

Excess lipids are stored in the form of neutral lipids in lipid droplets. The inability to convert excess lipids into neutral lipids during anaphase creates a lipid imbalance that perturbs the normal dynamics of cytokinesis molecules, causing a delay in cell separation.
Abstract
The neutral lipids steryl ester and triacylglycerol (TAG) are stored in the membrane-bound organelle lipid droplet (LD) in essentially all eukaryotic cells. It is unclear what physiological conditions require the mobilization or storage of these lipids. Here, we study the budding yeast mutant are1Δ are2Δ dga1Δ lro1Δ, which cannot synthesize the neutral lipids and therefore lacks LDs. This quadruple mutant is delayed at cell separation upon release from mitotic arrest. The cells have abnormal septa, unstable septin assembly during cytokinesis, and prolonged exocytosis at the division site at the end of cytokinesis. Lipidomic analysis shows a marked increase of diacylglycerol (DAG) and phosphatidic acid, the precursors for TAG, in the mutant during mitotic exit. The cytokinesis and separation defects are rescued by adding phospholipid precursors or inhibiting fatty acid synthesis, which both reduce DAG levels. Our results suggest that converting excess lipids to neutral lipids for storage during mitotic exit is important for proper execution of cytokinesis and efficient cell separation.
INTRODUCTION
Lipids are essential components in all life. They are synthesized and modified in the cell to provide energy, membranes, and signaling molecules. To avoid toxicity, cells convert excess lipids into the inert form of neutral lipids, predominantly triacylglycerol (TAG) and steryl ester (SE), which are stored in the membrane-bound organelle lipid droplets (LDs; Walther and Farese, 2012; Pol et al., 2014). The neutral lipids are synthesized within the endoplasmic reticulum membrane bilayer and organized into a lens that bulges out toward the cytosolic face of the endoplasmic reticulum to form nascent LDs. In yeast cells grown under normal conditions with glucose as the carbon source, equal amounts of TAG and SE are typically stored within the same LDs (Czabany et al., 2008). The acyltransferases Dga1 and Lro1 catalyze esterification of diacylglycerol (DAG) to generate TAG (Dahlqvist et al., 2000; Oelkers et al., 2000; Sorger and Daum, 2002), whereas Are1 and Are2 produce SE by sterol esterification (Zweytick et al., 2000). When all four of the neutral lipid-synthesizing enzymes are deleted in the yeast genome, the quadruple mutant (are1Δ are2Δ dga1Δ lro1Δ) fails to form LDs, supporting the idea that the biogenesis of LDs is coupled to the synthesis of neutral lipids (Sandager et al., 2002).
De novo TAG synthesis involves multiple fatty acid acylation steps. The two major precursors for TAG synthesis, phosphatidic acid (PA) and DAG, are also central intermediates for phospholipid synthesis (Athenstaedt and Daum, 1999; Kohlwein, 2010). PA can be either converted to DAG by the PA phosphatase Pah1, an orthologue of mammalian lipin (Carman and Han, 2006; Han et al., 2006; O’Hara et al., 2006), or used directly for the production of phospholipids via the intermediate cytidine diphosphate (CDP)–DAG, a precursor for phosphatidylinositol (PI), phosphatidylserine (PS), phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol, and cardiolipin (Carman and Zeimetz, 1996; Carman and Henry, 1999). In addition to the CDP-DAG pathway, membrane phospholipids can be made from DAG (Natter et al., 2005; Gibellini and Smith, 2010; Henry et al., 2012). The Kennedy pathway synthesizes CDP-choline or CDP-ethanolamine, which are ultimately attached to DAG to form PC or PE (Gibellini and Smith, 2010). Thus the metabolic flux of fatty acids can be incorporated into either phospholipids or the biologically inert TAG. Like other organelles, LDs can be taken up by autophagy pathway to mobilize their storage lipids (Wang, 2015). In addition, the surface of LDs harbors resident TAG lipases and SE hydrolases, which act to efficiently supply the cell with energy source and membrane phospholipids. Yeast cells have multiple TAG lipases, which exhibit substrate specificity for different TAG species. The consumption of neutral lipids may lead to the production of various types of phospholipids with specific lengths or types of the acyl chains, which may be used by cells for different purposes. However, very little is known about what physiological conditions involve neutral lipid metabolism and how the specific type of lipid product might control cellular functions.
The cellular membrane system undergoes dramatic remodeling during the cell division cycle to support nuclear division, organelle inheritance, and cell separation. The yeast nuclear envelope must double in size to accommodate its division, which is probably achieved by the continuous synthesis of phospholipids during the cell cycle (Arnone et al., 2013). After nuclear division, the physical separation of the cells, known as cytokinesis, also requires dynamic change and remodeling of the membrane (Weiss, 2012). This process involves temporally and spatially ordered events conducted by the sequential action of various molecules. It begins with ingression of the plasma membrane, which subsequently forms the cleavage furrow at the division plane to close the junction between the dividing cells. The final separation of cells requires targeting of secretory vesicles to the division site to deposit new membrane and enzymes, which remodel the extracellular matrix. The cell cycle events are likely coordinated with lipid metabolism. In fact, TAG synthesis and lipolysis are both subjected to cell cycle control. Pah1, the key enzyme in TAG and phospholipid synthesis, is phosphorylated and inactivated by the yeast cyclin-dependent kinase Cdk1/Cdc28 at the G2/M phase (Santos-Rosa et al., 2005), which promotes PA accumulation and phospholipid synthesis. In addition, Cdk1/Cdc28 phosphorylates and activates the major LD-localized TAG lipase Tgl4 at late G1 phase, which contributes to bud formation (Kurat et al., 2009). Given that the cell cycle, especially during mitosis to cytokinesis, involves many dynamic events of the membrane, it is conceivable that lipid metabolism from LDs might play a role in the lipid modification for remodeling membrane structure or generating lipid signaling molecules to govern the cell cycle transitions.
We are intrigued by the question of whether use or storage of neutral lipids in LDs might be involved in late cell cycle events. We tackled this question by the use of a LD-deficient quadruple mutant. Here we describe cytokinesis defects in this mutant after release from mitotic arrest. We examined the ultrastructure of the mutant by electron microscopy (EM) and followed the dynamics of cytokinesis molecules by fluorescence microscopy. Furthermore, we performed lipidomics analysis to understand how the lack of LDs might cause cell division defects.
RESULTS
LD deficiency affects cytokinesis
The cellular membrane system undergoes expansion, remodeling, and reorganization in order to achieve organelle inheritance and cell separation during eukaryotic cell division cycles. To investigate whether the neutral lipid reservoir LDs might contribute to this process in the budding yeast Saccharomyces cerevisiae, we examined mitosis in the quadruple mutant are1Δ are2Δ dga1Δ lro1Δ, which is unable to synthesize the neutral lipids TAG and SE and therefore lacks LDs. It was shown previously that the quadruple mutant grows normally under standard laboratory conditions, such as on yeast extract/peptone/dextrose (YPD) medium (Sandager et al., 2002). To examine carefully the late cell cycle stage, we first arrested the cells at mitosis with the microtubule inhibitor nocodazole. We then released the cells into synchronous cell cycle after removing nocodazole. Analysis of the DNA content showed that the shift of DNA peak from 2C to 1C was delayed in the quadruple mutant compared with that in the wild type (WT; Figure 1A), suggesting that mitosis or cell division was affected in the mutant. We scored mitotic cells based on the presence of a large bud by light microscopy. The population of large-budded WT cells gradually decreased after 30 min of release from mitotic arrest. At 60 min, ∼60% of WT had completed cell division, whereas ∼80% of the quadruple mutant remained large budded (Figure 1B), consistent with a delay in cell division in the mutant. Staining DNA with 4′,6-diamidino-2-phenylindole (DAPI) revealed that the fraction of cells with an undivided nucleus decreased over time after release from mitotic arrest, with only a small lag in the mutant compared with WT (Figure 1C). At 60 min, only ∼10% of the mutant cells were yet to finish anaphase (Figure 1C), indicating that the delay in cell division occurs after anaphase. A hallmark of anaphase is the ubiquitination and degradation of several mitotic regulators, including the anaphase inhibitor Pds1, the mitotic cyclin Clb2, and the activator for the anaphase ubiquitination machinery, Cdc20 (Thornton and Toczyski, 2006). We thus followed the degradation of these proteins during the time course of release from mitotic arrest. Western blot analysis showed that the levels of Pds1-8myc, Clb2, and Cdc20 declined with similar kinetics in WT and the mutant (Figure 1D), indicating that these proteins were degraded normally during the release from mitotic arrest. Therefore we conclude that the LD-deficient mutant undergoes normal mitosis but is probably delayed at or after cytokinesis.
