevity pathways, adding further complexity to understanding the aging process. In summary, young healthy cells maintain an epigenetic state that promotes the formation of a compact chromatin structure and precise regulation of all the basic biological processes. However, aging cells experience alterations in all aspects of the chromatin landscape, DNA accessibility, and ncRNA production, until a threshold of altered gene expression and compromised genomic integrity is crossed, and the cells finally succumb to a permanent halt in progression through the cell cycle. The reversible nature of epigenetic mechanisms makes it possible to restore or reverse some of these phenotypes to attain more youthful cells.
Whereas some of the molecular changes during aging can be categorized as causal to aging, other changes simply accompany the aging process. However, when characterizing the causes or consequences of aging, one has to carefully dissect the experimental findings because most of the relevant pathways are interconnected, making it difficult to separate the effects of the pro-aging pathways from bystander pathways. A concerted effort should be made to establish the hierarchical relationship among the relevant pathways to understand the exact causal network of aging and to derive effective therapies to counteract the aging process and age-associated diseases. There is still a great deal to learn about this complex biological process. Current progress and extensive utilization of next-generation sequencing techniques are enabling us to analyze age-associated changes at ever-increasing resolution. However, simultaneous gain-of-function and loss-of-function studies in different organisms are also crucial to understanding and providing evidence of the causal effects of certain pathways, to move beyond correlation analyses. A tour-de-force analysis of the effect of individually deleting 4700 yeast genes on replicative life span was recently completed, finding 237 genes whose deletion extends life span (146). However, we clearly have a lot more to learn about the aging process because 30 of those genes are so poorly understood that they have yet to be given functional names.
The continued combination of functional studies and molecular analyses in different age groups, different organisms, and different tissue types will hopefully provide the details necessary to comprehend this evolutionarily conserved fundamental process and to facilitate the development of therapeutic interventions to counteract age-induced complications. A central concept that is emerging is to develop epigenetic drugs, or even an epigenetic diet, with the goal to improve not only a single disease state but also multiple disorders of aging. Although an intriguing idea, caution should be taken when determining the specificity of epigenetic therapies because of the interconnected nature of mechanisms that regulate epigenomic information. Thus, the major challenges that will dominate the field in the near future will be to achieve a hierarchical understanding of how epigenomics affects the aging process and to understand the long-term effects of therapeutic interventions on the epigenome in an aging individual, given the interconnectivity of the epigenetic mechanisms.
Acknowledgments
Funding: This work was funded by grant RO1 CA95641 to J.K.T. Author contributions: S.P. and J.K.T. planned the review. S.P. wrote the review, and J.K.T. edited the review and made the figures. Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Brunet A., Berger S. L., Epigenetics of aging and aging-related disease. J. Gerontol. A Biol. Sci. Med. Sci. 69 (Suppl. 1), S17–S20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feser J., Tyler J., Chromatin structure as a mediator of aging. FEBS Lett. 585, 2041–2048 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kennedy B. K., Berger S. L., Brunet A., Campisi J., Cuervo A. M., Epel E. S., Franceschi C., Lithgow G. J., Morimoto R. I., Pessin J. E., Rando T. A., Richardson A., Schadt E. E., Wyss-Coray T., Sierra F., Geroscience: Linking aging to chronic disease. Cell 159, 709–713 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moskalev A., Aliper A., Smit-McBride Z., Buzdin A., Zhavoronkov A., Genetics and epigenetics of aging and longevity. Cell Cycle 13, 1063–1077 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campisi J., Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muñoz-Espín D., Serrano M., Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496 (2014). [DOI] [PubMed] [Google Scholar]
- 7.López-Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G., The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldberg A. D., Allis C. D., Bernstein E., Epigenetics: A landscape takes shape. Cell 128, 635–638 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Muñoz-Najar U., Sedivy J. M., Epigenetic control of aging. Antioxid. Redox Signal. 14, 241–259 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Sullivan R. J., Karlseder J., The great unravelling: Chromatin as a modulator of the aging process. Trends Biochem. Sci. 37, 466–476 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zane L., Sharma V., Misteli T., Common features of chromatin in aging and cancer: Cause or coincidence? Trends Cell Biol. 24, 686–694 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraga M. F., Ballestar E., Paz M. F., Ropero S., Setien F., Ballestar M. L., Heine-Suñer D., Cigudosa J. C., Urioste M., Benitez J., Boix-Chornet M., Sanchez-Aguilera A., Ling C., Carlsson E., Poulsen P., Vaag A., Stephan Z., Spector T. D., Wu Y.-Z., Plass C., Esteller M., Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U.S.A. 102, 10604–10609 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sargent M., Why twins age differently. Nature 464, 1130–1131 (2010). [Google Scholar]
- 14.Herskind A. M., McGue M., Holm N. V., Sørensen T. I., Harvald B., Vaupel J. W., The heritability of human longevity: A population-based study of 2872 Danish twin pairs born 1870–1900. Hum. Genet. 97, 319–323 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Poulsen P., Esteller M., Vaag A., Fraga M. F., The epigenetic basis of twin discordance in age-related diseases. Pediatr. Res. 61, 38R–42R (2007). [DOI] [PubMed] [Google Scholar]
- 16.Kucharski R., Maleszka J., Foret S., Maleszka R., Nutritional control of reproductive status in honeybees via DNA methylation. Science 319, 1827–1830 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Kornberg R. D., Chromatin structure: A repeating unit of histones and DNA. Science 184, 868–871 (1974). [DOI] [PubMed] [Google Scholar]
- 18.Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J., Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Maison C., Almouzni G., HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 5, 296–304 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Bitterman K. J., Medvedik O., Sinclair D. A., Longevity regulation in Saccharomyces cerevisiae: Linking metabolism, genome stability, and heterochromatin. Microbiol. Mol. Biol. Rev. 67, 376–399 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steinkraus K. A., Kaeberlein M., Kennedy B. K., Replicative aging in yeast: The means to the end. Annu. Rev. Cell Dev. Biol. 24, 29–54 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guarente L., Kenyon C., Genetic pathways that regulate ageing in model organisms. Nature 408, 255–262 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Harel I., Benayoun B. A., Machado B., Singh P. P., Hu C.-K., Pech M. F., Valenzano D. R., Zhang E., Sharp S. C., Artandi S. E., Brunet A., A platform for rapid exploration of aging and diseases in a naturally short-lived vertebrate. Cell 160, 1013–1026 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arancio W., Pizzolanti G., Genovese S. I., Pitrone M., Giordano C., Epigenetic involvement in Hutchinson-Gilford progeria syndrome: A mini-review. Gerontology 60, 197–203 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Burtner C. R., Kennedy B. K., Progeria syndromes and ageing: What is the connection? Nat. Rev. Mol. Cell Biol. 11, 567–578 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Tsurumi A., Li W., Global heterochromatin loss: A unifying theory of aging? Epigenetics 7, 680–688 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villeponteau B., The heterochromatin loss model of aging. Exp. Gerontol. 32, 383–394 (1997). [DOI] [PubMed] [Google Scholar]
