

Vascular smooth muscle cells (SMC) comprise the muscular layer of blood vessel walls and mediate arterial tone and blood pressure. These cells possess a remarkable plasticity that allows mature contractile myocytes to de-differentiate, enabling vessel growth and repair. This unusual ability to de-differentiate is especially important since SMC phenotypic modulation contributes to multiple cardiovascular pathologies, including atherosclerosis and restenosis post-angioplasty. In response to growth factors released at sites of injury, such as platelet-derived growth factor (PDGF), mature SMC can re-enter the cell cycle, become migratory, and de-differentiate to a synthetic phenotype capable of extensive extracellular matrix deposition for vascular repair (1). Much of the research on SMC phenotypic switching has focused on transcriptional regulation of the contractile (differentiated) versus synthetic (de-differentiated) phenotype. The new study by Fei and Cui et al in this issue (2) makes the exciting discovery that RNA editing, leading to changes in mRNA splicing, is an important mechanism underlying SMC phenotypic switching.
Differentiated SMC express a repertoire of smooth muscle (SM)-specific contractile proteins that give the cells their characteristic myocyte morphology and contractile properties. These include SM α-actin (ACTA2) and SM-myosin heavy chain (MYH11). These proteins are downregulated during SMC de-differentiation, which makes them a tangible biochemical readout of cellular differentiation status (1). A substantial body of work has demonstrated that contractile protein induction occurs largely at the level of transcriptional control, mediated by core factors including the CArG element binding protein serum response factor (SRF) and co-factor myocardin (3). However, early studies of cultured SMC suggested that post-transcriptional mechanisms may underlie the PDGF-induced downregulation of contractile proteins. Notably, Acta2 mRNA levels decreased after PDGF treatment at an even greater rate than with the transcriptional inhibitor Actinomycin D, and nuclear run-on indicated that nascent Acta2 mRNA synthesis did not decrease with PDGF treatment (4). Despite these initial observations, subsequent studies of contractile protein regulation in SMC have been largely at the transcriptional, and more recently, epigenetic levels (5,6).
The new work by Fei and Cui et al (2) revisited the question of how contractile protein mRNAs are regulated when SMC de-differentiate. Using a PCR approach, they found that nascent pre-mRNAs (unspliced transcripts detected using primers targeting introns) encoding Myh11 and Acta2 were sparse in mature, differentiated SMC isolated from healthy arteries, while the corresponding spliced, mature mRNAs (m-mRNA) were abundant. Conversely, PDGF-BB-induced de-differentiation of cultured SMC was accompanied by a downregulation of these m-mRNAs but a marked accumulation of the pre-mRNAs. The opposite trends were seen when SMC were induced to differentiate. Interestingly, cloning and sequencing of introns from Myh11 and Acta2 in de-differentiated SMC revealed that the pre-mRNA transcripts had undergone RNA editing (Figure).
ADAR1-mediated RNA editing is a new mechanism of contractile protein repression in SMC de-differentiation.
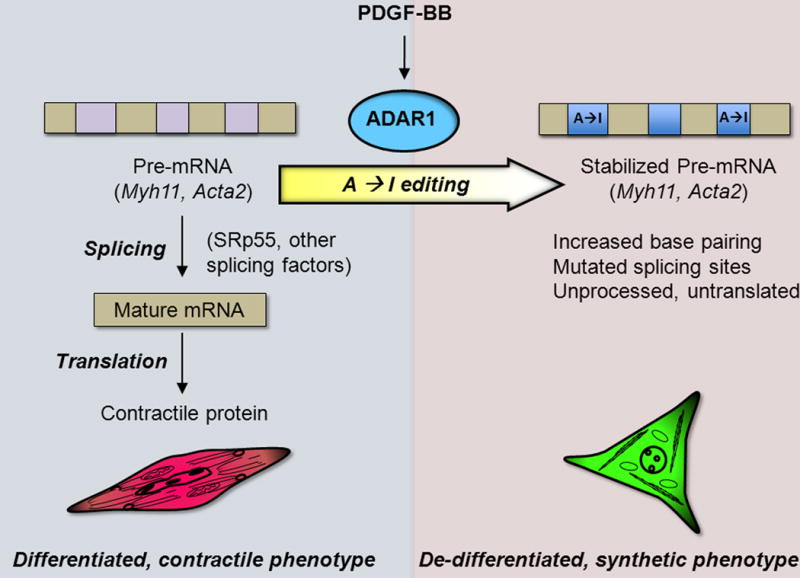
PDGF-BB promotes expression of ADAR1, which edits intronic adenosine (A) to inosine (I) in the contractile protein transcripts Mhy11 and Acta2. Splicing of these edited transcripts is inhibited through multiple potential mechanisms, leading to a reduced synthesis of the contractile proteins that characterize differentiated SMC.