FIGURE 1:

The LD-deficient mutant is delayed in cytokinesis. (A) Analysis of DNA content in WT and are1Δ are2Δ dga1Δ lro1Δ quadruple mutant after release from nocodazole arrest for the time indicated on the right. (B) Percentage of large-budded cells after release from nocodazole arrest. Mean ± SD from three experiments (n > 200). (C) The same cells as in B were stained with DAPI and scored for unseparated DNA mass. n.s., p > 0.05. (D) Cells released from mitotic arrest were processed for Western blot analysis of proteins indicated on the left. Actin serves as a loading control. The migration of molecular size standards is indicated on the right. (E) WT, are1Δ are2Δ, dga1Δ lro1Δ, and the quadruple mutant were scored for large-budded cells at 1 h after release from M phase. The data represent mean ± SD from five independent experiments. **p < 0.01, ***p < 0.001.
To determine which neutral lipid is important for cell division, we generated the double mutants are1Δ are2Δ and dga1Δ lro1Δ, which cannot synthesize SE and TAG, respectively. We found that after 1-h release from M phase, ∼48% of are1Δ are2Δ and 55% of dga1Δ lro1Δ remained large budded, values between those for WT (35%) and the quadruple mutant (79%) (Figure 1E). The result suggests that both double mutants have an intermediate degree of cell division defect compared with the quadruple mutant and that synthesis of both SE and TAG is important for cell division.
We next asked whether the quadruple mutant is defective in cytokinesis by directly examining the ultrastructure of cells by EM. In yeast, the ingression of plasma membrane at the division plane is concomitant with the synthesis of primary septum, an electron-translucent structure under EM (Figure 2A). The completion of primary septum is followed by the deposition of electron-dense secondary septum at both sides to form a trilaminar structure (Figure 2A). We analyzed cells at 1-h release from M phase and found that only a few (5 of 101) large-budded WT cells were at early cytokinesis with ingressing primary septum, all of which showed a symmetric pattern (Figure 2B). In addition, very few WT cells with complete septa (4 of 96) showed any structural abnormality (Figure 2B). However, approximately one-third (46 of 146) of the large-budded quadruple mutant cells were yet to complete the septa, and most of them showed asymmetric and/or multiple ingression of the primary septum (Figure 2B), indicating a defect in the early step of cytokinesis. In addition, most of the mutant cells with complete septa (77 of 100) showed curved or branched septa surrounding cytoplasmic materials, a structure known as lacuna, which was previously found in several cytokinesis mutants (Shaw et al., 1991; Cid et al., 1998; Schmidt et al., 2003). In addition, the cell wall at the division site often extended into mutant cells, forming a thickened cell wall and irregular cell surface (Figure 2B). The result confirms that the quadruple mutant is defective in forming cytokinetic apparatus. By removing the cell wall and the septum with Zymolyase, we found that the population of large-budded quadruple mutants at 1-h release was greatly reduced (unpublished data), indicative of a delay in cell separation. Therefore the structural abnormality of the septa might hinder its dissolution, affecting cell separation.
FIGURE 2:

The LD-deficient mutant displays abnormal structure at the cytokinesis region. (A) Schematic representation of cytokinesis in budding yeast. (B) Electron micrographs of the bud neck region in WT and the quadruple mutant cells at 1 h after release from M phase. White arrowhead, asymmetric ingression of the primary septum; red arrowhead, inclusion of cytoplasmic materials; yellow arrowhead, aberrant septa; white arrow, inward growth of the cell wall. Bar, 500 nm. The table shows the quantification of the structural abnormality in large-budded cells from the electron micrographs.
We also analyzed the cell cycle phenotype of LD deficiency in the BY4742 background strains. Similar to the results in W303 cells described earlier, we found that the BY4742 quadruple mutant was delayed in cell division upon release from nocodazole arrest (Supplemental Figure S1A). The mutant also displayed asymmetric ingression of the primary septum, aberrant septa, and the formation of lacuna in the EM study (Supplemental Figure S1B). These results support the idea that cytokinesis defects are common phenotypes of the LD-deficient mutant, irrespective of the strain background. Therefore we conclude that LDs play an important role in the proper separation of cells.
Septin is unstable during cytokinesis in the quadruple mutant
The molecular mechanism of cytokinesis has been studied in great detail in yeast. Cytokinesis involves a series of temporally and spatially ordered events. In budding yeast, the first cytokinesis molecules that accumulate at the mother–bud neck are the evolutionarily conserved septin proteins (Oh and Bi, 2011). The septins in vegetative yeast constitute five members (Cdc3, Cdc10, Cdc11, Cdc12, and Shs1), which assemble into a ring of filaments at the incipient bud site. The septin ring expands into an hourglass-like structure upon bud emergence and then splits into double rings at the onset of cytokinesis (Figure 2A; Vrabioiu and Mitchison, 2006). To investigate whether the quadruple mutant might affect septin, we tagged Cdc10 with green fluorescent protein (GFP) at the C-terminus and examined its localization every 15 min upon release from mitotic arrest. The protein resided at the mother–bud neck in both nocodazole-arrested WT and the quadruple mutant (Figure 3A). The population of cells containing Cdc10-GFP at the neck gradually decreased after release from the arrest—of interest, with a faster kinetics in the mutant than in the WT (Figure 3A). We hardly observed a Cdc10-GFP double ring at any time point, which is consistent with a previous finding that the W303 background strain is defective in septin double-ring formation due to a mutation in the septin-associated protein Bud4 (Voth et al., 2005; Wloka et al., 2011). In addition, the level of septin proteins is reduced in W303 cells at cytokinesis onset to a greater extent than is associated with normal ring splitting (Wloka et al., 2011). Consistently, our time-lapse imaging study showed that Cdc10-GFP signal intensity in WT W303 cells was reduced by ∼60% during mitotic exit without forming double rings (Figure 3B). Of interest, the signal intensity was reduced to <20% of the initial level in the quadruple mutant. The result suggests that septin assembly becomes unstable upon its structural reorganization at the time of ring splitting in the mutant.
FIGURE 3:
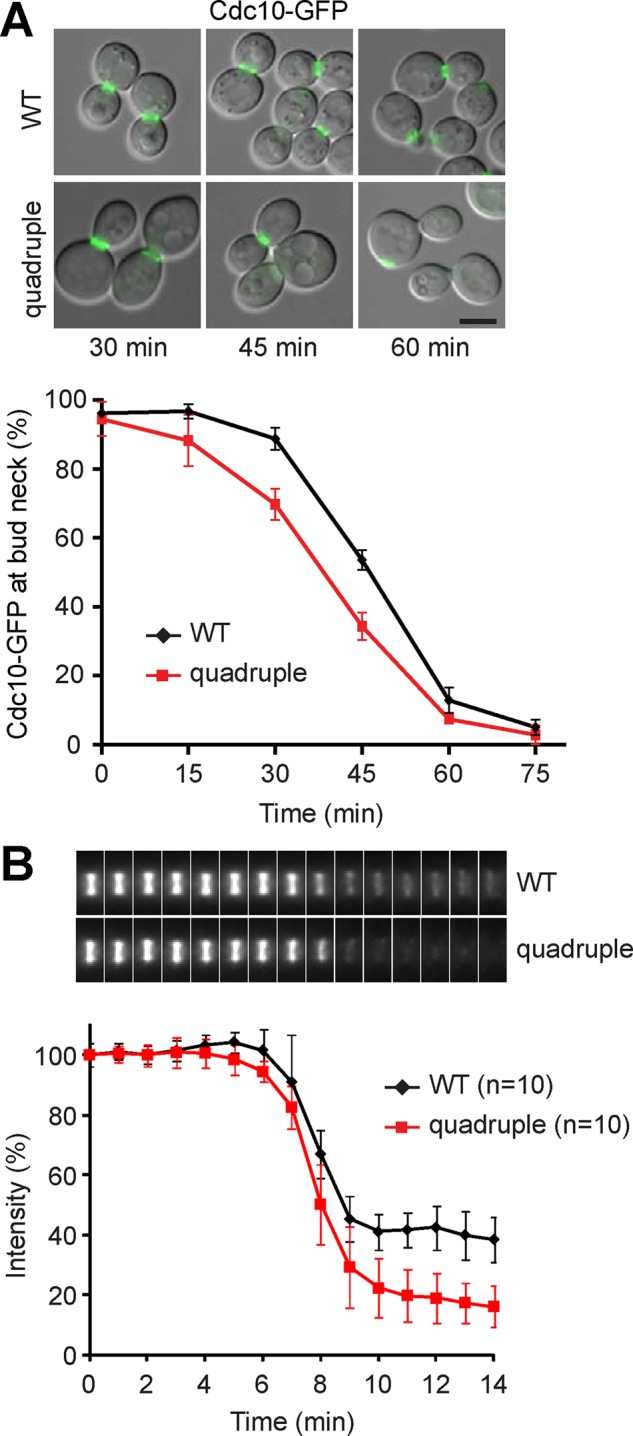
The septin assembly is unstable in the LD-deficient mutant at cytokinesis. (A) Localization of Cdc10-GFP in cells released from M phase at indicated times. Merged images of differential interference contrast (DIC) and Cdc10-GFP. Bar, 5 μm. The plot represents percentage of cells containing Cdc10-GFP at the bud neck after release from M phase; data (mean ± SD) from three independent experiments. (B) Montage of maximum projection images of Cdc10-GFP from representative cells corresponding to the time points shown in the plot below. The signal intensity of Cdc10-GFP at the bud neck relative to the first time point was plotted from 7 min before to 8 min after the signal declines during release from M phase.
