

Imaging of exocytic and endocytic proteins shows which are present at exocytic sites before, during, and after exocytosis in living cells. Rab proteins and SNARE modulators are lost, and dynamin, PIP2, and BAR-domain proteins are rapidly and transiently recruited, where they may modulate the nascent fusion pore.
Abstract
How and when the dozens of molecules that control exocytosis assemble in living cells to regulate the fusion of a vesicle with the plasma membrane is unknown. Here we image with two-color total internal reflection fluorescence microscopy the local changes of 27 proteins at single dense-core vesicles undergoing calcium-triggered fusion. We identify two broad dynamic behaviors of exocytic molecules. First, proteins enriched at exocytic sites are associated with DCVs long before exocytosis, and near the time of membrane fusion, they diffuse away. These proteins include Rab3 and Rab27, rabphilin3a, munc18a, tomosyn, and CAPS. Second, we observe a group of classical endocytic proteins and lipids, including dynamins, amphiphysin, syndapin, endophilin, and PIP2, which are rapidly and transiently recruited to the exocytic site near the time of membrane fusion. Dynamin mutants unable to bind amphiphysin were not recruited, indicating that amphiphysin is involved in localizing dynamin to the fusion site. Expression of mutant dynamins and knockdown of endogenous dynamin altered the rate of cargo release from single vesicles. Our data reveal the dynamics of many key proteins involved in exocytosis and identify a rapidly recruited dynamin/PIP2/BAR assembly that regulates the exocytic fusion pore of dense-core vesicles in cultured endocrine beta cells.
INTRODUCTION
Exocytosis is a fundamental process of eukaryotic cells in which the membrane of a cargo-loaded vesicle and the plasma membrane fuse (Jahn et al., 2003). The molecular control of exocytosis is complex, involving >25 proteins and an unknown number of lipids (Jahn and Fasshauer, 2012). Work over the last several decades has established that the core machinery for membrane fusion is a multipart assembly made up of the proteins syntaxin, SNAP25, and VAMP, collectively called the soluble N-ethylmaleimide–sensitive factor attachment protein receptors (SNAREs; Jahn and Fasshauer, 2012). These three proteins coil together, pulling the plasma membrane and vesicle membrane into close apposition to drive fusion. Although the SNAREs are sufficient to induce membrane fusion in vitro, dozens of other accessory proteins assemble together with the SNAREs to accelerate and regulate exocytosis in cells (Sudhof and Rothman, 2009; Jahn and Fasshauer, 2012).
Exocytosis can be divided into several distinct steps, including docking, priming, fusion, and recapture (Sudhof, 2004). Decades of biochemical, structural, genetic, and cellular work have contributed to a detailed understanding of the molecules involved in each of these stages. For example, vesicle-bound Rab GTPases are believed to control trafficking and docking (Wada et al., 1994; Geppert et al., 1997; Fukuda, 2008). The SNAREs and a collection of cofactors—tomosyn (Fujita et al., 1998; Hatsuzawa et al., 2003), munc18a (Hata et al., 1993; Voets et al., 2001), munc13 (Brose et al., 1995), Ca2+-dependent activator protein for secretion (CAPS; Loyet et al., 1998; Jockusch et al., 2007), and others—have been proposed to be critical to priming and fusion (Jahn and Fasshauer, 2012). The calcium sensor synaptotagmin is likely the trigger for fusion pore formation (Wang et al., 2001; Chapman, 2008; Sudhof, 2013). Phosphatidylinositol-4,5-bisphosphate (PIP2) and other lipids are central both biochemically and structurally to the process (Hay and Martin, 1993; Holz et al., 2000; Martin, 2015). It is intriguing that several classical endocytic proteins have been implicated in fusion pore regulation and membrane recapture. Dynamin, amphiphysin, and syndapin have been functionally connected to the regulation of the fusion pore (Tsuboi et al., 2004; Min et al., 2007; Llobet et al., 2008; Anantharam et al., 2011; Samasilp et al., 2012). Clathrin and dynamin have been linked with vesicle membrane recapture (Artalejo et al., 1995, 2002; Holroyd et al., 2002; Tsuboi et al., 2004; Anantharam et al., 2011). It is unclear, however, how, when, and whether these proteins are recruited directly to exocytic sites and how their localization might change during and after fusion. Visualizing the dynamic behavior of all these components at single sites of exocytosis is needed to fully understand this complex and highly regulated cellular process.
Here we image the behavior of 27 proteins at single sites of calcium-triggered exocytosis in living endocrine cells. Specifically, we image pairs of fluorescent proteins with two-color total internal reflection fluorescence microscopy (TIRF) in insulin-secreting INS-1 endocrine cells (Steyer and Almers, 2001; Taraska et al., 2003). By observing individual proteins and lipids both at steady state and over time in the same experimental system, we establish the dynamic behavior of the exocytic machinery and directly compare the relative temporal behaviors of the molecular components. Our data identify two broad classes of protein and lipid actions at exocytic sites: 1) proteins enriched at exocytic sites before exocytosis, including Rab proteins and SNARE modulators, that are lost from the site of fusion after exocytosis; and 2) a group of proteins and lipids involving dynamins that are recruited rapidly near the moment of fusion and appear to regulate the release of vesicle cargo. Together our data determine the local dynamics of proteins and lipids at exocytic sites before, during, and after membrane fusion and help to build a spatial and temporal model of the exocytic fusion machine.
RESULTS
We used INS-1 cells as a model system to track calcium-stimulated exocytosis (Hohmeier et al., 2000). These cultured endocrine cells are derived from pancreatic beta cells and release insulin and other hormones—for example, amylin and neuropeptide Y (NPY; Whim, 2011)—from dense-core vesicles (DCVs) in response to extracellular glucose, membrane depolarization, and increases in cytosolic calcium concentrations. We first studied the structure of INS-1 cells. Figure 1, A and B, shows a transmission electron microscope (EM) image of a thin section of an entire INS-1 cell. Numerous vesicles with electron-dense cores rest within a few nanometers of the plasma membrane. The size (170 ± 32 nm, n = 33) and shape of these vesicles are consistent with DCVs from these and other endocrine and neuroendocrine cells (Orci et al., 1977; Nakata and Hirokawa, 1992; Steyer et al., 1997; Toonen et al., 2006). To fluorescently label DCVs, we expressed NPY–green fluorescent protein (GFP), a cargo protein commonly used to mark the vesicle lumen (Lang et al., 1997) in INS-1 cells, using transient transfection. TIRF images of these cells showed dozens of diffraction-limited spots resting within a few hundred nanometers of the plasma membrane (Figure 1C).
FIGURE 1:

Dense core vesicles rest near the plasma membrane of INS-1 cells. (A) TEM image of a 80-nm cross-section of a cultured INS-1 cell. Several DCVs rest near the plasma membrane. Scale bar, 2 μm. (B) Enlarged area (cyan box in A) of the TEM image. Scale bar, 200 nm. (C) TIRF image of an INS-1 cell transfected with NPY-GFP. Scale bar, 2 μm. (D) Overlay of TIRF NPY-GFP image (magenta) and platinum replica TEM image (gray) of an INS-1 cell plasma membrane sheet. The image is 15.4 μm wide. Colored boxes are enlarged panels in E–G (scale bar, 200 nm). (E) A single DCV labeled with NPY-GFP enlarged from D (cyan box). (F) A clathrin-coated pit from D (green box). (G) Other labeled and unlabeled membrane structures found on the INS-1 cell membrane. The structure on the right is stained by NPY-GFP, and the larger structure on the left is not. Actin filaments are also visible (orange box).
To obtain a better physical picture of NPY-GFP–labeled vesicles and their cellular surroundings, we performed correlative light and platinum replica transmission electron microscopy (TEM; CLEM) on cells transfected with NPY-GFP (Heuser, 2000; Sochacki et al., 2012, 2014). Plasma membrane sheets were imaged with TIRF and subsequently prepared for TEM by platinum coating. Platinum replicas were then imaged, and the resulting TEM and fluorescence images were aligned (Figure 1D). CLEM revealed a wide variety of plasma membrane–associated vesicles, filaments, and endocytic structures (Figure 1, D–G). Prominent features of the plasma membrane included the dense network of cortical actin and clathrin-coated structures. Smooth, round vesicles were also prevalent. Figure 1D shows that NPY-GFP–labeled structures were small, smooth, spherical vesicles (172 ± 42 nm, n = 34) that match the diameter and shape of DCVs measured from thin-section TEM. Only a minority of all EM-visible vesicles, however, were marked with NPY-GFP. The remaining unlabeled vesicles at the plasma membrane could represent other vesicles types or DCVs formed before transfection (Corcoran et al., 1984). Thus, using transient transfection likely focuses our analysis on relatively newly packaged DCVs, which could have different behaviors than long-lived DCVs. Indeed, other groups have shown that DCVs exhibit distinct behaviors and localizations as a function of their age (Duncan et al., 2003; Tsuboi et al., 2010).
We first imaged the steady-state association of proteins with NPY-GFP–labeled DCVs. Many of the molecules involved in exocytosis are known (Jahn and Fasshauer, 2012), and we sought to quantify their steady-state colocalization with NPY-GFP–labeled DCVs in INS-1 cells. To determine colocalization, we expressed NPY-GFP and a second, red-labeled test protein in the same cells (Figure 2, A–C). We next used an unbiased quantitative correlation-based image mapping approach to determine the degree of association between the test, red-labeled protein and all visible NPY-labeled vesicles and randomized control images (Figure 2D; Larson et al., 2014). This method was previously developed to account for a wide range of expression levels and intracellular distributions and designed to quantitatively measure colocalization in an unbiased manner in cases that were otherwise difficult to evaluate (Larson et al., 2014).
