

Loss of VPS13 produces multiple phenotypes. This study implicates VPS13 in the function of membrane contact sites and suggests that different phenotypes of the mutant result from defects in different contact sites. In yeast, mutations found in the VPS13A gene of ChAc patients have specific defects in the mitochondrial aspect of VPS13 function.
Abstract
The Vps13 protein family is highly conserved in eukaryotic cells. Mutations in human VPS13 genes result in a variety of diseases, such as chorea acanthocytosis (ChAc), but the cellular functions of Vps13 proteins are not well defined. In yeast, there is a single VPS13 orthologue, which is required for at least two different processes: protein sorting to the vacuole and sporulation. This study demonstrates that VPS13 is also important for mitochondrial integrity. In addition to preventing transfer of DNA from the mitochondrion to the nucleus, VPS13 suppresses mitophagy and functions in parallel with the endoplasmic reticulum–mitochondrion encounter structure (ERMES). In different growth conditions, Vps13 localizes to endosome–mitochondrion contacts and to the nuclear–vacuole junctions, indicating that Vps13 may function at membrane contact sites. The ability of VPS13 to compensate for the absence of ERMES correlates with its intracellular distribution. We propose that Vps13 is present at multiple membrane contact sites and that separation-of-function mutants are due to loss of Vps13 at specific junctions. Introduction of VPS13A mutations identified in ChAc patients at cognate sites in yeast VPS13 are specifically defective in compensating for the lack of ERMES, suggesting that mitochondrial dysfunction might be the basis for ChAc.
INTRODUCTION
Yeast VPS13 is the founding member of a highly conserved gene family found in all eukaryotes. In humans, there are four VPS13 orthologues: VPS13A, B, C, and D (Velayos-Baeza et al., 2004). Mutations in VPS13A, B, and C are associated with the neurodegenerative disorder chorea acanthocytosis (ChAc), the developmental disorder Cohen syndrome, and Parkinson’s disease, respectively (Ueno et al., 2001; Kolehmainen et al., 2003; Lesage et al., 2016). Despite the importance and conservation of this family, the functions of the different Vps13 family proteins are elusive.
Most of our knowledge of VPS13 function comes from studies in Saccharomyces cerevisiae, in which there is just a single version of the gene. VPS13 was originally identified by a mutant that is defective in sorting the vacuolar protease carboxypeptidase Y (CPY) to the vacuole, resulting in secretion of CPY protein (Bankaitis et al., 1986). This sorting defect is due to a failure to transport the sorting receptor for CPY from the endosome to the Golgi (Brickner and Fuller, 1997). VPS13 is also required for the process of sporulation, which is independent of its function in vacuolar sorting (Brickner and Fuller, 1997; Enyenihi and Saunders, 2003; Nakanishi et al., 2007; Park and Neiman, 2012). A critical step in spore formation is the de novo synthesis of prospore membranes that encapsulate the haploid products of meiosis (Neiman, 2011; Park and Neiman, 2012). VPS13 is essential for prospore membrane formation, in which it promotes the presence of the lipids phosphatidylinositol (PtdIns)-4-phosphate and PtdIns-4,5-bisphosphate. This function appears to be conserved, as a reduction in PtdIns-4-phosphate levels at plasma membranes was also observed when VPS13A was knocked down by RNA interference in mammalian cell lines (Park and Neiman, 2012; Park et al., 2015). These results implicate VPS13 in some aspect of lipid metabolism, although whether VPS13 directly affects lipid phosphorylation or influences the levels of these lipids through a more indirect mechanism is not known.
Membrane contact sites are created where membranes from two different organelles form stable junctions (Helle et al., 2013). These sites may mediate transfer of lipids and ions between organelles (Henne et al., 2015; Olkkonen, 2015; Raiborg et al., 2015; Schrader et al., 2015). Many different protein complexes have been defined at membrane contact sites, which generally consist of 1) structural proteins that serve to link the membranes together and 2) transfer proteins that are recruited to the contact site to mediate exchange of material. Different contact sites can be functionally linked so that sites between one pair of organelles respond to changes in sites between a different pair of organelles (Elbaz-Alon et al., 2014, 2015). For instance, when subunits of the endoplasmic reticulum (ER)–mitochondrion contact site ERMES are mutated, a vacuolar–mitochondrial contact site called vCLAMP expands (Elbaz-Alon et al., 2014; Honscher et al., 2014). The combination of mutants in both ERMES and vCLAMP subunits is lethal, supporting the idea that the vCLAMP contact site expansion occurs to compensate for the loss of ERMES (Elbaz-Alon et al., 2014; Honscher et al., 2014).
The ERMES complex comprises the integral ER membrane protein Mmm1, the outer mitochondrial membrane proteins Mdm10 and Mdm34, and the soluble protein Mdm12 (Kornmann and Walter, 2010). Disruption of the genes encoding these proteins causes the loss of ERMES contact sites. Mutants deleted for MMM1 are viable but grow slowly. They rapidly lose mitochondrial DNA and cannot grow on nonfermentable carbon sources such as glycerol (Hobbs et al., 2001; Hanekamp et al., 2002). Deletion of VPS13 is synthetically lethal in combination with mutants in different subunits of the ERMES complex, including mmm1∆ (Costanzo et al., 2010; Hoppins et al., 2011; Lang et al., 2015). The requirement for VPS13 in the absence of ERMES suggests a third, distinct role for VPS13 in mitochondrial homeostasis, as well as a connection between VPS13 and membrane contact sites.
This study reports that the vps13∆ mutants display mitochondrial defects, identifying a role for VPS13 in mitochondrial homeostasis. Separation-of-function alleles were identified that affect both the function and localization of the protein. Altering the distribution of Vps13 results in different phenotypes, suggesting a model in which VPS13 functions at multiple membrane contact sites to promote different cellular processes. In addition, mutations found in ChAc patients, when introduced into the yeast VPS13, result in specific loss of the mitochondrial homeostasis function of VPS13, suggesting that the neurodegenerative symptoms of ChAc could be due to mitochondrial dysfunction.
RESULTS
VPS13 is important for mitochondrial integrity
Defects in mitochondrial integrity can be detected using an assay for mutants that exhibit an increased frequency of transfer of mitochondrial DNA (mtDNA) from the mitochondrion to the nucleus (defined as “mitochondrial escape”; Thorsness and Fox, 1993). The mitochondrial escape assay uses a strain in which the TRP1 gene is deleted from its chromosomal locus in the nucleus and inserted instead into the mitochondrial genome, rendering the cells auxotrophic for tryptophan. Transfer of the TRP1 gene from the mitochondrion to the nucleus allows appropriate expression, resulting in growth on medium lacking tryptophan. The MMM1 gene was previously identified by this screen (Hanekamp et al., 2002). An allele of VPS13, vps13-31 (previously yme3-1), showed high levels of Trp+ papillae in this assay, indicating a loss in mitochondrial integrity (Figure 1A). The vps13-31 mutation is a deletion of base pair 1565, creating a frameshift that truncates the protein from 3147 to 533 amino acids (Table 1). Consistent with vps13-31 being a null mutant, vps13∆ exhibited a similar high level of Trp+ papillae.
FIGURE 1:
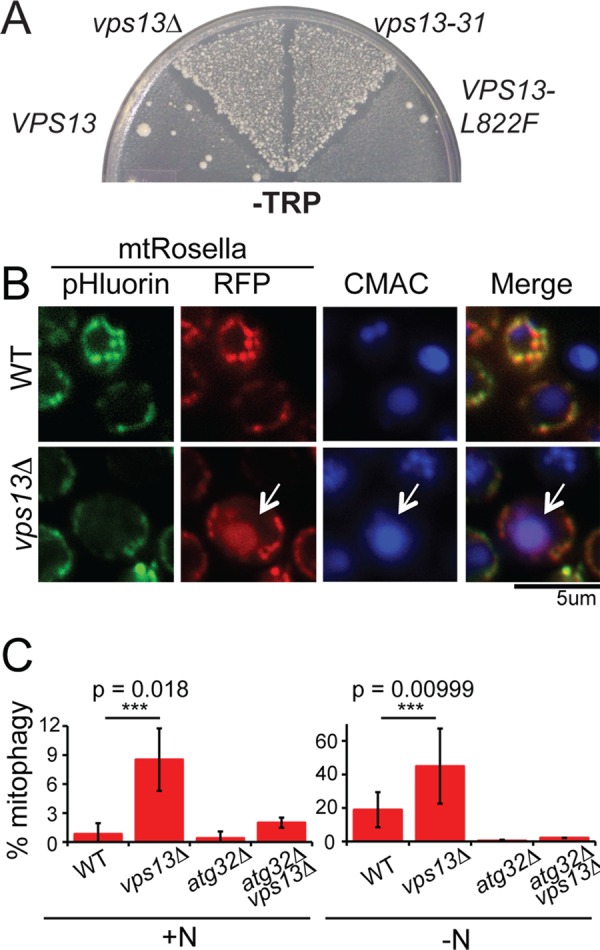
Mitochondrial phenotypes of vps13 alleles. (A) mtDNA escape assay. Wild-type WT; PTY44), vps13∆ (JKY2), vps13-31 (PTY66), and VPS13-L822F (MTY71) strains carrying a chromosomal TRP1 deletion, an insertion of TRP1 into the mitochondrial genome, and the indicated VPS13 alleles were grown as patches on complete ethanol/glycerol medium at 30°C for 2 d and then replica plated to minimal glucose medium lacking tryptophan (–Trp) to select for transfer of TRP1 to the nuclear DNA and incubated for 5 d at 30°C. The number of papillae growing in each sector reflects the frequency of transfer of mtDNA to the nucleus for that strain (Thorsness and Fox, 1993). (B) Mitophagy assay in WT (BY4741) and vps13∆ (KO1) cells grown in YPA expressing mtRosella, a fusion protein between a pH-sensitive GFP called pHluorin and RFP. Staining with the vacuolar lumen dye blue CMAC was used to determine the position of the vacuole. Scale bar, 5 μm. (C) Quantification of mitophagy assay in cells grown with or without nitrogen. The percentage of cells in the culture displaying mitophagy (indicated by red vacuolar fluorescence) was assessed in WT (BY4741), vps13∆ (KO1), atg32 (KO5), and vps13∆ atg32∆ (JSP461-1) cells expressing mtRosella and grown in the presence of nitrogen (+N) or the absence of nitrogen for 5.5 h to induce mitophagy (−N). The averages from three biological replicates are plotted with error bars representing the SD. At least 200 cells were analyzed for each strain in each replicate. Asterisks indicate a statistically significant difference as assessed by Student’s t test.
TABLE 1:
Sequence changes of VPS13 mutant alleles.
| VPS13 allele | Nucleotide change | Amino acid change |
|---|---|---|
| VPS13 | None | None |
| vps13-31 | G 1565∆ | Codon 533 stop |
| VPS13-P268R | C 803 G | Pro 268 Arg |
| VPS13-G718Ka | G 2152 T | Gly 718 Lys |
| VPS13-G718Ka | G 2152 T | Gly 718 Lys |
| VPS13-G718D | G 2153 A | Gly 718 Asp |
| VPS13-G820R | G 2458 C | Gly 820 Arg |
| VPS13-G820Db | G 2459 A | Gly 820 Asp |
| VPS13-L822F c | G 2466 T | Leu 822 Phe |
| VPS13-L984S | T 2951 C | Leu 984 Ser |
| VPS13-V1210E | T 3629 A | Val 1210 Glu |
| VPS13-G1245S | G 3733 A | Gly 1245 Ser |
| VPS13-N1467H | A 4399 C | Asn 1467 His |
| VPS13-A1512E | C 4535 A | Ala 1512 Glu |
aMutation was recovered in two independently isolated suppressor strains.
bOriginally designated YNT61-1 (Hanekamp et. al., 2002).
cOriginally designated YNT61-3 (Hanekamp et. al., 2002).
