

Summary
Systemic infection with Streptococcus pneumoniae is associated with a vigorous pro-inflammatory response to structurally complex cell wall fragments (PnCW) that are shed during cell growth and antibiotic-induced autolysis. Consistent with previous studies, inflammatory cytokine production induced by PnCW was dependent on TLR2 but independent of NOD2, a cytoplasmic NLR protein. However, in parallel with the pro-inflammatory response, we found that PnCW also induced prodigious secretion of anti-inflammatory IL-10 from macrophages. This response was dependent on TLR2, but also involved NOD2 as absence of NOD2-reduced IL-10 secretion in response to cell wall and translated into diminished downstream effects on IL-10-regulated target gene expression. PnCW-mediated production of IL-10 via TLR2 required RIPK2 a kinase required for NOD2 function, and MyD88 but differed from that known for zymosan in that ERK pathway activation was not detected. As mutations in NOD2 are linked to aberrant immune responses, the temporal and quantitative effects of activation of the TLR2-NOD2-RIPK2 pathway on IL-10 secretion may affect the balance between pro- and anti-inflammatory responses to Gram-positive bacteria.
Introduction
IL-10 secretion from T cells, macrophages and dendritic cells is an essential regulatory mechanism to temper excessive inflammatory responses (Murray, 2006). Mice lacking IL-10 are extremely sensitive to a wide variety of pro-inflammatory stimuli including LPS, and systemic infections with bacteria and parasites that provoke a strong inflammatory response (Murray, 2006). In all cases, IL-10 is required to restrain the inflammatory response and protect the host against the damaging effects of pro-inflammatory cytokines. Systemic infection with S. pneumoniae provokes a generalized inflammatory response linked with devastating neurological sequelae and a fatality rate of ~15% (Tuomanen et al., 1995). Administration of purified pneumococcal cell wall (PnCW) recapitulates the majority of the inflammatory responses observed in systemic S. pneumoniae infection, suggesting that the pro-inflammatory properties of PnCW are a primary driver of inflammatory pathology (Moreillon and Majcherczyk, 2003; Fillon et al., 2006; Orihuela et al., 2006). Because PnCW is mainly composed of peptidogly-can, teichoic acid, cell wall linked proteins and lipoteichoic acids, TLR2 is considered especially important in the process of PnCW detection by macrophages and den-dritic cells that are then stimulated to produce inflammatory cytokines (Yoshimura et al., 1999). IL-10 can modulate PnCW induced inflammation (Orihuela et al., 2006), but the mechanism leading to IL-10 production is entirely unclear.
Modulation of inflammation has been suggested to involve NOD2, a cytoplasmic protein of the larger NLR family (Inohara and Nunez, 2003; Inohara et al., 2005; Murray, 2005; Strober et al., 2006; Kanneganti et al., 2007). For example, single nucleotide polymorphisms in NOD2 have been observed at an increased frequency in Crohn’s disease (Lesage et al., 2002). However, several loss-of-function mice in the Nod2 gene do not have gut immunopathology when maintained under normal conditions (Pauleau and Murray, 2003; Kobayashi et al., 2005; Maeda et al., 2005; Mariathasan et al., 2006; Barreau et al., 2007). These data are consistent with the fact that some people have mutations in one or both NOD2 alleles but have no clinical inflammatory bowel disease (Hugot et al., 2007). Thus NOD2 likely plays a role in more complex pathways modulating pro-inflammatory responses (Strober et al., 2007; Xavier and Podolsky, 2007).
NOD2-deficient mice, their immune cells and human cells bearing predicted loss-of-function NOD2 alleles, are non-responsive to muramyl dipeptide (MDP), a ‘minimal’ component of bacterial peptidoglycan able to induce pro-inflammatory responses. A second pathway of MDP sensing appears to be involved in IL-1β processing and is mediated by NLR family members Cryopyrin (NLRP3) and Nalp1 (NLRP1) (Faustin et al., 2007; Pan et al., 2007; Marina-Garcia et al., 2008). At this stage it is unclear whether human cells bearing two copies of the NOD2 frame-shift mutation are completely non-responsive to MDP because the truncated mutant protein should still be expressed. Compared with other bacterial components such as LPS, peptidoglycan or CpG DNA, MDP is a very weak agonist of myeloid-derived cells, especially when considered by molar comparison. However, MDP synergizes with other TLR agonists to stimulate cytokine, chemokine and nitric oxide production. This effect is the basis of variants of the ‘MDP synergy’ assay, a common ‘readout’ of NOD2 function. In contrast to the lack of MDP responsiveness, macrophages from all NOD2-deficient mice have been generally reported to have ‘normal’ responses to highly defined TLR agonists. At present, the specific roles of MDP, the origin of MDP in mammalian infection systems and the link between NOD2 and MDP remain unresolved.
In the studies described here, we observed that TLR2 and NOD2 were together responsible for IL-10 production when macrophages were exposed to PnCW. By contrast, inflammatory cytokine production in response to PnCW was TLR2 dependent and NOD2-independent. Our studies reveal an unexpected pathway of signal integration in response to a complex microbial product that stimulates multiple signalling pathways in macrophages.