RNA editing typically targets regulatory regions of transcripts such as introns or UTRs while editing of coding sequences is less common (7). RNA editing has been implicated in lupus, cancer, and neurological disorders (8), but very little is known about RNA editing in cardiovascular diseases. One notable example is ApolipoproteinB (ApoB), an LDL component with a role in atherosclerosis. The two discrete ApoB isoforms are generated by tissue-specific Cytosine (C) to Uracil editing of a common precursor transcript (9). Additionally, a single nucleotide polymorphism has been detected in the human RNA editing enzyme ADARB2 that was associated with cardiometabolic risk factors including waist circumference, triglycerides, body mass index, and adiponectin levels, although potential mechanisms or edited transcripts underlying these effects are not known (10). As RNA editing in cardiovascular disease is a largely unexplored area, the present study suggests an important new avenue for research.
Fei and Cui et al (2) identified adenosine deaminase acting on RNA 1 (ADAR1) as the PDGF-induced editase acting on Mhy11 and Acta2. ADAR1 is a double-stranded RNA binding protein that post-transcriptionally substitutes inosine (I) for adenine (A), which alters RNA base pairing and subsequent secondary structure (7). Because inosine mimics guanine (G) to pair with cytosine (forming I:C pairs), multiple A to I edits can increase the stability of the mRNA. Structure-based modeling predictions indeed reveal decreased free energy of individual edited introns, the effect of which may be amplified due to editing of multiple introns within a transcript. The authors suggest several mechanisms by which RNA editing may alter mRNA maturation in de-differentiated SMC: 1) The altered I:C base pairing and enhanced stability may inhibit splicing; 2) editing generated mutations in binding sites for splicing factors (SRp55); and 3) editing shortened CA repeat regions that might also influence splicing. The relative contributions of these potential mechanisms remain to be determined.
ADAR1 upregulation and RNA editing was observed in both rat and human SMC and in multiple introns of two different contractile genes, MYH11 and ACTA2. Importantly, this phenomenon was also observed in response to vascular injury in two different in vivo models. ADAR1 expression was induced and the pre-mRNAs for Myh11 and Acta2 accumulated in rat carotid artery following balloon angioplasty, and the kinetics of this response were consistent with a role for RNA editing in mediating the contractile protein repression post-injury. ADAR1 homozygous knockout is lethal (7), but heterozygous deletion dramatically reduced neointimal hyperplasia in a mouse wire injury model, revealing a causal role for ADAR1 in phenotypic modulation in vivo. ADAR1 knockdown similarly promoted a contractile phenotype in cultured SMC. While SMC express both ADAR1 and ADAR2, only ADAR1 was regulated by PDGF, and ADAR2 did not appear to compensate for ADAR1 loss of function. Additional evidence implicated the editing and splicing mechanism, as an editing-deficient ADAR1 mutant or the general splicing inhibitor Isoginkgetin were unable to modulate contractile mRNA levels in SMC. Deciphering the signaling mechanisms by which ADAR1 is regulated by PDGF will be of interest, as will the question of whether there is a consensus motif that targets particular adenine for editing by ADAR1 in SMC mRNAs.
It is worth noting that the authors found no evidence for ADAR1-mediated editing in transcripts encoding other contractile proteins and genes that regulate SMC phenotype, including the transcriptional regulators SRF, myocardin, and Kruppel-like factor 4 (KLF4), suggesting that other mechanisms contribute to their regulation post-injury or PDGF treatment (2). Clearly, not all contractile protein mRNAs are regulated by editing, yet the phenotype of ADAR1 partial loss of function in vascular injury is dramatic. This suggests that this editing mechanism could potentially target additional genes that contribute to other aspects of SMC phenotypic switching, including proliferation, migration, and extracellular matrix deposition. miRNAs have also been implicated in regulation of SMC differentiation (6). As miRNA precursors can also undergo editing (7), it will be of interest to determine whether miRNA editing might influence SMC phenotype. In the future, next generation deep sequencing approaches could be employed to reveal the full spectrum of edited transcripts in phenotypically modulated SMC.