Although vps13 mutants show no respiration defects, that is, they are competent to grow on nonfermentable carbon sources, the mitochondrial escape phenotype suggests that mitochondria are more fragile and susceptible to breakage and DNA release (Campbell and Thorsness, 1998). These unstable mitochondria might therefore exhibit other phenotypes as well. Mitophagy is a specialized type of autophagy induced by nitrogen starvation in which mitochondria are degraded in the vacuole (Kanki et al., 2009; Okamoto et al., 2009). Mitophagy can be assayed using the marker mtRosella, which consists of a red fluorescent protein (RFP) and a pH-sensitive green fluorescent fusion protein (pHluorin) that is targeted to the mitochondrial matrix (Rosado et al., 2008; Mijaljica et al., 2011). In the mitochondrion, both moieties fluoresce, and overlapping green and red fluorescence is seen in the microscope. However, if the mitochondrion is degraded in the vacuole by autophagy, mtRosella is delivered to the vacuolar lumen. Vacuoles can be visualized with a blue fluorescent dye, 7-amino-4-chloromethylcoumarin (CMAC; Stefan and Blumer, 1999). Whereas mtRosella is resistant to vacuolar proteolysis, the lower pH of the vacuolar lumen specifically inhibits the green fluorescence (Mijaljica et al., 2011). Therefore red fluorescent vacuoles serve as an indicator of mitophagy.
VPS13 suppresses basal levels of mitophagy, as a fivefold increase in red fluorescing vacuoles was observed in vps13∆ cells grown in the presence of nitrogen (Figure 1, B and C; Kanki et al., 2009; Okamoto et al., 2009). As previously reported, the absence of nitrogen caused an increase in the fraction of wild-type cells exhibiting mitophagy (Kanki et al., 2009; Okamoto et al., 2009). The frequency of mitophagy was further increased threefold in vps13∆ (Figure 1C). VPS13 suppresses the canonical mitophagy pathway, as the increase in red-fluorescing vacuoles depended on ATG32, a gene that is specifically required for mitophagy (Figure 1C; Kanki et al., 2009; Okamoto et al., 2009). The requirement for VPS13 in preventing mitochondrial escape and mitophagy indicates a new role for VPS13 in promoting the structural integrity of mitochondria.
Dominant mutants in VPS13 suppress mmm1Δ
Deletion of MMM1 results in slow growth on glucose medium, abnormal mitochondrial morphology, and failure to grow on nonfermentable carbon sources (Hobbs et al., 2001; Figure 2). Further evidence of a role for VPS13 in mitochondrial function came from the discovery that several spontaneous dominant suppressors of the mmm1∆ growth defect are nonnull alleles of VPS13 (Hanekamp et al., 2002; Table 1 and Figure 2A). Mutants in MMM1 rapidly lose mtDNA (Hobbs et al., 2001). Therefore, to determine whether the VPS13 dominant suppressors bypass the requirement for MMM1 for respiration, it was necessary to first reintroduce mtDNA into the VPS13-X mmm1∆ deletion strains. This was done by crossing each strain to a ρ+ haploid, sporulating the diploids, and dissecting tetrads to get haploid segregants. VPS13-X mmm1∆ ρ+ strains were then tested for their ability to respire by looking at growth on the plates containing ethanol and glycerol as the sole carbon sources. All of the VPS13 alleles bypassed the MMM1 requirement for respiration, with growth rates ranging from slow to nearly wild type (Figure 2B). The mechanism by which these dominant mutants suppress MMM1 appears to be independent of mitochondrial integrity, as these mutants exhibited a low level of mitochondrial escape equivalent to wild type (e.g., VPS13-L822F is shown in Figure 1A).
FIGURE 2:
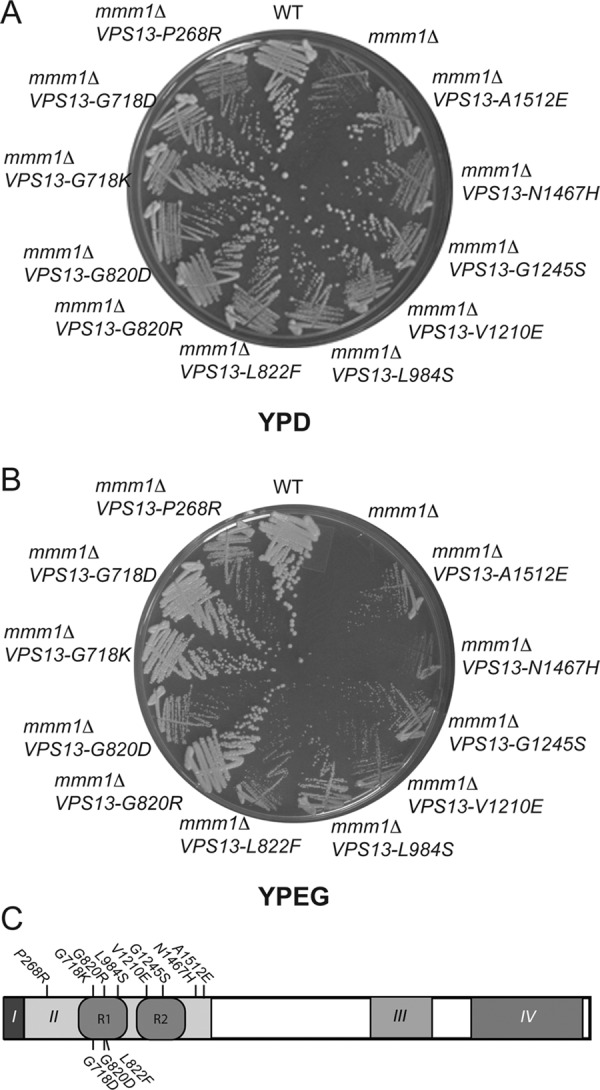
VPS13 suppressors of mmm1∆. Yeast strains containing mmm1∆ and various alleles of VPS13 were streaked for single colonies on the indicated medium and incubated from 3 to 5 d at 30°C. Strains: WT, PTY44; mmm1∆, THY23; mmm1∆ VPS13-A1512E, KWY114; mmm1∆ VPS13-N1467H, KWY76; mmm1∆ VPS13-G1245S, KWY105; mmm1∆ VPS13-V1210E, KWY75; mmm1∆ VPS13-L984S, KWY111; mmm1∆ VPS13-L822F, MTY79; mmm1∆ VPS13-G820R, KWY70; mmm1∆ VPS13-G820D, JHY14; mmm1∆ VPS13-G718K, KWY109; mmm1∆ VPS13-G718D, KWY131; mmm1∆ VPS13-P268R, KWY80. (A) Growth on 2% glucose medium (YPD). (B) Growth on 3% ethanol/3% glycerol medium (YPEG). (C) Location of VPS13 mutations that suppress the mmm1∆ growth defect. Conserved domains of Vps13 are indicated by roman numerals I–IV (not drawn to scale; Velayos-Baeza et al., 2004). I, N-chorein domain; II ,repeat containing domain (position of repeats shown by R1 and R2); III, DUF1162 domain; IV, C-terminal domain. Positions of the amino acid changes in the suppressor alleles are indicated (see also Table 2).
Vps13 family proteins have four major regions of conservation (Velayos-Baeza et al., 2004; Figure 2C). The highest level of conservation is found within the first 100 amino acids (aa) in what is termed the N-chorein domain (domain I in Figure 2C). This is followed by an ∼1200- aa conserved region (domain II), a DUF1162 domain of ∼300 aa (domain III), and then an ∼500-aa region of homology related to the Atg2 protein (domain IV; Velayos-Baeza et al., 2004). All of the suppressor alleles are missense mutations that localize within domain II (Figure 2C and Table 1). In the yeast Vps13, domain II includes two copies of a repeated region with a core P-X4-P-X13-17-G motif (Velayos-Baeza et al., 2004; shown as R1 and R2 in Figure 2C). Four of the independently isolated suppressors mutated a conserved glycine in one or the other of these repeats, and many of the remaining suppressor alleles fall within R1 or R2. Lang et al. (2015) recently reported the isolation of five different mutations in VPS13 that suppress the growth defect of mmm1∆ or deletions of other ERMES subunits. All five of these mutations fall into domain II as well. Thus alteration of this region of the protein seems particularly prone to giving rise to mmm1∆ suppressors. Both vps13∆ and mmm1∆ exhibit a mitochondrial escape phenotype, the two mutants are synthetically lethal, and gain-of-function alleles of VPS13 can bypass mmm1∆ for respiration. Taken together, these observations suggest that VPS13 acts in parallel to ERMES to support mitochondrial homeostasis.
Green fluorescent protein fusions create separation-of-function alleles of VPS13
A C-terminal fusion of Vps13 to green fluorescent protein (GFP; vps13-GFPC) exhibits wild-type levels of sporulation (Park and Neiman, 2012; Table 2). VPS13 and VPS10 (which encodes the CPY receptor in the vacuole) are both required for delivery of CPY to the vacuole (Bankaitis et al., 1986). In the absence of these genes, a fraction of the CPY is missorted at the Golgi and secreted (Bankaitis et al., 1986; Figure 3B). No secreted CPY was observed in the vps13-GFPC strain, however, indicating that this allele is functional for CPY sorting (Figure 3B).
TABLE 2:
Sporulation efficiency of different VPS13 alleles.
| Straina | Relevant genotype | Percentage sporulationb |
|---|---|---|
| JSYD1 | VPS13/VPS13 | 53 ± 8 |
| JSYD2 | vps13∆/vps13Δ | 0 ± 0 |
| JSYD3 | vps13∆/vps13-GFPC | 53 ± 4 |
| JSYD4 | vps13∆/VPS13-GFP1360 | 59 ± 1 |
| JSYD5 | vps13∆/GFPN-vps13 | 1 ± 1 |
| JSYD6 | vps13∆/vps13-L66P | 64 ± 1 |
| JSYD7 | vps13∆/vps13-C89K | 55 ± 3 |
| JSYD8 | vps13∆/vps13-L1107P | 40 ± 3 |
| JSYD9 | vps13∆/vps13-Y2702C | 48 ± 5 |
aHybrid diploids were created by mating haploids from the BY4741 background to the SK1 vps13∆ strain, HI27.
bAverages from at least three colonies are shown with the SD. At least 200 cells were scored for each colony.