We next looked at the BY4742 background strains, which have functional Bud4. We observed a transition of Cdc10-GFP hourglass to double ring after 45 min of release from mitotic arrest in both WT and the quadruple mutant (Supplemental Figure S2A). Time-lapse imaging analysis showed that Cdc10-GFP signal intensity was reduced by ∼25% in WT at the time of ring splitting, whereas the signal reduced by ∼35% in the quadruple mutant (Supplemental Figure S2B). Therefore the septin assembly is also less stable upon ring splitting in the BY4742 quadruple mutant.
Assembly and constriction of the actomyosin ring is intact in the quadruple mutant
The septins function as a scaffold for the recruitment of other cytokinesis molecules to the division site, including the actomyosin ring (Bi et al., 1998). The type II myosin encoded by MYO1 first forms a ring at the incipient bud site before bud emergence and then associates with actin to form the contractile actomyosin ring at anaphase. The contraction of the actomyosin ring at cytokinesis constricts the mother–bud neck and facilitates cell separation (Figure 2A; Bi et al., 1998; Lippincott and Li, 1998). We examined the localization of GFP-tagged Myo1 during release from mitotic arrest. Myo1-GFP resided at the mother–bud neck in nocodazole-arrested WT and quadruple mutant cells, and the signal contracted into a dot in some cells at 45 min of release from the arrest (Figure 4A). The population of cells containing Myo1-GFP at the bud neck decreased gradually after 30 min of release, with a lag in the quadruple mutant in comparison with WT (Figure 4A). Myo1-GFP largely moved to the new bud in WT at 60 and 75 min, whereas weak signal remained with a disorganized pattern at both sides of the neck in some mutant cells (Figure 4A), suggesting that the complete disassembly and dissociation of Myo1 from the cytokinesis site might be delayed in the mutant. Time-lapse imaging also showed that Myo1-GFP ring at the bud neck constricted into a dot in both WT and mutant, whereas the time interval between the initiation of Myo1 ring constriction and its disappearance from the neck was longer and more variable in the mutant than in the WT (Figure 4, B and C). Therefore assembly and constriction of the actomyosin ring are intact in the quadruple mutant, whereas there is a small delay in the disassembly and dissociation of the actomyosin ring at the end of cytokinesis.
FIGURE 4:

Assembly and constriction of the actomyosin ring is intact in the LD-deficient mutant. (A) Localization of Myo1-GFP in cells released from M phase at indicated times. Images of Myo1-GFP and DIC. Bar, 5 μm. The plot shows the percentage of cells containing Myo1-GFP at the bud neck after release from M phase, with data (mean ± SD) from three independent experiments. (B) Maximum projection images of Myo1-GFP in time series beginning with signal constriction and corresponding DIC images. White arrow, cell separation; white arrowhead, appearance of Myo1-GFP at the bud emergence site. Bar, 5 μm. (C) Time intervals between the initiation of Myo1-GFP constriction and its disappearance from the neck (black bar, mean ± SD; n = 43).
Exocyst complex persists at the neck in the quadruple mutant
At late cytokinesis, vesicle trafficking is directed toward the bud neck to deposit membrane and proteins at the division site (Figure 2A; Finger et al., 1998). The initial tethering of secretory vesicles with the plasma membrane is mediated by the evolutionarily conserved exocyst complex, followed by the fusion of membranes (He and Guo, 2009). Several exocyst subunits, including Exo84, are associated with the vesicle, whereas others reside at the targeting membrane to anchor exocyst complex to the plasma membrane (Boyd et al., 2004; He et al., 2007). We tagged Exo84 with GFP to study its dynamics during cytokinesis. At fixed time points after release from mitotic arrest, both WT and the quadruple mutant showed a transient increase of the population with Exo84-GFP signal at the mother–bud neck, peaking at 45 min for WT (Figure 5A). Of interest, the population of quadruple mutant with Exo84-GFP at the bud neck continued to increase until 60 min, when ∼70% of WT had relocated the protein to the tip of the emerging bud (Figure 5A). Consistently, time-lapse imaging of Exo84-GFP during M-phase release showed transient association of the protein at the mother–bud neck (Figure 5B). In WT, the signal often disappeared first from the mother side and relocated to the incipient bud site, showing a change in the direction of vesicle trafficking. However, Exo84-GFP in the quadruple mutant stayed longer at the mother–bud neck than in the WT (Figure 5B). The signal split before its intensity gradually decreased from both the mother and the bud (Figure 5B). We obtained essentially the same result with the exocyst subunit Exo70, which resides at the targeting membrane (unpublished data). These results show that the exocyst complex persists at the division site in the quadruple mutant. It raises the possibility that the anterograde traffic, that is, exocytosis, is up-regulated or that the microenvironment at the mother–bud neck favors secretory vesicle docking and fusion.
FIGURE 5:

Exocyst complex persists at the bud neck in the LD-deficient mutant. (A) Localization of Exo84-GFP in cells released from M phase at indicated times. Images of Exo84-GFP and DIC. The arrowhead points to the signal at a new bud. Bar, 5 μm. The plot shows the percentage of cells containing Exo84-GFP at the bud neck after release from M phase, with data (mean ± SD) from three independent experiments. WT at 90 min was not scored because many cells divided and became large-budded again. (B) Time series of maximum projection images of Exo84-GFP, starting from its appearance at the bud neck. The arrowhead denotes the signal at the incipient bud site. The duration of Exo84-GFP at the bud neck, from its arrival to disappearance at both sides of the neck, was scatter plotted (WT, n = 54; quadruple, n = 58). Black bars represent mean ± SD.
Retention of chitin synthase 2 at the division site in the quadruple mutant
The fusion of secretory vesicles at the division site delivers cargo proteins that are involved in the synthesis and remodeling of the extracellular matrix (the cell wall in yeast). One of the cargoes is chitin synthase 2 (Chs2), which synthesizes chitin to build the primary septum (Figure 2A; Shaw et al., 1991; VerPlank and Li, 2005). Chs2 is transiently targeted to the mother–bud neck during cytokinesis (Zhang et al., 2006) and then internalized by endocytosis for degradation in the vacuole at late stage of cytokinesis (Chuang and Schekman, 1996). As expected, we observed a transient increase of WT population with Chs2-GFP at the bud neck, peaking at 45 min of M-phase release (Figure 6A). The GFP signal disappeared from the neck and appeared in the vacuole at 60 and 75 min (Figure 6A), indicating that Chs2 has been internalized. The quadruple mutant population with Chs2-GFP at the bud neck was significantly higher than that in WT at 60 and 75 min (Figure 6A), paralleling the kinetics of Exo84-GFP localization at the neck. Time-lapse imaging analysis showed that the Chs2-GFP signal constricted during cytokinesis in both WT and the quadruple mutant (Figure 6B). However, the interval between the arrival of Chs2-GFP at the neck and its removal was longer and more variable in the mutant than in WT (Figure 6, B and C). The average duration of the protein at the neck was 6.4 min in WT, whereas the duration was highly variable, with an average of 8.1 min, in the quadruple mutant (Figure 6C). Together the results suggest that Chs2 persists at the cytokinesis site and that the internalization and degradation of Chs2 are delayed in the mutant.