FIGURE 2:

Correlation analysis with NPY-GFP measures protein colocalization with DCVs and identifies exocytic events in stimulated cells. (A–C) TIRF images of an INS-1 cell cotransfected with NPY-GFP (A) and mCherry-Rab27a (B). (C) Merged image. (D) Correlation analysis of 30 proteins with NPY-GFP–labeled DCVs in INS-1 cells. Cells are sorted based on their mean correlation values. Red boxes indicate proteins associated with DCVs, and gray boxes indicate proteins that are nonspecifically or not associated with DCVs. The box is the 25th–75th percentile range of data, and the whiskers are the SD. The solid bar is the median, and the small dash is the mean. The × marks above and below each data set are the 1st and 99th percentiles. (E, F) Exocytic fusion event analysis in the NPY-GFP channel from A (left) and the mCherry-Rab27a channel from B (right). Top, images of a single exocytic event over time. Middle, corresponding fluorescence intensity of the images. Bottom, average fluorescence intensity trajectory calculated from all events from the same cell. The SE of the data is small and shown in transparency.
Our imaging approach relies on the exogenous expression of fluorescently tagged proteins using transient transfection. We chose transient expression because it enabled us to rapidly screen a large subset of proteins, which would not have been feasible using lower-throughput methods such as genome-engineered cell lines (Ran et al., 2013). Furthermore, transient expression enables us to visualize dynamics that might otherwise be difficult to detect at low expression levels under either endogenous or lower-expression promoter control. Overexpression can be problematic, however, as protein levels could affect protein localization, dynamics, or other cellular behaviors. To test for possible overexpression effects on our analysis, we verified for 11 correlated proteins that there was no relationship between expression levels and correlation values (Supplemental Figure S1). Similar effects were seen in PC12 cells (Larson et al., 2014). With these experimental caveats in mind, these data identify key proteins and lipids that can specifically localize to DCVs, although we cannot fully exclude the possibility that the native behavior of the proteins could be modulated by expression levels or tagging.
We determined the steady-state correlation values for 30 proteins and analyzed >15,000 DCVs from 346 cells. Figure 2D plots the correlation values for each protein sorted according to their average values. Representative TIRF images for each protein are shown in Supplemental Figure S2. We see a wide range of correlation values, similar to previously measured values in PC12 cells (Larson et al., 2014). These data suggest that DCVs in these two cell types are similar in composition (Larson et al., 2014). At the two extremes, rabphilin3a has the highest correlation value at 0.62 and is strongly associated, whereas actin has a negative correlation value and is excluded from DCVs. To evaluate the data more quantitatively, we compared the NPY-GFP correlation values of each protein with the correlation data sets from every other protein using Student’s t test. The p values from this analysis are plotted in Supplemental Figure S3. We find a cluster of proteins that are not statistically different from values for nonspecific markers of the cytoplasm (mCherry) or membrane (farnesylated-mCherry). Highly correlated proteins that are strongly associated with DCVs were rabphilin3a, rab3a, rab27a, CAPS, syntaxin1a, munc18, tomosyn, αSNAP, VAMP2, and VAMP3. These proteins likely fall into three spatial groups: 1) proteins directly bound to the DCV membrane (Rab proteins, VAMP); 2) proteins directly bound to the plasma membrane beneath the docked DCV (syntaxin1a, munc18); and 3) accessory proteins likely interacting with the docking complex (tomosyn, CAPS).
To study the dynamic behavior of these proteins during exocytosis, we stimulated INS-1 cells by local superfusion with 10 μM calcium ionophore ionomycin. Ionomycin reproducibly triggers rapid and robust calcium-dependent exocytosis of DCVs (Suchard et al., 1982). Although we present data from cells stimulated with ionomycin, similar data were obtained with high-potassium stimulation of exocytosis (Supplemental Figure S4). Exocytosis was rarely seen in cells that were not stimulated. Single exocytic events were recognized by flashes of diffraction-limited NPY-GFP fluorescence and then complete loss of signal from the vesicle (Figure 2E; Taraska et al., 2003). The increase is due to neutralization of the acidic lumen of the vesicle and rapid diffusion of the pH-sensitive fluorophores toward the glass coverslip. Owing to the frame rate of our imaging, the exact moment of fusion is difficult to pinpoint, but we estimate fusion to occur within one frame before the maximum intensity change. All detectable fusion events were analyzed, and a majority occurred from vesicles with low diffusion coefficients (∼9 × 10−4 μm2/s) that were stably docked to the plasma membrane (Supplemental Figure S5).
By mapping the spatiotemporal coordinates of exocytic events as determined from the NPY-GFP channel onto the red fluorescence channel, we could then track the behavior of red-labeled test proteins at sites of fusion. We determined the intensity trajectories of these proteins by measuring the average fluorescence at the site of fusion, subtracting a local background, and normalizing the intensity. A representative example of a single event from a cell expressing NPY-GFP and mCherry-Rab27a is shown in Figure 2, E and F. In this example, Rab27a is quickly lost from the vesicle upon cargo release in both the single-event and average traces.
To identify general behaviors of the red-labeled proteins at sites of exocytosis in the noisy cellular environment, we averaged single events to generate average dynamic trajectories (see subsequent discussions of Figures 3–5). Each single trajectory was normalized so that events from different regions and cells could be combined, and we temporally aligned trajectories to the maximum intensity decrease before averaging them together (Taraska et al., 2003). The maximum intensity decrease in the NPY signal, marking the moment of luminal cargo release, was the most robust signal for time alignment, as it was invariantly observed in all exocytic cases, whereas sometimes the sharp increase in fluorescence was of variable amplitude due to our time resolution. Averaging these different types of trajectories gives a slow increase in NPY-GFP intensity before the sharp spike and then drop in fluorescence observed in the average trajectory in Figure 2E.
FIGURE 3:

(A) Rab proteins are stably associated with DCVs before exocytosis and rapidly diffuse away after exocytosis. (B) SNARE modulators are associated with DCVs before exocytosis and diffuse away rapidly after cargo release. Background-subtracted and normalized fluorescence intensity traces are shown for each protein. Top, average NPY-GFP signal for all events in each set (N = 98 for A, N = 118 for B) relative to fusion. Dashed line marks the zero time point. The mean fluorescence intensity is shown as a dark line, and the SE of the data set is shown in transparency for all traces. If the SE is not visible, it is smaller than the line. N = 50 for rab27a, 37 for rab3a, 31 for rabphilin3a, 31 for munc18a, 38 for tomosyn and 49 for CAPS.
FIGURE 4:
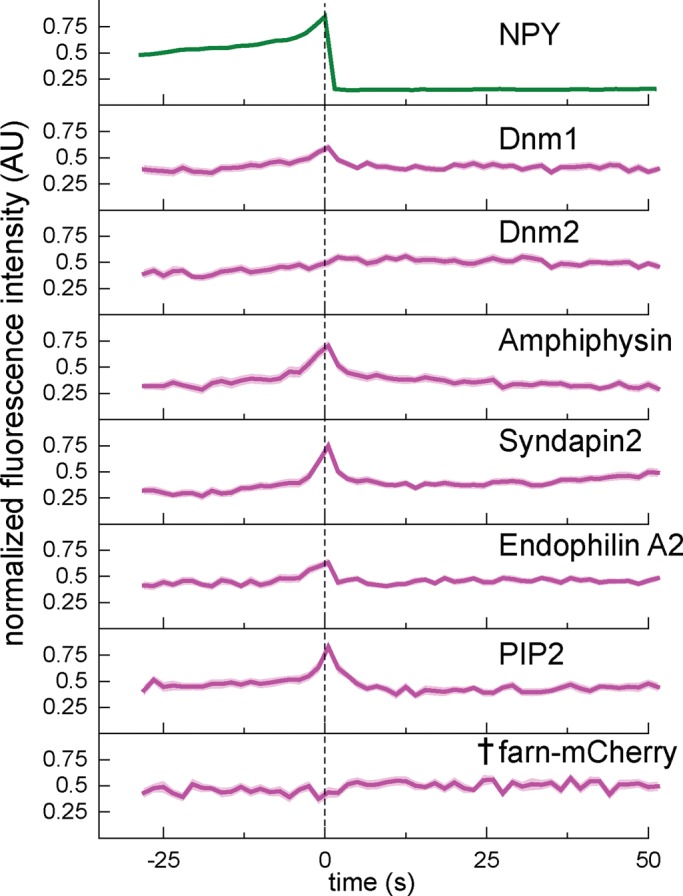
PIP2, dynamins, and several BAR domain–containing proteins are transiently recruited to the exocytic site near the time of membrane fusion. Background-subtracted and normalized fluorescence intensity traces for each protein indicated. Top, average NPY-GFP signal for all events (N = 200) as a relative standard of time. The dashed line marks the zero time point. The mean fluorescence intensity is shown as a dark line, and the SE of the data set is shown in transparency behind the data for all traces. The dagger indicates that there is not a statistically significant shift in intensity at t = 0 relative to the beginning of the trace, determined using t tests in Supplemental Figure S6. If the SE is not visible, it is smaller than the line. N = 50 for dnm-1, 38 for dnm-2, 34 for amphiphysin, 41 for syndapin2, 45 for endophilinA2, 30 for PIP2-sensor, and 30 for farn-mCherry.