FIGURE 3:
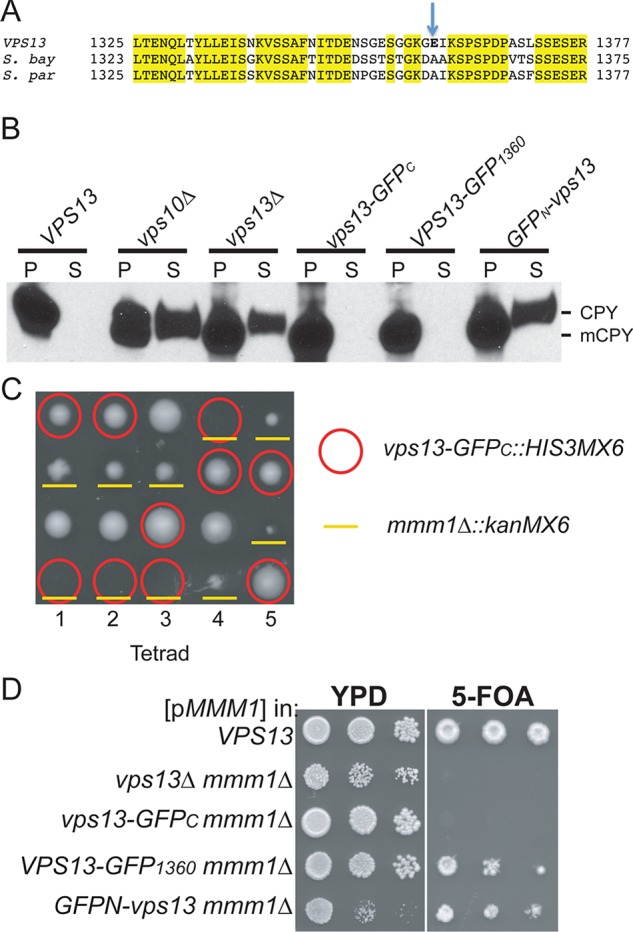
Phenotypes of different GFP-tagged VPS13 alleles. (A) Insertion site for the VPS13-GFP1360 allele. Alignment of the predicted sequence of a region of S. cerevisiae VPS13 with those from the closely related yeasts Saccharomyces bayanus and Saccharomyces paradoxus. GFP was integrated as an in-frame insertion immediately after the residue indicated by the arrow. (B) CPY sorting. Overnight cultures of WT (BY4741), vps10∆ (KO3), vps13∆ (KO1), vps13-GFPC (GCY1), VPS13-GFP1360 (JSP497), and GFPN-vps13 (JSP513) strains were separated by centrifugation into pellet (P) and supernatant (S) fractions. The proteins in the supernatant were precipitated with TCA, and the P and S samples were probed on a Western blot with anti-CPY antibodies. mCPY indicates the processed, vacuolar form of the protein. (C) Synthetic lethality of vps13-GFPC with mmm1∆ shown by tetrad analysis. An MMM1/mmm1∆::kanMX6 VPS13/vps13-GFPC::HIS3MX6 his3/his3 diploid, JSYD10, was sporulated and five tetrads dissected onto a YPD plate. Spores containing vps13-GFPC::HIS3MX6 are indicated by red circles, and mmm1∆::kanMX6 is indicated by yellow underlines. (D) Synthetic lethality of different VPS13 GFP fusions with mmm1∆ shown by plasmid loss. Haploid WT (BY4741), vps13Δ mmm1Δ (JSP443), vps13-GFPC mmm1Δ (JSP577), VPS13-GFP1360 mmm1Δ (LUKE3), or GFPN-vps13 mmm1∆ (JSP491) carrying pRS316-MMM1 strains were grown overnight at 30ºC in YPD. Tenfold serial dilutions were spotted onto YPD or SD complete medium containing 5-FOA and grown at 30ºC for 3 d.
In contrast, vps13-GFPC is synthetically lethal with mmm1∆, indicating that it lacks the VPS13 function that compensates for the lack of ERMES (Figure 3, C and D; Lang et al., 2015). This was demonstrated in two ways. First, a VPS13/vps13-GFPC mmm1∆::kanMX6/MMM1 double heterozygote was sporulated and the tetrads dissected. Given that VPS13 and MMM1 are unlinked, one-fourth of the spores should be double mutants. However, no viable vps13-GFPC mmm1∆::kanMX6 segregants were obtained from the dissection of 18 tetrads (Figure 3C; p <0.001, χ2 test). Second, vps13∆ mmm1∆ strains are viable when they contain a replicating plasmid carrying MMM1 and URA3 as a selectable marker (Figure 3D). Because loss of the MMM1 gene on the plasmid is lethal, vps13∆ mmm1∆/pRS316-MMM1 cells cannot grow on medium containing the drug 5-fluoroorotic acid (5-FOA), which kills cells containing URA3 (Boeke et al., 1987). Similarly, a vps13-GFPC mmm1∆/pRS316-MMM1 strain did not grow on 5-FOA (Figure 3D). In contrast, the MMM1 gene could be lost from the VPS13 control. Therefore fusion of GFP to the end of VPS13 creates an allele that is specifically defective in the ability of VPS13 to compensate for the loss of ERMES.
To create a fully functional GFP-tagged version of VPS13 for protein localization studies, GFP was either fused at the 5′ end of the gene (GFPN-vps13) or inserted in-frame between codons 1359 and 1360 of the gene (VPS13-GFP1360). This region of VPS13 was chosen because it encodes a patch of lower amino acid conservation, as determined by alignment of the S. cerevisiae VPS13 with orthologues from closely related yeasts (Figure 3A). The GFPN-vps13 allele displayed phenotypes complementary to that of vps13-GFPC: the GFPN-vps13 diploid did not sporulate (Table 2), CPY was secreted, similar to vps10∆ and vps13∆ (Figure 3B), and GFPN-vps13 was viable when combined with mmm1∆ (Figure 3D). By contrast, cells expressing VPS13-GFP1360, appeared wild type in all three assays (Figure 3 and Table 2). These results demonstrate that three different processes known to require VPS13—sporulation, vacuolar sorting, and viability in the absence of MMM1—are genetically separable, suggesting that they result from loss of different aspects of VPS13 function. Moreover, VPS13-GFP1360 seems to be fully functional, making it a useful reagent for localization studies.
Vps13 localizes to membrane contact sites
Like Vps13-GFPC, Vps13-GFP1360 localized to the prospore membrane in sporulating cells (Huh et al., 2003; Park and Neiman, 2012; Figure 4A). Consistent with studies using Vps13-GFPC in vegetative cells, Vps13-GFP1360 was present in foci of which ∼85% colocalized with the late endosomal marker Did2–monomeric RFP (mRFP), suggesting that Vps13 localizes primarily to the endosome (Figure 4B; Huh et al., 2003; Lottridge et al., 2006).
FIGURE 4:
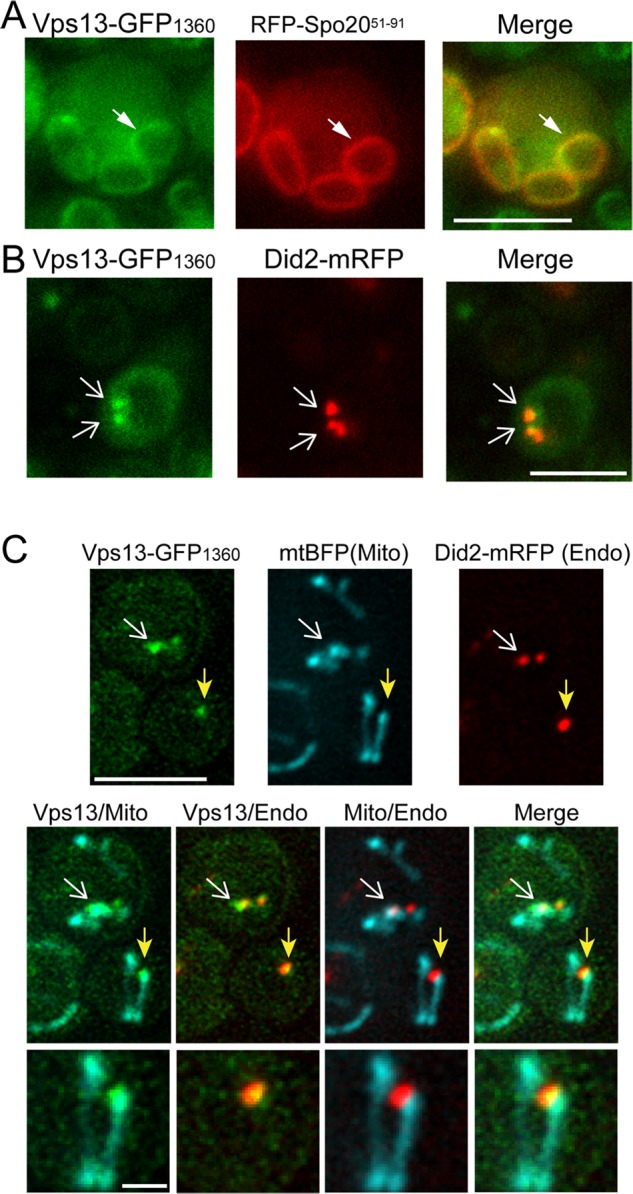
Localization of Vps13-GFP1360 in vegetative and sporulating cells. (A) Prospore membrane localization. The SK1 diploid JSP528 carrying pRS426-R20 was transferred to Spo medium for 6–8 h. The pRS426-R20 plasmid contains a protein fragment from Spo20 fused to RFP (RFP-Spo2051–91) that localizes to prospore membranes (Suda et al., 2007). White arrowheads indicate one prospore membrane of a developing tetrad. (B) Endosome localization. A strain expressing both VPS13-GFP1360 and a tagged version of the late endosome marker DID2-mRFP (JSP527) was grown to mid log phase in synthetic glucose medium and analyzed by fluorescence microscopy. White arrows indicate overlapping foci. Of 655 total Vps13-GFP1360 foci examined over four experiments, 561 displayed colocalization with Did2-mRFP. (C) Endosome and mitochondrial localization. Cells expressing Vps13-GFP1360, Did2-mRFP (Endo), and mtBFP (Mito; JSP512/pVT100U-mtBFP) were analyzed as in B. Arrows indicate sites where Vps13-GFP1360 colocalizes with both the endosomal and mitochondrial markers. Of 423 total Vps13-GFP1360 foci examined in two experiments, 97displayed colocalization with mtBFP. Of these 97, 87 also colocalized with Did2-mRFP. Scale bar, 5 μm. Bottom, higher magnification of the focus indicated by the yellow arrow. Scale bar, 1 μm.
Vps13-GFP1360 localization in a strain containing the mitochondrial marker mitochondrial blue fluorescent protein (mtBFP; Westermann and Neupert, 2000) revealed that ∼20% of the Vps13 puncta colocalized with mitochondria. When markers for Vps13, mitochondria, and endosomes were all present in the same cell, 91% of the Vps13GFP1360 foci that overlapped with mtBFP also overlapped with Did2-mRFP (Figure 4C). That is, a fraction of the Vps13 in the cells is localized at sites where endosomal and mitochondrial membranes are in close proximity.
The localization of the three markers at these sites characteristically shows overlap between the Vps13 signal and the mitochondrial signal and overlap between the endosomal signal and Vps13 but little or no overlap between the endosome and the mitochondrion (Figure 4C). That is, Vps13 sits at the interface between the endosome and mitochondrion, suggesting that it is a component of a contact site between the endosomal and mitochondrial membranes. To our knowledge, this is the first identification of a protein at endosome–mitochondrial contacts.
Vps13 moves to the prospore membrane in sporulation medium, in which acetate is the carbon source (Park and Neiman, 2012). VPS13 is important for mitochondrial homeostasis (Figure 1), and growth on nonfermentable carbon sources such as acetate requires mitochondria. Localization of Vps13-GFP1360 was therefore examined in cells grown in yeast extract/peptone/acetate (YPA) medium (Figure 5). When acetate was the sole carbon source, the localization of Vps13-GFP1360 was altered, with many cells displaying green fluorescence along one edge of the vacuole (Figure 5, A and B). This pattern is reminiscent of nuclear–vacuolar (NV) junctions, which are formed where the vacuole contacts the nuclear envelope (Pan et al., 2000). NV junctions are created by the interaction of the integral ER protein Nvj1 with the peripheral vacuolar membrane protein Vac8 (Pan et al., 2000). Vps13-GFP1360 colocalized with an Nvj1-3xmCherry marker protein in cells grown in YPA, demonstrating that Vps13 is present at NV junctions (Figure 5A), as was also observed by Lang et al. (2015). Thus the localization of Vps13 at different membrane contact sites varies, depending upon the carbon source.