Results
When S. pneumoniae grow in vivo, cell wall components are released into the host environment by several processes: cell wall turnover that occurs during normal growth, antibiotics that induce autolysis, and subpopulations that undergo spontaneous autolysis as a mechanism of interbacterial gene transfer. The latter process lead in part to the discovery of DNA as the source of genetic information. Released cell wall fragments stimulate a complex pathway of local and systemic inflammatory responses including neuronal damage and memory loss (Tuomanen et al., 1995). Because cell wall fragments derived from yeast (zymosan) induce robust IL-10 secretion from myeloid-derived cells (Dillon et al., 2004; 2006; Rogers et al., 2005; Slack et al., 2007), we tested if purified PnCW (Fillon et al., 2006) also induced IL-10 production from bone marrow-derived macrophages (BMDMs). PnCW induced previously unsuspected high amounts of IL-10 (Fig. 1). As purified PnCW is composed primarily of peptidoglycan, a TLR2 agonist, this response was predictably TLR2 dependent (Fig. 1). In parallel experiments we also observed that IL-10 and IL-10 mRNA production in response to PnCW was also dependent on NOD2 (Figs 1B, D and E). The reduction in IL-10 production from PnCW-stimulated NOD-deficient macrophages was not to the same extent as Tlr2−/− BMDMs but nevertheless highly reproducible. We also observed that BMDMs derived from Tlr2−/−;Nod2−/− mice secreted undetectable IL-10, demonstrating that TLR2 and NOD2 might function in the same or related pathways for IL-10 production (Figs 1B, D and E). By contrast, IL-10 production in response to LPS was indistinguishable in Tlr2−/−, Nod2−/− or Tlr2−/−;Nod2−/− cells (Fg. 1C). We tested if NOD2 was required for IL-10 production in response to Pam3CSK4, a synthetic lipid that signals via exclusively TLR2. We observed that Nod2−/− BMDMs produced IL-10 in response to Pam3CSK4 similar to control cells (data not shown). In contrast, Pam3CSK4 did not induce IL-10 in Tlr2−/− or Tlr2−/−;Nod2−/− cells as expected (data not shown). NOD2 was therefore not required for the ‘canonical’ TLR2 activation pathway stimulated by Pam3CSK4 but was important to the physiological PnCW stimulus.
Fig. 1. Combined signals from TLR2 and NOD2 are required for PnCW-induced IL-10 production.

A. Schematic representation of the composition of PnCW obtained by biochemical purification from whole lysed pneumococci. The upper part of the figure is cartoon of the bacteria cell membrane and wall indicating that the wall composed primarily of peptidoglycan, to which multiple proteins, lipopeptides and lipids are tethered, surrounds the cell membrane of pneumococci. Following purification, PnCW is predominantly peptidoglycan and teichoic acids. This fraction is particulate is nature, analogous to zymosan fractions from yeast cell walls.
B. IL-10 production from BMDMs stimulated with PnCW measured over time by ELISA. Results are combined from independent experiments (n = 7).
C. IL-10 production from LPS stimulation of BMDMs from the same genotypes used in (B).
D. Northern blotting analysis of IL-10 mRNA over time (h) following PnCW stimulation of BMDMs from the indicated genotypes. Data from Nod2−/−; Tlr2−/− BMDMs were obtained from a different part of the gel as shown the separation from the other part of the gel.
E. qRT-PCR analysis of IL-10 mRNA production following PnCW stimulation. Data points are averages of duplicate samples. Data are representative of three independent experiments.
F. Confocal microscopy to follow the fate of PnCW after contact with BMDMs. FITC-labelled PnCW was added to C57BL/6 BMDMs and followed over time. Representative images at times 0, 5, 24 and 48 h are shown. At each time, cultures were fixed and labelled with phalloidin to visualize actin. Note that the FITC signal is not visible until sufficient PnCW particle have been phagocytosed and concentrated inside macrophages (24 h). All images were collected using a 10× objective, except the 48 h time point (20×).
G. Higher power image of PnCW inside BMDMs at 48 h (40× objective).
Because PnCW is a complex macromolecule, we tested its cellular fate after exposure to BMDMs. We visualized PnCW fragments in macrophages through the use of FITC-labelled PnCW (Fig. 1F and G). Macrophages consumed all the PnCW over a period of ~24 h and thereafter, the PnCW remained internalized and apparently unchanged over 7 days when the experiments were terminated. Comparing these data with the kinetics of cytokine secretion, we suspect that macrophages are stimulated with early, external contact with PnCW. By contrast, phagocytosed PnCW appeared to persist within macrophages. Further experiments will be necessary to determine if macrophages can generate MDP from the PnCW and any signalling connections between NOD2 and in-dwelling PnCW.
We next asked how broadly NOD2 influenced overall macrophage transcriptional responses to PnCW. We performed transcriptome analysis of BMDMs derived from background-matched C57BL/6 Nod2+/+ or Nod2−/− mice stimulated for 0, 1 or 4 h with PnCW at a concentration and conditions identical to that used for the experiments in Fig. 1. We analysed the data first by principal component analysis (data not shown) and then by ordering gene expression according to degree of induction relative to the untreated (0 h) time point data (Table 1). We observed that the absence of NOD2 had limited effects on the overall inflammatory response to PnCW (Table 1, Fig. 2A) consistent with the notion that other receptors, especially TLR2, mediate a strong cellular activation by PnCW. Of the mRNAs whose expression was altered, we noted that IL-10 mRNA expression was reduced in NOD2-deficient cells relative to controls consistent with the ELISA and mRNA data shown in Fig. 1. Because IL-10 has essential autocrine-paracrine inhibitory effects on activated macrophages, we next asked if a panel of known downstream target genes of IL-10 signalling (Lang et al., 2002; El Kasmi et al., 2007) were affected by the reduction of IL-10 after PnCW stimulation of NOD2-deficient macrophages. We observed that expression of multiple IL-10 target genes was lowered relative to controls in NOD2-deficient macrophages at 4 h post stimulation (Fig. 2B). This is consistent with the data that PnCW stimulates IL-10 production in a NOD2-dependent way that leads, in part, to autocrine-paracrine downstream effects on IL-10-responsive gene expression.
Table 1.