FIGURE 5:

Disruption of dynamin-1 and dynamin-2 amphiphysin binding impairs recruitment to exocytic sites. (A) Dynamin-1 point mutants were made to disrupt interaction with PIP2 (K535A), amphiphysin (833-838A), syndapin (S774E,S778E), or GTPase activity (K44A). (B) Dynamin-2 mutations were made to disrupt interaction with amphiphysin and other BAR-domain proteins (dPRD) or PIP2 (K535A). Background-subtracted and normalized fluorescence intensity traces are shown for each protein. Top, average NPY-GFP signal for all events in A (N = 135) as a relative standard of time. The dashed line marks the zero time point. The mean fluorescence intensity is shown as a dark line, and the SE of the data set is shown in transparency behind the data for all traces. If the SE is not visible, it is smaller than the line. Dagger indicates that there is not a statistically significant shift in intensity at t = 0 relative to the beginning of the trace, determined using t tests in Supplemental Figure S6. N = 50 for Dnm1, 29 for Dnm1-K44A, 43 for Dnm1-S774E-S778E, 39 for Dnm1-K535A, 46 for Dnm1-833-838A, 38 for Dnm2, 18 for Dnm2-dPRD, and 33 for Dnm2-K535A.
Although averaging enables us to detect subtle and stereotypical changes in fluorescence in the cellular environment, there are several important considerations to bear in mind. First, the absolute amplitude of the averaged traces is difficult to interpret quantitatively because of the normalization procedure. Consequently the normalized intensity of Rab27a decays to only 0.25 even though we observe no evidence of significant protein retention after exocytosis (single trace, Figure 2F and Supplemental Figure S6). Second, because averaging might obscure heterogeneity in DCV behaviors, we tested whether we could match single-vesicle correlation values with exocytic behaviors. Specifically we tested whether vesicles that underwent fusion had correlation values different from those of steady-state DCVs (Figure 2). We did not, however, detect any substantial differences between vesicles that underwent fusion and their steady-state prefusion values. We hypothesize that this observation might arise because vesicle priming could result from conformational changes in the proteins rather than specific changes in protein components or concentrations. Finally, because we imaged all proteins relative to NPY-GFP and not to each another, we cannot report whether certain combinations of exocytic factors might be enriched at specific DCVs. Here both our correlation analysis and dynamic imaging analysis focus on average DCV behavior, and thus we cannot conclusively show that a large group of proteins coexists or functionally interacts at individual DCVs even though they display similar average dynamic behaviors at DCVs.
To evaluate the statistical significance of the dynamic protein changes we observed at exocytic sites, we performed t tests on the proteins we imaged (Supplemental Figure S6). We calculated an average baseline intensity for each single trajectory by averaging the first 10 frames of the trajectory and then performed a Student’s t test between this baseline value and every other time point across all individual trajectories for that protein. The p values are plotted against time in Supplemental Figure S6, and we interpret p < 0.05 to suggest that the average intensity at that data point in the trajectory is statistically distinguishable from the average baseline intensity before fusion. We use this statistical method to evaluate whether fluorescence fluctuations in average intensity trajectories represent meaningful deviations and therefore protein or lipid recruitments or losses from the site of exocytosis.
We visualized the dynamics of 27 proteins at single sites of exocytosis (1071 events from 154 cells; n values for individuals constructs are given in Supplemental Figure S3B and figure legends). As mentioned earlier, the functionality of tagged proteins is a general concern, but we observed no evidence that our introduced proteins impaired exocytosis or induced morphological changes to the cells or vesicles. Similar numbers of exocytic events were observed across all proteins tested (Supplemental Figure S3B), and no expressed protein caused failure of exocytosis.
The dynamics of Rab proteins—lipidated GTPases located on the cytoplasmic face of the vesicle membrane proposed to be involved in vesicle docking—is shown in Figure 3 (Sudhof, 2004). Rab3a, Rab27a, and rabphilin3a, an effector that binds Rab3a, all showed similar behaviors at exocytosis (Figure 3A). Each was lost rapidly from the vesicle membrane upon cargo release. The average decay kinetics from these Rab proteins was similar, suggesting that these proteins in general diffuse away from vesicles with similar kinetics. The modest increase in mCherry after fusion is consistent with previous observations and is likely due to cytosolic mCherry filling the space vacated by the exocytic protein machinery (Taraska et al., 2003).
Because Rab membrane interactions are important to their function, we examined their diffusion behaviors more closely. Rab27a appeared to be unique, in that it clearly diffused laterally away from the vesicle into the plane of plasma membrane as a spreading wave (Supplemental Figure S7). We calculated a diffusion coefficient of ∼0.1 μm2/s, which is consistent with the diffusion of a membrane-bound protein (Knowles et al., 2010; Honigmann et al., 2014). For other Rabs, despite having similar decay kinetics, we did not observe strong evidence of lateral diffusion, suggesting that loss of these proteins might reflect some degree of detachment from the membrane and diffusion into the cytosol. Regardless of the mechanisms of their loss, our data clearly show that Rabs are lost from the vesicle upon cargo release.
After vesicles dock with the plasma membrane, SNARE proteins are believed to assemble and prime under the control of specific SNARE-binding proteins (Sudhof and Rothman, 2009). Next we considered the dynamics of these SNARE modulators before and after exocytosis. Most of the SNARE modulators had stable fluorescence in the seconds preceding the start of fusion. Several were dramatically lost from the vesicle site upon cargo release (Figure 3B). Munc18a diffuses away from the vesicle within seconds of membrane fusion. Surprisingly, we found that tomosyn is present and stable on the vesicle up until near the time of fusion and then diffuses away. CAPS also remained stably associated with DCVs until within seconds of membrane fusion and then diffused away. We did not observe substantial recruitment or decay of complexin and synaptotagmin upon exocytosis, although both proteins are suggested to have functional roles in membrane fusion (Martens et al., 2007; An et al., 2010; Rao et al., 2014; Supplemental Figure S6). Our correlation analysis confirmed that these proteins are associated with DCVs, although with relatively low values. Complexin and synaptotagmin are diffusely distributed, which contributes to high background fluorescence, and we suspect that this high background may mask our ability to detect small changes in their localization at exocytic sites. It is possible the particular isoforms—synaptotagmin-1 and complexin-2—that we used are either not properly processed or are simply not the primary physiological isoforms of these proteins for INS cells. Indeed, work from other groups has shown that other synaptotagmin isoforms are very important in DCV exocytosis (Rao et al., 2014). Taken together, our data suggest that munc18a, tomosyn, and CAPS remain associated with the vesicle until exocytosis and, like Rabs, are lost and rapidly diffuse away when the fusion pore is formed.
On calcium stimulation, the SNARE proteins are proposed to assemble and pull the vesicular and plasma membrane together to initiate fusion (Jahn and Fasshauer, 2012). We observed only minor intensity changes of the three SNARE proteins at sites of exocytosis (Supplemental Figure S6). Of the three SNARES, VAMP showed the largest intensity changes and appears to be rapidly lost into the membrane at fusion, which is consistent with previous work (Allersma et al., 2004). SNARE proteins are extremely abundant in cells, and we suspect that high expression levels make it difficult to see subtle dynamic changes in their local concentration upon exocytosis (Lang et al., 2001; Barg et al., 2010; Knowles et al., 2010).
Several studies have linked the dynamin GTPases to the latter stages of exocytosis (Tsuboi et al., 2004; Min et al., 2007; Anantharam et al., 2011), and we sought to visualize when and where dynamin is recruited relative to cargo release in INS-1 cells. We found that both dynamin-1 and dynamin-2 are recruited to exocytic sites near the moment of fusion. Dynamin-1 peaks at NPY-GFP release and then decays rapidly, whereas dynamin-2 remains associated for a longer time (Figure 4). Dynamin localization has been shown to depend on both PIP2 and BAR-domain proteins (Schmid and Frolov, 2011; Ferguson and De Camilli, 2012). Thus we visualized these factors to test whether their dynamics mirrored the behavior of dynamin. Strikingly, we observed a large transient increase in the lipid PIP2 near the moment of NPY release (Figure 4). PIP2 was visualized using the pleckstrin homology (PH) domain of PLC-δ4 (Kabachinski et al., 2014). The same pattern of recruitment was observed with the higher-affinity PH domain of PLC-δ1 (Supplemental Figure S6; Suh et al., 2006). We did not observe any recruitment of the control lipid marker farnesylated mCherry (denoted by dagger in Figure 4; intensity at t = 0 was not significantly different from average baseline fluorescence before fusion; see t tests in Supplemental Figure S6). Similarly, we observed a strong transient recruitment of the BAR-domain proteins amphiphysin, syndapin2, and endophilinA2 (Figure 4). We did not observe recruitment of the related BAR domain–containing protein endophilinA1 or endophilinB1, suggesting that this recruitment is not a general property of curvature-sensing or -generating proteins (Supplemental Figure S6; Daumke et al., 2014). We assessed the significance of recruitment using t tests as described earlier, which showed that for dynamin-1, dynamin-2, PIP-sensor, amphiphysin, endophilinA2, and syndapin2, the intensity increase around the time of fusion (time 0 and surrounding time points) was statistically distinguishable from the average intensity before fusion (Supplemental Figure S6). Together our data support the hypothesis that the dynamins (Tsuboi et al., 2004; Min et al., 2007; Anantharam et al., 2011), PIP2 (Hay and Martin, 1993; Holz et al., 2000; Martin, 2015), and specific BAR-domain proteins (Llobet et al., 2008; Samasilp et al., 2012) are not associated with DVCs at steady state but are transiently recruited to exocytic sites near the moment of membrane fusion.