FIGURE 6:
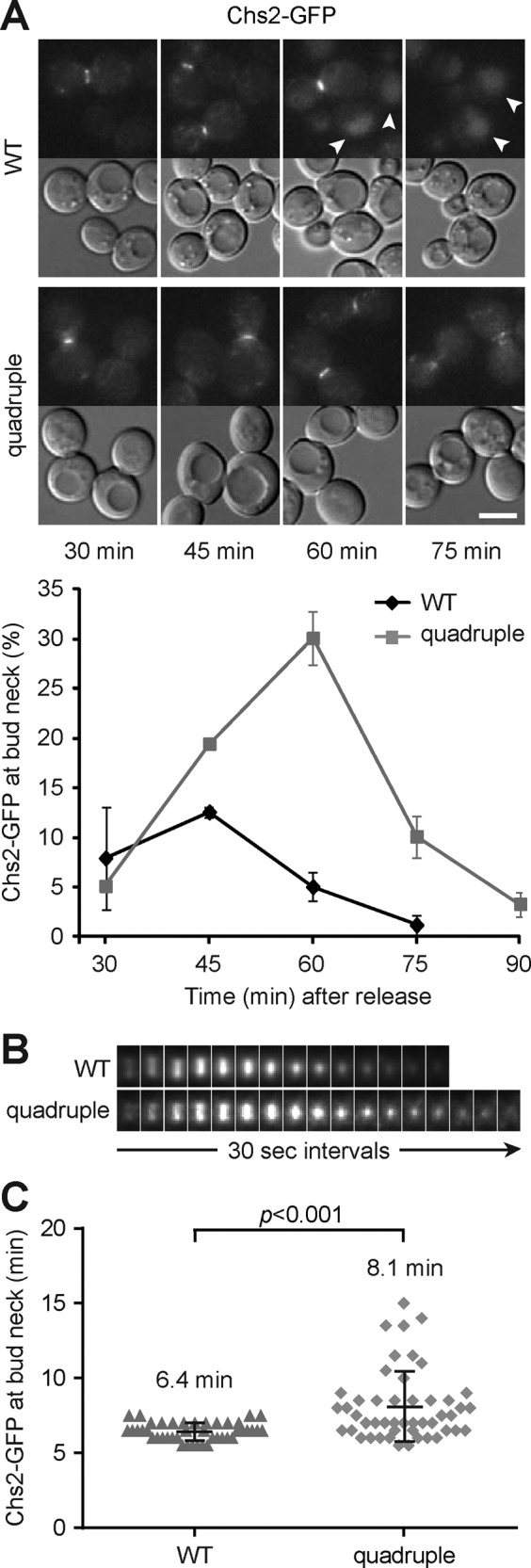
Chs2 is retained at the bud neck in the LD-deficient mutant. (A) Chs2-GFP and DIC images of cells released from M phase at indicated times. Arrowheads point to the signal in the vacuole. Bar, 5 μm. The plot shows percentage of cells with Chs2-GFP at the neck after release from M phase, with data (mean ± SD) from three independent experiments. (B) Montage of maximum projection images of Chs2-GFP at the neck from representative cells in the time series. (C) The scatter plot shows the duration of Chs2-GFP at the neck (WT, n = 43; quadruple, n = 49). Black bars represents mean ± SD.
We also analyzed Chs1, the chitin synthase responsible for the repair of cell wall at the cell junction to prevent daughter cell lysis during cytokinesis (Cabib et al., 1989). We tagged Chs1 with 3GFP to enhance its visualization. Chs1-3GFP appeared as a punctate signal around the cell periphery during the time course of mitotic arrest and release in both WT and the quadruple mutant (Supplemental Figure S3). It indicates that anterograde and retrograde trafficking of Chs1 is probably constant during mitotic exit and is not altered in the quadruple mutant. Consistent with the proper repair of cell wall during cytokinesis, we did not observe a lytic phenotype in the quadruple mutant after release from mitotic arrest (unpublished data).
From the analysis of key factors in cytokinesis, we conclude that the quadruple mutant has at least two alterations: unstable septin assembly and prolonged exocytosis during cytokinesis. To support the notion of prolonged exocytosis at the division site, we examined the formin protein Bni1, which localizes to the site of polarized growth, including the bud neck during the late stage of cytokinesis (Ozaki-Kuroda et al., 2001). We found that the population of cells with Bni1-3GFP at the bud neck was higher in the quadruple mutant than in WT at 45–75 min after release from mitotic arrest (Supplemental Figure S4). This result suggests that the signal for polarized growth at the bud neck persists longer in the mutant.
The quadruple mutant has altered lipid profile during cytokinesis
To understand how the lack of LDs or lipid perturbation in the quadruple mutant might contribute to cytokinesis defects, we analyzed the lipidome of cells that were at log phase, arrested at mitosis, or released from mitosis for 1 h. As expected, liquid chromatography showed that TAG was absent in the quadruple mutant under all conditions (Figure 7A). Of interest, the relative levels of TAG were increased in WT cells at M phase compared with that in log phase and further increased upon M-phase release (Figure 7A and Supplemental Figure S5), indicating continuous synthesis and storage of TAG from mitotic arrest to mitotic exit. In addition, we observed most dramatic difference in the levels of DAG and PA, the precursors for TAG, between WT and the quadruple mutant during mitotic exit. The major DAG species, 34:2 (total length of acyl chain:unsaturated bond) and 36:2, were elevated by greater than threefold in the quadruple mutant compared with WT (Figure 7B and Supplemental Figure S6), whereas the less abundant species with shorter acyl chains, 32:1 and 34:1, were modestly decreased in the mutant under all conditions (Supplemental Figure S6). Similarly, the major PA species, 32:2 and 34:2, were markedly increased in the mutant during mitotic exit (Figure 7C and Supplemental Figure S6). The accumulation of PA and DAG in the mutant during mitotic exit also supports that TAG is otherwise synthesized at this stage.
FIGURE 7:

The LD-deficient mutant displays an altered lipidome during mitotic exit. (A) Chromatograms of DAG and TAG that eluted at 6.4–10 min during liquid chromatography. The graphs from cells grown exponentially (log phase), arrested at mitosis with nocodazole (M phase), or released from the arrest for 1 h (M release) are overlaid. The lipid species in the peaks are indicated above. See also Supplemental Figure S5. (B, C) Relative levels of various DAG (B) and PA (C) species in cells at log phase (L), M phase (M), or release from M phase for 1 h (R). The data from three technical repeats of three independent experiments were normalized to WT at log phase for each species and are shown as mean ± SD. See also Supplemental Figure S6. (D) BODIPY 493/503 signal in WT and the quadruple mutant at log phase, M phase, and release from M phase for 1 h. Bar, 5 μm. (E) Mean ± SD of LD numbers in WT at various stages from three independent experiments. ***p < 0.001. (F) Mean ± SD of LD size in WT at various stages from three independent experiments.
We also visualized the neutral lipids by staining the cell with BODIPY493/503 dye. The result showed an increased number of LDs in cells arrested at mitosis compared with that in log-phase cells (Figure 7, D and E). The number of LDs increased further after 1 h of release from the arrest (Figure 7, D and E), although the LD size remained constant under all of these conditions (Figure 7F). The generation of new LDs during mitotic arrest and release is consistent with the accumulation of TAG found in the lipidomic analysis. As expected, BODIPY staining did not give a discrete pattern in the quadruple mutant, due to the lack of LDs (Figure 7D). Nevertheless, the overall signal intensity was markedly increased in the mutant after mitotic release (Figure 7D), consistent with the accumulation of DAG.
Among the major phospholipids, we found an overall increase of PI 34:2 and 36:2 in the mutant under all conditions, whereas other types of phospholipids showed only a small difference between WT and the mutant (Supplemental Figure S6). We conclude that the inability to synthesize TAG has a greater effect on the levels of DAG and PA than on phospholipids during mitotic exit.
Modulating lipids rescues the cytokinesis defects
Lipid imbalance in the quadruple mutant might be the cause of cytokinesis defects. We thus modulated the cellular lipids by adding inositol and choline to promote PI and PC synthesis at the consumption of PA and DAG, respectively. The addition of choline or inositol during M-phase release reduced the population of the large-budded quadruple mutant at 1 h of release (Figure 8A), indicating partial suppression of cytokinesis phenotypes. Adding both choline and inositol further decreased the large-budded cells to nearly WT level (Figure 8A), suggesting an additive effect. However, adding choline and inositol during mitotic arrest failed to restore cell division (Figure 8A), implying that the cytokinesis defects are probably caused by lipid imbalance generated during anaphase. Of interest, adding the fatty acid synthase inhibitor cerulenin during M-phase release also restored cell division of the quadruple mutant to nearly WT level (Figure 8B), indicating that synthesis of fatty acids during anaphase contributes to the cytokinesis defect in the quadruple mutant. In addition, the addition of choline, inositol, and cerulenin restored normal kinetics of Exo84-GFP at the bud neck of the quadruple mutant, with a gradual decrease of the cell population containing the protein at the neck from 45–75 min of M-phase release (Figure 8C). Consistently, EM study showed that the supplements rescued the structural abnormality at the division site in the quadruple mutant (Figure 8D). Whereas a small fraction of cells (30 of 147 for choline and inositol supplements and 28 of 158 for cerulenin addition) still contained aberrant septa and cytoplasmic inclusions, the defects were much more minor compared with that without any additional treatment during M-phase release (Figure 8D). Furthermore, lipidomic analysis showed that the levels of DAG were slightly reduced in the quadruple mutant supplemented with choline or inositol alone during M-phase arrest, whereas the addition of both or cerulenin further reduced the level (Figure 8E). PA levels were also reduced slightly by the addition of choline or inositol alone and further reduced by the addition of both, whereas the levels were increased by cerulenin (Figure 8E). Together these results indicate that lipid imbalance, in particular the increase of DAG during anaphase, is probably the main cause of the cytokinesis defect in the LD-deficient mutant.