Gene expression increased by PnCW treatment.
| Gene | Rank (WT) | Fold (4 h) | Rank (Nod2−/−) | Fold (4 h) | P (time) | P (genotype) |
|---|---|---|---|---|---|---|
| Gm 1960 | 1 | 222.14 | 1 | 205.56 | 1.41E-13 | 0.5314 |
| Irg1 | 2 | 124.62 | 2 | 145.62 | 4.32E-11 | 0.8515 |
| Cxcl1 | 3 | 102.85 | 4 | 111.82 | 2.17E-11 | 0.3880 |
| Tnf | 4 | 82.04 | 3 | 124.60 | 1.02E-11 | 0.8967 |
| Cxcl1 | 5 | 78.06 | 5 | 102.79 | 4.75E-11 | 0.3036 |
| Ccl3 | 6 | 77.61 | 6 | 76.38 | 4.39E-11 | 0.9913 |
| Saa3 | 7 | 76.27 | 8 | 54.01 | 2.37E-14 | 0.1997 |
| Ptgs2 | 8 | 57.72 | 16 | 29.27 | 4.13E-09 | 0.9574 |
| Ccl4 | 9 | 54.47 | 14 | 32.23 | 3.61E-12 | 0.6201 |
| Il1b | 10 | 51.93 | 15 | 32.00 | 8.30E-07 | 0.7485 |
| Gpr109a | 11 | 51.73 | 10 | 37.09 | 7.80E-14 | 0.2307 |
| Slc7a11 | 12 | 51.72 | 19 | 24.34 | 4.11E-14 | 0.6030 |
| Ptgs2 | 13 | 51.63 | 46 | 16.03 | 8.02E-08 | 0.1154 |
| Il1a | 14 | 45.04 | 50 | 15.37 | 6.39E-07 | 0.7571 |
| Il6 | 15 | 44.03 | 11 | 35.24 | 1.02E-06 | 0.3773 |
| Socs3 | 16 | 43.81 | 13 | 32.65 | 2.46E-11 | 0.2271 |
| Socs3 | 17 | 40.92 | 17 | 26.16 | 1.79E-11 | 0.4451 |
| Fpr1 | 18 | 38.41 | 26 | 21.24 | 2.66E-09 | 0.8110 |
| Tnfaip3 | 19 | 36.91 | 20 | 24.25 | 6.30E-09 | 0.4238 |
| Cxcl2 | 20 | 36.81 | 7 | 60.84 | 6.79E-14 | 0.9650 |
| BB548587 | 21 | 36.54 | 71 | 11.55 | 1.85E-06 | 0.7213 |
| Tnfsf9 | 22 | 35.17 | 64 | 12.63 | 5.17E-08 | 0.9962 |
| Socs3 | 23 | 35.10 | 18 | 25.02 | 1.24E-12 | 0.2296 |
| Ccl2 | 24 | 34.01 | 21 | 23.20 | 1.41E-09 | 0.5926 |
| Ccl7 | 25 | 33.98 | 9 | 42.02 | 8.91E-10 | 0.3433 |
| Fpr-rs2 | 26 | 31.15 | 27 | 21.21 | 3.19E-12 | 0.6983 |
| Il1rn | 27 | 29.49 | 69 | 11.71 | 2.74E-10 | 0.0624 |
| Ets2 | 28 | 29.25 | 49 | 15.50 | 3.77E-09 | 0.5326 |
| Ccrl2 | 29 | 29.13 | 30 | 19.91 | 2.75E-08 | 0.7250 |
| LOC622976 | 30 | 28.83 | 12 | 33.45 | 6.43E-12 | 0.1185 |
| Il1rn | 31 | 28.08 | 32 | 18.99 | 4.52E-11 | 0.1198 |
| Tnfaip3 | 32 | 27.38 | 24 | 21.82 | 1.42E-10 | 0.8902 |
| Zc3h12c | 33 | 25.12 | 23 | 22.49 | 7.12E-10 | 0.3216 |
| Ccl12 | 34 | 25.12 | 59 | 13.15 | 1.25E-08 | 0.2671 |
| Cdc42ep2 | 35 | 24.16 | 44 | 16.31 | 1.03E-08 | 0.7966 |
| Gpr84 | 36 | 23.46 | 31 | 19.54 | 3.15E-10 | 0.2886 |
| Il1rn | 37 | 23.35 | 73 | 11.35 | 9.55E-12 | 0.1113 |
| Nr4a3 | 38 | 22.46 | 48 | 15.73 | 2.80E-08 | 0.7223 |
| Ccl5 | 39 | 22.15 | 36 | 18.02 | 1.01E-07 | 0.2819 |
| Ch25h | 40 | 21.83 | 65 | 12.59 | 9.66E-11 | 0.2134 |
| Cd40 | 41 | 21.42 | 66 | 12.25 | 2.74E-11 | 0.8623 |
| Cd40 | 42 | 21.31 | 54 | 13.89 | 6.89E-12 | 0.3615 |
| Zc3h12c | 43 | 21.07 | 28 | 21.05 | 3.52E-10 | 0.7250 |
| Clec4e | 44 | 20.75 | 29 | 20.40 | 2.51E-09 | 0.9522 |
| Malt1 | 45 | 20.22 | 33 | 18.56 | 5.46E-12 | 0.8940 |
| Hivep3 | 46 | 19.93 | 22 | 22.60 | 3.95E-15 | 0.4636 |
| Ptges | 47 | 19.70 | 53 | 14.23 | 2.23E-10 | 0.2554 |
| Cd69 | 48 | 19.60 | 34 | 18.55 | 3.57E-07 | 0.5920 |
| Cd40 | 49 | 19.48 | 60 | 13.08 | 2.69E-12 | 0.7930 |
| Traf1 | 50 | 19.18 | 43 | 16.55 | 5.61E-08 | 0.2824 |
Probe sets from the top 50 induced targets in PnCW-stimulated control cells are ordered 1 through 50 based on fold induction relative to the untreated control cells at 4 h. The correlative rank of genes expressed in PnCW-stimulated NOD2-deficient macrophages relative to the control ranking is listed along with the P-values (ANOVA) by time relative to unstimulated cells and by genotype.