To determine the hierarchy of associations in this complex, we examined how mutant dynamins behaved at single sites of fusion. We observed that the GTPase mutant dynamin-1 K44A was still recruited rapidly during exocytosis (Figure 5). Similar data were obtained for a mutant of dynamin-1 (S774E/S778E) that does not bind to syndapin (Figure 5). Figure 5 shows that the K535A mutation in dynamin-1’s PH domain, which blocks PIP2 binding, was also recruited, but with a smaller relative spike, to wild-type dynamin. The same mutation to block PIP2 binding in dynamin-2 appeared to have no effect on dynamin-2 recruitment. Critically, the dynamin-1 mutant that is unable to bind to amphiphysin (833-838A) was not recruited to vesicles at exocytosis (Figure 5; Supplemental Figure S6 shows p > 0.05 at time 0, denoted by a dagger in Figure 5). Deletion of the PRD domain from dynamin-2, which blocks its interaction with amphiphysin and other BAR-domain proteins, also blocked its recruitment to exocytic sites (intensity actually fluctuates down at fusion, and p > 0.05 at time 0, denoted by a dagger in Figure 5). Collectively these data indicate that dynamin-1 is directed to exocytic sites by association through its PRD domain with the SH3 domain of the BAR protein amphiphysin. This interaction is stabilized by binding of dynamin-1’s PH domain with PIP2 at the vesicle site. Dynamin-2 appears to be recruited primarily through interaction with a binding partner, likely a BAR-domain protein, via its PRD domain. Syndapin may play a role in dynamin-2 recruitment or be functionally important at exocytic sites in a capacity other than dynamin recruitment. In addition, these data indicate that the GTPase activity of dynamin-1 is not required for its recruitment at fusion.
We next investigated dynamin function at exocytic sites by measuring the effect of dynamin perturbation on vesicle cargo release. Fluorescently tagged dynamins can functionally replace endogenous dynamins (Doyon et al., 2011; Taylor et al., 2011; Grassart et al., 2014). We transfected cells with tissue plasminogen activator (tPA) tagged with GFP to visualize DCV cargo release patterns. tPA-GFP is a long-lived vesicular cargo protein that is released slowly from DCVs (Taraska et al., 2003; Weiss et al., 2014). We identified tPA-GFP exocytic events by their dramatic increase in fluorescence upon fusion, caused by neutralization of the acidic vesicle lumen, followed by exponential fluorescence decay as tPA-GFP molecules leave the vesicle lumen with τ = 44.2 s (Figure 6 and Supplemental Figure S8). We cotransfected cells with dynamin-1 or dynamin-2 mutants to investigate how these affect cargo release patterns. Dynamin-1-K535A, which is recruited less strikingly to exocytic sites than is wild-type (WT) dynamin-1, showed substantially faster (τ = 23.6 s; 187% faster than WT decays; Supplemental Figure S8) tPA-GFP decay after fusion pore opening. Other dynamin-1 mutants (S774E/S778E, τ = 32.8 s; K44A, τ = 37.8 s; and 833-838A, τ = 28.6 s) caused faster tPA-GFP release from vesicles, although the effect was not as pronounced as with the K535A mutant. Given that the K535A mutant does not fully block recruitment of dynamin to exocytic sites, this mutant likely imposes a dominant-negative phenotype to disrupt fusion pore regulation. The K535A protein is localized to exocytic sites but cannot interact with lipid, thereby blocking the action of endogenous dynamins and leading to the more pronounced phenotype we observe. Alternatively, we found that perturbation of dynamin-2 binding to PIP2 had no effect on tPA-GFP release (τ = 40.0 s), whereas deletion of the dynamin-2 PRD domain enhanced cargo release (τ = 30.9 s; 143% faster than WT decays; Figure 6 and Supplemental Figure S8). These results suggest that, in addition to their distinct localization patterns, the effect on cargo release of dynamin-1 and dynamin-2 may also be distinct at exocytic sites. Dynamin-1-K535A appears to act as a dominant-negative mutation and perturb cargo release, whereas loss of transgenic dynamin-1 from exocytic sites, with dynamin-1-833-838A, has no effect on release. Conversely, dynamin-2-K535A has no effect on cargo release, and only loss of dynamin-2 from exocytic sites, with dynamin-2-dPRD, enhances cargo release.
FIGURE 6:

Disruption of dynamin-1 or dynamin-2 binding interactions enhances vesicle cargo release. Exocytic events were visualized using tPA-GFP, a long-lived vesicle cargo protein. Plots show mean tPA-GFP fluorescence intensity, background subtracted and normalized, from exocytic sites in WT cells (black) or cells transfected with indicated dynamin mutant proteins (red). Vertical red hash marks overlaid on the plot denote where p < 0.05 for t test comparison between the mutant and WT trajectories. The SE of the data set is shown in transparency around the trace. N = 15 for Dnm1-K535A, 21 for Dnm1-833-838A, 43 for Dnm1-S774E-S778E, 10 for Dnm1-K44A. 57 for Dnm2-K535A, and 25 for Dnm2-dPRD.
Depleting endogenous dynamin-2 also affected DCV cargo release kinetics (Figure 7). First, we confirmed that both dynamin-1 and dynamin-2 are expressed in INS-1 cells with Western blotting and immunofluorescence (Figure 7, A and B), although we detected dynamin-1 at low expression levels only. This observation might be due to proteolysis of dynamin-1, which we also suspect is the reason we do not observe a higher–molecular weight band, corresponding to tagged protein, in positive control cells expressing mCherry-tagged dynamin-1. Small interfering RNA (siRNA) treatment against dynamin-1 had only a very modest effect on dynamin-1 levels in INS-1 cells, as measured by immunofluorescence (Figure 7C), and consequently there was no effect of this treatment on tPA-GFP release kinetics. It is possible that with a more effective knockdown against dynamin-1 we might have observed an effect on tPA-GFP cargo release. Alternately, it might be that mutant dynamin-1-K535A simply has a stronger dominant-negative effect on cargo release than the removal or reduction of dynamin-1 from exocytic sites. Indeed, our dynamin-1 mutation data support such a hypothesis; dynamin-1-K535A, which is still mildly recruited to exocytic sites, has a deleterious effect on cargo release, whereas dynamin-1-833-838A, which is not recruited to exocytic sites, has no effect (Figure 6). However, knockdown of dynamin-2 resulted in substantially faster tPA-GFP decay, which is consistent with our mutational analysis of dynamin-2 at exocytic sites, specifically that lowering dynamin-2 levels at exocytic sites enhances cargo release.
FIGURE 7:

INS-1 cells express dynamin-1 and dynamin-2 and knockdown of dynamin-2 enhances cargo release. (A) Western blots on WT INS-1 cell lysates or INS-1 cell lysates transfected with dynamin-1– or dynamin-2–mCherry constructs. Dynamin-2 is abundant in INS-1 lysates. Dynamin-1 is detectable as full length (∼100 kDa) and proteolytic fragments. (B) Immunofluorescence staining for endogenous dynamin-1 or dynamin-2 in INS-1 cells. Again, dynamin-2 is abundant, but dynamin-1 is also detectable. (C) Quantification of siRNA-induced knockdown of dynamin-2 (p = 0.0125). Fluorescence intensity in transfected (KD) versus nontransfected (dnm2) cells in the same samples. (D) Mean tPA-GFP fluorescence intensity, background subtracted and normalized, from exocytic sites in WT cells (black) or cells transfected with siRNA against dynamin-2 (red); N = 74. The SE of the data set is shown in transparency around the trace.
It is surprising that when we cotransfected cells with siRNA against dynamin-1 and dynamin-2, we found no effect on tPA-GFP release kinetics. Owing to the importance of the dynamins in this and other cellular processes, we hypothesize that cells may compensate for depletion of both dynamins, but not dynamin-2 only, by up-regulating other mechanisms to control exocytosis and endocytosis. Indeed, dynamin-1 and dynamin-2 double-knockout cells remain viable, likely due to compensatory activity by dynamin-3 (Park et al., 2013). Depletion of dynamins in both animal and cell model systems has been shown to affect insulin secretion (Min et al., 2007; Fan et al., 2015). Our results, both the effects of dynamin depletion and distinct localization patterns for dynamin-1 and dynamin-2 (Figure 4), support recent hypotheses for distinct functional roles of dynamin-1 and dynamin-2 in cells (Reis et al., 2015).
After exocytosis, we observed a modest but significant recruitment of clathrin at sites of exocytosis that lasted ∼1 min (Supplemental Figure S6). Clathrin densely populates the plasma membrane of INS-1 cells and PC12 cells, and we cannot exclude the possibility of nonspecific association of clathrin at sites no longer occupied by the assembled DCV (Sochacki et al., 2012). However, the clathrin enhancement is substantially greater than what we observe for nonspecific cytoplasmic marker mCherry, plasma membrane lipid marker farnesylated mCherry, and many other cytoplasmic proteins (Supplemental Figure S6). We also observe a second phase of syndapin2 recruitment tens of seconds after exocytosis (Figure 4). These data suggest the possibility of localized but modest recruitment of specific endocytic proteins to sites of DCV exocytosis several seconds after membrane fusion (Grynszpan-Winograd, 1971; Bittner et al., 2013).