- 28.Haithcock E., Dayani Y., Neufeld E., Zahand A. J., Feinstein N., Mattout A., Gruenbaum Y., Liu J., Age-related changes of nuclear architecture in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 102, 16690–16695 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larson K., Yan S.-J., Tsurumi A., Liu J., Zhou J., Gaur K., Guo D., Eickbush T. H., Li W. X., Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLOS Genet. 8, e1002473 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smeal T., Claus J., Kennedy B., Cole F., Guarente L., Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell 84, 633–642 (1996). [DOI] [PubMed] [Google Scholar]
- 31.Guarente L., Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N. Engl. J. Med. 364, 2235–2244 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Haigis M. C., Sinclair D. A., Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oberdoerffer P., Michan S., McVay M., Mostoslavsky R., Vann J., Park S.-K., Hartlerode A., Stegmuller J., Hafner A., Loerch P., Wright S. M., Mills K. D., Bonni A., Yankner B. A., Scully R., Prolla T. A., Alt F. W., Sinclai D. A.r, SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907–918 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kennedy B. K., Gotta M., Sinclair D. A., Mills K., McNabb D. S., Murthy M., Pak S. M., Laroche T., Gasser S. M., Guarente L., Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell 89, 381–391 (1997). [DOI] [PubMed] [Google Scholar]
- 35.Oberdoerffer P., Sinclair D. A., The role of nuclear architecture in genomic instability and ageing. Nat. Rev. Mol. Cell Biol. 8, 692–702 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi T., Regulation of ribosomal RNA gene copy number and its role in modulating genome integrity and evolutionary adaptability in yeast. Cell. Mol. Life Sci. 68, 1395–1403 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sinclair D. A., Guarente L., Extrachromosomal rDNA circles—A cause of aging in yeast. Cell 91, 1033–1042 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Goldman R. D., Shumaker D. K., Erdos M. R., Eriksson M., Goldman A. E., Gordon L. B., Gruenbaum Y., Khuon S., Mendez M., Varga R., Collins F. S., Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. U.S.A. 101, 8963–8968 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scaffidi P., Misteli T., Lamin A-dependent nuclear defects in human aging. Science 312, 1059–1063 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shumaker D. K., Dechat T., Kohlmaier A., Adam S. A., Bozovsky M. R., Erdos M. R., Eriksson M., Goldman A. E., Khuon S., Collins F. S., Jenuwein T., Goldman R. D., Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. U.S.A. 103, 8703–8708 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang W., Li J., Suzuki K., Qu J., Wang P., Zhou J., Liu X., Ren R., Xu X., Ocampo A., Yuan T., Yang J., Li Y., Shi L., Guan D., Pan H., Duan S., Ding Z., Li M., Yi F., Bai R., Wang Y., Chen C., Yang F., Li X., Wang Z., Aizawa E., Goebl A., Devi Soligalla R., Reddy P., Rodriguez Esteban C., Tang F., Liu G.-H., Carlos Izpisua Belmonte J., Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 348, 1160–1163 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Narita M., Nuñez S., Heard E., Narita M., Lin A. W., Hearn S. A., Spector D. L., Hannon G. J., Lowe S. W., Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113, 703–716 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Chandra T., Ewels P. A., Schoenfelder S., Furlan-Magaril M., Wingett S. W., Kirschner K., Thuret J.-Y., Andrews S., Fraser P., Reik W., Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 10, 471–483 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Cecco M., Criscione S. W., Peckham E. J., Hillenmeyer S., Hamm E. A., Manivannan J., Peterson A. L., Kreiling J. A., Neretti N., Sedivy J. M., Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 12, 247–256 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sedivy J. M., Banumathy G., Adams P. D., Aging by epigenetics—A consequence of chromatin damage? Exp. Cell Res. 314, 1909–1917 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dang W., Steffen K. K., Perry R., Dorsey J. A., Johnson F. B., Shilatifard A., Kaeberlein M., Kennedy B. K., Berger S. L., Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459, 802–807 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feser J., Truong D., Das C., Carson J. J., Kieft J., Harkness T., Tyler J. K., Elevated histone expression promotes life span extension. Mol. Cell 39, 724–735 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu Z., Chen K., Xia Z., Chavez M., Pal S., Seol J.-H., Chen C.-C., Li W., Tyler J. K., Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 28, 396–408 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen K., Hu Z., Xia Z., Zhao D., Li W., Tyler J. K., The overlooked fact: Fundamental need for spike-in control for virtually all genome-wide analyses. Mol. Cell. Biol. 36, 662–667 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lesur I., Campbell J. L., The transcriptome of prematurely aging yeast cells is similar to that of telomerase-deficient cells. Mol. Biol. Cell 15, 1297–1312 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu L., Cheung T. H., Charville G. W., Hurgo B. M., Leavitt T., Shih J., Brunet A., Rando T. A., Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 4, 189–204 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCormick M. A., Delaney J. R., Tsuchiya M., Tsuchiyama S., Shemorry A., Sim S., Chou A. C., Ahmed U., Carr D., Murakami C. J., Schleit J., Sutphin G. L., Wasko B. M., Bennett C. F., Wang A. M., Olsen B., Beyer R. P., Bammler T. K., Prunkard D., Johnson S. C., Pennypacker J. K., An E., Anies A., Castanza A. S., Choi E., Dang N., Enerio S., Fletcher M., Fox L., Goswami S., Higgins S. A., Holmberg M. A., Hu D., Hui J., Jelic M., Jeong K.-S., Johnston E., Kerr E. O., Kim J., Kim D., Kirkland K., Klum S., Kotireddy S., Liao E., Lim M., Lin M. S., Lo W. C., Lockshon D., Miller H. A., Moller R. M., Muller B., Oakes J., Pak D. N., Jun Peng Z., Pham K. M., Pollard T. G., Pradeep P., Pruett D., Rai D., Robison B., Rodriguez A. A., Ros B., Sage M., Singh M. K., Smith E. D., Snead K., Solanky A., Spector B. L., Steffen K. K., Nga Tchao B., Ting M. K., Vander Wende H., Wang D., Linnea Welton K., Westman E. A., Brem R. B., Liu X.-., Suh Y., Zhou Z., Kaeberlein M., Kennedy B. K., A comprehensive analysis of replicative lifespan in 4,698 single-gene deletion strains uncovers conserved mechanisms of aging. Cell Metab. 22, 895–906 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee C., Longo V., Dietary restriction with and without caloric restriction for healthy aging. F1000Res. 5, pii: F1000 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McColl G.; Supported by the American Federation for Aging Research, Killilea D. W., Hubbard A. E., Vantipalli M. C., Melov S., Lithgow G. J., Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J. Biol. Chem. 283, 350–357 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ni Z., Ebata A., Alipanahiramandi E., Lee S. S., Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell 11, 315–325 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Sullivan R. J., Kubicek S., Schreiber S. L., Karlseder J., Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 17, 1218–1225 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ivanov A., Pawlikowski J., Manoharan I., van Tuyn J., Nelson D. M., Rai T. S., Shah P. P., Hewitt G., Korolchuk V. I., Passos J. F., Wu H., Berger S. L., Adams P. D., Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 202, 129–143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu B., Yip R., Zhou Z., Chromatin remodeling, DNA damage repair and aging. Curr. Genomics 13, 533–547 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mah L.-J., El-Osta A., Karagiannis T. C., γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 24, 679–686 (2010). [DOI] [PubMed] [Google Scholar]