Fig. 2. Absence of NOD2 has a limited effect on PnCW-induced inflammatory transcription.

A. Select inflammatory targets induced by PnCW in control or NOD2-deficient BMDMs. Data for each target mRNA is plotted as the average difference signal intensity. Data are averages from independent (n = 4) Affymetrix arrays performed per genotype and per time. No significant differences (paired t-test) were detected between genotypes. Complete data sets for this experiment have been deposited in the GEO database.
B. Expression of IL-10 target genes is affected in the absence of NOD2 function. Signal intensity for IL-10 (left most in the panel) and selected genes whose expression is dependent, in part, on autocrine-paracrine IL-10 production. *P < 0.05, paired t-test comparing 4 h time points between control and Nod2−/− BMDMs (n = 4).
By contrast to the requirement for both TLR2 and NOD2 in IL-10 production, we observed that all tested pro-inflammatory genes induced by PnCW were dependent on TLR2 (Fig. 3), but independent of NOD2 as shown by the microarray data (Table 1). These results are consistent with the notion that TLR2 is the primary pathway by which macrophages detect PnCW and that NOD2 plays a downstream role in fine tuning the signalling response, in this case by regulating IL-10 production.
Fig. 3.

Pro-inflammatory cytokine transcription in response to PnCW is dependent on TLR2. BMDMs from the indicated genotypes were stimulated with PnCW for the times shown. Cytokine mRNAs were measured by qRT-PCR. Data are representative of three independent experiments.
NOD2 has been reported to form heterotypic complexes with another CARD domain protein, RIPK2 (Kobayashi et al., 2002; Park et al., 2007; Hasegawa et al., 2008). Like NOD2-deficient cells, macrophages lacking RIPK2 cannot respond to MDP, suggesting that NOD2 and RIPK2 function in the same pathway for MDP responsiveness (Park et al., 2007). Furthermore, RIPK2 is also required for NOD1 function (which does not ‘sense’ MDP), suggesting that RIPK2 is genetically and biochemically linked to both NOD1 and NOD2 activity (Park et al., 2007). We therefore used macrophages derived from two strains of Ripk2−/− mice and mice lacking both NOD1 and NOD2 (Nod1−/−;Nod2−/−) to probe if IL-10 production was inhibited. We observed by ELISA and qRT-PCR analysis that RIPK2-deficient cells had a similar phenotype to NOD2-deficient BMDMs (Fig. 4) in terms of reduced IL-10 production in response to PnCW. Furthermore, macrophages from Nod1−/−;Nod2−/− mice, which have been reported to have similar phenotypes to Ripk2−/− macrophages (Park et al., 2007), also had a deficiency in IL-10 production when stimulated with PnCW. Finally, when introduced into Nod2−/− macrophages by retroviral-mediated transduction, human NOD2 rescued IL-10 in response to PnCW and MDP responsiveness (Fig. 4E–G). Analogous studies with NOD1 were not possible because human NOD1 producer lines do not make virus at sufficiently high titre to infect stem cells. Collectively, these data provide evidence that NOD2 and RIPK2 function in the same pathway that mediated IL-10 production in response to PnCW.
Fig. 4. IL-10 expression in response to PnCW is dependent on the RIPK2 pathway.

A. BMDMs from Ripk2−/− or Nod1−/−; Nod2−/− mice were stimulated with PnCW and IL-10 measured over time by ELISA.
B. As in (A), but following LPS stimulation.
C and D. Analysis of BMDMs from an independent Ripk2 mutant strain showing IL-10 and IL-10 mRNA produced in response to PnCW requires RIPK2.
E and F. Reconstitution of Nod2−/− hematopoietic stem cells with human or mouse NOD2 cDNAs rescues IL-10 production in response to PnCW. Stem cells from Nod2−/− mice were infected with retroviruses containing hNOD2 or mNOD2 cDNAs and selected by cell sorting for YFP and plated into media containing CSF-1 to drive differentiation into macrophages. After differentiation, macrophages were stimulated with LPS or LPS and MDP to measure the MDP synergy with TLR4 signalling (E) or IFN-γ signalling (F) or IL-10 production after PnCW treatment (G). Data are representation of two independent infection studies. In the case of mNOD2, sufficient cells were obtained to perform the MDP-IFN-γ synergy assay only.
Zymosan, a complex cell wall fraction from yeast, has been shown to induce IL-10 production by the TLR2-DECTIN1-SYK pathway and the TLR2-DECTIN1-ERK pathway (Dillon et al., 2004; 2006; Rogers et al., 2005; Slack et al., 2007). We therefore asked if PnCW also stimulated ERK activation with the idea that ERK could be a common downstream mediator of multiple TLR2-driven pathways, including the TLR2-NOD2 pathway. However, we observed that PnCW induced low or negligible ERK phosphorylation (Fig. 5). Therefore, PnCW seems to stimulate IL-10 production via pathways distinct from those induced by zymosan, even though both the DECTIN1-SYK and NOD2-RIPK2 pathways depend on TLR2. Finally, IL-10 production in response to PnCW was dependent on MyD88 (Fig. 5E), suggesting that the TLR2-MyD88 pathway is absolutely necessary for PnCW-induced IL-10 production. MyD88 was not, however, required for MDP sensing (Fig. 5F) as expected as shown by the use of a sensitive MDP stimulation assay where nitric oxide production is measured in response to MDP and IFN-γ (Totemeyer et al., 2006). By contrast, MDP sensing was dependent on NOD2.
Fig. 5. PnCW activation of BMDMs does not activate the ERK or p38 pathways but requires MyD88 signalling.

A. C57BL/6 BMDMs were stimulated with Pam3CSK4, LPS or PnCW over time and ERK phosphorylation measured by immunoblotting for phosphorylated forms of ERK or reprobed for total ERK amounts.