FIGURE 5:

Vps13 localization in acetate medium. (A) Vps13-GFP1360 localization in cells (JSP552) growing in YPA medium. The NV junction was marked using Nvj1-3xmCherry. White arrows point to foci containing both Vps13-GFP1360 and Nvj1-3xmCherry. (B) Vps13-GFP1360 localization in NV junction mutants. VPS13-GFP1360 (JSP497), VPS13-GFP1360 nvj1∆ (JSP541), and VPS13-GFP1360 vac8∆ (JSP545) cells were grown in YPA and stained with FM4-64 and Hoechst to mark the vacuolar membranes and nuclei, respectively. White arrows indicate GFP fluorescence at the NV junction. (C) Quantification of the localization pattern shown in B. Percentages are the averages of four experiments (at least 100 cells scored in each experiment). Error bars indicate the SD. (D) NVJ1 and VAC8 are codependent for localization to the NV junction. Nvj1-GFP localization was examined in WT (GCY3) and vac8∆ (JSP555) cells, and Vac8-GFP was examined in WT (GCY4) and nvj1∆ (JSP547). Cells were labeled with FM4-64 to visualize the vacuolar membrane. (E) The length of NV junctions as marked by Nvj1-GFP fluorescence was measured in VPS13 (GCY3) and vps13∆ (JSP536) cells grown in YPA. Insets, merged images of green (Nvj1-GFP) and red (FM4-64) fluorescence for a representative cell from each culture. The average length of the GFP fluorescence at the NV junction was plotted. The horizontal line indicates the median value, the boxes represent three quartiles, and the whiskers denote the range of values. More than 790 cells in each strain were measured. Asterisks indicate a significant difference (p < 0.0001 as calculated by Student’s t test). Scale bars, 5 μm.
Interaction of Nvj1 with Vac8 is necessary to recruit other proteins to the NV junction (Kvam and Goldfarb, 2004; Kvam et al., 2005). To determine whether NVJ1 or VAC8 is necessary to recruit Vps13, we examined the localization of Vps13-GFP1360 in nvj1∆ and vac8∆ cells. Cells were stained with both FM4-64 and Hoechst to visualize the vacuolar membrane and nucleus, respectively. The position of the NV junction is defined as the site where the nucleus abuts the vacuolar membrane (Pan et al., 2000). Vps13-GFP1360 was concentrated at the interface of the vacuole and nucleus to comparable degrees in wild-type, nvj1∆, and vac8∆ strains (Figure 5. B and C). This result is in contrast to a recent report that Vps13 localization to the NV junction depends on NVJ1 (Lang et al., 2015). This discrepancy is not due to a problem with the assay, as delocalization of Nvj1 and Vac8 was observed in vac8∆ and nvj1∆ strains, respectively (Figure 5D; Pan et al., 2000). Our data indicate that Vps13 is recruited to the nuclear envelope–vacuole interface independently of the canonical NV junction architectural proteins.
Different membrane contact sites can respond to one another (Elbaz-Alon et al., 2014, 2015; Honscher et al., 2014). That is, loss of one membrane contact site produces compensatory expansion of other sites. When wild-type and vps13∆ cells grown in YPA were compared, the region of Nvj1 localization was more extensive in the vps13∆ mutant (Figure 5E). Measurement of the length of the Nvj1-GFP fluorescence along the nuclear envelope–vacuole interface in wild type and vps13∆ confirmed that Nvj1-containing junctions were longer in the vps13∆ cells (Figure 5E). Thus loss of VPS13 can lead to expansion of Nvj1-containing NV junctions, perhaps to compensate for the loss of Vps13-containing junctions.
The intracellular distribution of Vps13 correlates with function
The vps13-GFPC mutant is synthetically lethal with mmm1∆, and, conversely, dominant mutations such as VPS13-G718K suppress the mmm1∆ growth phenotype. To determine whether changes in Vps13 localization are correlated with these differences in activity, we examined the localization of Vps13-GFPC, Vps13-GFP1360, and Vps13G718K-GFP1360 in YPA-grown cells. The localization patterns within individual cells were classified into four different categories: NV junctions only; NV junctions and cytoplasmic foci (“dots”); cytoplasmic foci only; and cells in which the signal was diffuse in the cytosol or only in the vacuolar lumen (the latter suggesting degradation of the fusion protein; Figure 6A). The Vps13-GFPC protein localized more extensively than Vps13-GFP1360 to NV junctions and was not present in cytoplasmic dots, suggesting that the absence of “dots” may be responsible for the synthetic lethal phenotype observed between vps13-GFPC and mmm1∆. In contrast, the number of cells with only dots was increased by introduction of the G718K mutation compared with Vps13-GFP1360 (Figure 6B). The localization of Vps13-GFPC containing G718K was similarly shifted away from NV junctions to cells with dots. Furthermore, the introduction of the G718K mutation into vps13-GFPC suppressed the synthetic lethality with mmm1∆ (Figure 6C). Lang et al. (2015) observed a similar result using a different VPS13 mutant. We conclude that the Vps13-GFPC protein fails to reach some location at which its activity is critical to compensate for the loss of ERMES and that mutations in the region around G718 redistribute Vps13 back to this location.
FIGURE 6:

The effect of the GFPC tag and the G718K mutation on Vps13 localization. Cells expressing VPS13-GFP1360 (JSP497) or vps13-GFPC (GCY1), VPS13-G718K-GFP1360 (JSP531), or VPS13-G718K -GFPC (JSP556) were grown in YPA, and the pattern of Vps13-GFP distribution in individual cells was categorized. (A) Representative cells for each pattern. Scale bar, 5 μm. (B) The distribution of cells in each category in the four different strains. Average percentages from three experiments (at least 200 cells scored in each). Error bars indicate the SD. (C) Suppression of the vps13-GFPC mmm1∆ synthetic lethal phenotype by the G718K mutation. Serial dilutions of WT (BY4741), vps13-GFPC mmm1∆ (JSP577), and vps13-G718K-GFPC mmm1∆ (JSP590) carrying pRS316-MMM1 were spotted on YPD or SD complete medium with 5-FOA.
Human disease–associated VPS13A mutations introduced into yeast VPS13 are synthetically lethal with mmm1∆
To elucidate why mutations in VPS13A in humans cause a neurodegenerative disorder, it would be helpful to determine which VPS13-dependent processes are affected by these mutations. Most of the identified VPS13A mutations are deletions or splice junction mutations that likely generate null alleles; however, six missense alleles have been identified in patient samples (Rampoldi et al., 2001; Dobson-Stone et al., 2002; Tomiyasu et al., 2011). Alignment of Vps13A with yeast Vps13 identified cognate residues in the yeast protein for four of the amino acids changed by the missense mutations (Figure 7A). We then characterized VPS13 alleles in which these cognate amino acids were individually substituted with the amino acids encoded in the VPS13A disease alleles (L66P, C89K, L1107P, Y2702C; Figure 7B) for effects on sporulation, vacuolar sorting, and synthetic lethality with mmm1∆.
FIGURE 7:
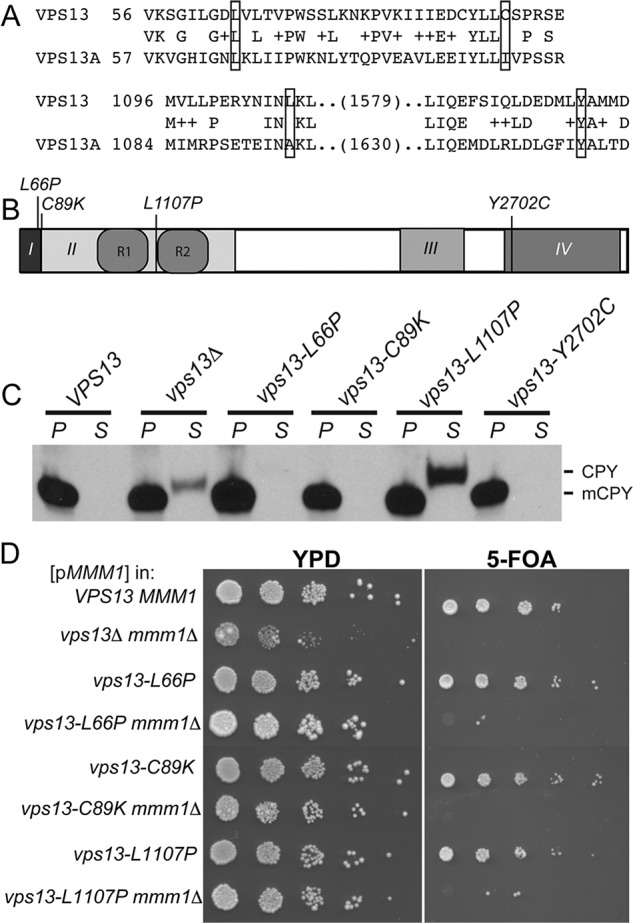
Phenotypes of human VPS13A ChAc mutations introduced into yeast VPS13. (A) Alignment of the Vps13 amino acid sequence with Vps13A. Residues that were mutated are boxed. The cognate amino acid substitutions encoded by the mutations are as follows: Vps13A L67P = Vps13 L66P; Vps13A I90K = Vps13 C89K; Vps13A A1095P = Vps13 L1107P; and Vps13A Y2721C = Vps13 Y2702C. (B) Vps13 domain structure showing the position of the disease alleles in yeast Vps13. Amino acid changes listed above are the position of the yeast VPS13 residues. (C) CPY sorting in the vps13 mutants containing the cognate vps13A disease mutations performed as in Figure 3A. Strains assayed: WT (BY4741) vps13∆ (KO1), vps13-L66P (RP201), vps13-C89K (RP205), vps13-L1107P (RP202), and vps13-Y2702C (RP203). mCPY indicates the mobility of the processed, vacuolar form of the protein. (D) Assay for synthetic lethality between vps13 mutants containing the cognate vps13A disease mutations and mmm1∆, assayed as in Figure 3C. Strains: WT (BY4741), vps13Δ mmm1Δ (JSP446), vps13-L66P (RP201), vps13-L66P mmm1∆ (RP301), vps13-C89K (RP205), vps13-C89K mmm1∆ (JSP582), vps13-L1107P (RP202), and vps13-L1107P mmm1∆ (JSP585).
No obvious phenotypes were observed for VPS13-Y2702C, indicating that this mutation does not alter the function of the yeast protein for these processes. All of the mutants were proficient for sporulation (Table 2). The vps13-L1107P strain showed a defect in CPY sorting, whereas vps13-L66P and C89K behaved like wild type (Figure 7C). The phenotype in common for vps13-L66P, vps13-C89K, and vps13-L1107P was synthetic lethality with mmm1∆ (Figure 7D). Thus the inability to compensate for the loss of ERMES is associated with missense mutants that cause disease in humans.
The G718K mutation rescues the synthetic lethality of vps13-L66P and mmm1∆
The vps13-L66P mutant is phenotypically similar to vps13-GFPC in being specifically synthetically lethal with mmm1∆. Like vps13-GFPC, Vps13-L66P localizes primarily to NV junctions (Figure 8, A and B). The synthetic lethal interaction between vps13-GFPC and mmm1∆ can be suppressed by the relocalization of Vps13-GFPC to NV junctions by the G718K mutation (Figure 6). The addition of G718K to vps13-L66P also redistributed the protein within the cells and suppressed the synthetic lethal phenotype with mmm1∆ (Figure 8, B and C). Thus the distribution of Vps13 within the cell correlates with its ability to promote mitochondrial homeostasis.