FIGURE 8:
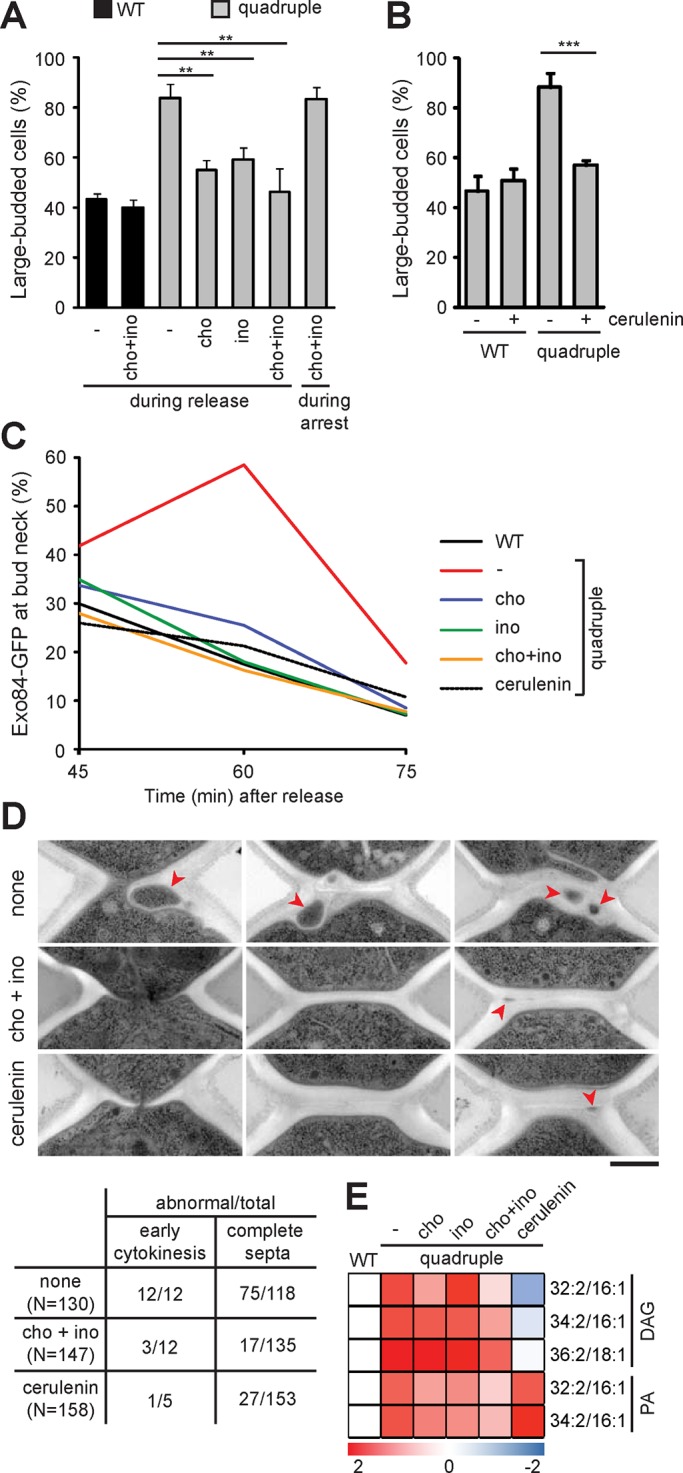
Modulating the lipidome rescues cytokinesis defects in the LD-deficient mutant. (A) Large-budded cells were scored after release from M phase for 1 h; the plot shows mean ± SD from three independent experiments. Choline (cho) and/or inositol (ino) were added after the release or during the arrest as indicated. (B) Large-budded cells were scored after release from M phase for 1 h with or without the addition of cerulenin; the plot shows mean ± SD from three independent experiments. (C) Percentage of cells containing Exo84-GFP at the bud neck after release from M phase at indicated times in the presence of choline, inositol, or cerulenin. (D) Electron micrographs of quadruple mutant released from M phase for 1 h without any supplementation (none), with choline and inositol, or with cerulenin. Red arrowhead, cytoplasmic inclusions. Bar, 500 nm. The table shows quantification of the abnormality. (E) The log2-transformed fold change of lipids in the quadruple mutant under each condition relative to WT after 1-h release from M phase. The heat map shows the data, with the color scale indicated below.
DISCUSSION
In this study, we used yeast mutants that lack the enzymes responsible for the synthesis of neutral lipids TAG and SE to investigate the function of LDs in mitosis. We showed that the quadruple mutant cannot efficiently separate the mother and the daughter after release from mitotic arrest. The mutants display abnormal ultrastructures at the division site, reminiscent of known cytokinesis mutants. Imaging analysis of key cytokinesis molecules revealed unstable septin assembly during cytokinesis and prolonged exocytosis at the division site in the mutant. Of importance, WT and the quadruple mutant show different lipidomes—in particular, a marked increase of PA and DAG levels in the mutant during mitotic exit. The lipid imbalance is likely the cause of cytokinesis defects because the phenotypes can be rescued by the addition of choline, inositol, or the fatty acid synthase inhibitor cerulenin during mitotic exit. Our results suggest a model in which the inability to convert excess lipids into TAG and SE for storage in LDs causes a lipid imbalance that affects dynamic events in cytokinesis (Figure 9).
FIGURE 9:
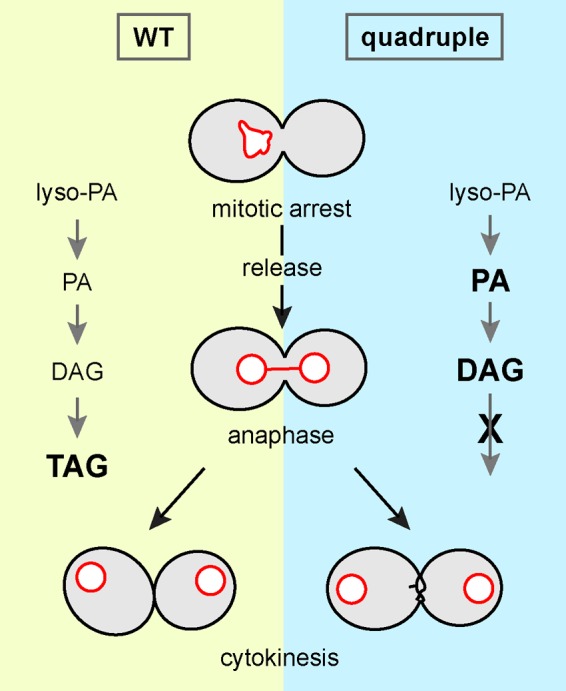
Model for the cytokinesis defect in the LD-deficient mutant. The inability to convert excess lipids into neutral lipids for storage in LDs during anaphase causes lipid imbalance, including accumulation of DAG and PA, which affects cytokinesis and cell separation.
We showed cytokinesis defects in the quadruple mutant of two yeast background strains, W303 and BY4742. Both mutants are delayed in cell division upon release from mitotic arrest. They also share the phenotypes of asymmetric ingression of primary septum, aberrant septa, and the inclusion of cytoplasmic materials in the septa. The W303 background strain contains a mutation in the septin-associated protein Bud4 (Voth et al., 2005), causing a defect in septin double-ring formation and a reduction of septin proteins at the division site after cytokinesis onset (Wloka et al., 2011). We also observed reduction of Cdc10-GFP signal at the bud neck in W303 cells during mitotic exit, with the signal further decreased in the W303 quadruple mutant. Similarly, Cdc10-GFP signal in the BY4742 background strain was lower in the quadruple mutant than in WT after ring splitting. Therefore the septin assembly after reorganization into double ring is sensitive to lipid imbalance in the quadruple mutant. The loss of septin from the division site might account at least in part for asymmetric primary septum, as reported for cells lacking Cdc10 or Bud4 (Wloka et al., 2011). The additional Bud4 mutation likely renders the W303 background cells more sensitive to lipid perturbation, causing a higher frequency of abnormal septa and separation delay than in BY4742 background cells.