B. A similar experiment to (A) was performed using BMDMs from the genotypes indicated on the right.
C. The same lysates from (B) were probed for phospho-p38. Note that PnCW does not activate ERK or p38 in any of these experiments.
D. BMDMs from control, Nod2−/−, Tlr2−/− or MyD88−/− mice were stimulated with PnCW for 8 h and IL-10 measured by ELISA. Data are average ± SD from independent samples (n = 3).
E. BMDMs were stimulated with MDP or MDP + IFN-γ overnight and nitrites measured by the Griess assay. Stimulation with IFN-γ alone did not produce detectable nitrites (data not shown).
Discussion
Our data suggest that NOD2 is a component of a signalling module that regulates IL-10 production in a stimulus-specific and cell type-specific way (Fig. 6). Activation of TLR2-MyD88 by Pam3CSK4 leads to ERK activation and IL-10 induction. In contrast, activation of TLR2-MyD88 by PnCW invokes NOD2 and RIPK2 and bypasses ERK activation to produce IL-10. Activation of TLR2 and DECTIN by zymosan leads to ERK activation and IL-10 induction. It is well accepted that IL-10 is essential for regulating the amplitude of most, if not all inflammatory responses in vivo. Therefore, mutant forms of NOD2 may be a component of homeostatic regulation of IL-10 in myeloid-derived cells. Notably, Netea and colleagues have demonstrated that human monocyte-derived macrophages and dendritic cells isolated from people bearing NOD2 mutations have reduced IL-10 production in response to peptidoglycan and MDP (Netea et al., 2004; 2005; Kullberg et al., 2008).
Fig. 6.
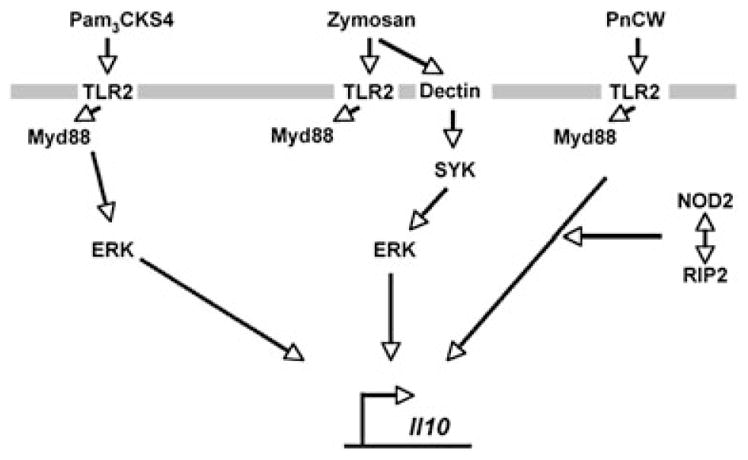
Model of IL-10 production myeloid lineage cells (macrophages and DCs) stimulated by agents that activate TLR2, alone or in combination with other cell surface molecules.
We found parallels between PnCW stimulation of IL-10 production and the activation of dendritic cells by zymosan, a large fragmentary remnant of yeast cell walls. In both cases, immune cells are exposed to particulate fractions of cell walls of varying sizes and displaying a variety of different sugars, lipids and structural biopolymers. Zymosan stimulates IL-10 production by a MyD88-independent pathway involving TLR2, DECTIN1, SYK and ERK (Dillon et al., 2004; 2006; Rogers et al., 2005; Slack et al., 2007). PnCW stimulation of IL-10 also requires TLR2, but does not seem to activate substantial ERK phosphorylation (Fig. 5). Instead, MyD88, NOD2 and RIPK2 are required. The PnCW and zymosan recognition pathways present a considerable contrast to the activation of immune cells with Pam3CSK4 through TLR2, a pathway that has an absolute requirement for MyD88 but not NOD2. Therefore, there are different signalling routes to IL-10 production following TLR2 activation (Fig. 6). Similarly, additional diversity of signalling for IL-10 production has been observed for receptors such as the LPS-mediated pathway that requires TLR4 and p38 (Park et al., 2005), and the immune complex pathway that enhances IL-10 production via FcR ligation (Edwards et al., 2006). BTK also plays a key role in stimulus-specific IL-10 production: the absence of BTK leads to a decrease in IL-10 produced from macrophages and a corresponding increase in inflammatory mediators normally blocked by IL-10 (e.g. IL-12, IL-6) (Kawakami et al., 2006; Schmidt et al., 2006). However, the reduced amounts of IL-10 made in the absence of BTK, like the absence of NOD2, is insufficient to initiate the severe inflammatory bowel disease observed in the complete absence of IL-10 (Schmidt et al., 2006) (L.M. and P.J.M., unpubl. data). Therefore, NOD2 and BTK both seem to function to tune stimulus-specific IL-10 production.
We found that neither TLR2 nor NOD2 were absolutely required for PnCW stimulation of IL-10 production. IL-10 was made, although in much lower amounts, in the absence of either TLR2 or NOD2. However, cells lacking both TLR2 and NOD2 had a complete abrogation of IL-10 production. These data suggest that TLR2 and NOD2 have a genetic interaction. A similar finding has been made in the ECOVA gut inflammation system where pathology observed in the absence of NOD2 was reversed in the combined deficiency of both NOD2 and TLR2 (Watanabe et al., 2006). It is however, premature to suggest that TLR2 and NOD2 function in a linear pathway where NOD2 is downstream of TLR2. Instead, we suggest that cross-talk between the TLR2 and NOD2 pathways occurs in a stimulus- and cell-specific context. Cross-talk is not revealed when TLR2 is stimulated with Pam3CSK4 but instead is exposed by agents such as PnCW and peptidoglycan, at least in the BMDMs used here. This idea may help resolve previous reports that suggested that NOD2 was not involved in TLR2 signalling (Kobayashi et al., 2005): dependence on both stimulus and cell type is needed to expose differences among pathways. Recent data from human monocytes bearing NOD2 mutations has also revealed a complex interplay between NOD2 and TLR2 (Borm et al., 2008).