- 60.Sedelnikova O. A., Horikawa I., Redon C., Nakamura A., Zimonjic D. B., Popescu N. C., Bonner W. M., Delayed kinetics of DNA double-strand break processing in normal and pathological aging. Aging Cell 7, 89–100 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Sedelnikova O. A., Horikawa I., Zimonjic D. B., Popescu N. C., Bonner W. M., Barrett J. C., Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 6, 168–170 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Garcia A. M., Calder R. B., Dollé M. E. T., Lundell M., Kapahi P., Vijg J., Age- and temperature-dependent somatic mutation accumulation in Drosophila melanogaster. PLOS Genet. 6, e1000950 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vijg J., Dollé M. E. T., Large genome rearrangements as a primary cause of aging. Mech. Ageing Dev. 123, 907–915 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Kaya A., Lobanov A. V., Gladyshev V. N., Evidence that mutation accumulation does not cause aging in Saccharomyces cerevisiae. Aging Cell 14, 366–371 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.MacRae S. L., Croken M. M., Calder R. B., Aliper A., Milholland B., White R. R., Zhavoronkov A., Gladyshev V. N., Seluanov A., Gorbunova V., Gorbunova V., Zhang Z. D., Vijg J., DNA repair in species with extreme lifespan differences. Aging 7, 1171–1184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Cecco M., Criscione S. W., Peterson A. L., Neretti N., Sedivy J. M., Kreiling J. A., Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging 5, 867–883 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dennis S., Sheth U., Feldman J. L., English K. A., Priess J. R., C. elegans germ cells show temperature and age-dependent expression of Cer1, a Gypsy/Ty3-related retrotransposon. PLOS Pathog. 8, e1002591 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li W., Prazak L., Chatterjee N., Gruninger S., Krug L., Theodorou D., Dubnau J., Activation of transposable elements during aging and neuronal decline in Drosophila. Nat. Neurosci. 16, 529–531 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maxwell P. H., Burhans W. C., Curcio M. J., Retrotransposition is associated with genome instability during chronological aging. Proc. Natl. Acad. Sci. U.S.A. 108, 20376–20381 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang J., Geesman G. J., Hostikka S. L., Atallah M., Blackwell B., Lee E., Cook P. J., Pasaniuc B., Shariat G., Halperin E., Dobke M., Rosenfeld M. G., King Jordan I., Lunyak V. V., Inhibition of activated pericentromeric SINE/Alu repeat transcription in senescent human adult stem cells reinstates self-renewal. Cell Cycle 10, 3016–3030 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wood J. G., Helfand S. L., Chromatin structure and transposable elements in organismal aging. Front. Genet. 4, 274 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Van Meter M., Kashyap M., Rezazadeh S., Geneva A. J., Morello T. D., Seluanov A., Gorbunova V., SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun. 5, 5011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sedivy J. M., Kreiling J. A., Neretti N., De Cecco M., Criscione S. W., Hofmann J. W., Zhao X., Ito T., Peterson A. L., Death by transposition–the enemy within? Bioessays 35, 1035–1043 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Criscione S. W., Zhang Y., Thompson W., Sedivy J. M., Neretti N., Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics 15, 583 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee E., Iskow R., Yang L., Gokcumen O., Haseley P., Luquette L. J. III, Lohr J. G., Harris C. C., Ding L., Wilson R. K., Wheeler D. A., Gibbs R. A., Kucherlapati R., Lee C., Kharchenko P. V., Park P. J., Landscape of somatic retrotransposition in human cancers. Science 337, 967–971 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reilly M. T., Faulkner G. J., Dubnau J., Ponomarev I., Gage F. H., The role of transposable elements in health and diseases of the central nervous system. J. Neurosci. 33, 17577–17586 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tan H., Qurashi A., Poidevin M., Nelson D. L., Li H., Jin P., Retrotransposon activation contributes to fragile X premutation rCGG-mediated neurodegeneration. Hum. Mol. Genet. 21, 57–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kamakaka R. T., Biggins S., Histone variants: Deviants? Genes Dev. 19, 295–310 (2005). [DOI] [PubMed] [Google Scholar]
- 79.Skene P. J., Henikoff S., Histone variants in pluripotency and disease. Development 140, 2513–2524 (2013). [DOI] [PubMed] [Google Scholar]
- 80.Filipescu D., Muller S., Almouzni G., Histone H3 variants and their chaperones during development and disease: Contributing to epigenetic control. Annu. Rev. Cell Dev. Biol. 30, 615–646 (2014). [DOI] [PubMed] [Google Scholar]
- 81.Duarte L. F., Young A. R. J., Wang Z., Wu H.-A., Panda T., Kou Y., Kapoor A., Hasson D., Mills N. R., Ma’ayan A., Narita M., Bernstein E., Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun. 5, 5210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maze I., Wenderski W., Noh K. M., Bagot R. C., Tzavaras N., Purushothaman I., Elsasser S. J., Guo Y., Ionete C., Hurd Y. L., Tamminga C. A., Halene T., Farrelly L., Soshnev A. A., Wen D., Rafii S., Birtwistle M. R., Akbarian S., Buchholz B. A., Blitzer R. D., Nestler E. J., Yuan Z.-F., Garcia B. A., Shen L., Molina H., David Allis C., Critical role of histone turnover in neuronal transcription and plasticity. Neuron 87, 77–94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Adams P. D., Remodeling of chromatin structure in senescent cells and its potential impact on tumor suppression and aging. Gene 397, 84–93 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang R., Poustovoitov M. V., Ye X., Santos H. A., Chen W., Daganzo S. M., Erzberger J. P., Serebriiskii I. G., Canutescu A. A., Dunbrack R. L., Pehrson J. R., Berger J. M., Kaufman P. D., Adams P. D., Formation of macroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 8, 19–30 (2005). [DOI] [PubMed] [Google Scholar]
- 85.Gaspar-Maia A., Qadeer Z. A., Hasson D., Ratnakumar K., Leu N. A., Leroy G., Liu S., Costanzi C., Valle-Garcia D., Schaniel C., Lemischka I., Garcia B., Pehrson J. R., Bernstein E., MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat. Commun. 4, 1565 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pasque V., Halley-Stott R. P., Gillich A., Garrett N., Gurdon J. B., Epigenetic stability of repressed states involving the histone variant macroH2A revealed by nuclear transfer to Xenopus oocytes. Nucleus 2, 533–539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kreiling J. A., Tamamori-Adachi M., Sexton A. N., Jeyapalan J. C., Munoz-Najar U., Peterson A. L., Manivannan J., Rogers E. S., Pchelintsev N. A., Adams P. D., Sedivy J. M., Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell 10, 292–304 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen H., Ruiz P. D., McKimpson W. M., Novikov L., Kitsis R. N., Gamble M. J., MacroH2A1 and ATM play opposing roles in paracrine senescence and the senescence-associated secretory phenotype. Mol. Cell 59, 719–731 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kozlowski M., Ladurner A. G., ATM, MacroH2A.1, and SASP: The checks and balances of cellular senescence. Mol. Cell 59, 713–715 (2015). [DOI] [PubMed] [Google Scholar]