This new work (2) implicates the novel mechanism of PDGF-induced RNA editing in SMC and importantly reveals that inhibition of ADAR1 activity may have therapeutic potential for restenosis and other pathologies of misregulated SMC phenotypic modulation including atherosclerosis, aneurysm, pulmonary hypertension, transplant vasculopathy, hypertension, and others. Recent exciting studies have shown that SMC plasticity extends beyond the contractile to synthetic transition such that SMC can also transdifferentiate toward a macrophage-like phenotype that has implications for atherosclerotic plaque formation and stability (11,12). It will be of interest to determine whether inflammatory stimuli can also regulate RNA editing in SMC and whether this editing is involved in transdifferentiation as well. Additionally, in vivo studies of an Acta2 promoter-reporter gene in a mouse model revealed that transcriptional mechanisms do contribute to ACTA2 downregulation post-injury in vivo (13), and KLF4 has been implicated as a key transcription factor that contributes to SMC phenotypic modulation (12). It is likely that transcriptional and post-transcriptional mechanisms are coordinately regulated, perhaps with distinct kinetics, to control vascular remodeling responses.
Acknowledgments
K.A.M. is supported by NIH grants R01HL091013, 1R01HL118430, and 1R01HL119529, A.B. is supported by an NSF Graduate Research Fellowship DGE-1122492, and R.L. is supported by the David Richmond Fellowship. We thank Dr. James Cooper for assistance with figure preparation.
Footnotes
Disclosures The authors declare no conflicts of interest.
References
- 1.Shi N, Chen SY. Smooth muscle cell differentiation: Model systems, regulatory mechanisms, and vascular diseases. J Cell Physiol. 2016;231(4):777–87. doi: 10.1002/jcp.25208. [DOI] [PubMed] [Google Scholar]
- 2.Fei J, Cui X, Wang J, Dong K, Chen SY. ADAR1-Mediated RNA Editing, A Novel Mechanism Controlling Phenotypic Modulation of Vascular Smooth Muscle Cells. Circ Res. 2016 doi: 10.1161/CIRCRESAHA.116.309003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miano JM. Myocardin in biology and disease. J Biomed Res. 2015 Jan;29(1):3–19. doi: 10.7555/JBR.29.20140151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corjay MH, Blank RS, Owens GK. Platelet-derived growth factor-induced destabilization of smooth muscle alpha-actin mRNA. Journal of cellular physiology. 1990;145:391–7. doi: 10.1002/jcp.1041450302. [DOI] [PubMed] [Google Scholar]
- 5.Liu R, Leslie KL, Martin KA. Epigenetic regulation of smooth muscle cell plasticity. Biochim Biophys Acta. 2015 Apr;1849(4):448–53. doi: 10.1016/j.bbagrm.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miano JM, Long X. The short and long of noncoding sequences in the control of vascular cell phenotypes. Cell Mol Life Sci. 2015 Sep;72(18):3457–88. doi: 10.1007/s00018-015-1936-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol. 2016 Feb;17(2):83–96. doi: 10.1038/nrm.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maas S. Gene regulation through RNA editing. Discovery medicine. 2010;10:379–86. [PubMed] [Google Scholar]
- 9.Whitfield AJ, Barrett PH, van Bockxmeer FM, Burnett JR. Lipid disorders and mutations in the APOB gene. Clin Chem. 2004 Oct;50(10):1725–32. doi: 10.1373/clinchem.2004.038026. [DOI] [PubMed] [Google Scholar]
- 10.Oguro R, Kamide K, Katsuya T, Akasaka H, Sugimoto K, Congrains A, Arai Y, Hirose N, Saitoh S, Ohishi M, Miura T, Rakugi H. A single nucleotide polymorphism of the adenosine deaminase, RNA-specific gene is associated with the serum triglyceride level, abdominal circumference, and serum adiponectin concentration. Exp Gerontol. 2012 Feb;47(2):183–7. doi: 10.1016/j.exger.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Cherepanova OA, Gomez D, Shankman LS, Swiatlowska P, Williams J, Sarmento OF, Alencar GF, Hess DL, Bevard MH, Greene ES, Murgai M, Turner SD, Geng YJ, Bekiranov S, Connelly JJ, Tomilin A, Owens GK. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat Med. 2016 Jun;22(6):657–65. doi: 10.1038/nm.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015 Jun;21(6):628–37. doi: 10.1038/nm.3866. Epub 2015 May 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hendrix JA, Wamhoff BR, McDonald OG, Sinha S, Yoshida T, Owens GK. 5′ CArG degeneracy in smooth muscle alpha-actin is required for injury-induced gene suppression in vivo. J Clin Invest. 2005 Feb;115(2):418–27. doi: 10.1172/JCI22648. [DOI] [PMC free article] [PubMed] [Google Scholar]