Using transcriptome profiling, we also found that the absence of NOD2 affected a surprisingly low number of mRNAs when BMDMs were stimulated with PnCW. These data are in keeping with published studies suggesting that loss of NOD2 has minimal effects on pro-inflammatory TLR signalling (Pauleau and Murray, 2003; Kobayashi et al., 2005; Park et al., 2007). However, PnCW is an example of a pathogen ‘signal’ that can activate multiple pathways including the TLR2 pathway. It is not surprising that NOD2 plays limited roles in the overall transcriptional response as the mammalian immune system most likely has developed multiple, overlapping ways to recognize and respond to such a complex material. Nevertheless, the finding that IL-10 was one of the most affected mRNAs, along with a cohort of IL-10-regulated genes, suggests that further work is warranted to determine the role of NOD2 in IL-10 production in humans bearing mutations in NOD2.
It is tempting to speculate that IL-10 regulation has key functional significance in chronic inflammatory states such as the inflammatory bowel diseases. Most researchers agree that common pathways of inflammatory mediator production, arrived at by a multitude of genetic and environmental mechanisms, drive disease and are the central focus of therapeutic intervention through anticytokine antagonists. IL-10 plays an essential regulatory role in controlling the products of the common inflammatory pathway: IL-12, TNF-α, IL-6, etc. Therefore, small disturbances in temporal, anatomic or quantitative IL-10 amounts may translate into greater inflammation.
Experimental procedures
Mice
Nod2−/− mice have been previously described (Pauleau and Murray, 2003). Nod2−/−;Tlr2−/− mice on a C57BL/6 background (n = 5 generations) have been described (Watanabe et al., 2006). Tlr2−/− mice were purchased from the Jackson Laboratories. Mice were bred and genotyped according to published protocols. For some experiments, Nod2−/− mice were backcrossed 10 generations to the C57BL/6 background. All animal experiments were performed in accordance with protocols governed by the St Jude Animal Care and Use Committee (P.J. M., Principal Investigator). Nod1−/−;Nod2−/− and Rip2k−/− mice have been described previously (Chin et al., 2002; Kobayashi et al., 2002; Park et al., 2007).
Cell preparation and stimulus conditions
Bone marrow-derived macrophages were isolated and cultured as described (Lang et al., 2002). Macrophages were stimulated with highly purified PnCW prepared as described previously (Tuomanen et al., 1985a,b). Briefly, S. pneumoniae unencapsulated strain R6 were grown in C+Y medium, bacteria were boiled in SDS, mechanically broken by shaking with acid-washed glass beads, and treated sequentially with DNase, RNase, trypsin, LiCl, EDTA and acetone. PnCW was confirmed to be free of protein by analysis for non-cell wall amino acids using mass spectrometry. The absence of contaminating endotoxin was confirmed first by the Limulus test (Associates of Cape Cod) and then by ELISA measurement of hIL-8 production by HEK 293 cells transfected with plasmids designed to express TLR4 and MD2 (a generous gift of Dr Doug Golenbock, UMass) in the presence of purified PnCW. For fluorescence microscopy experiments, BMDMs were labelled with Phalloidin and 4′,6-diamidino-3-phenylindole (DAPI), a nuclear stain. PnCW was directly labelled with 1 mg ml−1 FITC (Sigma-Aldrich) solution in carbonate buffer (pH 9.2) for 1 h at room temperature in the dark and washed twice with PBS containing calcium and magnesium (Gosink et al., 2000).
ELISA
ELISA was performed as previously described (El Kasmi et al., 2006). Capture and detection antibodies used were purchased from BD Pharmingen. Detection limits for the ELISAs were 50 pg ml−1 (IL-10) and 10 pg ml−1 (IL-12p40).
Northern blotting, RT-PCR and immunoblotting
Total RNA was isolated from primary macrophages using Trizol. Reverse transcription (qRT-PCR) was performed as described previously using primers and probes (Applied Biosciences) specific for each cytokine mRNA (Lang et al., 2002). Immunoblotting was performed using the following rabbit polyclonal antibodies: antiphospho-p42, p44 ERK and anti-phospho-p38 from Cell Signaling Technology used at 1:500 final dilution. Anti-p42, p44 ERK and anti-p38 were from the same source and were used at a final concentration of 1:1000.
Transcriptome analysis
RNA samples from BMDM cultures representing four PnCW preparations, two genotypes (wild-type and the NOD2 deficient) and three time points (0, 1 and 4 h) were arrayed on Affymetrix GeneChip analysis murine 430v2GeneChip arrays. Signal was acquired with MAS5.0 software and natural log transformed [ln(signal + 20)] to stabilize variance and better conform the data to the normal distribution. Principal components analysis (Partek 6.1) revealed a batch effect which, being orthogonal to hour and genotype, was removed by capturing and mean adjusting residuals of a one-way ANOVA-based on PnCW batch (STATA/SE 9.2). The batch-corrected transformed signal was tested for exposure time and genotype effects in a two-way ANOVA using Partek. The P-values for the time effect were corrected for multiple comparisons using the false discovery rate (Benjamini et al., 2001). Fold change calculations were based on the geometric means of the batch corrected data between the 4 h and 0 h time points for each genotype. Array data has been submitted to the GEO database (accession GSE8960).