- 90.Bannister A. J., Kouzarides T., Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kouzarides T., Chromatin modifications and their function. Cell 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Tan M., Luo H., Lee S., Jin F., Yang J. S., Montellier E., Buchou T., Cheng Z., Rousseaux S., Rajagopal N., Lu Z., Ye Z., Zhu Q., Wysocka J., Ye Y., Khochbin S., Ren B., Zhao Y., Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tsuchiya M., Dang N., Kerr E. O., Hu D., Steffen K. K., Oakes J. A., Kennedy B. K., Kaeberlein M., Sirtuin-independent effects of nicotinamide on lifespan extension from calorie restriction in yeast. Aging Cell 5, 505–514 (2006). [DOI] [PubMed] [Google Scholar]
- 94.Hachinohe M., Hanaoka F., Masumoto H., Hst3 and Hst4 histone deacetylases regulate replicative lifespan by preventing genome instability in Saccharomyces cerevisiae. Genes Cells 16, 467–477 (2011). [DOI] [PubMed] [Google Scholar]
- 95.D. W. Lamming, D. A. Sinclair, The regulation of lifespan by sirtuins in Saccharomyces cerevisiae, dissertation, Harvard University (2008). [Google Scholar]
- 96.Liu W. H.,Churchill M. E. A., Histone transfer among chaperones. Biochem. Soc. Trans. 40, 357–363 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Williams S. K., Truong D., Tyler J. K., Acetylation in the globular core of histone H3 on lysine-56 promotes chromatin disassembly during transcriptional activation. Proc. Natl. Acad. Sci. U.S.A. 105, 9000–9005 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xu F., Zhang K., Grunstein M., Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell 121, 375–385 (2005). [DOI] [PubMed] [Google Scholar]
- 99.Kaeberlein M., McVey M., Guarente L., The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guarente L., Calorie restriction and sirtuins revisited. Genes Dev. 27, 2072–2085 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wątroba M., Szukiewicz D., The role of sirtuins in aging and age-related diseases. Adv. Med. Sci. 61, 52–62 (2016). [DOI] [PubMed] [Google Scholar]
- 102.Lin Y.-., Lu J.-., Zhang J., Walter W., Dang W., Wan J., Tao S.-C., Qian J., Zhao Y., Boeke J. D., Berger S. L., Zhu H., Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell 136, 1073–1084 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim S., Benguria A., Lai C.-Y., Jazwinski S. M., Modulation of life-span by histone deacetylase genes in Saccharomyces cerevisiae. Mol. Biol. Cell 10, 3125–3136 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rogina B., Helfand S. L., Frankel S., Longevity regulation by Drosophila Rpd3 deacetylase and caloric restriction. Science 298, 1745 (2002). [DOI] [PubMed] [Google Scholar]
- 105.Frankel S., Woods J., Ziafazeli T., Rogina B., RPD3 histone deacetylase and nutrition have distinct but interacting effects on Drosophila longevity. Aging 7, 1112–1129 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ryu S. H., Kang K., Yoo T., Joe C. O., Chung J. H., Transcriptional repression of repeat-derived transcripts correlates with histone hypoacetylation at repetitive DNA elements in aged mice brain. Exp. Gerontol. 46, 811–818 (2011). [DOI] [PubMed] [Google Scholar]
- 107.Peleg S., Sananbenesi F., Zovoilis A., Burkhardt S., Bahari-Javan S., Agis-Balboa R. C., Cota P., Wittnam J. L., Gogol-Doering A., Opitz L., Salinas-Riester G., Dettenhofer M., Kang H., Farinelli L., Chen W., Fischer A., Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 328, 753–756 (2010). [DOI] [PubMed] [Google Scholar]
- 108.Wood J. G., Hillenmeyer S., Lawrence C., Chang C., Hosier S., Lightfoot W., Mukherjee E., Jiang N., Schorl C., Brodsky A. S., Neretti N., Helfand S. L., Chromatin remodeling in the aging genome of Drosophila. Aging Cell 9, 971–978 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang C. M., Tsai S. N., Yew T. W., Kwan Y. W., Ngai S. M., Identification of histone methylation multiplicities patterns in the brain of senescence-accelerated prone mouse 8. Biogerontology 11, 87–102 (2010). [DOI] [PubMed] [Google Scholar]
- 110.McCauley B. S., Dang W., Histone methylation and aging: Lessons learned from model systems. Biochim. Biophys. Acta 1839, 1454–1462 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bochkis I. M., Przybylski D., Chen J., Regev A., Changes in nucleosome occupancy associated with metabolic alterations in aged mammalian liver. Cell Rep. 9, 996–1006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cheung I., Shulha H. P., Jiang Y., Matevossian A., Wang J., Weng Z., Akbarian S., Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 107, 8824–8829 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shulha H. P., Cheung I., Guo Y., Akbarian S., Weng Z., Coordinated cell type–specific epigenetic remodeling in prefrontal cortex begins before birth and continues into early adulthood. PLOS Genet. 9, e1003433 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sun D., Luo M., Jeong M., Rodriguez B., Xia Z., Hannah R., Wang H., Le T., Faull K. F., Chen R., Gu H., Bock C., Meissner A., Göttgens B., Darlington G. J., Li W., Goodell M. A., Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 14, 673–688 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hamilton B., Dong Y., Shindo M., Liu W., I.Odell, Ruvkun G., Lee S. S., A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 19, 1544–1555 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Greer E. L., Maures T. J., Hauswirth A. G., Green E. M., Leeman D. S., Maro G. S., Han S., Banko M. R., Gozani O., Brunet A., Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 466, 383–387 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Han S., Brunet A., Histone methylation makes its mark on longevity. Trends Cell Biol. 22, 42–49 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li L., Greer C., Eisenman R. N., Secombe J., Essential functions of the histone demethylase lid. PLOS Genet. 6, e1001221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jin C., Li J., Green C. D., Yu X., Tang X., Han D., Xian B., Wang D., Huang X., Cao X., Yan Z., Hou L., Liu J., Shukeir N., Khaitovich P., Chen C. D., Zhang H., Jenuwein T., Han J.-D. J., Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab. 14, 161–172 (2011). [DOI] [PubMed] [Google Scholar]
- 120.Maures T. J., Greer E. L., Hauswirth A. G., Brunet A., The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell 10, 980–990 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Geisler S. J., Paro R., Trithorax and Polycomb group-dependent regulation: A tale of opposing activities. Development 142, 2876–2887 (2015). [DOI] [PubMed] [Google Scholar]
- 122.Siebold A. P., Banerjee R., Tie F., Kiss D. L., Moskowitz J., Harte P. J., Polycomb repressive complex 2 and trithorax modulate Drosophila longevity and stress resistance. Proc. Natl. Acad. Sci. U.S.A. 107, 169–174 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu B., Wang Z., Zhang L., Ghosh S., Zheng H., Zhou Z., Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat. Commun. 4, 1868 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sen P., Dang W., Donahue G., Dai J., Dorsey J., Cao X., Liu W., Cao K., Perry R., Lee J. Y., Wasko B. M., Carr D. T., He C., Robison B., Wagner J., Gregory B. D., Kaeberlein M., Kennedy B. K., Boeke J. D., Berger S. L., H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 29, 1362–1376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pu M., Ni Z., Wang M., Wang X., Wood J. G., Helfand S. L., Yu H., Lee S. S., Trimethylation of Lys36 on H3 restricts gene expression change during aging and impacts life span. Genes Dev. 29, 718–731 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rangaraju S., Solis G. M., Thompson R. C., Gomez-Amaro R. L., Kurian L., Encalada S. E., Niculescu A. B. III, Salomon D. R., Petrascheck M., Suppression of transcriptional drift extends C. elegans lifespan by postponing the onset of mortality. Elife 4, e08833 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.McCormick M. A., Mason A. G., Guyenet S. J., Dang W., Garza R. M., Ting M. K., Moller R. M., Berger S. L., Kaeberlein M., Pillus L., La Spada A. R., Kennedy B. K., The SAGA histone deubiquitinase module controls yeast replicative lifespan via Sir2 interaction. Cell Rep. 8, 477–486 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cao J., Yan Q., Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front. Oncol. 2, 26 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Aalfs J. D., Kingston R. E., What does ‘chromatin remodeling’ mean? Trends Biochem. Sci. 25, 548–555 (2000). [DOI] [PubMed] [Google Scholar]