FIGURE 8:
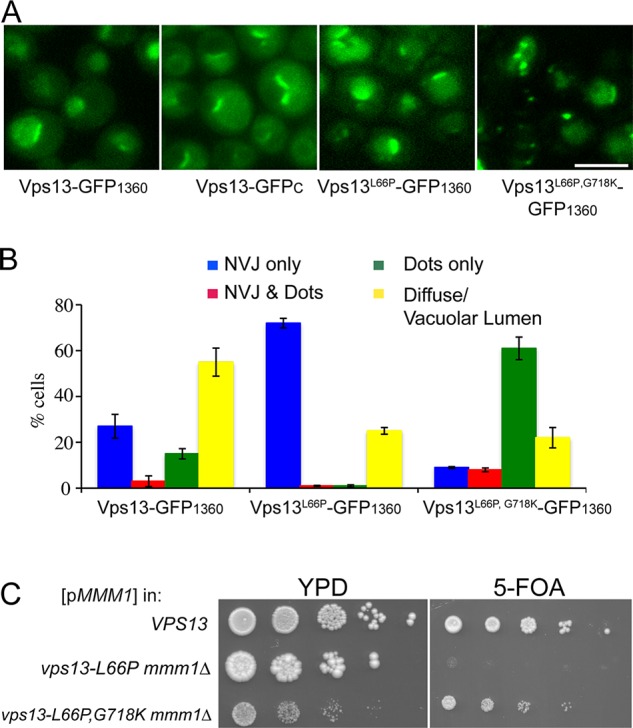
Effect of the G718K mutation on vps13-L66P phenotypes (A) Representative images of cells expressing VPS13-GFP1360 (JSP497), vps13-GFPC (GCY1), vps13-L66P-GFP1360 (JSP560), or vps13-L66P, G718K-GFP1360 (JSP563) grown in YPA medium. Scale bar, 5 μm. (B) Quantification of the localization patterns for proteins in A. Data for Vps13-GFP1360 are from Figure 6B. Average percentages from three experiments (at least 200 cells scored in each). The error bars indicate the SD. (C) Synthetic lethality with mmm1∆. Tenfold serial dilutions of WT (BY4741), vps13-L66P mmm1∆ (RP301), and vps13-L66P,G718K mmm1∆ (JSP533) cells carrying pRS316-MMM1 were spotted onto either YPD or SD complete with 5-FOA.
DISCUSSION
VPS13 is important for mitochondrial integrity
The synthetic lethality between null alleles of VPS13 and components of the ERMES complex suggests that VPS13 is somehow involved in mitochondrial function, but this connection could be indirect. This work demonstrates that VPS13 promotes mitochondrial integrity even when ERMES function is intact. VPS13 is required to prevent mitochondrial DNA from being transferred to the nucleus, an indicator of mitochondrial stability. Furthermore, VPS13 is required to prevent autophagy of mitochondria by the vacuole, a phenomenon that may be stimulated by less stable mitochondria. Whether the contribution of VPS13 to mitochondrial integrity is structural or is the result of a more indirect effect, for example, on lipid levels as is observed in sporulating cells, remains to be determined. What is clear is that VPS13 is required for normal mitochondrial function.
Vps13 acts at membrane contact sites
There are several different types of membrane contact sites in a cell that allow communication between different organelles. Synthetic lethal interactions between components in different membrane contact sites indicate that these junctions are sometimes able to compensate for each other. For example, vCLAMP mutants are synthetically lethal with ERMES mutants (Elbaz-Alon et al., 2014; Honscher et al., 2014). Lang et al. (2015) found that Vps13 localizes to vacuole–mitochondria contact sites, perhaps as a component of vCLAMP, which may explain the requirement of VPS13 for viability in the absence of ERMES. Our identification of Vps13 at endosome–mitochondrion contacts indicates a second possible site through which VPS13 may act in parallel to ERMES.
Vps13 localizes to three different membrane contact sites—the NV junctions, endosome–mitochondria junctions, and vacuole-mitochondria junctions—and perhaps additional contact sites between endosomes and other organelles (Figure 9). We propose that in wild-type cells, these different membrane contact sites compete to recruit Vps13. When VPS13 is deleted, the protein is lost from all of the contact sites, leading to pleiotropic phenotypes. Because some of the Vps13-containing contact sites act in parallel to ERMES, vps13∆ cells depend on ERMES for survival. Mutations or conditions that change VPS13 distribution can phenocopy aspects of the deletion phenotype. For example, redistribution of the protein toward NV junctions depletes it from endosome–mitochondria junctions and renders the cell dependent on ERMES for growth. Conversely, redistribution of Vps13 so that it is enriched at mitochondrial contacts might increase the flow of materials into mitochondria through these routes, thereby making ERMES dispensable.
FIGURE 9:

Model for Vps13 intracellular distribution. In vegetative cells, Vps13 (orange circles) is found at membrane contact sites between different organelles (purple rectangles). At these sites, Vps13 acts to enhance exchange of materials, likely phospholipids, between the organellar membranes.
Contact sites between different combinations of organelles have been identified in both yeast and mammalian cells (Toulmay and Prinz, 2011; Schrader et al., 2015). Although functions of these different membrane junctions are conserved in different organisms, many of the protein constituents are not. For example, no orthologues of either the ERMES complex or the NV junction complex protein Nvj1 have been found in metazoans. These lineage-specific complexes may therefore be more recent evolutionary innovations. In contrast, the VPS13 family is strongly conserved, suggesting a fundamental function in eukaryotic cells. The relatively modest phenotypes of vps13∆ in mitotically dividing cells is consistent with the idea that Vps13-mediated junctions are redundant with other membrane contact sites. In this view, Vps13-mediated junctions are important as “back-up” systems but are only primarily responsible for mediating interorganelle traffic at a few locations and conditions such as sporulation. Contact sites generally consist of both structural proteins that act to hold the two organelles together and transfer proteins such as oxysterol-binding protein or Lam-family enzymes (Murley et al., 2015; Olkkonen, 2015). Very large size (>300 kDa) is a conserved feature of Vps13 family proteins. It is possible that the protein is large enough to provide both the tethering function and lipid transfer activity, thereby creating a functional membrane contact site on its own.
Whether VPS13 functions solely at membrane contact sites is not clear. With respect to sporulation, VPS13 is required for wild-type levels of the lipids PtdIns-4-phosphate and PtdIns-4,5-bisphosphate in the prospore membrane (Park and Neiman, 2012). One possibility is that VPS13 directly regulates the kinases and/or phosphatases that generate these different types of lipids. An alternative explanation is that Vps13-mediated contact sites are necessary to generate wild-type levels of PtdIns in the prospore membrane and, indirectly, PtdIns phosphate levels as well.
Regulation of Vps13 distribution
The presence of Vps13 at multiple locations raises the question of how the distribution of Vps13 is controlled. Vps13 localization varies both with growth medium and developmental stage. For sporulating cells, relocalization of Vps13 to prospore membranes requires the sporulation-specific gene SPO71 (Park et al., 2013). On induction, Spo71 binds to Vps13 and recruits it to the prospore membrane. Ectopic expression of SPO71 changes Vps13 localization in vegetative cells (Park et al., 2013). It may also be the case that different Vps13-binding proteins in vegetative cells recruit Vps13 to different sites of action. By extension, alleles that alter the function VPS13 may result from changes in interactions with specific binding proteins. Separation-of-function alleles of Vps13 may result from a failure to interact with specific binding proteins. For example, it is striking that all the mmm1∆ suppressor mutations in VPS13 have been found in domain II of the protein. One possibility is that these alleles have increased interactions with proteins at the mitochondrial–endosome interface. However, it seems unlikely that so many different mutations could increase affinity. A related possibility is that Vps13 is recruited to the NV junction through interactions between some protein and domain II. Mutations that alter the structure of domain II might reduce recruitment to the NV junction (or other some other site), making more Vps13 protein available for function at mitochondrial junctions. Similarly, alleles specifically defective in the mitochondrial homeostasis aspect of Vps13 function, such as vps13-L66P and vps13-GFPC, might interfere with binding to proteins that recruit Vps13 to the mitochondrion–endosome interface.
An alternative possibility is that the different point mutants affect levels of the protein rather than interactions with specific targeting proteins. For instance, the L66P mutation might simply lower the amount of protein, and mitochondrial homeostasis may be the activity most sensitive to Vps13 levels. Conversely, G718K might stabilize the protein, making more available to promote mitochondrial integrity. We have not been able to directly compare the levels of the proteins. However, if differences in protein stability were the basis for different phenotypes, it would still be consistent with the model that different subcellular locations compete for a limiting pool of Vps13.
The canonical NV junction is created by interaction of Nvj1 with Vac8. This interaction tethers the membranes, and, in addition, Nvj1 serves to recruit other proteins of the junction that are directly involved in material transport (Kvam and Goldfarb, 2004; Kvam et al., 2005). However, contrary to a previous report (Lang et al., 2015), Vps13 localization to the nuclear–vacuole interface is independent of NVJ1 and VAC8, and Nvj1 junctions appear to expand in the absence of VPS13. Taken together, these observations suggest that Vps13 is part of an alternative NV junction complex that perhaps has overlapping functions with the Nvj1-based junctions.
VPS13 and human disease
The finding that three different VPS13A disease-associated mutations are specifically defective in mitochondrial homeostasis has significant implications for VPS13-related human diseases. This correlation suggests that the ChAc disorder arises due to a specific defect in the mitochondrial function of VPS13A. Tagging of the C-terminus of VPS13 with GFP also specifically interferes with the mitochondrial homeostasis function, suggesting that the end of the protein is important for this activity. This idea is supported by the finding of a VPS13A mutation from ChAc patients that truncates the last 30 aa of the protein (Tomiyasu et al., 2011).
This interpretation rests on the inference that the mitochondrial homeostasis function of VPS13 is conserved in VPS13A. Although the localization of Vps13A has been reported in different cell types, none of these studies observed Vps13A on mitochondria (Kurano et al., 2007; Hayashi et al., 2012). However, localization may depend on the cell type or growth conditions. Given that ChAc is a neurodegenerative disease, it will be important to examine the possible association of Vps13A with mitochondria in mature neurons.
If mitochondrial dysfunction is the basis for the symptoms of ChAc, this would make ChAc similar to several other neurodegenerative disorders, such as Parkinson’s disease, in which mitochondrial dysfunction is a common basis (Burte et al., 2015). Moreover, defects in mitochondrial membrane contact sites have been suggested to be a common pathology in a variety of neurodegenerative disorders (Krols et al., 2016). Thus an underlying mitochondrial defect would be consistent with this aspect of ChAc.
MATERIALS AND METHODS
Yeast strains and media
Yeast strains used for this study are listed in Table 3. Unless otherwise mentioned, standard yeast media and genetic techniques were used (Rose and Fink, 1990). Strains carrying different GFP fusions and VPS13 point mutations were generated using a two-step clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 technique. For example, to create strain JSP497 carrying VPS13-GFP1360, the Saccharomyces kluyveri HIS3 gene was first amplified from pFA6a-HIS3MX6 (Longtine et al., 1998) with primers that incorporate flanking sequences that can direct integration of the HIS3 cassette between nucleotides 4079 and 4080 of the VPS13 coding region. The amplified fragment was transformed into strain BY4741, and His+ transformants were screened by PCR to identify strains carrying the insertion (VPS13::HIS3MX6int1360). The coding region for GFP was then amplified with primers that create an in-frame insertion of the GFP sequence between codons 1359 and 1360 of VPS13. The VPS13::HIS3MX6int1360 strain was cotransformed with this amplified GFP and pRS425-Cas9-SkHIS3-381, a LEU2-marked plasmid expressing both Cas9, and a guide RNA targeting the S. kluyveri HIS3 sequence (Jin et al., 2015). Leu+ transformants were screened for auxotrophy for histidine, and in-frame insertion of the GFP sequence was confirmed by PCR and DNA sequencing. Insertion of GFP at the same position in VPS13 was used to create strains JSP499-1, JSP560, and LUKE3 and was accomplished as described for JSP497, except that the starting strains were BY4742, RP201, and KO4 respectively. Similarly, VPS13-GFP1360 was introduced into the SK1 strains AN117-4B and AN117-16D as described, and then these strains were mated to generate the diploid JSP528. The N-terminal GFPN-VPS13 fusion in strains JSP490 and JSP513 was also created using CRISPR/Cas9, but in these cases, the HIS3MX6 cassette and then GFP were targeted immediately upstream of the start codon. The starting strains were AN117-16D and BY4742, respectively. Strain JSP495 carrying DID2::mRFP was created by transforming strain GCY2 with pRS425-Cas9-SkHIS3-381 to introduce a double-strand break into the HIS3MX6 cassette 3′ of the GFP sequence as well as a PCR fragment carrying the mRFP coding region flanked by sequences targeting mRFP to the 3′ end of DID2. Recombinational repair of the break resulted in replacement of the entire GFP-HISMX6 insertion with mRFP.