Retroviral-mediated transduction of stem cells
Retroviral transduction of murine bone marrow cells was performed as described previously (El Kasmi et al., 2006; Holst et al., 2006a,b). For the transduction experiments, we used fetal liver stem cells isolated from E14 pregnant Nod2−/− female mice. Fetal liver cells were cultured for 48 h in complete DMEM with 20% FBS, 20 ng ml−1 murine IL-3, 50 ng ml−1 human IL-6 and 50 ng ml−1 murine stem cell factor (BioSource International). Stem cells were subsequently co-cultured for a further 48 h with irradiated (1200 rad) retroviral producer cell lines plus 6 mg ml−1 polybrene and cytokines as previously detailed. Non-adherent, transduced cells were collected, red and re-suspended in PBS with 2% FBS with 20 U ml−1 heparin. Cells were sorted by gating on forward and side scatter, and YFP positivity. Cells were subsequently plated out with 500 ng ml−1 Fungizone and 25 mg ml−1 gentamicin in complete DMEM with 20% FBS and L cell conditioned medium as a source of CSF-1. Following differentiation, mature cells were measured by flow cytometry for YFP along with surface markers (CD11b, F4/80) for macrophages.
Acknowledgments
We thank Doug Golenbock (UMass) for the gift of plasmids expressing TLR4 and MD2, Neal Silverman (UMass) for advice on detecting LPS contamination using the IL-8 ELISA, Cristel Thomas for initial work on retroviral constructs to express NOD cDNAs and Paul Dempsey and Genhong Cheng for RIPK2-deficient bone marrow samples. This work was supported by the American Lebanese Syrian Associated Charities (ALSAC), NIH CORE grant P30 CA21765, and NIH grants AI062921 to P.M. and DK61707 to G. Núñez.
References
- Barreau F, Meinzer U, Chareyre F, Berrebi D, Niwa-Kawakita M, Dussaillant M, et al. CARD15/NOD2 is required for Peyer’s patches homeostasis in mice. PLoS ONE. 2007;2:e523. doi: 10.1371/journal.pone.0000523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- Borm ME, van Bodegraven AA, Mulder CJ, Kraal G, Bouma G. The effect of NOD2 activation on TLR2-mediated cytokine responses is dependent on activation dose and NOD2 genotype. Genes Immun. 2008;9:274–278. doi: 10.1038/gene.2008.9. [DOI] [PubMed] [Google Scholar]
- Chin AI, Dempsey PW, Bruhn K, Miller JF, Xu Y, Cheng G. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature. 2002;416:190–194. doi: 10.1038/416190a. [DOI] [PubMed] [Google Scholar]
- Dillon S, Agrawal A, Van Dyke T, Landreth G, McCauley L, Koh A, et al. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J Immunol. 2004;172:4733–4743. doi: 10.4049/jimmunol.172.8.4733. [DOI] [PubMed] [Google Scholar]
- Dillon S, Agrawal S, Banerjee K, Letterio J, Denning TL, Oswald-Richter K, et al. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J Clin Invest. 2006;116:916–928. doi: 10.1172/JCI27203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. 2006;80:1298–1307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kasmi KC, Holst J, Coffre M, Mielke L, de Pauw A, Lhocine N, et al. General nature of the STAT3-activated anti-inflammatory response. J Immunol. 2006;177:7880–7888. doi: 10.4049/jimmunol.177.11.7880. [DOI] [PubMed] [Google Scholar]
- El Kasmi KC, Smith AM, Williams L, Neale G, Panopolous A, Watowich SS, et al. Cutting edge: a transcriptional repressor and corepressor induced by the STAT3-regulated anti-inflammatory signaling pathway. J Immunol. 2007;179:7215–7219. doi: 10.4049/jimmunol.179.11.7215. [DOI] [PubMed] [Google Scholar]
- Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25:713–724. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- Fillon S, Soulis K, Rajasekaran S, Benedict-Hamilton H, Radin JN, Orihuela CJ, et al. Platelet-activating factor receptor and innate immunity: uptake of Gram-positive bacterial cell wall into host cells and cell-specific pathophysiology. J Immunol. 2006;177:6182–6191. doi: 10.4049/jimmunol.177.9.6182. [DOI] [PubMed] [Google Scholar]
- Gosink KK, Mann ER, Guglielmo C, Tuomanen EI, Masure HR. Role of novel choline binding proteins in virulence of Streptococcus pneumoniae. Infect Immun. 2000;68:5690–5695. doi: 10.1128/iai.68.10.5690-5695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Nunez G, Inohara N. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J. 2008;27:373–383. doi: 10.1038/sj.emboj.7601962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst J, Vignali KM, Burton AR, Vignali DA. Rapid analysis of T-cell selection in vivo using T cell-receptor retrogenic mice. Nat Methods. 2006a;3:191–197. doi: 10.1038/nmeth858. [DOI] [PubMed] [Google Scholar]
- Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, Vignali DA. Generation of T-cell receptor retrogenic mice. Nat Protoc. 2006b;1:406–417. doi: 10.1038/nprot.2006.61. [DOI] [PubMed] [Google Scholar]
- Hugot JP, Zaccaria I, Cavanaugh J, Yang H, Vermeire S, Lappalainen M, et al. Prevalence of CARD15/NOD2 mutations in Caucasian healthy people. Am J Gastroenterol. 2007;102:1259–1267. doi: 10.1111/j.1572-0241.2007.01149.x. [DOI] [PubMed] [Google Scholar]
- Inohara C, McDonald C, Nunez G. NOD-LRR proteins: role in host–microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74:355–383. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3:371–382. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549–559. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Inagaki N, Salek-Ardakani S, Kitaura J, Tanaka H, Nagao K, et al. Regulation of dendritic cell maturation and function by Bruton’s tyrosine kinase via IL-10 and Stat3. Proc Natl Acad Sci USA. 2006;103:153–158. doi: 10.1073/pnas.0509784103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA, et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416:194–199. doi: 10.1038/416194a. [DOI] [PubMed] [Google Scholar]
