

Although stress-specific factors increase ATP demand for cellular growth under stressful conditions in yeast, increased ATP demand for cellular maintenance underpins a general stress response and is responsible for the onset of overflow metabolism.
Abstract
Yeast cell factories encounter physical and chemical stresses when used for industrial production of fuels and chemicals. These stresses reduce productivity and increase bioprocess costs. Understanding the mechanisms of the stress response is essential for improving cellular robustness in platform strains. We investigated the three most commonly encountered industrial stresses for yeast (ethanol, salt, and temperature) to identify the mechanisms of general and stress-specific responses under chemostat conditions in which specific growth rate–dependent changes are eliminated. By applying systems-level analysis, we found that most stress responses converge on mitochondrial processes. Our analysis revealed that stress-specific factors differ between applied stresses; however, they are underpinned by an increased ATP demand. We found that when ATP demand increases to high levels, respiration cannot provide sufficient ATP, leading to onset of respirofermentative metabolism. Although stress-specific factors increase ATP demand for cellular growth under stressful conditions, increased ATP demand for cellular maintenance underpins a general stress response and is responsible for the onset of overflow metabolism.
INTRODUCTION
The budding yeast Saccharomyces cerevisiae is widely used as a microbial cell factory in the biotechnology industry for the production of various chemicals, ranging from endogenous metabolites, such as ethanol, to heterologous metabolites, such as advanced biofuels (Caspeta and Nielsen, 2013; Nielsen, 2015). Industrial processes impose severe stresses, however, both physical and chemical, that reduce the catalytic efficiency of this cell factory. Therefore it is critical for the economic viability of industrial processes to engineer yeast strains that not only produce novel bioproducts but also can withstand multiple stresses while maintaining high productivity. For these reasons, there is much interest in identifying the mechanisms of stress response and, in particular, evaluating whether exposure to different types of stress causes common phenotypic responses, as this would make it possible to engineer yeast strains with enhanced robustness.
Common stresses in the biotechnology industry are caused by toxicity of the product (e.g., ethanol), components of the medium (e.g., salts and glucose), the physical environment (e.g., temperature and pH), hydrostatic pressure, or nutrient limitations (Belloch et al., 2008; Graf et al., 2009; Zhao and Bai, 2009; Jullesson et al., 2015). Yeast is most widely used for the production of ethanol, which is by far the most dominant biofuel used today (Caspeta and Nielsen, 2013). In this process, yeast is exposed to relatively high temperatures, high initial glucose concentrations (causing high osmolarity), high salt concentrations, and, at the end of the fermentation, very high ethanol concentrations. We were therefore interested in quantifying the phenotypic responses of yeast to these three stress factors—temperature, osmolarity, and ethanol.
Previously the stress response of yeast has been studied in batch experiments by comparing various stress conditions at the transcriptional level (Gasch et al., 2000; Causton et al., 2001). However, with this experimental design, many responses are masked due to dynamic growth conditions (O’Duibhir et al., 2014). Industrial stresses have also been studied as an adaptive response using adaptive laboratory evolution (Cakar et al., 2005; Ding et al., 2010; Dhar et al., 2011; Caspeta et al., 2014). Most of these stress studies, however, are stand-alone experiments, which makes it difficult to identify the general stress response encountered in a typical industrial process. Moreover, only limited overlap was found between genes required for stress survival and stress-induced genes (Gibney et al., 2013). This indicates that the previously reported general stress response studies are rather elusive and the identified phenotypic responses are likely caused by changes in the specific growth rate (Gasch et al., 2000; Brauer et al., 2008; Gerosa et al., 2013; O’Duibhir et al., 2014; Kanshin et al., 2015). Therefore it is important to distinguish between transient stress responses and stress adaptation. Under transient stress responses, usually studied under batch conditions, stress is applied as a shock, and it is likelier that cells initiate a common survival or buffering system to cope with the extreme environmental conditions. In the case of an adapted stress conditions in which stress is applied smoothly or cells have time to adapt to the stressor in a continuous culture experiments—for example, chemostats—it is possible to separate effects of growth rate and the applied stressor (Olz et al., 1993; Postmus et al., 2008, 2011, 2012).
To overcome the combinatorial effects of a stressor and changes in the specific growth rate of cells, we used an experimental design in which we used glucose-limited chemostat cultures and monitored the adapted stress response for multiple stresses in a gradual manner to enable identification of the key factors that are driven solely by the applied stress. This experimental design provided us with triplicate data points that allowed us to make solid conclusions regarding the response to three types of stress—ethanol, osmolarity, and temperature. Furthermore, our approach to studying multiple stresses allowed us to identify mechanism(s) of general and stress-specific responses in yeast and examine the molecular changes behind the initiation of overflow metabolism in yeast.