DISCUSSION
The multiprotein assembly that drives regulated exocytosis in eukaryotic cells is complex. More than 20 proteins and lipids are involved (Jahn et al., 2003; Jahn and Fasshauer, 2012). This macromolecular machine is likely dynamic (Gandasi and Barg, 2014). Here we mapped the dynamic, temporal behavior of proteins and lipid probes at single sites of exocytosis in endocrine cells. Our data produce a global, sequential map of protein and lipid dynamics during calcium-triggered exocytosis and help to build a comprehensive model for how proteins and lipids behave during exocytosis.
From our data, we identify two general dynamic behaviors for proteins and lipids involved in exocytosis. One is that exocytic proteins enriched at DCVs before exocytosis are lost from the site of exocytosis upon vesicle fusion. Of importance, we did not observe the recruitment of exocytic factors in the moments preceding membrane fusion. At the time resolution of our experiment, loss occurred with similar kinetics for all proteins, suggesting concerted loss of DCV docking and fusion machinery. The relatively low time resolution here, however, likely obscures more complex, multiple kinetic behaviors as molecules diffuse away from exocytic sites.
In this study, we map single proteins associated with DCVs. Specific subpopulations of DCVs within the cell could associate with distinct collections of these proteins. For example, although both CAPS and tomosyn are strongly associated with DCVs on average, it is possible that these proteins are bound to unique vesicle pools and are not themselves colocalized. Indeed, different synaptotagmin isoforms were shown to associate with distinct subsets of DCVs in chromaffin cells (Rao et al., 2014). Regardless of what subset of exocytic components is enriched at any particular DCV, our data suggest that they are lost from the DCV completely after fusion and diffuse away with broadly similar kinetics. Previous studies have been at odds over exocytic protein behavior postfusion; some suggest that exocytic components are lost upon fusion (Gandasi and Barg, 2014), whereas others suggest retention of proteins at exocytic sites (Ceridono et al., 2011; Bittner et al., 2013).
Unexpectedly, tomosyn strongly associated with some vesicles close to the time of fusion. Tomosyn has been shown to form fusion-incompetent SNARE complexes with syntaxin and SNAP25 and is a potent blocker of exocytosis (Hatsuzawa et al., 2003). Our data, however, show that vesicles with large amounts of tomosyn appear to be fusion active. These data support models in which tomosyn’s role at exocytic sites may be more nuanced (Ashery et al., 2009). We hypothesize that tomosyn could block excess t-SNAREs on the plasma membrane and focus fusogenic activity to only a handful of SNAREs (Takamori et al., 2006; Sieber et al., 2007; van den Bogaart et al., 2010; Sinha et al., 2011). This might be necessary, given the relatively large membrane/vesicle contact area for DCVs.
The possible gap between the assembly of the exocytic machinery and its activity in membrane fusion is in contrast to other dynamic macromolecular machines in the cell. For example, in the formation clathrin-coated pits, dozens of components assemble and disassemble in a continuous pattern of discrete steps over the course of vesicle budding (Taylor et al., 2011; Kukulski et al., 2012). Perhaps most exocytic protein assembly is coupled close to DCV docking and occurs on time scales longer than that of calcium stimulation. This would provide a waiting vesicle pool that is poised to react rapidly to calcium stimulation (Lang, 2003). Furthermore, the loss of proteins involved in vesicle docking and fusion provides evidence against models of exocytosis in which the exocytic fusion protein complex on DCVs remains intact after membrane fusion (Ceridono et al., 2011; Bittner et al., 2013). Although we observe that some components of this machinery are lost from DCVs in INS-1 cells at exocytosis, it is possible that the dense vesicle core remains behind for subsequent recapture (Taraska et al., 2003). This would fit with cavicapture models of exocytosis in PC12 and chromaffin cells, where some vesicle components are lost, but the vesicle cavity remains and the membrane can reseal (Taraska et al., 2003; Perrais et al., 2004). Furthermore, we observe no evidence of exocytic hotspots or preferred docking sites on the plasma membrane defined by the stable accumulation of exocytic factors that remain there postfusion.
The second general dynamic behavior we observed involved a group of classical endocytic proteins and lipids that are rapidly recruited to exocytic sites near the time of membrane fusion, where they likely modulate cargo release. Dynamins, PIP2, and the BAR- domain proteins endophilin A2, amphiphysin, and syndapin all transiently associated with exocytic sites in this manner. Biochemical and electrophysiological studies demonstrated that dynamins and amphiphysin have important roles in modulating exocytosis (Artalejo et al., 1995; Holroyd et al., 2002; Anantharam et al., 2010, 2011). These components dynamically increase at exocytic sites near the time when the fusion pore opens. With the exception of dynamin-2, which appears to remain associated for longer times, dynamin-1 and the other molecules are quickly lost. At our time resolution, protein loss appears to occur with similar kinetics; however, future work is necessary to determine whether these components are recruited and lost with distinct kinetics. The parallel recruitment of these molecules to exocytic sites, their well-established direct binding interactions, and mutagenesis studies performed here suggest a straightforward hypothesis that these molecules form a complex at sites of exocytosis. Indeed, we determined that dynamin-1 is localized to the nascent pore by collective interactions with the BAR-domain protein amphiphysin and the lipid PIP2 (Figure 5; Schmid and Frolov, 2011; Ferguson and De Camilli, 2012). It was previously shown that stimulation with calcium can increase PIP2 synthesis in cells, and it was further hypothesized that PIP2 might be locally synthesized by phosphoinositide kinases localized on the DCV membrane (Eberhard and Holz, 1991; Holz and Axelrod, 2002). Given that PIP2 is important for dynamin recruitment and is known to critically interact with a variety of other factors, localized PIP2 synthesis represents an intriguing means by which DCVs could regulate cargo release. Future work is needed to identify the potential kinases responsible for the burst of PIP2. Amphiphysin and other BAR-domain proteins could localize to the high membrane curvature of the nascent fusion pore. We also observed that depletion of endogenous dynamin-2 leads to faster cargo release. On the basis of their distinct recruitment patterns and depletion effects, we speculate that the two dynamin isoforms may act in distinct functional roles. We hypothesize that dynamin-1 could stabilize the nascent fusion pore, and dynamin-2 could mediate longer–time scale fusion pore expansion. In support of this idea, previous work showed that dynamins have distinct functional properties both in vitro and in vivo (Liu et al., 2011; Reis et al., 2015). These results suggest that these classical endocytic proteins could serve another function at exocytic sites to help stabilize and modulate the fusion pore (Zhang and Jackson, 2010; Schmid and Frolov, 2011; Daumke et al., 2014). Such protein and lipid machinery may be critical in endocrine and neuroendocrine DCV systems with complex and modulatory cargo release patterns (Perrais et al., 2004; Anantharam et al., 2010).
In conclusion, our data show that many exocytic components involved in docking, priming, and fusion are present at exocytic sites before fusion. These static components are lost from sites of exocytosis when cargo is released. Another class of proteins and lipids is rapidly and transiently recruited to exocytic sites near the time of membrane fusion. These factors likely modulate the fusion pore and directly regulate the release of vesicle cargo.
MATERIALS AND METHODS
Plasmids, solutions, and cells
INS-1 832/13 cells were maintained in RPMI-1640 without phenol red (10% fetal bovine serum, 1% penicillin/streptomycin, 11.1 mM glucose, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 2 mM glutamine, 1 mM pyruvate, and 50 μM β-mercaptoethanol) at 37°C at 5% CO2. Cells were used until approximately passage number 80. For experiments, cells were plated on 25-mm #1.5 coverslips. Coverslips were prepared by incubation for approximately 10 min with 0.1% poly-l-lysine solution and then rinsed thoroughly with medium. Cells were transfected using 1 μg of each plasmid with Lipofectamine 2000 according to the manufacturer’s instructions and imaged 24–48 h posttransfection. All imaging was performed at 28°C in imaging buffer (10 mM HEPES, 130 mM NaCl, 2.8 mM KCl, 5 mM CaCl2, 1 mM MgCl2, and 10 mM glucose at pH 7.4). Exocytosis was stimulated using 10 μM ionomycin in imaging buffer or stimulation buffer (10 mM HEPES, 50 mM NaCl, 105 mM KCl, 5 mM CaCl2, 1 mM MgCl2, and 1 mM NaH2PO4 at pH 7.4) applied using a μFlow perfusion system (ALA Scientific Instruments, Farmingdale, NY) with a 100-μm-diameter perfusion tip positioned over the cell. A complete list of the plasmids used is given in Supplemental Figure S3. Chemicals were from Sigma-Aldrich (St. Louis, MO), cell culture reagents and Lipofectamine from Life Technologies (Carlsbad, CA), and fixatives from Electron Microscopy Sciences (Hatfield, PA).
TIRF microscopy
TIRF microscopy was performed as previously described (Sochacki et al., 2012; Larson et al., 2014). Briefly, an inverted fluorescence microscope (IX-81; Olympus, Center Valley, PA) with a 100×/1.45 numerical aperture objective (Olympus) was used for TIRF imaging. Fluorescence was excited alternatively with 488- or 561-nm laser lines that were combined and passed through a 488/561DM filter cube (Semrock, Rochester, NY). Emission was spectrally separated using a 565DCXR dichroic mirror and projected side by side on an electron-multiplying charge-coupled device camera (DU 897; Andor, Belfast, UK) with a DualView image splitter (Photometrics, Tucson, AZ) containing 525Q/50 and 605Q/55 filters. The green and red images were superimposed in postprocessing by acquiring an image of 100-nm yellow-green beads (Invitrogen) that were visible in both channels, mapping the position of six beads in both channels, and then superimposing the channels using projective image transform. This protocol was performed each day before experimental images were recorded. Bead imaging was also used to confirm the uniformity and quality of the TIRF illumination field before each experiment.