The completion of cell separation requires targeted exocytosis at the division site, which provides membranes and proteins to seal the mother–daughter junction and remodel extracellular matrix. We observed persistence of the exocyst complex and its cargo, Chs2, at the division site in the quadruple mutant. The prolonged exocytosis could produce excess membrane and septum materials, which might account for the aberrant septa and irregular cell surface at the division site that we found in the EM studies. It is possible that the secretory vesicles are continuously targeted to the division site or that recycling of the exocyst complex from the site is affected. Chs2 is transiently localized to the division site to synthesize the primary septum, followed by its internalization by endocytosis and degradation in the vacuole in WT. However, internalization and degradation of Chs2 appear to be delayed in the mutant. It is possible that lipid imbalance might affect the signal that switches exocytosis to endocytosis at the end of cytokinesis.
We found cytokinesis defects in the quadruple mutant upon release from mitotic arrest. However, the mutant cells undergoing unperturbed cell cycles neither form cluster of cells nor display any aberrant structure at the bud neck region in EM studies (unpublished results), indicating that cytokinesis is intact. Thus the cytokinesis phenotype is probably associated with prolonged mitosis. The cellular membrane system must expand during mitosis in order to partition into newly divided cells. Prolonged mitosis is known to cause overproliferation of the membrane system, as phospholipid synthesis continues during mitotic arrest (Witkin et al., 2012). It is possible that excess lipids generated during prolonged mitosis should be converted to neutral lipids for storage. In support of this notion, our lipidomics analysis reveals that WT cells arrested at mitosis accumulate more TAG than cells at log phase. It is striking that the levels of TAG are further increased in WT cells released from mitotic arrest. It is likely that Pah1, previously phosphorylated and inactivated by Cdc28 in mitosis (Santos-Rosa et al., 2005), is reactivated to convert PA to DAG for TAG synthesis once Cdc28 activity declines. Mitotic exit might also trigger additional signal that shifts phospholipid synthesis to lipid storage in LDs. As expected from this notion, the quadruple mutant accumulates more PA and DAG, especially the most abundant species, than WT during mitotic exit. Both PA and DAG favor the membrane leaflet with negative curvature to promote membrane fission and fusion (Roth, 2008). Therefore the increased PA and DAG in the quadruple mutant might enhance exocytosis, which could explain the persistence of exocyst complex at the cytokinesis site. The addition of choline and inositol promotes PC and PI synthesis at the expense of DAG and PA. Indeed, the levels of PA and DAG were significantly reduced in quadruple mutant cells that were supplemented with both choline and inositol during release from mitotic arrest. This treatment rescues the cytokinesis defects and restores the normal dynamics of the exocyst complex at the division site, suggesting that the increased PA or DAG might be the cause of the defects. Furthermore, addition of the fatty acid synthase inhibitor cerulenin also rescues cytokinesis defects. Intriguingly, cerulenin significantly reduces DAG while it increases PA levels in the mutant cells during mitotic exit. It remains a possibility that inhibiting fatty acid synthesis might activate phospholipase D, which converts PC into PA, or it might inhibit conversion of PA to DAG due to a shortage of fatty acids. The correlation between restoring cytokinesis and down-regulating DAG suggests that the elevated level of DAG might be more detrimental to cytokinesis or that reducing DAG alone is sufficient to restore cytokinesis. However, we cannot rule out the possibility that other lipid species might affect cytokinesis by destabilizing septin assembly and delaying the directed exocytosis at the bud neck at the end of telophase. Together our results suggest that the inability to synthesize neutral lipids for storage during mitotic exit causes a lipid imbalance that affects membrane properties, leading to cytokinesis defects.
Our study suggests that successful cytokinesis after mitotic arrest particularly requires lipid flux to LDs. Antimitotic agents such as taxanes and vinca alkaloids are extensively used in cancer chemotherapy. Like nocodazole used in our study, these drugs perturb microtubule assembly or dynamics and arrest the cell cycle at mitosis. Instead of permanent arrest, cells exposed to the drugs may undergo cell death, exit mitosis without division, or divide unequally (Gascoigne and Taylor, 2009). This last-cited “mitotic slippage” produces aneuploidy, which can lead to further chromosome instability and genetic alteration in cancer cells, causing failure in the cancer treatment. Our finding that lipid imbalance impairs cytokinesis upon release from mitotic arrest provides an opportunity for a new chemotherapy that combines the antimitotic agents and inhibitors perturbing lipid homeostasis. By interfering with cell separation during mitotic slippage, this combinatorial strategy might reduce drug resistance during chemotherapy.
MATERIALS AND METHODS
Yeast strains and cultures
Carboxyl-terminal GFP tagging and gene deletion were generated by PCR-mediated integration (Longtine et al., 1998). Carboxyl-terminal 8myc tagging of Pds1 was done as described previously (Cheng and Chen, 2010). For tagging Cdc10 with GFP in the BY4742 background strains, we constructed a plasmid containing the carboxyl-terminal 700 base pairs of CDC10 sequence at SacI-HindIII, GFP at ClaI-SalI, and CYC1 terminator sequence at XhoI-KpnI in pRS406. The plasmid was linearized with BglII for integration at the CDC10 locus. For carboxyl-terminal 3GFP tagging, 3GFP sequence was inserted at BamHI-NotI, followed by HIS3 at NotI-SacII in a pBluescript II SK plasmid. The carboxyl-terminal 991 base pairs of BNI1 sequence without the stop codon were inserted in-frame with GFP at HindIII-BamHI, and 421 base pairs of sequence downstream of the stop codon were inserted at SacI. Similarly, the carboxyl-terminal 666 base pairs of CHS1 sequence without the stop codon were inserted at HindIII-BamHI, and 553 base pairs of downstream sequence were inserted at SacI. The plasmids were linearized with HindIII for 3GFP tagging at BNI1 or CHS1. All strains used in this study are listed in Supplemental Table S1. For M-phase arrest, cells were grown in YPD to OD600 = 0.4–0.5 and then treated with 15 μg/ml nocodazole (Sigma-Aldrich, St. Louis, MO) at 30ºC for 2 or 2.5 h for W303 and BY4742 background strains, respectively. M-phase cells were then washed three times with water and resuspended in synthetic complete medium with 2% dextrose. Nocodazole arrest was performed in YPD because the drug does not produce mitotic arrest in synthetic complete medium. For the experiments in Figure 8, cells were supplemented with a final concentration of 1 mM choline (Sigma-Aldrich) from 1 M stock in water, 75 μM myoinositol (Sigma-Aldrich) from 100 mM stock in water, or 10 μg/ml cerulenin (Sigma-Aldrich) from 10 mg/ml stock in dimethyl sulfoxide during the last hour of nocodazole arrest or during the release from M-phase arrest.
Western blots
Cell lysates were prepared as described (Cheng and Chen, 2010). The samples were resolved by SDS–PAGE and transferred to nitrocellulose membrane for blotting with anti-Myc (9E10; Covance, Princeton, NJ), anti-Cdc20 (sc-6731; Santa Cruz Biotechnology, Dallas, TX), anti-yClb2 (sc-9071; Santa Cruz Biotechnology), and anti-actin (MAB1501; Chemicon, Billerica, MA).
Light microscopy
Single-frame still images were taken using a Plan-Neofluar 100×/numerical aperture (NA) 1.3 lens and a MicroMAX 5-MHz charge-coupled device (CCD) camera (Princeton Instruments, Acton, MA) on a Zeiss Axioskop2 fluorescence microscope (Carl Zeiss, Jena, Germany) equipped with MetaMorph Imaging System (version 6.2; Universal Imaging, Bedford Hills, NY). Z-stack still images were acquired with a Plan-Apochromat 100×/NA1.4 lens on a Zeiss Observer Z1 microscope using an AxioCam MR CCD camera and AxioVision, release 4.8, software (Carl Zeiss). All images were processed by ImageJ software (rsb.info.nih.gov/ij/). For time-lapse imaging, cells were immobilized on a layer of 2% agarose made in synthetic complete medium. The coverslip was sealed on the slide with Vaseline/lanolin/paraffin wax in equal parts. Images were taken using a 100×/NA 1.4 lens and a Cascade II 512 electron-multiplying CCD camera (Photometrics, Tucson, AZ) on an Olympus IX71 fluorescence microscope controlled by the DeltaVision system (Applied Precision-GE Healthcare Life Sciences, Pittsburgh, PA). Z-stacks of 10 optical sections with spacing of 0.5 μm were collected and processed by Softworx software (Applied Precision-GE Healthcare Life Sciences). For measuring Cdc10 fluorescence intensity, the average intensity at the bud neck region of maximum projection images was subtracted with the background intensity in the nearby cytoplasm and then normalized to the measurement of the first frame in the time series. To visualize neutral lipids, BODIPY 493/503 (Thermo Fisher Scientific, Waltham, MA) was added to cells at 50 μg/ml and incubated in the dark at room temperature for 10 min, followed by two washes with water. Z-stacks of 13 optical sections at 0.4-μm intervals were collected. The number of lipid droplets in each cell was counted manually from each Z-stack, and the size of lipid droplets was determined using the granularity module of MetaMorph NX1.1 (Molecular Devices, Sunnyvale, CA).