TABLE 3:
Strains used in this study.
| Name | Genotype | Source |
|---|---|---|
| BY4741a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho | Winzeler et al. (1999) |
| BY4742a | MATα his3∆0 leu2∆0 lys2∆0 ura3∆0 ho | Winzeler et al. (1999) |
| JHY14b,c,d | MATα lys2 ura3-52 leu2-3112 trp1-∆1 mmm1-∆1::LEU2 VPS13-G820D [ρ+, TRP1] | This study |
| JKY2b | MATα lys2 ura3-52 leu2-3112 trp1-∆1 vps13∆::KAN-MX4 [ρ+, TRP1] | This study |
| KWY70b | MATα lys2 ura3-52 leu2-3112 trp1-∆1 mmm1-∆1::LEU2 VPS13-G820R [ρ+, TRP1] | This study |
| KWY75b | MATα lys2 ura3-52 leu2-3112 trp1-∆1 mmm1-∆1::LEU2 VPS13-V1210E [ρ+, TRP1] | This study |
| KWY76b | MATα lys2 ura3-52 leu2-3112 trp1-∆1 mmm1-∆1::LEU2 VPS13-N1467H [ρ+, TRP1] | This study |
| KWY80b | MATα lys2 ura3-52 leu2-3112 trp1-∆1 mmm1-∆1::LEU2 VPS13-P268R [ρ+, TRP1] | This study |
| KWY105b | MATα lys2 ura3-52 leu2-3112 trp1-∆1 mmm1-∆1::LEU2 VPS13-G1245S [ρ+, TRP1] | This study |
| KWY109b | MATα lys2 ura3-52 leu2-3112 trp1-∆1 mmm1-∆1::LEU2 VPS13-G718K [ρ+, TRP1] | This study |
| KWY111b | MATα lys2 ura3-52 leu2-3112 trp1-∆1 mmm1-∆1::LEU2 VPS13-L984S [ρ+, TRP1] | This study |
| KWY114b | MATα lys2 ura3-52 leu2-3112 trp1-∆1 mmm1-∆1::LEU2 VPS13-A1512E [ρ+, TRP1] | This study |
| KWY131b | MATα lys2 ura3-52 leu2-3112 trp1-∆1 mmm1-∆1::LEU2 VPS13-G718D [ρ+, TRP1] | This study |
| MTY71b,c,d | MATα lys2 ura3-52 leu2-3112 trp1-∆1 VPS13-L822F [ρ+, TRP1] | This study |
| MTY79b,c,d | MATα lys2 ura3-52 leu2-3112 trp1-∆1 mmm1-∆1::LEU2 VPS13-L822F [ρ+, TRP1] | This study |
| PTY33b | MATa ade2 ura3-52 leu2-3112 trp1-∆1 [ρ+, TRP1] | Thorsness and Fox (1993) |
| PTY44b | MATα lys2 ura3-52 leu2-3112 trp1-∆1 [ρ+, TRP1] | Thorsness and Fox (1993) |
| PTY66b,d | MATα lys2 ura3-52 leu2-3112 trp1-∆1 vps13-31 [ρ+, TRP1] | Thorsness and Fox (1993) |
| THY23b | MATα lys2 ura3-52 leu2-3112 trp1-Δ1 mmm1-Δ1::LEU2 [ρo] | Hanekamp et al. (2002) |
| JSYD10 | MATα leu2 his3∆ MET15 ura3 arg4-NspI lys2 trp1::hisG ho | This study |
| MATa leu2∆0 his3∆0 met15∆0 ura3∆0 ARG4 LSY2 TRP1 ho | ||
| mmm1∆::kanMX6 VPS13 | ||
| MMM1 vps13-GFPc::HIS3MX6 | ||
| GCY1a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho vps13-GFPc::HIS3MX6 | Huh et al. (2003) |
| GCY2a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho DID2::GFPc::HIS3MX6 | Huh et al. (2003) |
| GCY3a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho NVJ1::GFP::HIS3MX6 | Huh et al. (2003) |
| GCY4a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho VAC8::GFP::HIS3MX6 | Huh et al. (2003) |
| KO1a | MATa his3∆0 leu2∆0 met15∆0 ura3∆ ho vps13∆::kanMX6 ho | Winzeler et al. (1999) |
| KO3a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 vps10∆::kanMX6 ho | Winzeler et al. (1999) |
| KO4a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho mmm1∆::kanMX6/pRS316-MMM1 | Winzeler et al. (1999) |
| KO5a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho atg32∆::kanMX6 | Winzeler et al. (1999) |
| JSP461-1a | MATa his3∆0 leu2∆ ura3∆ ho vps13∆::HIS3MX6 atg32∆::kanMX6 | This study |
| JSP497a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho VPS13-GFP1360 | This study |
| JSP513a | MATα his3∆0 leu2∆0 lys2∆0 ura3∆0 ho GFPN-vps13 | This study |
| RP201a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho vps13-L66P | This study |
| RP202a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho vps13-L1107P | This study |
| RP203a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho vps13-Y2702C ho | This study |
| RP205a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho vps13-C89K | This study |
| LUKE3a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho VPS13-GFP1360 mmm1∆::kanMX6 ho/pRS316-MMM1 | This study |
| JSP499-1a | MATα his3∆0 leu2∆0 lys2∆0 ura3∆0 ho VPS13-GFP1360 | This study |
| JSP527a | MATa leu2∆0 his3∆0 LYS2 met15∆0 ura3∆0 ho DID2::mRFP VPS13 | This study |
| MATα leu2∆0 his3∆0 lys2∆0 MET15 ura3∆0 ho DID2 VPS13::GFP1360 | ||
| JSP512a | MATa his3∆0 leu2∆0 lys2∆0 ura3∆0 ho VPS13-GFP1360 DID2::mRFP | This study |
| JSP541a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho VPS13-GFP1360 nvj1Δ::kanMX6 | This study |
| JSP545a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho VPS13-GFP1360 vac8∆::kanMX6 | This study |
| JSP547a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho NVJ1::GFP::HIS3MX6 vac8∆::kanMX6 | This study |
| JSP555a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho VAC8::GFP::HIS3MX6 nvj1∆::kanMX6 | This study |
| JSP495a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho DID2::mRFP | This study |
| JSP536a | MATa his3∆0 leu2∆0 met15∆0 ura3∆0 ho vps13∆::kanMX6 NVJ1::GFP::HIS3MX6 | This study |
| JSP531a | MATα his3∆0 leu2∆0 lys2∆0 ura3∆0 ho VPS13-G718K-GFP1360 | This study |
| JSP549a | MATα his3∆0 leu2∆0 lys2∆0 ura3∆0 ho VPS13-G718K | This study |
| JSP556a | MATα his3∆0 leu2∆0 lys2∆0 ura3∆0 ho vps13-G718K-GFPc::HIS3MX6 | This study |
| JSP560a | MATa his3∆1 leu2∆0 met 15∆0 ura3∆0 ho vps13-L66P-GFP1360 | This study |
| JSP563a | MATa his3∆1 leu2∆0 met 15∆0 ura3∆0 ho vps13-L66P G718K-GFP1360 | This study |
| JSP590 | MATa his3∆0 leu2∆0 ura3∆0 lys2∆0 ho VPS13-G718K-GFPc::HIS3MX6 mmm1∆::kanMX6/pRS316-MMM1 | This study |
| JSP577 | MATa his3 leu2 ura3 lys2 ho vps13-GFPc::HIS3MX6 mmm1∆::kanMX6 /pRS316-MMM1 | This study |
| JSP442 | MATα his3∆ arg4-NspI leu2 lys2 trp1::hisG ura3 mmm1∆::kanMX6 | This study |
| JSP443 | MATa leu2 trp1::hisG ura3 his3∆ ho vps13∆::HIS3MX6 mmm1∆::kanMX6 /pRS316-MMM1 | This study |
| JSP491 | MATα ura3 leu2 trp1::hisG his3∆ lys2 ho GFPN-vps13 mmm1∆::kanMX6 | This study |
| JSP552 | MATa his3 leu2 lys2 ura3 ho VPS13::GFP1360 PADH1::NVJ1::3xmCherry::hphNT1 | This study |
| JSP446 | MATα his3 leu2 trp1::hisG ura3 ho vps13∆::HIS3MX6 mmm1∆::kanMX6 PDIT1-lacZ::LEU2 /pRS316-MMM1 | This study |
| JSP582 | MATa his3 ura3 vps13-C89K mmm1∆::kanMX6/ pRS316-MMM1 | This study |
| JSP585 | MATα his3 leu2 ura3 trp1::hisG ho vps13-L1107P mmm1∆::kanMX6 /pRS316-MMM1 | This study |
| RP301e | MATa ura3 leu2 trp1-hisG his3∆ lys2 ho∆::LYS2 VPS13-L66P mmm1∆::kanMX6/ pRS316-MMM1 | This study |
| JSP533e | MATa ura3 leu2 trp1-hisG his3∆ lys2 ho::LYS2 vps13-L66P G718K mmm1∆::kanMX6/pRS316-MMM1 | This study |
| JSP417e | MATα his3∆SK arg4-NspI leu2 ura3 trp1::hisG ho vps13∆::HIS3MX6 | This study |
| JSP441 | MATα his3∆ leu2 trp1::hisG ura3 ho mmm1∆::kanMX6 /pRS316-MMM1 | This study |
| JSP490e | MATa ura3 leu2 trp1::hisG his3∆ lys2 ho::LYS2 GFPN-vps13 | This study |
| CUY9002 | MATα leu2-3112 ura3-52 his3-Δ200 trp1-∆101 lys2-801 suc2-∆9 GAL natMX4-PTEF1-GFP-VPS39 PADH1-NVJ1-3xmCherry-hphNT1 | Honscher et al. (2014) |
| HI27e | MATα ura3 his3∆SK trp1::hisG arg4-NspI lys2 ho∆::LYS2 rme1::LEU2 leu2 vps13∆::HIS3MX6 ho | Nakanishi et al. (2007) |
| HI28e | MATa ura3 his3∆SK trp1::hisG lys2 ho∆::LYS2 leu2 vps13∆::HIS3MX6 | Nakanishi et al. (2007) |
| AN117-16De | MATa ura3 leu2 trp1::hisG his3∆SK lys2 ho∆::LYS2 | Neiman et al. (2000) |
| JSP528e | MATa leu2 VPS13-GFP1360 his3∆SK arg4-NspI ura3 trp1::hisG lys2 ho:: LYS2 | This study |
| MATα LEU2 VPS13-GFP1360 his3∆SK ARG4 ura3 trp1::hisG lys2 ho:: LYS2 | ||
| JSYD1f | MATa leu2∆0 his3∆0 met15∆0 ura3∆0 TRP1 ARG4 LYS2 ho | This study |
| MATα leu2 his3∆SK MET15 ura3 trp1::hisG arg4-NspI lys2 ho∆::LYS2 | ||
| RME1 VPS13 | ||
| rme1::LEU2 VPS13 | ||
| JSYD2f | Same as JSYD1 only vps13∆::kanMX6 | This study |
| vps13∆::HIS3MX6 | ||
| JSYD3f | Same as JSYD1 only vps13-GFPc::HIS3MX6 | This study |
| vps13∆::HIS3MX6 | ||
| JSYD4f | Same as JSYD1 only VPS13-GFP1360 | This study |
| vps13∆::HIS3MX6 | ||
| JSYD5f | Same as JSYD1 only GFPN-vps13 | This study |
| vps13∆::HIS3MX6 | ||
| JSYD6f | Same as JSYD1 only vps13-L66P | This study |
| vps13∆::HIS3MX6 | ||
| JSYD7f | Same as JSYD1 only vps13-C89K | This study |
| vps13∆::HIS3MX6 | ||
| JSYD8f | Same as JSYD1 only vps13-L1107P | This study |
| vps13∆::HIS3MX6 | ||
| JSYD9f | Same as JSYD1 only vps13-Y2702C | This study |
| vps13∆::HIS3MX6 |
aIsogenic with the BY4741 strain background.
bStrains are isogenic and were derived from D273-10B (Sherman, 1963).
cThe mitochondrial genotype is bracketed.
dThe vps13-31 allele was previously designated yme3-1 (Thorsness and Fox, 1993). The VPS13-G820D allele was previously designated YNT61-1 (Hanekamp et al., 2002). The VPS13-L822F allele had four previous designations; YNT61-2, YNT61-3, YNT61-4, and YNT61-5 (Hanekamp et al., 2002).
eIsogenic with the SK1 background.
fStrains are hybrids derived by crossing haploids from the BY4741 and SK1 background.