Images were acquired with IQ2 software (Andor) successively in the green and then the red channels with an exposure time of 500 ms and a 500-ms pause between pairs of images. Pixel size was 160 nm.
Image analysis
All analysis was performed using custom MATLAB (MathWorks, Natick, MA) scripts and ImageJ. Correlation analysis was performed as previously described (Larson et al., 2014). Exocytic events were identified according to the criteria described in the text by visual inspection of stimulated cell movies in the green (NPY-GFP) channel. Event coordinates were recorded in ImageJ. All identifiable events were analyzed. MATLAB scripts extracted a raw intensity mean (Fcenter) from a 480-nm square box centered on the coordinates and a mean background value from a 1440-nm square surround area (Fsurround); this was performed for both channels. The traces were then temporally aligned to the frame before the maximum intensity decrease of the NPY-GFP channel (t = 0 in our trajectories). We aligned to the maximum intensity decrease because this was the most robust feature of the NPY-GFP signal at exocytic events. At the time resolution used here, the transient brightening of NPY-GFP is not always observed, but the rapid decrease of fluorescence intensity was observed in all traces. Individual trajectories were background subtracted and normalized over 0–1 by
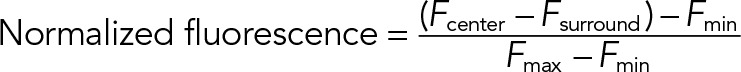 |
where Fmin is the minimum fluorescence intensity over the Fcenter background-subtracted trace and Fmax is the maximum. Average trajectories and SEs were calculated from the normalized trajectories.
tPA-GFP release assay
Cells were transfected with tPA-GFP and a red-tagged protein of interest or siRNA, imaged, and superfused as described. Exocytic events were hand selected as dramatic brightening in the green channel, and these coordinates were used in image analysis as described. Events were time aligned to the maximum intensity increase, as this is the most robust feature of tPA-GFP exocytic events.
Immunofluorescence and siRNA
Cells were transfected with SMARTpool siRNAs (Dharmacon, GE Healthcare Life Sciences, Pittsburgh, PA) against dynamin-1 (140694) or dynamin-2 (25751) according to the manufacturer’s instructions using Lipofectamine 2000. Cells were cultured with siRNAs for 24–72 h before imaging experiments. For immunofluorescence, cells were fixed with 2% paraformaldehyde (PFA) in imaging buffer for 20 min at room temperature. Cells were permeabilized with 0.5% Triton X-100 for 2 min and then incubated in blocking buffer (3% bovine serum albumin, 0.2% Triton X-100 in phosphate-buffered saline [PBS]) for 1 h at room temperature or overnight at 4°C. Primary antibodies for dynamin-1 (1:300, ab52611; Abcam, Cambridge, UK) or dynamin-2 (1:1000, sc-6400; Santa Cruz Biotechnology, Dallas, TX) were added to samples in blocking buffer for 1 h at room temperature or overnight at 4°C. Samples were washed and incubated with secondary antibodies (anti-rabbit or anti-goat Alexa 647–conjugated secondary antibodies, 1:1000) for 30 min at z before washing and postfixing for 20 min at room temperature in 2% PFA. siRNA depletion was estimated from images by measuring mean fluorescence intensity in whole-cell regions in siRNA-transfected cells versus nontransfected cells in the same field of view.
Western blotting
Cells were harvested, pelleted, and lysed with incubation in RIPA buffer (1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS in PBS with Complete Protease inhibitor tablet [Roche, Basel, Switzerland] and 100 μM phenylmethylsulfonyl fluoride added) for 30 min on ice with intermittent vortexing. Cell debris was pelleted by centrifuging the lysate at 15,000 rpm for 10 min. Lysate protein content was quantified with a bicinchoninic acid assay (BCA-1; Sigma-Aldrich), and 20 μg of protein was loaded per lane on a 10–20% Tris-Glycine Novex gel (Invitrogen). The gel was transferred to a polyvinylidene fluoride membrane and blocked in 10% Omniblock in Tris-buffered saline with 0.2% Tween-20 overnight at 4°C. Blots were incubated in primary antibody (dynamin-1, 1:1000, ab52611; dynamin-2, 1:1000, sc-6400; actin, 1:1000, sc-1615) for 1 h at room temperature, washed, and incubated in secondary antibody (dynamin-1, 1:1000, anti-rabbit; dynamin-2, 1:2500, anti-goat; actin, 1:5000, anti-goat) before extensive washing and treatment with enhanced chemiluminescence kit and exposure to film.
Electron microscopy
For thin-section TEM, cells were fixed in 2.5% glutaraldehyde (GA) and 1% PFA for 20 min at room temperature. Secondary fixation with 1% osmium and staining with 1% uranyl acetate was performed before dehydration and infiltration with Epon LX-112. Sections of 80 nm were made and imaged on a JEOL 1400 TEM with an AMT XR-111 camera (Advanced Microscopy Techniques, Woburn, MA).
For CLEM, cells were plated on standard coverslips as described, transfected with NPY-GFP, and processed 1 d later (Sochacki et al., 2014). Cells were rinsed briefly in stabilization buffer (70 mM KCl, 30 mM HEPES, 5 mM MgCl2, 3 mM ethylene glycol tetraacetic acid, pH 7.4) twice and then unroofed by sonication in 2% PFA in stabilization buffer (Heuser, 2000). Sonication was performed within 10 s after being placed in PFA with a Branson Sonifier 450 (Branson Ultrasonics, Danbury, CT; 1/8-inch-tapered tip, 5 mm above the coverslip, single 300-ms pulse, lowest output setting). Cells were fixed for 20 min in 2% PFA. After fixation, cells were stained for 1 h with phalloidin–Alexa 647 (Invitrogen) according to the manufacturer’s instructions. TIRF microscopy was used to image a 15 × 15 image montage of a large field of unroofed cells. After fluorescence imaging, the coverslips were marked with a diamond objective marker and fixed in 2% GA in PBS overnight at 4°C. They were stained with freshly made 0.1% tannic acid for 20 min, rinsed thoroughly in water, stained with 0.1% uranyl acetate for 20 min, rinsed again, and then dehydrated with increasing concentrations of ethanol (5 min each of 15, 30, 50, 70, 80, 90, and 3× 100%). The coverslips were then critical-point dried with a Tousimis Samdri-795 (Tousimis, Rockville, MD). The samples were cut down to the area of interest with a diamond scribe and coated with platinum-carbon and carbon in a JEOL JFD-V freeze-fracture device (JEOL, Tokyo, Japan). The platinum-carbon was rotary coated at 17°, and the carbon was coated at 90°.
The replica was lifted from the coverslip with 5% hydrofluoric acid and placed onto a 75-mesh copper TEM grid coated with Formvar and carbon. Transmission electron microscopy was performed in a JEOL 1400 using SERIALEM software (D. Mastronarde, University of Colorado, Boulder, CO) to create montages at a magnification of 15,000×. The phalloidin fluorescence image was used to map the fluorescence image onto the EM image in Photoshop (Adobe, San Jose, CA). This allowed correlation of NPY fluorescence to specific vesicles. The coordinates of three NPY-positive vesicles in fluorescence and EM were used to quantitatively map the fluorescence image onto the EM image using a nearest-neighbor two-dimensional affine transformation in MATLAB. This maintains the pixel information from the original fluorescence image rather than interpolating to get an artificially smoothed fluorescence image.
Supplementary Material
Acknowledgments
We thank J. Silver, J. Hammer, and all members of the Taraska lab for critical reading of the manuscript and S. L. Shyng (Oregon Health & Science University, Portland, OR) for INS-1/832-13 cells. We also thank Patricia Connelly of the National Heart, Lung, and Blood Institute Electron Microscopy Core for help with thin-section TEM. J.W.T. is supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health.
Abbreviations used:
- DCV
dense-core vesicle
- GFP
green fluorescent protein
- NPY
neuropeptide Y
- PIP2
phosphatidylinositol-4,5-bisphosphate
- TIRF
total internal reflection fluorescence.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-01-0057) on June 15, 2016.