- 130.Clapier C. R., Cairns B. R., The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 78, 273–304 (2009). [DOI] [PubMed] [Google Scholar]
- 131.Narlikar G. J., Sundaramoorthy R., Owen-Hughes T., Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 154, 490–503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pegoraro G., Kubben N., Wickert U., Göhler H., Hoffmann K., Misteli T., Ageing-related chromatin defects through loss of the NURD complex. Nat. Cell Biol. 11, 1261–1267 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.De Vaux V., Pfefferli C., Passannante M., Belhaj K., von Essen A., Sprecher S. G., Müller F., Wicky C., The Caenorhabditis elegans LET-418/Mi2 plays a conserved role in lifespan regulation. Aging Cell 12, 1012–1020 (2013). [DOI] [PubMed] [Google Scholar]
- 134.Dang W., Sutphin G. L., Dorsey J. A., Otte G. L., Cao K., Perry R. M., Wanat J. J., Saviolaki D., Murakami C. J., Tsuchiyama S., Robison B., Gregory B. D., Vermeulen M., Shiekhattar R., Johnson F. B., Kennedy B. K., Kaeberlein M., Berger S. L., Inactivation of yeast Isw2 chromatin remodeling enzyme mimics longevity effect of calorie restriction via induction of genotoxic stress response. Cell Metab. 19, 952–966 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Moynahan M. E., Jasin M., Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 11, 196–207 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Riedel C. G., Dowen R. H., Lourenco G. F., Kirienko N. V., Heimbucher T., West J. A., Bowman S. K., Kingston R. E., Dillin A., Asara J. M., Ruvkun G., DAF-16 employs the chromatin remodeller SWI/SNF to promote stress resistance and longevity. Nat. Cell Biol. 15, 491–501 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jung M., Pfeifer G. P., Aging and DNA methylation. BMC Biol. 13, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bjornsson H. T., Sigurdsson M. I., Fallin M. D., Irizarry R. A., Aspelund T., Cui H., Yu W., Rongione M. A., Ekström T. J., Harris T. B., Launer L. J., Eiriksdottir G., Leppert M. F., Sapienza C., Gudnason V., Feinberg A. P., Intra-individual change over time in DNA methylation with familial clustering. JAMA 299, 2877–2883 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bollati V., Schwartz J., Wright R., Litonjua A., Tarantini L., Suh H., Sparrow D., Vokonas P., Baccarelli A., Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech. Ageing Dev. 130, 234–239 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bormann F., Rodríguez-Paredes M., Hagemann S., Manchanda H., Kristof B., Gutekunst J., Raddatz G., Haas R., Terstegen L., Wenck H., Kaderali L., Winnefeld M., Lyko F., Reduced DNA methylation patterning and transcriptional connectivity define human skin aging. Aging Cell 15, 563–571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Christensen B. C., Houseman E. A., Marsit C. J., Zheng S., Wrensch M. R., Wiemels J. L., Nelson H. H., Karagas M. R., Padbury J. F., Bueno R., Sugarbacker D. J., Yeh R.-F., Wiencke J. K., Kelsey K. T., Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLOS Genet. 5, e1000602 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Horvath S., DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jintaridth P., Mutirangura A., Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol. Genomics 41, 194–200 (2010). [DOI] [PubMed] [Google Scholar]
- 144.Zampieri M., Ciccarone F., Calabrese R., Franceschi C., Burkle A., Caiafa P., Reconfiguration of DNA methylation in aging. Mech. Ageing Dev. 151, 60–70 (2015). [DOI] [PubMed] [Google Scholar]
- 145.Liu L., van Groen T., Kadish I., Li Y., Wang D., James S. R., Karpf A. R., Tollefsbol T. O., Insufficient DNA methylation affects healthy aging and promotes age-related health problems. Clin. Epigenetics 2, 349–360 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wei L., Liu B., Tuo J., Shen D., Chen P., Li Z., Liu X., Ni J., Dagur P., Sen H. N., Jawad S., Ling D., Park S., Chakrabarty S., Meyerle C., Agron E., Ferris F. L. III, Chew E. Y., Philip McCoy J., Blum E., Francis P. J., Klein M. L., Guymer R. H., Baird P. N., Chan C.-C., Nussenblatt R. B., Hypomethylation of the IL17RC promoter associates with age-related macular degeneration. Cell Rep. 2, 1151–1158 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang Z., Deng C., Lu Q., Richardson B., Age-dependent DNA methylation changes in the ITGAL (CD11a) promoter. Mech. Ageing Dev. 123, 1257–1268 (2002). [DOI] [PubMed] [Google Scholar]
- 148.Cedar H., Bergman Y., Programming of DNA methylation patterns. Annu. Rev. Biochem. 81, 97–117 (2012). [DOI] [PubMed] [Google Scholar]
- 149.Weidner C. I., Wagner W., The epigenetic tracks of aging. Biol. Chem. 395, 1307–1314 (2014). [DOI] [PubMed] [Google Scholar]
- 150.Avrahami D., Li C., Zhang J., Schug J., Avrahami R., Rao S., Stadler M. B., Burger L., Schübeler D., Glaser B., Kaestner K. H., Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved β cell function. Cell Metab. 22, 619–632 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Beerman I., Bock C., Garrison B. S., Smith Z. D., Gu H., Meissner A., Rossi D. J., Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 12, 413–425 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yuan T., Jiao Y., de Jong S., Ophoff R. A., Beck S., Teschendorff A. E., An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging. PLOS Genet. 11, e1004996 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Heyn H., Li N., Ferreira H. J., Moran S., Pisano D. G., Gomez A., Diez J., Sanchez-Mut J. V., Setien F., Carmona F. J., Puca A. A., Sayols S., Pujana M. A., Serra-Musach J., Iglesias-Platas I., Formiga F., Fernandez A. F., Fraga M. F., Heath S. C., Valencia A., Gut I. G., Wang J., Esteller M., Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. U.S.A. 109, 10522–10527 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Weidner C. I., Lin Q., Koch C. M., Eisele L., Beier F., Ziegler P., Bauerschlag D. O., Jöckel K. H., Erbel R., Mühleisen T. W., Zenke M., Henrik Brümmendorf T., Wagner W., Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 15, R24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bocker M. T., Hellwig I., Breiling A., Eckstein V., Ho A. D., Lyko F., Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood 117, e182–e189 (2011). [DOI] [PubMed] [Google Scholar]