- Kullberg BJ, Ferwerda G, de Jong DJ, Drenth JP, Joosten LA, Van der Meer JW, Netea MG. Crohn’s disease patients homozygous for the 3020insC NOD2 mutation have a defective NOD2/TLR4 cross-tolerance to intestinal stimuli. Immunology. 2008;123:600–605. doi: 10.1111/j.1365-2567.2007.02735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- Lesage S, Zouali H, Cezard JP, Colombel JF, Belaiche J, Almer S, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70:845–857. doi: 10.1086/339432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, et al. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Marina-Garcia N, Franchi L, Kim YG, Miller D, McDonald C, Boons GJ, Nunez G. Pannexin-1-mediated intracellular delivery of muramyl dipeptide induces caspase-1 activation via cryopyrin/NLRP3 independently of Nod2. J Immunol. 2008;180:4050–4057. doi: 10.4049/jimmunol.180.6.4050. [DOI] [PubMed] [Google Scholar]
- Moreillon P, Majcherczyk PA. Proinflammatory activity of cell-wall constituents from Gram-positive bacteria. Scand J Infect Dis. 2003;35:632–641. doi: 10.1080/00365540310016259. [DOI] [PubMed] [Google Scholar]
- Murray PJ. NOD proteins: an intracellular pathogen-recognition system or signal transduction modifiers? Curr Opin Immunol. 2005;17:352–358. doi: 10.1016/j.coi.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr Opin Pharmacol. 2006;6:379–386. doi: 10.1016/j.coph.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Netea MG, Kullberg BJ, de Jong DJ, Franke B, Sprong T, Naber TH, et al. NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn’s disease. Eur J Immunol. 2004;34:2052–2059. doi: 10.1002/eji.200425229. [DOI] [PubMed] [Google Scholar]
- Netea MG, Ferwerda G, de Jong DJ, Jansen T, Jacobs L, Kramer M, et al. Nucleotide-binding oligomerization domain-2 modulates specific TLR pathways for the induction of cytokine release. J Immunol. 2005;174:6518–6523. doi: 10.4049/jimmunol.174.10.6518. [DOI] [PubMed] [Google Scholar]
- Orihuela CJ, Fillon S, Smith-Sielicki SH, El Kasmi KC, Gao G, Soulis K, et al. Cell wall-mediated neuronal damage in early sepsis. Infect Immun. 2006;74:3783–3789. doi: 10.1128/IAI.00022-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Mathison J, Fearns C, Kravchenko VV, Da Silva Correia J, Hoffman HM, et al. MDP-induced interleukin-1beta processing requires Nod2 and CIAS1/NALP3. J Leukoc Biol. 2007;82:177–183. doi: 10.1189/jlb.1006627. [DOI] [PubMed] [Google Scholar]
- Park JH, Kim YG, McDonald C, Kanneganti TD, Hasegawa M, Body-Malapel M, et al. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol. 2007;178:2380–2386. doi: 10.4049/jimmunol.178.4.2380. [DOI] [PubMed] [Google Scholar]
- Park JM, Greten FR, Wong A, Westrick RJ, Arthur JS, Otsu K, et al. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis –CREB and NF-kappaB as key regulators. Immunity. 2005;23:319–329. doi: 10.1016/j.immuni.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Pauleau AL, Murray PJ. Role of nod2 in the response of macrophages to Toll-like receptor agonists. Mol Cell Biol. 2003;23:7531–7539. doi: 10.1128/MCB.23.21.7531-7539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers NC, Slack EC, Edwards AD, Nolte MA, Schulz O, Schweighoffer E, et al. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity. 2005;22:507–517. doi: 10.1016/j.immuni.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Schmidt NW, Thieu VT, Mann BA, Ahyi AN, Kaplan MH. Bruton’s tyrosine kinase is required for TLR-induced IL-10 production. J Immunol. 2006;177:7203–7210. doi: 10.4049/jimmunol.177.10.7203. [DOI] [PubMed] [Google Scholar]
- Slack EC, Robinson MJ, Hernanz-Falcon P, Brown GD, Williams DL, Schweighoffer E, et al. Syk-dependent ERK activation regulates IL-2 and IL-10 production by DC stimulated with zymosan. Eur J Immunol. 2007;37:1600–1612. doi: 10.1002/eji.200636830. [DOI] [PubMed] [Google Scholar]
- Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol. 2006;6:9–20. doi: 10.1038/nri1747. [DOI] [PubMed] [Google Scholar]
- Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totemeyer S, Sheppard M, Lloyd A, Roper D, Dowson C, Underhill D, et al. IFN-gamma enhances production of nitric oxide from macrophages via a mechanism that depends on nucleotide oligomerization domain-2. J Immunol. 2006;176:4804–4810. doi: 10.4049/jimmunol.176.8.4804. [DOI] [PubMed] [Google Scholar]
- Tuomanen EI, Austrian R, Masure HR. Pathogenesis of pneumococcal infection. N Engl J Med. 1995;332:1280–1284. doi: 10.1056/NEJM199505113321907. [DOI] [PubMed] [Google Scholar]
- Tuomanen E, Tomasz A, Hengstler B, Zak O. The relative role of bacterial cell wall and capsule in the induction of inflammation in pneumococcal meningitis. J Infect Dis. 1985a;151:535–540. doi: 10.1093/infdis/151.3.535. [DOI] [PubMed] [Google Scholar]
- Tuomanen E, Liu H, Hengstler B, Zak O, Tomasz A. The induction of meningeal inflammation by components of the pneumococcal cell wall. J Infect Dis. 1985b;151:859–868. doi: 10.1093/infdis/151.5.859. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Kitani A, Murray PJ, Wakatsuki Y, Fuss IJ, Strober W. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity. 2006;25:473–485. doi: 10.1016/j.immuni.2006.06.018. [DOI] [PubMed] [Google Scholar]
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163:1–5. [PubMed] [Google Scholar]