References
- Allersma MW, Wang L, Axelrod D, Holz RW. Visualization of regulated exocytosis with a granule-membrane probe using total internal reflection microscopy. Mol Biol Cell. 2004;15:4658–4668. doi: 10.1091/mbc.E04-02-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An SJ, Grabner CP, Zenisek D. Real-time visualization of complexin during single exocytic events. Nat Neurosci. 2010;13:577–583. doi: 10.1038/nn.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharam A, Bittner MA, Aikman RL, Stuenkel EL, Schmid SL, Axelrod D, Holz RW. A new role for the dynamin GTPase in the regulation of fusion pore expansion. Mol Biol Cell. 2011;22:1907–1918. doi: 10.1091/mbc.E11-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharam A, Onoa B, Edwards RH, Holz RW, Axelrod D. Localized topological changes of the plasma membrane upon exocytosis visualized by polarized TIRFM. Eur J Cell Biol. 2010;188:415–428. doi: 10.1083/jcb.200908010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artalejo CR, Elhamdani A, Palfrey HC. Sustained stimulation shifts the mechanism of endocytosis from dynamin-1-dependent rapid endocytosis to clathrin- and dynamin-2-mediated slow endocytosis in chromaffin cells. Proc Natl Acad Sci USA. 2002;99:6358–6363. doi: 10.1073/pnas.082658499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artalejo CR, Henley JR, McNiven MA, Palfrey HC. Rapid endocytosis coupled to exocytosis in adrenal chromaffin cells involves Ca2+, GTP, and dynamin but not clathrin. Proc Natl Acad Sci USA. 1995;92:8328–8332. doi: 10.1073/pnas.92.18.8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashery U, Bielopolski N, Barak B, Yizhar O. Friends and foes in synaptic transmission: the role of tomosyn in vesicle priming. Trends Neurosci. 2009;32:275–282. doi: 10.1016/j.tins.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barg S, Knowles MK, Chen X, Midorikawa M, Almers W. Syntaxin clusters assemble reversibly at sites of secretory granules in live cells. Proc Natl Acad Sci USA. 2010;107:20804–20809. doi: 10.1073/pnas.1014823107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner MA, Aikman RL, Holz RW. A nibbling mechanism for clathrin-mediated retrieval of secretory granule membrane after exocytosis. J Biol Chem. 2013;288:9177–9188. doi: 10.1074/jbc.M113.450361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, Hofmann K, Hata Y, Sudhof TC. Mammalian homologues of Caenorhabditis elegans unc-13 gene define novel family of C2-domain proteins. J Biol Chem. 1995;270:25273–25280. doi: 10.1074/jbc.270.42.25273. [DOI] [PubMed] [Google Scholar]
- Ceridono M, Ory S, Momboisse F, Chasserot-Golaz S, Houy S, Calco V, Haeberle AM, Demais V, Bailly Y, Bader MF, et al. Selective recapture of secretory granule components after full collapse exocytosis in neuroendocrine chromaffin cells. Traffic. 2011;12:72–88. doi: 10.1111/j.1600-0854.2010.01125.x. [DOI] [PubMed] [Google Scholar]
- Chapman ER. How does synaptotagmin trigger neurotransmitter release. Annu Rev Biochem. 2008;77:615–641. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- Corcoran JJ, Wilson SP, Kirshner N. Flux of catecholamines through chromaffin vesicles in cultured bovine adrenal medullary cells. J Biol Chem. 1984;259:6208–6214. [PubMed] [Google Scholar]
- Daumke O, Roux A, Haucke V. BAR domain scaffolds in dynamin-mediated membrane fission. Cell. 2014;156:882–892. doi: 10.1016/j.cell.2014.02.017. [DOI] [PubMed] [Google Scholar]
- Doyon JB, Zeitler B, Cheng J, Cheng AT, Cherone JM, Santiago Y, Lee AH, Vo TD, Doyon Y, Miller JC, et al. Rapid and efficient clathrin-mediated endocytosis revealed in genome-edited mammalian cells. Nat Cell Biol. 2011;13:331–337. doi: 10.1038/ncb2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan RR, Greaves J, Wiegand UK, Matskevich I, Bodammer G, Apps DK, Shipston MJ, Chow RH. Functional and spatial segregation of secretory vesicle pools according to vesicle age. Nature. 2003;422:176–180. doi: 10.1038/nature01389. [DOI] [PubMed] [Google Scholar]
- Eberhard DA, Holz RW. Calcium promotes the accumulation of polyphosphoinositides in intact and permeabilized bovine adrenal chromaffin cells. Cell Mol Neurobiol. 1991;11:357–370. doi: 10.1007/BF00713279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, Ji C, Wu Y, Ferguson SM, Tamarina N, Philipson LH, Lou X. Dynamin 2 regulates biphasic insulin secretion and plasma glucose homeostasis. J Clin Invest. 2015;125:4026–4041. doi: 10.1172/JCI80652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol. 2012;13:75–88. doi: 10.1038/nrm3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Shirataki H, Sakisaka T, Asakura T, Ohya T, Kotani H, Yokoyama S, Nishioka H, Matsuura Y, Mizoguchi A, et al. Tomosyn: a syntaxin-1-binding protein that forms a novel complex in the neurotransmitter release process. Neuron. 1998;20:905–915. doi: 10.1016/s0896-6273(00)80472-9. [DOI] [PubMed] [Google Scholar]
- Fukuda M. Regulation of secretory vesicle traffic by Rab small GTPases. Cell Mol Life Sci. 2008;65:2801–2813. doi: 10.1007/s00018-008-8351-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandasi NR, Barg S. Contact-induced clustering of syntaxin and munc18 docks secretory granules at the exocytosis site. Nat Commun. 2014;5:3914. doi: 10.1038/ncomms4914. [DOI] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Stevens CF, Sudhof TC. The small GTP-binding protein Rab3A regulates a late step in synaptic vesicle fusion. Nature. 1997;387:810–814. doi: 10.1038/42954. [DOI] [PubMed] [Google Scholar]
- Grassart A, Cheng AT, Hong SH, Zhang F, Zenzer N, Feng Y, Briner DM, Davis GD, Malkov D, Drubin DG. Actin and dynamin2 dynamics and interplay during clathrin-mediated endocytosis. Eur J Cell Biol. 2014;205:721–735. doi: 10.1083/jcb.201403041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynszpan-Winograd O. Morphological aspects of exocytosin in the adrenal medulla. Philos Trans R Soc Lond B Biol Sci. 1971;261:291–292. doi: 10.1098/rstb.1971.0058. [DOI] [PubMed] [Google Scholar]
- Hata Y, Slaughter CA, Sudhof TC. Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin. Nature. 1993;366:347–351. doi: 10.1038/366347a0. [DOI] [PubMed] [Google Scholar]
- Hatsuzawa K, Lang T, Fasshauer D, Bruns D, Jahn R. The R-SNARE motif of tomosyn forms SNARE core complexes with syntaxin 1 and SNAP-25 and down-regulates exocytosis. J Biol Chem. 2003;278:31159–31166. doi: 10.1074/jbc.M305500200. [DOI] [PubMed] [Google Scholar]
- Hay JC, Martin TF. Phosphatidylinositol transfer protein required for ATP-dependent priming of Ca(2+)-activated secretion. Nature. 1993;366:572–575. doi: 10.1038/366572a0. [DOI] [PubMed] [Google Scholar]
- Heuser J. The production of “cell cortices” for light and electron microscopy. Traffic. 2000;1:545–552. doi: 10.1034/j.1600-0854.2000.010704.x. [DOI] [PubMed] [Google Scholar]
- Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 2000;49:424–430. doi: 10.2337/diabetes.49.3.424. [DOI] [PubMed] [Google Scholar]
- Holroyd P, Lang T, Wenzel D, De Camilli P, Jahn R. Imaging direct, dynamin-dependent recapture of fusing secretory granules on plasma membrane lawns from PC12 cells. Proc Natl Acad Sci USA. 2002;99:16806–16811. doi: 10.1073/pnas.222677399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz RW, Axelrod D. Localization of phosphatidylinositol 4,5-P(2) important in exocytosis and a quantitative analysis of chromaffin granule motion adjacent to the plasma membrane. Ann NY Acad Sci. 2002;971:232–243. doi: 10.1111/j.1749-6632.2002.tb04467.x. [DOI] [PubMed] [Google Scholar]
- Holz RW, Hlubek MD, Sorensen SD, Fisher SK, Balla T, Ozaki S, Prestwich GD, Stuenkel EL, Bittner MA. A pleckstrin homology domain specific for phosphatidylinositol 4, 5-bisphosphate (PtdIns-4,5-P2) and fused to green fluorescent protein identifies plasma membrane PtdIns-4,5-P2 as being important in exocytosis. J Biol Chem. 2000;275:17878–17885. doi: 10.1074/jbc.M000925200. [DOI] [PubMed] [Google Scholar]
- Honigmann A, Mueller V, Ta H, Schoenle A, Sezgin E, Hell SW, Eggeling C. Scanning STED-FCS reveals spatiotemporal heterogeneity of lipid interaction in the plasma membrane of living cells. Nat Commun. 2014;5:5412. doi: 10.1038/ncomms6412. [DOI] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490:201–207. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Lang T, Sudhof TC. Membrane fusion. Cell. 2003;112:519–533. doi: 10.1016/s0092-8674(03)00112-0. [DOI] [PubMed] [Google Scholar]
- Jockusch WJ, Speidel D, Sigler A, Sorensen JB, Varoqueaux F, Rhee JS, Brose N. CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell. 2007;131:796–808. doi: 10.1016/j.cell.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Kabachinski G, Yamaga M, Kielar-Grevstad DM, Bruinsma S, Martin TF. CAPS and Munc13 utilize distinct PIP2-linked mechanisms to promote vesicle exocytosis. Mol Biol Cell. 2014;25:508–521. doi: 10.1091/mbc.E12-11-0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles MK, Barg S, Wan L, Midorikawa M, Chen X, Almers W. Single secretory granules of live cells recruit syntaxin-1 and synaptosomal associated protein 25 (SNAP-25) in large copy numbers. Proc Natl Acad Sci USA. 2010;107:20810–20815. doi: 10.1073/pnas.1014840107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukulski W, Schorb M, Kaksonen M, Briggs JA. Plasma membrane reshaping during endocytosis is revealed by time-resolved electron tomography. Cell. 2012;150:508–520. doi: 10.1016/j.cell.2012.05.046. [DOI] [PubMed] [Google Scholar]