Various point mutant alleles of VPS13 were introduced using the CRISPR/Cas9 strategy. The codon to be mutated was first replaced by the HIS3MX6 cassette, and then cotransformation of pRS425-Cas9-SkHIS3-381 with a 75-mer, single-stranded oligonucleotide carrying the mutated codon was performed. The exception was the vps13-C89K mutation, in which the rescue fragment was amplified by PCR from pRS426-VPS13C89K (plasmid construction described later). Transformants were screened for loss of the HIS3 marker, and then the mutated region was amplified by PCR and sequenced to confirm the proper integration of the mutation. For strains JSP531, JSP533, JSP549, and JSP563 (VPS13-G718K), the starting strains were JSP499-1, RP301, BY4742, and JSP560, respectively. For strains RP201, RP202, RP203, and RP205 carrying disease alleles, the starting strain was BY4741. For strain RP301, the starting strain was AN117-16D.
Strains JSP536, JSP541, JSP545, JSP547, and JSP555 carrying kanMX6-marked deletions of VPS13, NVJ1, or VAC8 were generated using flanking primers to amplify the deletion allele from genomic DNA of the appropriate strains in the yeast knockout collection (Winzeler et al., 1999). The amplified knockout fragments were transformed into strains GCY3, GCY4, JSP497, and GCY1, transformants were selected on plates containing G418, and the knockouts were confirmed by colony PCR. Similarly, the C-terminal GFP fusion to VPS13 was introduced into strain JSP549 by first amplifying the 3′ end of the VPS13 gene, along with the GFP coding region and the HIS3 selectable marker, from genomic DNA prepared from the VPS13-GFPc fusion in the yeast GFP-tagged collection (Huh et al., 2003). Transformants were selected on synthetic defined medium (SD) –His plates and proper integration confirmed by PCR, generating strains JSP556 and JSP557.
JSP417 is a segregant from a cross of AN117-16D and HI27. JSP441, JSP442, and JSP443 are segregants from a cross between JSP417 and KO4. JSP446 is a segregant from a cross between JSP441 and HI28 carrying PDIT1-lacZ::LEU2. JSP491 is a segregant from a cross between JSP442 and JSP490. JSP552 is a segregant from a cross between CUY9002 and JSP497. JSP577 is a segregant from a cross between JSP441 and GCY1. JSP582 and JSP585 are segregants from crosses between JSP446 and RP205 or RP202, respectively. JSP590 is a segregant from a cross between JSP557 and KO4. JSP512 is a segregant from a cross between JSP495 and JSP499-1, and JSP527 was created by mating two segregants from this same cross.
Diploids JSYD1 to JSYD9 were generated as follows: BY4741 (VPS13) was mated to AN117-4B (VPS13) to create JSYD1. Haploids KO1 (vps13Δ), GCY1 (vps13-GPFC), JSP497 (VPS13-GFP-1360), RP201 (vps13-L66P), RP205 (vps13-C89K), RP202 (vps13-L1107P), and RP203 (vps13-Y2702C) were crossed with HI27 (vps13Δ), resulting in JSYD2 to JSYD4 and JSYD6 to JSYD9, respectively. JSYD5 was generated by a cross of JSP513 (GFPN-vps13) and HI28 (vps13Δ).
Suppressors of the mmm1∆ slow-growth phenotype were isolated from independent streaks of single colonies of THY23 (mmm1∆) on rich-glucose medium. Initial isolates were colony purified by streaking three times onto rich-glucose medium. These isolates were then backcrossed to an isogenic wild type (PTY33), sporulated, and subjected to tetrad analysis. Strains JHY14, KWY70, KWY75, KWY76, KWY80, KWY105, KWY109, KWY111, KWY114, KWY131, and MTY79 are segregants from those crosses carrying both mmm1∆ and the suppressor mutation.
Plasmids
pRS426-VPS13-C89K was generated using site-directed mutagenesis. Briefly, a PCR fragment carrying the first 2345 base pairs of the VPS13 coding region (VPS13fragA) was cloned into the EcoRV site of pBluescript KS(+). Site-directed mutagenesis was performed in pBluescript KS(+)-VPS13fragA using overlapping primers carrying the C89K (TGC-to-AAA) mutation, generating pBluescript KS(+)-VPS13fragA-C89K. The mutation was confirmed by sequencing performed by the Stony Brook University DNA Sequencing Facility. pRS426-VPS13-C89K was finally constructed by homologous recombination of overlapping fragments of VPS13fragA-C89K and other VPS13 coding regions in S. cerevisiae as described in Park and Neiman (2012). Plasmid pRS316-MMM1 was generated by subcloning a 2.1-kb BclI fragment of yeast genomic DNA containing MMM1 into the BamHI site of the CEN/ARS/URA3 yeast shuttle vector, pRS316. This plasmid was previously given the name pRSMY6 (Hanekamp et al., 2002). pVT100U-mtBFP was constructed by subcloning a HindIII-mtBFP-XhoI fragment from pYES-mtBFP (Westermann and Neupert, 2000) into HindIII/XhoI-digested expression vector pVT100U (Vernet et al., 1987). Plasmids pBN-mtRosella and pRS426-R20 are described elsewhere (Bockler and Westermann, 2014).
Sporulation assays
Diploid cells were sporulated on Spo medium (2% agar, 1% potassium acetate, 0.05% yeast extract, and 0.05% glucose) at 30°C for 3–5 d. Three independent diploids from each cross were sporulated, and at least 200 cells were counted in each culture for the sporulation efficiency measurement.
For growth assay on plates, 3 μl of 10-fold serial dilutions were spotted onto yeast extract/peptone/glucose (YPD) rich medium and SD plus complete amino acids with 0.08% 5-FOA. These plates were incubated at 30°C until visible individual colonies were seen.
Western blot analysis
Exponentially growing cells (2 ml) were spun down to separate cell pellets containing internal cellular CPY and medium supernatants containing extracellular CPY. To prepare intracellular samples, cell pellets were resuspended in a solution of 100 μl of distilled water and 100 μl of 0.2 M NaOH and incubated for 5 min at room temperature. After pelleting the cells (6900 × g, 1 min), 50 μl of 1× SDS sample buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 0.1 mg/ml bromophenol blue, 0.36 M β-mercaptoethanol) was added to the pellets and the samples boiled for 5 min. To prepare the extracellular samples, 100% trichloroacetic acid (TCA) was added to the medium supernatants to a final concentration of 20% TCA. After 30 min of incubation on ice, the mixtures were centrifuged at 4°C at 16,000 × g for 15 min to precipitate proteins from the supernatants. The precipitants were washed with 300 μl of cold acetone and resuspended in 20 μl of 1× SDS sample buffer. Protein samples were fractionated using a 10% SDS-polyacrylamide gel, transferred to nitrocellulose, and probed with a 1:1000 dilution of monoclonal anti-CPY antibody (Life Technologies, Carlsbad, CA). The secondary antibody, horseradish peroxidase–conjugated sheep anti-mouse (GE Healthcare, Little Chalfont, UK), was used at a 1:2500 dilution, and the signal was detected using the ECL kit (Bio-Rad, Hercules, CA).
Mitophagy assay
Cells were grown overnight in either YPA or synthetic medium containing 2% galactose, 2% raffinose, and 0.1% glucose and lacking leucine to select for the mtRosella plasmid (pBN-mtRosella; Bockler and Westermann, 2014). Cells were washed with distilled water and diluted to a similar cell density of (1–2) × 106 cells/ml into either YPA from the YPA overnight culture or nitrogen starvation medium (0.17% yeast nitrogen base without amino acids and without ammonium sulfate; 2% glucose) from the synthetic medium overnight culture. The diluted cells were incubated at 30°C for 5.5 h and then examined by fluorescence microscopy.
Microscopy
Live-cell imaging was performed using either a Zeiss Axioplan2 microscope (Carl Zeiss, Thornwood, NY) with a Zeiss mRM AxioCam or a Zeiss Observer.Z1 microscope with an attached Orca II ERG camera (Hamamatsu, Bridgewater, NJ). Zeiss AxioVision 4.8 and ZEN 2012 (Blue edition) software were used to acquire images.
To visualize the vacuolar membrane, vacuolar lumen, and nucleus in live cells, FM4-64, blue CMAC, and Hoechst 33342 (all obtained from ThermoFisher, Waltham, MA) were used, respectively. FM4-64 was added for a final concentration of 2 μg/ml in live cells and incubated at 30°C for 1 h. Hoechst was then added to a final concentration of 3.3 μM, and the cells were incubated for an additional 10 min. Cells stained with FM4-64 and Hoechst were washed once in phosphate-buffered saline and observed by fluorescence microscopy. Blue CMAC was added to a final concentration of 100 μM to live cells and incubated at room temperature for 15–30 min. Cells were then washed once in fresh medium and observed by fluorescence microscopy.
To quantify the colocalization of Vps13-GFP1360 and endosomes, strains JSP512 and JSP527, expressing both Vps13-GFP1360 and Did2-mRFP were examined The percentage colocalization was defined as the fraction of GFP foci displaying overlap with RFP as defined by the presence of yellow (mixed red/green) signal. The percentage reported is the average of four independent experiments with at least 100 GFP foci counted in each experiment. The frequency of Vps13-GFP1360 localization at endosome–mitochondrial junctions was determined in JSP512/pVT100-mtBFP possessing all three markers (VPS13-GFP1360, DID2-mRFP, and mtBFP). The GFP foci overlapping with mRFP foci were first identified as described, and the fraction of these foci that also overlapped with the mitochondrial BFP signal was determined. The percentage reported is the average of two independent experiments with 200 GFP foci scored in both experiments.
Acknowledgments
We thank members of the Stony Brook Yeast Center for helpful discussions. This work was supported by National Institutes of Health Grants R01 GM072540 to A.M.N. and R01 GM050717 to N.M.H.
Abbreviations used:
- ChAc
chorea acanthocytosis
- CPY
carboxypeptidase Y
- ERMES
ER-mitochondrion encounter site
- mtDNA
mitochondrion DNA
- YPA
yeast extract/peptone/acetate.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-02-0112) on June 8, 2016.