- 156.Maegawa S., Hinkal G., Kim H. S., Shen L., Zhang L., Zhang J., Zhang N., Liang S., Donehower L. A., Issa J.-P. J., Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 20, 332–340 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Teschendorff A. E., Menon U., Gentry-Maharaj A., Ramus S. J., Weisenberger D. J., Shen H., Campan M., Noushmehr H., Bell C. G., Maxwell A. P., Savage D. A., Mueller-Holzner E., Marth C., Kocjan G., Gayther S. A., Jones A., Beck S., Wagner W., Laird P. W., Jacobs I. J., Widschwendter M., Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 20, 440–446 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.McClay J. L., Aberg K. A., Clark S. L., Nerella S., Kumar G., Xie L. Y., Hudson A. D., Harada A., Hultman C. M., Magnusson P. K., Sullivan P. F., Van Den Oord E. J. C. G., A methylome-wide study of aging using massively parallel sequencing of the methyl-CpG-enriched genomic fraction from blood in over 700 subjects. Hum. Mol. Genet. 23, 1175–1185 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Raddatz G., Hagemann S., Aran D., Söhle J., Kulkarni P. P., Kaderali L., Hellman A., Winnefeld M., Lyko F., Aging is associated with highly defined epigenetic changes in the human epidermis. Epigenetics Chromatin 6, 36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Koch C. M., Joussen S., Schellenberg A., Lin Q., Zenke M., Wagner W., Monitoring of cellular senescence by DNA-methylation at specific CpG sites. Aging Cell 11, 366–369 (2012). [DOI] [PubMed] [Google Scholar]
- 161.Ions L. J., Wakeling L. A., Bosomworth H. J., Hardyman J. E. J., Escolme S. M., Swan D. C., Valentine R. A., Mathers J. C., Ford D., Effects of Sirt1 on DNA methylation and expression of genes affected by dietary restriction. Age 35, 1835–1849 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wakeling L. A., Ions L. J., Escolme S. M., Cockell S. J., Su T., Dey M., Hampton E. V., Jenkins G., Wainwright L. J., McKay J. A., Ford D., SIRT1 affects DNA methylation of polycomb group protein target genes, a hotspot of the epigenetic shift observed in ageing. Hum. Genomics 9, 14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Finkel T., Serrano M., Blasco M. A., The common biology of cancer and ageing. Nature 448, 767–774 (2007). [DOI] [PubMed] [Google Scholar]
- 164.Gautrey H. E., van Otterdijk S. D., Cordell H. J.; Newcastle 85+ Study Core Team, Mathers J. C., Strathdee G., DNA methylation abnormalities at gene promoters are extensive and variable in the elderly and phenocopy cancer cells. FASEB J. 28, 3261–3272 (2014). [DOI] [PubMed] [Google Scholar]
- 165.ENCODE Project Consortium, Birney E., Stamatoyannopoulos J. A., Dutta A., Guigo R., Gingeras T. R., Margulies E. H., Weng Z., Snyder M., Dermitzakis E. T., Thurman R. E., Kuehn M. S., Taylor C. M., Neph S., Koch C. M., Asthana S., Malhotra A., Adzhubei I., Greenbaum J. A., Andrews R. M., Flicek P., Boyle P. J., Cao H., Carter N. P., Clelland G. K., Davis S., Day N., Dhami P., Dillon S. C., Dorschner M. O., Fiegler H., Giresi P. G., Goldy J., Hawrylycz M., Haydock A., Humbert R., James K. D., Johnson B. E., Johnson E. M., Frum T. T., Rosenzweig E. R., Karnani N., Lee K., Lefebvre G. C., Navas P. A., Neri F., Parker S. C., Sabo P. J., Sandstrom R., Shafer A., Vetrie D., Weaver M., Wilcox S., Yu M., Collins F. S., Dekker J., Lieb J. D., Tullius T. D., Crawford G. E., Sunyaev S., Noble W. S., Dunham I., Denoeud F., Reymond A., Kapranov P., Rozowsky J., Zheng D., Castelo R., Frankish A., Harrow J., Ghosh S., Sandelin A., Hofacker I. L., Baertsch R., Keefe D., Dike S., Cheng J., Hirsch H. A., Sekinger E. A., Lagarde J., Abril J. F., Shahab A., Flamm C., Fried C., Hackermüller J., Hertel J., Lindemeyer M., Missal K., Tanzer A., Washietl S., Korbel J., Emanuelsson O., Pedersen J. S., Holroyd N., Taylor R., Swarbreck D., Matthews N., Dickson M. C., Thomas D. J., Weirauch M. T., Gilbert J., Drenkow J., Bell I., Zhao X., Srinivasan K. G., Sung W. K., Ooi H. S., Chiu K. P., Foissac S., Alioto T., Brent M., Pachter L., Tress M. L., Valencia A., Choo S. W., Choo C. Y., Ucla C., Manzano C., Wyss C., Cheung E., Clark T. G., Brown J. B., Ganesh M., Patel S., Tammana H., Chrast J., Henrichsen C. N., Kai C., Kawai J., Nagalakshmi U., Wu J., Lian Z., Lian J., Newburger P., Zhang X., Bickel P., Mattick J. S., Carninci P., Hayashizaki Y., Weissman S., Hubbard T., Myers R. M., Rogers J., Stadler P. F., Lowe T. M., Wei C. L., Ruan Y., Struhl K., Gerstein M., Antonarakis S. E., Fu Y., Green E. D., Karaöz U., Siepel A., Taylor J., Liefer L. A., Wetterstrand K. A., Good P. J., Feingold E. A., Guyer M. S., Cooper G. M., Asimenos G., Dewey C. N., Hou M., Nikolaev S., Montoya-Burgos J. I., Löytynoja A., Whelan S., Pardi F., Massingham T., Huang H., Zhang N. R., Holmes I., Mullikin J. C., Ureta-Vidal A., Paten B., Seringhaus M., Church D., Rosenbloom K., Kent W. J., Stone E. A.; NISC Comparative Sequencing Program, Baylor College of Medicine Human Genome Sequencing Center, Washington University Genome Sequencing Center, Broad Institute, Children’s Hospital Oakland Research Institute, Batzoglou S., Goldman N., Hardison R. C., Haussler D., Miller W., Sidow A., Trinklein N. D., Zhang Z. D., Barrera L., Stuart R., King D. C., Ameur A., Enroth S., Bieda M. C., Kim J., Bhinge A. A., Jiang N., Liu J., Yao F., Vega V. B., Lee C. W., Ng P., Shahab A., Yang A., Moqtaderi Z., Zhu Z., Xu X., Squazzo S., Oberley M. J., Inman D., Singer M. A., Richmond T. A., Munn K. J., Rada-Iglesias A., Wallerman O., Komorowski J., Fowler J. C., Couttet P., Bruce A. W., Dovey O. M., Ellis P. D., Langford C. F., Nix D. A., Euskirchen G., Hartman S., Urban A. E., Kraus P., Van Calcar S., Heintzman N., Kim T. H., Wang K., Qu C., Hon G., Luna R., Glass C. K., Rosenfeld M. G., Aldred S. F., Cooper S. J., Halees A., Lin J. M., Shulha H. P., Zhang X., Xu M., Haidar J. N., Yu Y., Ruan Y., Iyer V. R., Green R. D., Wadelius C., Farnham P. J., Ren B., Harte R. A., Hinrichs A. S., Trumbower H., Clawson H., Hillman-Jackson J., Zweig A. S., Smith K., Thakkapallayil A., Barber G., Kuhn R. M., Karolchik D., Armengol L., Bird C. P., de Bakker P. I., Kern A. D., Lopez-Bigas N., Martin J. D., Stranger B. E., Woodroffe A., Davydov E., Dimas A., Eyras E., Hallgrímsdóttir I. B., Huppert J., Zody M. C., Abecasis G. R., Estivill X., Bouffard G. G., Guan X., Hansen N. F., Idol J. R., Maduro V. V., Maskeri B., McDowell J. C., Park M., Thomas P. J., Young A. C., Blakesley R. W., Muzny D. M., Sodergren E., Wheeler D. A., Worley K. C., Jiang H., Weinstock G. M., Gibbs R. A., Graves T., Fulton R., Mardis E. R., Wilson R. K., Clamp M., Cuff J., Gnerre S., Jaffe D. B., Chang J. L., Lindblad-Toh K., Lander E. S., Koriabine M., Nefedov M., Osoegawa K., Yoshinaga Y., Zhu B., de Jong P. J., Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wilusz J. E., Sunwoo H., Spector D. L., Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 23, 1494–1504 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Djebali S., Davis C. A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., Xue C., Marinov G. K., Khatun J., Williams B. A., Zaleski C., Rozowsky J., Röder M., Kokocinski F., Abdelhamid R. F., Alioto T., Antoshechkin I., Baer M. T., Bar N. S., Batut P., Bell K., Bell I., Chakrabortty S., Chen X., Chrast J., Curado J., Derrien T., Drenkow J., Dumais E., Dumais J., Duttagupta R., Falconnet E., Fastuca M., Fejes-Toth K., Ferreira P., Foissac S., Fullwood M. J., Gao H., Gonzalez D., Gordon A., Gunawardena H., Howald C., Jha S., Johnson R., Kapranov P., King B., Kingswood C., Luo O. J., Park E., Persaud K., Preall J. B., Ribeca P., Risk B., Robyr D., Sammeth M., Schaffer L., See L.-H., Shahab A., Skancke J., Maria Suzuki A., Takahashi H., Tilgner H., Trout D., Walters N., Wang H., Wrobel J., Yu Y., Ruan X., Hayashizaki Y., Harrow J., Gerstein M., Hubbard T., Reymond A., Antonarakis S. E., Hannon G., Giddings M. C., Ruan Y., Wold B., Carninci P., Guigó R., Gingeras T. R., Landscape of transcription in human cells. Nature 489, 101–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hangauer M. J., Vaughn I. W., McManus M. T., Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLOS Genet. 9, e1003569 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Prasanth K. V., Spector D. L., Eukaryotic regulatory RNAs: An answer to the ‘genome complexity’ conundrum. Genes Dev. 21, 11–42 (2007). [DOI] [PubMed] [Google Scholar]