- Lang J. PIPs and pools in insulin secretion. Trends Endocrinol Metab. 2003;14:297–299. doi: 10.1016/s1043-2760(03)00136-x. [DOI] [PubMed] [Google Scholar]
- Lang T, Bruns D, Wenzel D, Riedel D, Holroyd P, Thiele C, Jahn R. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 2001;20:2202–2213. doi: 10.1093/emboj/20.9.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Wacker I, Steyer J, Kaether C, Wunderlich I, Soldati T, Gerdes HH, Almers W. Ca2+-triggered peptide secretion in single cells imaged with green fluorescent protein and evanescent-wave microscopy. Neuron. 1997;18:857–863. doi: 10.1016/s0896-6273(00)80325-6. [DOI] [PubMed] [Google Scholar]
- Larson BT, Sochacki KA, Kindem JM, Taraska JW. Systematic spatial mapping of proteins at exocytic and endocytic structures. Mol Biol Cell. 2014;25:2084–2093. doi: 10.1091/mbc.E14-02-0771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YW, Neumann S, Ramachandran R, Ferguson SM, Pucadyil TJ, Schmid SL. Differential curvature sensing and generating activities of dynamin isoforms provide opportunities for tissue-specific regulation. Proc Natl Acad Sci USA. 2011;108:E234–E242. doi: 10.1073/pnas.1102710108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llobet A, Wu M, Lagnado L. The mouth of a dense-core vesicle opens and closes in a concerted action regulated by calcium and amphiphysin. Eur J Cell Biol. 2008;182:1017–1028. doi: 10.1083/jcb.200807034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyet KM, Kowalchyk JA, Chaudhary A, Chen J, Prestwich GD, Martin TF. Specific binding of phosphatidylinositol 4,5-bisphosphate to calcium-dependent activator protein for secretion (CAPS), a potential phosphoinositide effector protein for regulated exocytosis. J Biol Chem. 1998;273:8337–8343. doi: 10.1074/jbc.273.14.8337. [DOI] [PubMed] [Google Scholar]
- Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–1208. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- Martin TF. PI(4,5)P-binding effector proteins for vesicle exocytosis. Biochim Biophys Acta. 2015;1851:785–793. doi: 10.1016/j.bbalip.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min L, Leung YM, Tomas A, Watson RT, Gaisano HY, Halban PA, Pessin JE, Hou JC. Dynamin is functionally coupled to insulin granule exocytosis. J Biol Chem. 2007;282:33530–33536. doi: 10.1074/jbc.M703402200. [DOI] [PubMed] [Google Scholar]
- Nakata T, Hirokawa N. Organization of cortical cytoskeleton of cultured chromaffin cells and involvement in secretion as revealed by quick-freeze, deep-etching, and double-label immunoelectron microscopy. J Neurosci. 1992;12:2186–2197. doi: 10.1523/JNEUROSCI.12-06-02186.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L, Perrelet A, Friend DS. Freeze-fracture of membrane fusions during exocytosis in pancreatic B-cells. Eur J Cell Biol. 1977;75:23–30. doi: 10.1083/jcb.75.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park RJ, Shen H, Liu L, Liu X, Ferguson SM, De Camilli P. Dynamin triple knockout cells reveal off target effects of commonly used dynamin inhibitors. J Cell Sci. 2013;126:5305–5312. doi: 10.1242/jcs.138578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrais D, Kleppe IC, Taraska JW, Almers W. Recapture after exocytosis causes differential retention of protein in granules of bovine chromaffin cells. J Physiol. 2004;560:413–428. doi: 10.1113/jphysiol.2004.064410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao TC, Passmore DR, Peleman AR, Das M, Chapman ER, Anantharam A. Distinct fusion properties of synaptotagmin-1 and synaptotagmin-7 bearing dense core granules. Mol Biol Cell. 2014;25:2416–2427. doi: 10.1091/mbc.E14-02-0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis CR, Chen PH, Srinivasan S, Aguet F, Mettlen M, Schmid SL. Crosstalk between Akt/GSK3beta signaling and dynamin-1 regulates clathrin-mediated endocytosis. EMBO J. 2015;34:2132–2146. doi: 10.15252/embj.201591518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samasilp P, Chan SA, Smith C. Activity-dependent fusion pore expansion regulated by a calcineurin-dependent dynamin-syndapin pathway in mouse adrenal chromaffin cells. J Neurosci. 2012;32:10438–10447. doi: 10.1523/JNEUROSCI.1299-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid SL, Frolov VA. Dynamin: functional design of a membrane fission catalyst. Annu Rev Cell Dev Biol. 2011;27:79–105. doi: 10.1146/annurev-cellbio-100109-104016. [DOI] [PubMed] [Google Scholar]
- Sieber JJ, Willig KI, Kutzner C, Gerding-Reimers C, Harke B, Donnert G, Rammner B, Eggeling C, Hell SW, Grubmuller H, et al. Anatomy and dynamics of a supramolecular membrane protein cluster. Science. 2007;317:1072–1076. doi: 10.1126/science.1141727. [DOI] [PubMed] [Google Scholar]
- Sinha R, Ahmed S, Jahn R, Klingauf J. Two synaptobrevin molecules are sufficient for vesicle fusion in central nervous system synapses. Proc Natl Acad Sci USA. 2011;108:14318–14323. doi: 10.1073/pnas.1101818108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sochacki KA, Larson BT, Sengupta DC, Daniels MP, Shtengel G, Hess HF, Taraska JW. Imaging the post-fusion release and capture of a vesicle membrane protein. Nat Commun. 2012;3:1154. doi: 10.1038/ncomms2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sochacki KA, Shtengel G, van Engelenburg SB, Hess HF, Taraska JW. Correlative super-resolution fluorescence and metal-replica transmission electron microscopy. Nat Methods. 2014;11:305–308. doi: 10.1038/nmeth.2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyer JA, Almers W. A real-time view of life within 100 nm of the plasma membrane. Nat Rev. Mol Cell Biol. 2001;2:268–275. doi: 10.1038/35067069. [DOI] [PubMed] [Google Scholar]
- Steyer JA, Horstmann H, Almers W. Transport, docking and exocytosis of single secretory granules in live chromaffin cells. Nature. 1997;388:474–478. doi: 10.1038/41329. [DOI] [PubMed] [Google Scholar]
- Suchard SJ, Lattanzio FA, Jr, Rubin RW, Pressman BC. Stimulation of catecholamine secretion from cultured chromaffin cells by an ionophore-mediated rise in intracellular sodium. Eur J Cell Biol. 1982;94:531–539. doi: 10.1083/jcb.94.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–690. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science. 2006;314:1454–1457. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Taraska JW, Perrais D, Ohara-Imaizumi M, Nagamatsu S, Almers W. Secretory granules are recaptured largely intact after stimulated exocytosis in cultured endocrine cells. Proc Natl Acad Sci USA. 2003;100:2070–2075. doi: 10.1073/pnas.0337526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MJ, Perrais D, Merrifield CJ. A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PLoS Biol. 2011;9:e1000604. doi: 10.1371/journal.pbio.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen RF, Kochubey O, de Wit H, Gulyas-Kovacs A, Konijnenburg B, Sorensen JB, Klingauf J, Verhage M. Dissecting docking and tethering of secretory vesicles at the target membrane. EMBO J. 2006;25:3725–3737. doi: 10.1038/sj.emboj.7601256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi T, Kitaguchi T, Karasawa S, Fukuda M, Miyawaki A. Age-dependent preferential dense-core vesicle exocytosis in neuroendocrine cells revealed by newly developed monomeric fluorescent timer protein. Mol Biol Cell. 2010;21:87–94. doi: 10.1091/mbc.E09-08-0722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi T, McMahon HT, Rutter GA. Mechanisms of dense core vesicle recapture following “kiss and run” (“cavicapture”) exocytosis in insulin-secreting cells. J Biol Chem. 2004;279:47115–47124. doi: 10.1074/jbc.M408179200. [DOI] [PubMed] [Google Scholar]
- van den Bogaart G, Holt MG, Bunt G, Riedel D, Wouters FS, Jahn R. One SNARE complex is sufficient for membrane fusion. Nat Struct Mol Biol. 2010;17:358–364. doi: 10.1038/nsmb.1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Sudhof TC, Neher E, Verhage M. Munc18–1 promotes large dense-core vesicle docking. Neuron. 2001;31:581–591. doi: 10.1016/s0896-6273(01)00391-9. [DOI] [PubMed] [Google Scholar]
- Wada K, Mizoguchi A, Kaibuchi K, Shirataki H, Ide C, Takai Y. Localization of rabphilin-3A, a putative target protein for Rab3A, at the sites of Ca(2+)-dependent exocytosis in PC12 cells. Biochem Biophys Res Commun. 1994;198:158–165. doi: 10.1006/bbrc.1994.1023. [DOI] [PubMed] [Google Scholar]
- Wang CT, Grishanin R, Earles CA, Chang PY, Martin TF, Chapman ER, Jackson MB. Synaptotagmin modulation of fusion pore kinetics in regulated exocytosis of dense-core vesicles. Science. 2001;294:1111–1115. doi: 10.1126/science.1064002. [DOI] [PubMed] [Google Scholar]
- Weiss AN, Anantharam A, Bittner MA, Axelrod D, Holz RW. Lumenal protein within secretory granules affects fusion pore expansion. Biophys J. 2014;107:26–33. doi: 10.1016/j.bpj.2014.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whim MD. Pancreatic beta cells synthesize neuropeptide Y and can rapidly release peptide co-transmitters. PLoS One. 2011;6:e19478. doi: 10.1371/journal.pone.0019478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Jackson MB. Membrane bending energy and fusion pore kinetics in Ca(2+)-triggered exocytosis. Biophys J. 2010;98:2524–2534. doi: 10.1016/j.bpj.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.