REFERENCES
- Bankaitis VA, Johnson LM, Emr SD. Isolation of yeast mutants defective in protein targeting to the vacuole. Proc Natl Acad Sci USA. 1986;83:9075–9079. doi: 10.1073/pnas.83.23.9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockler S, Westermann B. Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev Cell. 2014;28:450–458. doi: 10.1016/j.devcel.2014.01.012. [DOI] [PubMed] [Google Scholar]
- Boeke JD, Trueheart J, Natsoulis G, Fink GR. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 1987;154:164–175. doi: 10.1016/0076-6879(87)54076-9. [DOI] [PubMed] [Google Scholar]
- Brickner JH, Fuller RS. SOI1 encodes a novel, conserved protein that promotes TGN-endosomal cycling of Kex2p and other membrane proteins by modulating the function of two TGN localization signals. J Cell Biol. 1997;139:23–36. doi: 10.1083/jcb.139.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. 2015;11:11–24. doi: 10.1038/nrneurol.2014.228. [DOI] [PubMed] [Google Scholar]
- Campbell CL, Thorsness PE. Escape of mitochondrial DNA to the nucleus in yme1 yeast is mediated by vacuolar-dependent turnover of abnormal mitochondrial compartments. J Cell Sci. 1998;111:2455–2464. doi: 10.1242/jcs.111.16.2455. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson-Stone C, Danek A, Rampoldi L, Hardie RJ, Chalmers RM, Wood NW, Bohlega S, Dotti MT, Federico A, Shizuka M, et al. Mutational spectrum of the CHAC gene in patients with chorea-acanthocytosis. Eur J Hum Genet. 2002;10:773–781. doi: 10.1038/sj.ejhg.5200866. [DOI] [PubMed] [Google Scholar]
- Elbaz-Alon Y, Eisenberg-Bord M, Shinder V, Stiller SB, Shimoni E, Wiedemann N, Geiger T, Schuldiner M. Lam6 regulates the extent of contacts between organelles. Cell Rep. 2015;12:7–14. doi: 10.1016/j.celrep.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz-Alon Y, Rosenfeld-Gur E, Shinder V, Futerman AH, Geiger T, Schuldiner M. A dynamic interface between vacuoles and mitochondria in yeast. Dev Cell. 2014;30:95–102. doi: 10.1016/j.devcel.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Enyenihi AH, Saunders WS. Large-scale functional genomic analysis of sporulation and meiosis in Saccharomyces cerevisiae. Genetics. 2003;163:47–54. doi: 10.1093/genetics/163.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanekamp T, Thorsness MK, Rebbapragada I, Fisher EM, Seebart C, Darland MR, Coxbill JA, Updike DL, Thorsness PE. Maintenance of mitochondrial morphology is linked to maintenance of the mitochondrial genome in Saccharomyces cerevisiae. Genetics. 2002;162:1147–1156. doi: 10.1093/genetics/162.3.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Kishida M, Nishizawa Y, Iijima M, Koriyama C, Nakamura M, Sano A, Kishida S. Subcellular localization and putative role of VPS13A/chorein in dopaminergic neuronal cells. Biochem Biophys Res Commun. 2012;419:511–516. doi: 10.1016/j.bbrc.2012.02.047. [DOI] [PubMed] [Google Scholar]
- Helle SC, Kanfer G, Kolar K, Lang A, Michel AH, Kornmann B. Organization and function of membrane contact sites. Biochim Biophys Acta. 2013;1833:2526–2541. doi: 10.1016/j.bbamcr.2013.01.028. [DOI] [PubMed] [Google Scholar]
- Henne WM, Liou J, Emr SD. Molecular mechanisms of inter-organelle ER-PM contact sites. Curr Opin Cell Biol. 2015;35:123–130. doi: 10.1016/j.ceb.2015.05.001. [DOI] [PubMed] [Google Scholar]
- Hobbs AE, Srinivasan M, McCaffery JM, Jensen RE. Mmm1p, a mitochondrial outer membrane protein, is connected to mitochondrial DNA (mtDNA) nucleoids and required for mtDNA stability. J Cell Biol. 2001;152:401–410. doi: 10.1083/jcb.152.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honscher C, Mari M, Auffarth K, Bohnert M, Griffith J, Geerts W, van der Laan M, Cabrera M, Reggiori F, Ungermann C. Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev Cell. 2014;30:86–94. doi: 10.1016/j.devcel.2014.06.006. [DOI] [PubMed] [Google Scholar]
- Hoppins S, Collins SR, Cassidy-Stone A, Hummel E, Devay RM, Lackner LL, Westermann B, Schuldiner M, Weissman JS, Nunnari J. A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J Cell Biol. 2011;195:323–340. doi: 10.1083/jcb.201107053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Jin L, Zhang K, Xu Y, Sternglanz R, Neiman AM. Sequestration of mRNAs modulates the timing of translation during meiosis in budding yeast. Mol Cell Biol. 2015;35:3448–3458. doi: 10.1128/MCB.00189-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009;17:98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolehmainen J, Black GC, Saarinen A, Chandler K, Clayton-Smith J, Traskelin AL, Perveen R, Kivitie-Kallio S, Norio R, Warburg M, et al. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet. 2003;72:1359–1369. doi: 10.1086/375454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B, Walter P. ERMES-mediated ER-mitochondria contacts: molecular hubs for the regulation of mitochondrial biology. J Cell Sci. 2010;123:1389–1393. doi: 10.1242/jcs.058636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krols M, van Isterdael G, Asselbergh B, Kremer A, Lippens S, Timmerman V, Janssens S. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. 2016:505–523. doi: 10.1007/s00401-015-1528-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, Agemura A, Izumo S, Sano A. In vivo distribution and localization of chorein. Biochem Biophys Res Commun. 2007;353:431–435. doi: 10.1016/j.bbrc.2006.12.059. [DOI] [PubMed] [Google Scholar]
- Kvam E, Gable K, Dunn TM, Goldfarb DS. Targeting of Tsc13p to nucleus-vacuole junctions: a role for very-long-chain fatty acids in the biogenesis of microautophagic vesicles. Mol Biol Cell. 2005;16:3987–3998. doi: 10.1091/mbc.E05-04-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvam E, Goldfarb DS. Nvj1p is the outer-nuclear-membrane receptor for oxysterol-binding protein homolog Osh1p in Saccharomyces cerevisiae. J Cell Sci. 2004;117:4959–4968. doi: 10.1242/jcs.01372. [DOI] [PubMed] [Google Scholar]
- Lang AB, John Peter AT, Walter P, Kornmann B. ER-mitochondrial junctions can be bypassed by dominant mutations in the endosomal protein Vps13. J Cell Biol. 2015;210:883–890. doi: 10.1083/jcb.201502105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, Cormier-Dequaire F, Hassoun SM, Pujol C, Ciura S, et al. Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am J Hum Genet. 2016;98:500–513. doi: 10.1016/j.ajhg.2016.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lottridge JM, Flannery AR, Vincelli JL, Stevens TH. Vta1p and Vps46p regulate the membrane association and ATPase activity of Vps4p at the yeast multivesicular body. Proc Natl Acad Sci USA. 2006;103:6202–6207. doi: 10.1073/pnas.0601712103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijaljica D, Prescott M, Devenish RJ. A fluorescence microscopy assay for monitoring mitophagy in the yeast Saccharomyces cerevisiae. J Vis Exp. 2011:pii 2779. doi: 10.3791/2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murley A, Sarsam RD, Toulmay A, Yamada J, Prinz WA, Nunnari J. Ltc1 is an ER-localized sterol transporter and a component of ER-mitochondria and ER-vacuole contacts. J Cell Biol. 2015;209:539–548. doi: 10.1083/jcb.201502033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H, Suda Y, Neiman AM. Erv14 family cargo receptors are necessary for ER exit during sporulation in Saccharomyces cerevisiae. J Cell Sci. 2007;120:908–916. doi: 10.1242/jcs.03405. [DOI] [PubMed] [Google Scholar]
- Neiman AM. Sporulation in the budding yeast Saccharomyces cerevisiae. Genetics. 2011;189:737–765. doi: 10.1534/genetics.111.127126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiman AM, Katz L, Brennwald PJ. Identification of domains required for developmentally regulated SNARE function in Saccharomyces cerevisiae. Genetics. 2000;155:1643–1655. doi: 10.1093/genetics/155.4.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Olkkonen VM. OSBP-related protein family in lipid transport over membrane contact sites. Lipid Insights. 2015;8:1–9. doi: 10.4137/LPI.S31726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Roberts P, Chen Y, Kvam E, Shulga N, Huang K, Lemmon S, Goldfarb DS. Nucleus-vacuole junctions in Saccharomyces cerevisiae are formed through the direct interaction of Vac8p with Nvj1p. Mol Biol Cell. 2000;11:2445–2457. doi: 10.1091/mbc.11.7.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Halegoua S, Kishida S, Neiman AM. A conserved function in phosphatidylinositol metabolism for mammalian Vps13 family proteins. PLoS One. 2015;10:e0124836. doi: 10.1371/journal.pone.0124836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Neiman AM. VPS13 regulates membrane morphogenesis during sporulation in Saccharomyces cerevisiae. J Cell Sci. 2012;125:3004–3011. doi: 10.1242/jcs.105114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Okumura Y, Tachikawa H, Neiman AM. SPO71 encodes a developmental stage-specific partner for VPS13 in Saccharomcyes cerevisiae. Eukaryot Cell. 2013;12:1530–1537. doi: 10.1128/EC.00239-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Wenzel EM, Stenmark H. ER-endosome contact sites: molecular compositions and functions. EMBO J. 2015;34:1848–1858. doi: 10.15252/embj.201591481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampoldi L, Dobson-Stone C, Rubio JP, Danek A, Chalmers RM, Wood NW, Verellen C, Ferrer X, Malandrini A, Fabrizi GM, et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat Genet. 2001;28:119–120. doi: 10.1038/88821. [DOI] [PubMed] [Google Scholar]
- Rosado CJ, Mijaljica D, Hatzinisiriou I, Prescott M, Devenish RJ. Rosella: a fluorescent pH-biosensor for reporting vacuolar turnover of cytosol and organelles in yeast. Autophagy. 2008;4:205–213. doi: 10.4161/auto.5331. [DOI] [PubMed] [Google Scholar]
- Rose MD, Fink GR. Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- Schrader M, Godinho LF, Costello JL, Islinger M. The different facets of organelle interplay-an overview of organelle interactions. Front Cell Dev Biol. 2015;3:56. doi: 10.3389/fcell.2015.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. Respiration-deficient mutants of yeast. I. Genetics. Genetics. 1963;48:375–385. doi: 10.1093/genetics/48.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan CJ, Blumer KJ. A syntaxin homolog encoded by VAM3 mediates down-regulation of a yeast G protein-coupled receptor. J Biol Chem. 1999;274:1835–1841. doi: 10.1074/jbc.274.3.1835. [DOI] [PubMed] [Google Scholar]
- Suda Y, Nakanishi H, Mathieson EM, Neiman AM. Alternative modes of organellar segregation during sporulation in Saccharomyces cerevisiae. Eukaryot Cell. 2007;6:2009–2017. doi: 10.1128/EC.00238-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsness PE, Fox TD. Nuclear mutations in Saccharomyces cerevisiae that affect the escape of DNA from mitochondria to the nucleus. Genetics. 1993;134:21–28. doi: 10.1093/genetics/134.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomiyasu A, Nakamura M, Ichiba M, Ueno S, Saiki S, Morimoto M, Kobal J, Kageyama Y, Inui T, Wakabayashi K, et al. Novel pathogenic mutations and copy number variations in the VPS13A gene in patients with chorea-acanthocytosis. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:620–631. doi: 10.1002/ajmg.b.31206. [DOI] [PubMed] [Google Scholar]
- Toulmay A, Prinz WA. Lipid transfer and signaling at organelle contact sites: the tip of the iceberg. Curr Opin Cell Biol. 2011;23:458–463. doi: 10.1016/j.ceb.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Maruki Y, Nakamura M, Tomemori Y, Kamae K, Tanabe H, Yamashita Y, Matsuda S, Kaneko S, Sano A. The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat Genet. 2001;28:121–122. doi: 10.1038/88825. [DOI] [PubMed] [Google Scholar]
- Velayos-Baeza A, Vettori A, Copley RR, Dobson-Stone C, Monaco AP. Analysis of the human VPS13 gene family. Genomics. 2004;84:536–549. doi: 10.1016/j.ygeno.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Vernet T, Dignard D, Thomas DY. A family of yeast expression vectors containing the phage f1 intergenic region. Gene. 1987;52:225–233. doi: 10.1016/0378-1119(87)90049-7. [DOI] [PubMed] [Google Scholar]
- Westermann B, Neupert W. Mitochondria-targeted green fluorescent proteins: convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast. 2000;16:1421–1427. doi: 10.1002/1097-0061(200011)16:15<1421::AID-YEA624>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]