- 170.Cech T. R., Steitz J. A., The noncoding RNA revolution—Trashing old rules to forge new ones. Cell 157, 77–94 (2014). [DOI] [PubMed] [Google Scholar]
- 171.Costa F. F., Non-coding RNAs: Meet thy masters. Bioessays 32, 599–608 (2010). [DOI] [PubMed] [Google Scholar]
- 172.Esteller M., Non-coding RNAs in human disease. Nat. Rev. Genet. 12, 861–874 (2011). [DOI] [PubMed] [Google Scholar]
- 173.Szafranski K., Abraham K. J., Mekhail K., Non-coding RNA in neural function, disease, and aging. Front. Genet. 6, 87 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Saka K., Ide S., Ganley A. R. D., Kobayashi T., Cellular senescence in yeast is regulated by rDNA noncoding transcription. Curr. Biol. 23, 1794–1798 (2013). [DOI] [PubMed] [Google Scholar]
- 175.Mori M. A., Raghavan P., Thomou T., Boucher J., Robida-Stubbs S., Macotela Y., Russell S. J., Kirkland J. L., Blackwell T. K., Kahn C. R., Role of microRNA processing in adipose tissue in stress defense and longevity. Cell Metab. 16, 336–347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ibíñez-Ventoso C., Yang M., Guo S., Robins H., Padgett R. W., Driscoll M., Modulated microRNA expression during adult lifespan in Caenorhabditis elegans. Aging Cell 5, 235–246 (2006). [DOI] [PubMed] [Google Scholar]
- 177.Kato M., Chen X., Inukai S., Zhao H., Slack F. J., Age-associated changes in expression of small, noncoding RNAs, including microRNAs, in C. elegans. RNA 17, 1804–1820 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Boehm M., Slack F., A developmental timing microRNA and its target regulate life span in C. elegans. Science 310, 1954–1957 (2005). [DOI] [PubMed] [Google Scholar]
- 179.de Lencastre A., Pincus Z., Zhou K., Kato M., Lee S. S., Slack F. J., MicroRNAs both promote and antagonize longevity in C. elegans. Curr. Biol. 20, 2159–2168 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pincus Z., Smith-Vikos T., Slack F. J., MicroRNA predictors of longevity in Caenorhabditis elegans. PLOS Genet. 7, e1002306 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Jung H. J., Suh Y., MicroRNA in aging: From discovery to biology. Curr. Genomics 13, 548–557 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Li N., Bates D. J., An J., Terry D. A., Wang E., Up-regulation of key microRNAs, and inverse down-regulation of their predicted oxidative phosphorylation target genes, during aging in mouse brain. Neurobiol. Aging 32, 944–955 (2011). [DOI] [PubMed] [Google Scholar]
- 183.Noren Hooten N. N., Abdelmohsen K., Gorospe M., Ejiogu N., Zonderman A. B., Evans M. K., microRNA expression patterns reveal differential expression of target genes with age. PLOS One 5, e10724 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhang J., Liu Q., Zhang W., Li J., Li Z., Tang Z., Li Y., Han C., Hall S. H., Zhang Y., Comparative profiling of genes and miRNAs expressed in the newborn, young adult, and aged human epididymides. Acta Biochim. Biophys. Sin. 42, 145–153 (2010). [DOI] [PubMed] [Google Scholar]
- 185.Liu N., Landreh M., Cao K., Abe M., Hendriks G.-J., Kennerdell J. R., Zhu Y., Wang L.-S., Bonini N. M., The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 482, 519–523 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wang W.-X., Huang Q., Hu Y., Stromberg A. J., Nelson P. T., Patterns of microRNA expression in normal and early Alzheimer’s disease human temporal cortex: White matter versus gray matter. Acta Neuropathol. 121, 193–205 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wang X., Liu P., Zhu H., Xu Y., Ma C., Dai X., Huang L., Liu Y., Zhang L., Qin C., miR-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer’s disease, inhibits bcl2 translation. Brain Res. Bull. 80, 268–273 (2009). [DOI] [PubMed] [Google Scholar]
- 188.Lee J., Padhye A., Sharma A., Song G., Miao J., Mo Y.-Y., Wang L., Kemper J. K., A pathway involving farnesoid X receptor and small heterodimer partner positively regulates hepatic sirtuin 1 levels via microRNA-34a inhibition. J. Biol. Chem. 285, 12604–12611 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Meier I., Fellini L., Jakovcevski M., Schachner M., Morellini F., Expression of the snoRNA host gene gas5 in the hippocampus is upregulated by age and psychogenic stress and correlates with reduced novelty-induced behavior in C57BL/6 mice. Hippocampus 20, 1027–1036 (2010). [DOI] [PubMed] [Google Scholar]
- 190.Monnier P., Martinet C., Pontis J., Stancheva I., Ait-Si-Ali S., Dandolo L., H19 lncRNA controls gene expression of the imprinted gene network by recruiting MBD1. Proc. Natl. Acad. Sci. U.S.A. 110, 20693–20698 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Fu V. X., Dobosy J. R., Desotelle J. A., Almassi N., Ewald J. A., Srinivasan R., Berres M., Svaren J., Weindruch R., Jarrard D. F., Aging and cancer-related loss of insulin-like growth factor 2 imprinting in the mouse and human prostate. Cancer Res. 68, 6797–6802 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Abdelmohsen K., Panda A., Kang M.-J., Xu J., Selimyan R., Yoon J.-H., Martindale J. L., De S., Wood W. H. III, Becker K. G., Gorospe M., Senescence-associated lncRNAs: Senescence-associated long noncoding RNAs. Aging Cell 12, 890–900 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kour S., Rath P. C., Long noncoding RNAs in aging and age-related diseases. Ageing Res. Rev. 26, 1–21 (2015). [DOI] [PubMed] [Google Scholar]
- 194.Johnson R., Long non-coding RNAs in Huntington’s disease neurodegeneration. Neurobiol. Dis. 46, 245–254 (2012). [DOI] [PubMed] [Google Scholar]
- 195.Soldati C., Bithell A., Johnston C., Wong K.-Y., Stanton L. W., Buckley N. J., Dysregulation of REST-regulated coding and non-coding RNAs in a cellular model of Huntington’s disease. J. Neurochem. 124, 418–430 (2013). [DOI] [PubMed] [Google Scholar]
- 196.Castel S. E., Martienssen R. A., RNA interference in the nucleus: Roles for small RNAs in transcription, epigenetics and beyond. Nat. Rev. Genet. 14, 100–112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Cohen A. L., Jia S., Noncoding RNAs and the borders of heterochromatin. Wiley Interdiscip. Rev. RNA 5, 835–847 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Greer E. L., Maures T. J., Ucar D., Hauswirth A. G., Mancini E., Lim J. P., Benayoun B. A., Shi Y., Brunet A., Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479, 365–371 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lim J. P., Brunet A., Bridging the transgenerational gap with epigenetic memory. Trends Genet. 29, 176–186 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Huypens P., Sass S., Wu M., Dyckhoff D., Tschöp M., Theis F., Marschall S., Hrabě de Angelis M., Beckers J., Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat. Genet. 48, 497–499 (2016). [DOI] [PubMed] [Google Scholar]
- 201.Li Y., Daniel M., Tollefsbol T. O., Epigenetic regulation of caloric restriction in aging. BMC Med. 9, 98 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Vaquero A., Reinberg D., Calorie restriction and the exercise of chromatin. Genes Dev. 23, 1849–1869 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wood S. H., van Dam S., Craig T., Tacutu R., O’Toole A., Merry B. J., de Magalhães J. P., Transcriptome analysis in calorie-restricted rats implicates epigenetic and post-translational mechanisms in neuroprotection and aging. Genome Biol. 16, 28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Blagosklonny M. V., Calorie restriction: Decelerating mTOR-driven aging from cells to organisms (including humans). Cell Cycle 9, 683–688 (2010). [DOI] [PubMed] [Google Scholar]
- 205.Ingram D. K., Roth G. S., Calorie restriction mimetics: Can you have your cake and eat it, too? Ageing Res. Rev. 20, 46–62 (2015). [DOI] [PubMed] [Google Scholar]
- 206.Ingram D. K., Zhu M., Mamczarz J., Zou S., Lane M. A., Roth G. S., deCabo R., Calorie restriction mimetics: An emerging research field. Aging Cell 5, 97–108 (2006). [DOI] [PubMed] [Google Scholar]