Electron microscopy
Yeast cells were grown and treated as described, and 10 OD600 units of cells were concentrated by centrifugation and immediately cryofixed by high-pressure freezing (High Pressure Freezer EM HPM100; Leica, Wetzlar, Germany). Frozen samples of vitrification were transferred under liquid nitrogen to a freeze substitution and low-temperature embedding system (EM AFS2; Leica). Freeze substitution was performed in 2% osmium tetroxide (Electron Microscopy Sciences [EMS], Hatfield, PA) and 0.1% uranyl acetate (SPI-CHEM, West Chester, PA) in acetone (Merck, Kenilworth, NJ) at −85°C for 66 h, −50°C for 10 h, −20°C for 10 h, and 0°C for 6 h, with the temperature increased at 5°C/h between steps. The samples were then washed by acetone and subjected to infiltration with an increasing gradient (5, 10, 25, 50, 75, and 100%) of Spurr’s resin (EMS) for 2 h to overnight each. After polymerization at 70°C for 48 h, the blocks were cut into 70- to 90-nm ultrathin sections on the ultramicrotome (EM UC6; Leica) with a diamond knife (Ultra 45°, DiATOME, Biel, Switzerland). Sections were collected onto 0.4% formvar (EMS)-coated slot grids (EMS) and imaged at 120 kV on a transmission electron microscope (Tecnai G2 Spirit TWIN; FEI, Hillsboro, OR) using a digital CCD camera (832 Orius SC1000B; Gatan, Pleasanton, CA). The images were processed with Photoshop software (CS5; Adobe, San Jose, CA).
Lipidomic analysis
Lipid extraction was performed as described (Folch et al., 1957). Briefly, 40 OD600 units of cells were collected and lysed by bead beating in methanol and then in additional chloroform. Linoleic acid-d4 (Cayman Chemical, Ann Arbor, MI) was included in the lipid extraction step to serve as an internal standard for subsequent lipid quantification. The samples were centrifuged to remove debris, and the supernatants were supplemented with sodium chloride. After centrifugation, the aqueous phase was removed from the top, and the organic phase containing lipids was dried by the use of a SpeedVac (Thermo Fisher Scientific). The lipids were then dissolved in chloroform/methanol (2:1). For lipid profiling, a linear ion trap-orbitrap mass spectrometer (Orbitrap Elite; Thermo Fisher Scientific, Bremen, Germany) coupled online with an ultrahigh-performance liquid chromatography system (ACQUITY UPLC; Waters, Milford, MA) was used. For liquid chromatography-mass spectrometry analysis, solvent A with 10 mM ammonium acetate, pH 5.0, and 40% acetonitrile in the aqueous phase and solvent B with 10 mM ammonium acetate, pH 5.0, and 10% acetonitrile in isopropyl alcohol were used as the mobile phases for liquid chromatography separation. The lipids were separated online with a reverse-phase column (CSH C18, 1.8 μm, 2.1 mm × 100 mm; Waters) at a flow rate of 450 μl/min using a gradient of 40–99.5% solvent B at 0–10 min. The total chromatography separation time for each of the analyses was 14 min. The mass spectrometer was operated in positive- and negative-ion modes and set to one full Fourier transform–mass spectrometry scan (mass-to-charge ratio m/z = 100–1200, resolution 30,000). The data were analyzed with Xcalibur 2.2 software (Thermo Scientific, Waltham, MA). DAG, TAG, and PC were detected by the positive-ion mode, and PA, PE, and PI were detected by the negative-ion mode. Peak areas were calculated by QuanBrowser for all lipid species, which were determined by their respective retention times on chromatography and by their m/z according to the Lipidomics Gateway (www.lipidmaps.org). The mass tolerance for peak areas quantification was within ±10 mDa. The calculated peak areas representing abundance levels for each lipid species were then normalized to linoleic acid as internal control (monitored under the negative-ion mode).
Supplementary Material
Acknowledgments
We thank the Imaging and EM Core Facility of the Institute of Molecular Biology for assistance in microscopy studies and Yu-Ching Wu of the Metabolomics Core of the Institute of Plant and Microbial Biology for help in lipidomics analysis. This work was supported by grants from the Ministry of Science and Technology and Academia Sinica.
Abbreviations used:
- CDP
cytidine diphosphate
- DAG
diacylglycerol
- DAPI
4′,6-diamidino-2-phenylindole
- DIC
differential interference contrast
- EM
electron microscopy
- LD
lipid droplet
- PA
phosphatidic acid
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PI
phosphatidylinositol
- PS
phosphatidylserine
- SE
steryl ester
- TAG
triacylglycerol
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-02-0106) on June 15, 2016.
REFERENCES
- Arnone JT, Walters AD, Cohen-Fix O. The dynamic nature of the nuclear envelope: lessons from closed mitosis. Nucleus. 2013;4:261–266. doi: 10.4161/nucl.25341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athenstaedt K, Daum G. Phosphatidic acid, a key intermediate in lipid metabolism. Eur J Biochem. 1999;266:1–16. doi: 10.1046/j.1432-1327.1999.00822.x. [DOI] [PubMed] [Google Scholar]
- Bi E, Maddox P, Lew DJ, Salmon ED, McMillan JN, Yeh E, Pringle JR. Involvement of an actomyosin contractile ring in Saccharomyces cerevisiae cytokinesis. J Cell Biol. 1998;142:1301–1312. doi: 10.1083/jcb.142.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd C, Hughes T, Pypaert M, Novick P. Vesicles carry most exocyst subunits to exocytic sites marked by the remaining two subunits, Sec3p and Exo70p. J Cell Biol. 2004;167:889–901. doi: 10.1083/jcb.200408124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabib E, Sburlati A, Bowers B, Silverman SJ. Chitin synthase 1, an auxiliary enzyme for chitin synthesis in Saccharomyces cerevisiae. J Cell Biol. 1989;108:1665–1672. doi: 10.1083/jcb.108.5.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman GM, Han GS. Roles of phosphatidate phosphatase enzymes in lipid metabolism. Trends Biochem Sci. 2006;31:694–699. doi: 10.1016/j.tibs.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman GM, Henry SA. Phospholipid biosynthesis in the yeast Saccharomyces cerevisiae and interrelationship with other metabolic processes. Prog Lipid Res. 1999;38:361–399. doi: 10.1016/s0163-7827(99)00010-7. [DOI] [PubMed] [Google Scholar]
- Carman GM, Zeimetz GM. Regulation of phospholipid biosynthesis in the yeast Saccharomyces cerevisiae. J Biol Chem. 1996;271:13293–13296. doi: 10.1074/jbc.271.23.13293. [DOI] [PubMed] [Google Scholar]
- Cheng YL, Chen RH. The AAA-ATPase Cdc48 and cofactor Shp1 promote chromosome bi-orientation by balancing Aurora B activity. J Cell Sci. 2010;123:2025–2034. doi: 10.1242/jcs.066043. [DOI] [PubMed] [Google Scholar]
- Chuang JS, Schekman RW. Differential trafficking and timed localization of two chitin synthase proteins, Chs2p and Chs3p. J Cell Biol. 1996;135:597–610. doi: 10.1083/jcb.135.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid VJ, Adamikova L, Cenamor R, Molina M, Sanchez M, Nombela C. Cell integrity and morphogenesis in a budding yeast septin mutant. Microbiology. 1998;144:3463–3474. doi: 10.1099/00221287-144-12-3463. [DOI] [PubMed] [Google Scholar]
- Czabany T, Wagner A, Zweytick D, Lohner K, Leitner E, Ingolic E, Daum G. Structural and biochemical properties of lipid particles from the yeast Saccharomyces cerevisiae. J Biol Chem. 2008;283:17065–17074. doi: 10.1074/jbc.M800401200. [DOI] [PubMed] [Google Scholar]