RESULTS
Macroscopic phenotypic characterization
We studied the response of the yeast S. cerevisiae to three industrially important stress factors—ethanol, osmolarity, and temperature—in a gradual manner at a constant specific growth rate (Figure 1, A and B). Each stress condition was studied at three levels (low, medium, and high) until a near-maximal value was reached that can be maintained at a specific growth rate of 0.1 h−1. Quantitative transcriptome data were obtained using RNA sequencing and used to generate condition-dependent genome-scale metabolic models (Agren et al., 2014; Supplemental Figure S1). The condition-specific genome-scale metabolic models were then used for flux balance analysis (FBA) to calculate intracellular fluxes. Among other fluxes, for the first time, total maintenance energy was estimated for the studied stress conditions.
FIGURE 1:

The stress response of the yeast S. cerevisiae was studied in chemostat cultivations at a constant dilution rate (0.1 h−1) in a gradual manner. (A) Heat, osmotic conditions (NaCl), and ethanol were increased in three steps from reference conditions to the highest pretested value. (B) Physiology in terms of biomass yield (blue line, c-mol [dry biomass] c-mol−1 [consumed substrate]), specific oxygen consumption rate (red dashed line, mmol/g [DW] h), specific CO2 production rate (green dashed line, mmol/g [DW] h), and specific ethanol production rate (black line, mmol/g [DW] h). Additional maintenance energy (blue bars, mmol ATP/g [DW] h) was calculated using FBA on condition-specific genome-scale models. (C) Transcriptome data used for the PCA; increasing circle size indicates the increase in studied stress conditions, and stress is color coded accordingly: ethanol is green, osmosis is blue, and temperature is red. (D) Bar chart showing the importance of the PC components. (E) Venn diagram representing the overlap of significantly changed genes (p < 0.001); numbers in parentheses indicate greater-than-twofold change.
Our experimental design enabled us to evaluate stress responses at different exposure levels. From the macroscopic measurements, we observed clear differences in phenotypic response. Thus the cells coconsumed glucose and ethanol at different levels of ethanol stress and produced glycerol in response to osmotic stress conditions, whereas at high temperature, the cells shifted to respirofermentative metabolism (Figure 1B and Supplemental Table S1). Despite observed differences for the three different types of stress, we identified one common response in all of the studied conditions, namely a decrease in biomass yield with increasing exposure to any of the three types of stress. Supported by FBA calculations, the decreasing biomass yield was directly related to an increased maintenance cost for the cells (Figure 1B). Because increased maintenance could be caused by various factors, we performed a more thorough analysis of the RNA sequencing (RNAseq) data.
Transcriptional response
To obtain a general overview of the RNAseq data, we first performed a hierarchical clustering analysis. The most distinct transcriptional pattern was found for the osmotic stress experiments (Supplemental Figure S2). Low- and medium-temperature conditions clustered together with the reference, showing only a small number of changes in their profile (Supplemental Figure S3). High temperature clustered with high ethanol, as well as other ethanol conditions, a phenomenon also observed by others (Piper et al., 1994; Auesukaree et al., 2009). When the same data were separated on a principal component (PC) analysis plot, the more stressful conditions clustered to the first quarter of the graph and were separated from the reference conditions by both the first and second PCs (Figure 1, C and D). Of interest, independent of whether ethanol was consumed or produced, all of the conditions in which ethanol was present in the environment were separated by the first PC, which described 63% of the occurring changes. Using data clustering and gene enrichment analysis, we found that these changes accounted for mainly oxidation-reduction processes, NAD(P)H regulation, and ergosterol biosynthesis pathways (p < 0.001). In addition, some pentose phosphate pathway genes (GND2, TKL2) exhibited strong down-regulation at the transcriptome level for those conditions. The second PC, which was responsible for an additional 13% of the changes, was characterized by significant changes in the tricarboxylic acid (TCA) cycle, transporter activity, and mitochondrion. Samples in the first quadrant of the plot, which represented the highest stress (especially high ethanol and temperature), were affected by strong down-regulation of the glyoxylate shunt, arginine metabolism, and gluconeogenesis. The third PC (unpublished data) indicated similarities between higher osmotic stress and low ethanol conditions. These similarities were associated with similar patterns in the transmembrane transporter genes.