- 207.Hubbard B. P., Sinclair D. A., Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 35, 146–154 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Pearson K. J., Baur J. A., Lewis K. N., Peshkin L., Price N. L., Labinskyy N., Swindell W. R., Kamara D., Minor R. K., Perez E., Jamieson H. A., Zhang Y., Dunn S. R., Sharma K., Pleshko N., Woollett L. A., Csiszar A., Ikeno Y., Le Couteur D., Elliott P. J., Becker K. G., Navas P., Ingram D. K., Wolf N. S., Ungvari Z., Sinclair D. A., de Cabo R., Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 8, 157–168 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Howitz K. T., Bitterman K. J., Cohen H. Y., Lamming D. W., Lavu S., Wood J. G., Zipkin R. E., Chung P., Kisielewski A., Zhang L.-L., Scherer B., Sinclair D. A., Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196 (2003). [DOI] [PubMed] [Google Scholar]
- 210.Cao D., Wang M., Qiu X., Liu D., Jiang H., Yang N., Xu R.-M., Structural basis for allosteric, substrate-dependent stimulation of SIRT1 activity by resveratrol. Genes Dev. 29, 1316–1325 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Dai H., Case A. W., Riera T. V., Considine T., Lee J. E., Hamuro Y., Zhao H., Jiang Y., Sweitzer S. M., Pietrak B., Schwartz B., Blum C. A., Disch J. S., Caldwell R., Szczepankiewicz B., Oalmann C., Ng P. Y., White B. H., Casaubon R., Narayan R., Koppetsch K., Bourbonais F., Wu B., Wang J., Qian D., Jiang F., Mao C., Wang M., Hu E., Wu J. C., Perni R. B., Vlasuk G. P., Ellis J. L., Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat. Commun. 6, 7645 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Gertz M., Nguyen G. T. T., Fischer F., Suenkel B., Schlicker C., Fränzel B., Tomaschewski J., Aladini F., Becker C., Wolters D., Steegborn C., A molecular mechanism for direct sirtuin activation by resveratrol. PLOS One 7, e49761 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Hubbard B. P., Gomes A. P., Dai H., Li J., Case A. W., Considine T., Riera T. V., Lee J. E., S. Y. E, Lamming D. W., Pentelute B. L., Schuman E. R., Stevens L. A., Ling A. J. Y., Armour S. M., Michan S., Zhao H., Jiang Y., Sweitzer S. M., Blum C. A., Disch J. S., Yee Ng P., Howitz K. T., Rolo A. P., Hamuro Y., Moss J., Perni R. B., Ellis J. L., Vlasuk G. P., Sinclair D. A., Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339, 1216–1219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lakshminarasimhan M., Rauh D., Schutkowski M., Steegborn C., Sirt1 activation by resveratrol is substrate sequence-selective. Aging 5, 151–154 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Minor R. K., Baur J. A., Gomes A. P., Ward T. M., Csiszar A., Mercken E. M., Abdelmohsen K., Shin Y. K., Canto C., Scheibye-Knudsen M., Krawczyk M., Irusta P. M., Martín-Montalvo A., Hubbard B. P., Zhang Y., Lehrmann E., White A. A., Price N. L., Swindell W. R., Pearson K. J., Becker K. G., Bohr V. A., Gorospe M., Egan J. M., Talan M. I., Auwerx J., Westphal C. H., Ellis J. L., Ungvari Z., Vlasuk G. P., Elliott P. J., Sinclair D. A., de Cabo R., SRT1720 improves survival and healthspan of obese mice. Sci. Rep. 1, 70 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Anisimov V. N., Metformin: Do we finally have an anti-aging drug? Cell Cycle 12, 3483–3489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Cabreiro F., Au C., Leung K.-Y., Vergara-Irigaray N., Cochemé H. M., Noori T., Weinkove D., Schuster E., Greene N. D. E., Gems D., Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 153, 228–239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Onken B., Driscoll M., Metformin induces a dietary restriction–like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN-1. PLOS One 5, e8758 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Martin-Montalvo A., Mercken E. M., Mitchell S. J., Palacios H. H., Mote P. L., Scheibye-Knudsen M., Gomes A. P., Ward T. M., Minor R. K., Blouin M. J., Schwab M., Pollak M., Zhang Y., Yu Y., Becker K. G., Bohr V. A., Ingram D. K., Sinclair D. A., Wolf N. S., Spindler S. R., Bernier M., de Cabo R., Metformin improves healthspan and lifespan in mice. Nat. Commun. 4, 2192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Bannister C. A., Holden S. E., Jenkins-Jones S., Morgan C. L., Halcox J. P., Schernthaner G., Mukherjee J., Currie C. J., Can people with type 2 diabetes live longer than those without? A comparison of mortality in people initiated with metformin or sulphonylurea monotherapy and matched, non-diabetic controls. Diabetes Obes. Metab. 16, 1165–1173 (2014). [DOI] [PubMed] [Google Scholar]
- 221.de Kreutzenberg S. V., Ceolotto G., Cattelan A., Pagnin E., Mazzucato M., Garagnani P., Borelli V., Bacalini M. G., Franceschi C., Fadini G. P., Avogaro A., Metformin improves putative longevity effectors in peripheral mononuclear cells from subjects with prediabetes. A randomized controlled trial. Nutr. Metab. Cardiovasc. Dis. 25, 686–693 (2015). [DOI] [PubMed] [Google Scholar]
- 222.Eisenberg T., Knauer H., Schauer A., Buttner S., Ruckenstuhl C., Carmona-Gutierrez D., Ring J., Schroeder S., Magnes C., Antonacci L., Fussi H., Deszcz L., Hartl R., Schraml E., Criollo A., Megalou E., Weiskopf D., Laun P., Heeren G., Breitenbach M., Grubeck-Loebenstein B., Herker E., Fahrenkrog B., Fröhlich K.-U., Sinner F., Tavernarakis N., Minois N., Kroemer G., Made F., Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314 (2009). [DOI] [PubMed] [Google Scholar]
- 223.Mariño G., Pietrocola F., Eisenberg T., Kong Y., Malik S. A., Andryushkova A., Schroeder S., Pendl T., Harger A., Niso-Santano M., Zamzami N., Scoazec M., Durand S., Enot D. P., Fernández Á. F., Martins I., Kepp O., Senovilla L., Bauvy C., Morselli E., Vacchelli E., Bennetzen M., Magnes C., Sinner F., Pieber T., López-Otín C., Chiara Maiuri M., Codogno P., Andersen J. S., Hill J. A., Madeo F., Kroemer G., Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell 53, 710–725 (2014). [DOI] [PubMed] [Google Scholar]
- 224.Eisenberg T., Schroeder S., Andryushkova A., Pendl T., Kuttner V., Bhukel A., Marino G., Pietrocola F., Harger A., Zimmermann A., Moustafa T., Sprenger A., Jany E., Büttner S., Carmona-Gutierrez D., Ruckenstuhl C., Ring J., Reichelt W., Schimmel K., Leeb T., Moser C., Schatz S., Kamolz L.-P., Magnes C., Sinner F., Sedej S., Fröhlich K.-U., Juhasz G., Pieber T. R., Dengjel J., Sigrist S. J., Kroemer G., Madeo F., Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab. 19, 431–444 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Gelino S., Hansen M., Autophagy—An emerging anti-aging mechanism. J. Clin. Exp. Pathol. (Suppl. 4), pii: 006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Madeo F., Tavernarakis N., Kroemer G., Can autophagy promote longevity? Nat. Cell Biol. 12, 842–846 (2010). [DOI] [PubMed] [Google Scholar]
- 227.Füllgrabe J., Lynch-Day M. A., Heldring N., Li W., Struijk R. B., Ma Q., Hermanson O., Rosenfeld M. G., Klionsky D. J., Joseph B., The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 500, 468–471 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ng F., Tang B. L., Sirtuins’ modulation of autophagy. J. Cell. Physiol. 228, 2262–2270 (2013). [DOI] [PubMed] [Google Scholar]
- 229.Morselli E., Maiuri M. C., Markaki M., Megalou E., Pasparaki A., Palikaras K., Criollo A., Galluzzi L., Malik S. A., Vitale I., Michaud M., Madeo F., Tavernarakis N., Kroemer G., Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 1, e10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Roos C. M., Zhang B., Palmer A. K., Ogrodnik M. B., Pirtskhalava T., Thalji N. M., Hagler M., Jurk D., Smith L. A., Casaclang-Verzosa G., Zhu Y., Schafer M. J., Tchkonia T., Kirkland J. L., Miller J. D., Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 10.1111/acel.12458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]