- Dahlqvist A, Stahl U, Lenman M, Banas A, Lee M, Sandager L, Ronne H, Stymne S. Phospholipid:diacylglycerol acyltransferase: an enzyme that catalyzes the acyl-CoA-independent formation of triacylglycerol in yeast and plants. Proc Natl Acad Sci USA. 2000;97:6487–6492. doi: 10.1073/pnas.120067297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger FP, Hughes TE, Novick P. Sec3p is a spatial landmark for polarized secretion in budding yeast. Cell. 1998;92:559–571. doi: 10.1016/s0092-8674(00)80948-4. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Gascoigne KE, Taylor SS. How do anti-mitotic drugs kill cancer cells. J Cell Sci. 2009;122:2579–2585. doi: 10.1242/jcs.039719. [DOI] [PubMed] [Google Scholar]
- Gibellini F, Smith TK. The Kennedy pathway—de novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life. 2010;62:414–428. doi: 10.1002/iub.337. [DOI] [PubMed] [Google Scholar]
- Han GS, Wu WI, Carman GM. The Saccharomyces cerevisiae Lipin homolog is a Mg2+-dependent phosphatidate phosphatase enzyme. J Biol Chem. 2006;281:9210–9218. doi: 10.1074/jbc.M600425200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Guo W. The exocyst complex in polarized exocytosis. Curr Opin Cell Biol. 2009;21:537–542. doi: 10.1016/j.ceb.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Xi F, Zhang X, Zhang J, Guo W. Exo70 interacts with phospholipids and mediates the targeting of the exocyst to the plasma membrane. EMBO J. 2007;26:4053–4065. doi: 10.1038/sj.emboj.7601834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry SA, Kohlwein SD, Carman GM. Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics. 2012;190:317–349. doi: 10.1534/genetics.111.130286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlwein SD. Triacylglycerol homeostasis: insights from yeast. J Biol Chem. 2010;285:15663–15667. doi: 10.1074/jbc.R110.118356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurat CF, Wolinski H, Petschnigg J, Kaluarachchi S, Andrews B, Natter K, Kohlwein SD. Cdk1/Cdc28-dependent activation of the major triacylglycerol lipase Tgl4 in yeast links lipolysis to cell-cycle progression. Mol Cell. 2009;33:53–63. doi: 10.1016/j.molcel.2008.12.019. [DOI] [PubMed] [Google Scholar]
- Lippincott J, Li R. Sequential assembly of myosin II, an IQGAP-like protein, and filamentous actin to a ring structure involved in budding yeast cytokinesis. J Cell Biol. 1998;140:355–366. doi: 10.1083/jcb.140.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Natter K, Leitner P, Faschinger A, Wolinski H, McCraith S, Fields S, Kohlwein SD. The spatial organization of lipid synthesis in the yeast Saccharomyces cerevisiae derived from large scale green fluorescent protein tagging and high resolution microscopy. Mol Cell Proteomics. 2005;4:662–672. doi: 10.1074/mcp.M400123-MCP200. [DOI] [PubMed] [Google Scholar]
- Oelkers P, Tinkelenberg A, Erdeniz N, Cromley D, Billheimer JT, Sturley SL. A lecithin cholesterol acyltransferase-like gene mediates diacylglycerol esterification in yeast. J Biol Chem. 2000;275:15609–15612. doi: 10.1074/jbc.C000144200. [DOI] [PubMed] [Google Scholar]
- Oh Y, Bi E. Septin structure and function in yeast and beyond. Trends Cell Biol. 2011;21:141–148. doi: 10.1016/j.tcb.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hara L, Han GS, Peak-Chew S, Grimsey N, Carman GM, Siniossoglou S. Control of phospholipid synthesis by phosphorylation of the yeast lipin Pah1p/Smp2p Mg2+-dependent phosphatidate phosphatase. J Biol Chem. 2006;281:34537–34548. doi: 10.1074/jbc.M606654200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki-Kuroda K, Yamamoto Y, Nohara H, Kinoshita M, Fujiwara T, Irie K, Takai Y. Dynamic localization and function of Bni1p at the sites of directed growth in Saccharomyces cerevisiae. Mol Cell Biol. 2001;21:827–839. doi: 10.1128/MCB.21.3.827-839.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pol A, Gross SP, Parton RG. Review: biogenesis of the multifunctional lipid droplet: lipids, proteins, and sites. J Cell Biol. 2014;204:635–646. doi: 10.1083/jcb.201311051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth MG. Molecular mechanisms of PLD function in membrane traffic. Traffic. 2008;9:1233–1239. doi: 10.1111/j.1600-0854.2008.00742.x. [DOI] [PubMed] [Google Scholar]
- Sandager L, Gustavsson MH, Stahl U, Dahlqvist A, Wiberg E, Banas A, Lenman M, Ronne H, Stymne S. Storage lipid synthesis is non-essential in yeast. J Biol Chem. 2002;277:6478–6482. doi: 10.1074/jbc.M109109200. [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Leung J, Grimsey N, Peak-Chew S, Siniossoglou S. The yeast lipin Smp2 couples phospholipid biosynthesis to nuclear membrane growth. EMBO J. 2005;24:1931–1941. doi: 10.1038/sj.emboj.7600672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Varma A, Drgon T, Bowers B, Cabib E. Septins, under Cla4p regulation, and the chitin ring are required for neck integrity in budding yeast. Mol Biol Cell. 2003;14:2128–2141. doi: 10.1091/mbc.E02-08-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JA, Mol PC, Bowers B, Silverman SJ, Valdivieso MH, Duran A, Cabib E. The function of chitin synthases 2 and 3 in the Saccharomyces cerevisiae cell cycle. J Cell Biol. 1991;114:111–123. doi: 10.1083/jcb.114.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorger D, Daum G. Synthesis of triacylglycerols by the acyl-coenzyme A:diacyl-glycerol acyltransferase Dga1p in lipid particles of the yeast Saccharomyces cerevisiae. J Bacteriol. 2002;184:519–524. doi: 10.1128/JB.184.2.519-524.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton BR, Toczyski DP. Precise destruction: an emerging picture of the APC. Genes Dev. 2006;20:3069–3078. doi: 10.1101/gad.1478306. [DOI] [PubMed] [Google Scholar]
- VerPlank L, Li R. Cell cycle-regulated trafficking of Chs2 controls actomyosin ring stability during cytokinesis. Mol Biol Cell. 2005;16:2529–2543. doi: 10.1091/mbc.E04-12-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth WP, Olsen AE, Sbia M, Freedman KH, Stillman DJ. ACE2, CBK1, and BUD4 in budding and cell separation. Eukaryot Cell. 2005;4:1018–1028. doi: 10.1128/EC.4.6.1018-1028.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrabioiu AM, Mitchison TJ. Structural insights into yeast septin organization from polarized fluorescence microscopy. Nature. 2006;443:466–469. doi: 10.1038/nature05109. [DOI] [PubMed] [Google Scholar]
- Walther TC, Farese RV., Jr Lipid droplets and cellular lipid metabolism. Annu Rev Biochem. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CW. Lipid droplets, lipophagy, and beyond. Biochim Biophys Acta. 2015;1861:793–805. doi: 10.1016/j.bbalip.2015.12.010. [DOI] [PubMed] [Google Scholar]
- Weiss EL. Mitotic exit and separation of mother and daughter cells. Genetics. 2012;192:1165–1202. doi: 10.1534/genetics.112.145516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin KL, Chong Y, Shao S, Webster MT, Lahiri S, Walters AD, Lee B, Koh JL, Prinz WA, Andrews BJ, et al. The budding yeast nuclear envelope adjacent to the nucleolus serves as a membrane sink during mitotic delay. Curr Biol. 2012;22:1128–1133. doi: 10.1016/j.cub.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wloka C, Nishihama R, Onishi M, Oh Y, Hanna J, Pringle JR, Krauss M, Bi E. Evidence that a septin diffusion barrier is dispensable for cytokinesis in budding yeast. Biol Chem. 2011;392:813–829. doi: 10.1515/BC.2011.083. [DOI] [PubMed] [Google Scholar]
- Zhang G, Kashimshetty R, Ng KE, Tan HB, Yeong FM. Exit from mitosis triggers Chs2p transport from the endoplasmic reticulum to mother-daughter neck via the secretory pathway in budding yeast. J Cell Biol. 2006;174:207–220. doi: 10.1083/jcb.200604094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweytick D, Leitner E, Kohlwein SD, Yu C, Rothblatt J, Daum G. Contribution of Are1p and Are2p to steryl ester synthesis in the yeast Saccharomyces cerevisiae. Eur J Biochem. 2000;267:1075–1082. doi: 10.1046/j.1432-1327.2000.01103.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.