The strong similarities between ethanol and high-temperature conditions can be explained by exposure of the cells to ethanol in these experiments (produced for the high-temperature condition and partly consumed for the ethanol conditions). Down-regulation of the entire branch chain amino acid (BCAA) pathway, as well as arginine and ergosterol biosynthesis, was the most evident response for the four different conditions in which ethanol was consumed or produced and separated on the first PC (Figure 1C). Because ergosterol and the ratio change between saturated and unsaturated fatty acids are reported to be the major components that influence ethanol tolerance by controlling the fluidity of the membrane and preventing interdigitation (Ma and Liu, 2010; Vanegas et al., 2012; Dong et al., 2015; Wang et al., 2015), we analyzed this further. Genes involved in ergosterol biosynthesis were significantly down-regulated (Figure 2), but, unexpectedly, we found a significant increase in intracellular ergosterol levels under conditions in which ethanol was present in the environment (Supplemental Figure S4). In the case of osmotic stress, in which ethanol was not present in the environment, the levels of ergosterol and gene expression in the ergosterol synthesis pathway were unchanged (Figure 2). Ergosterol plays a crucial role in membrane fluidity, and its biosynthesis seems to be tightly regulated to retain a desired concentration within cells. In yeast cells, the AMP-activated kinase Snf1 plays an important role in controlling flux toward lipids during growth on ethanol, both at the transcriptional level and through inactivation of Acc1 by phosphorylation. In mammalian cells, the Snf1 analogue AMP-activated protein kinase also controls the activity of 3-hydroxy-3-methylglutaryl-CoA reductase Hmg1 by phosphorylation, one of the key flux-controlling enzymes of the sterol biosynthetic pathway. This can explain the paradox between down-regulation of ERG genes and elevated levels of sterol, as in the presence of ethanol, the flux toward acetyl-CoA is increased, allowing for increased flux through the pathway even when there is lower expression of the enzymes (Supplemental Figure S5). The reduced expression of the ERG genes is consistent with finding of negative feedback control of gene expression in ergosterol biosynthesis through the transcription factor Upc2 (Caspeta et al., 2014; Yang et al., 2015). Alternatively, ergosterol degradation could be lower under conditions in which ethanol is present in the environment. However, little is known about the ergosterol turnover in yeast.
FIGURE 2:

Changes in gene expression for the studied stress conditions. Transcript intensities at the reference condition and log2-fold changes compared with the reference condition for selected genes. ETC, electron transport chain; see Supplemental Table S2 for gene name abbreviations.
General stress response
Among the highest stress conditions studied for ethanol, osmotic, and temperature stress, 297 genes showed differential expression overlap (p < 0.001) under all three conditions, in which only 63 had an expression change of greater than twofold (Figure 1E). Enrichment analysis of commonly differentially expressed genes indicated rearrangements mainly in oxidoreductase activity, pointing to an imbalance in NAD(P)H production (p < 0.001; Supplemental Figure S6). Additional changes were detected in the biosynthesis of amino acids. Given that the transcriptional environmental stress response (ESR) was identified previously mainly under batch conditions (Gasch et al., 2000), we were interested to examine the overlap between that data set and our stress adaptation experiments. Combining 94 conditions reported in Gasch et al. (2000) with our results on stress responses, we saw limited overlap (Supplemental Figure S7). Fewer than 8% of the genes determined as ESR (59 of 780 ESR genes compared) showed significant differential expression in our data set. Moreover, the majority of detected changes were in the opposite direction to their batch experiments (Supplemental Figure S7). It is of interest that temperature-dependent experiments under chemostat conditions by Gasch et al. (2000) showed a similar profile as the stress experiments reported here. This indicates that previously reported transcriptional ESR is strongly biased toward growth rate–dependent genes, a phenomenon always found in stress experiments under batch conditions.
We next used dosage-dependent data for the most significantly changed genes in terms of their fold change and abundance for covariance analysis, which provides an excellent measure of the significance of a transcriptional change. Covariance scores, which were calculated separately for each stress (ethanol, osmotic, and temperature), revealed the highest common effect for the biosynthesis of amino acids (mainly BCAA and arginine), αKG metabolism, GSH metabolism, and pyruvate metabolism (enrichment p < 0.01; Supplemental Table S3). In addition, among the most significantly regulated genes, heat-shock proteins (HSPs; and genes for HSP-related proteins SSA3 and SSE2) and ergosterol pathway genes were changed most significantly. These changes can be considered to be a common stress response to address increased maintenance. However, the initial cause of increased maintenance seemed to depend on stress condition.
Ethanol increases membrane fluidity
Ethanol is the main by-product of the yeast fermentation processes and the main growth-inhibiting factor in yeast-based industrial applications. At low ethanol concentrations (20 g/l), all of the ethanol supplemented to the medium was fully consumed together with glucose, and the culture was under carbon limitation (Figure 1A). However, already at the median ethanol concentration (40 g/l), ethanol consumption decreased (approximately two-thirds of the consumption detected at the low ethanol concentration of 20 g/l; Figure 1A), and a large amount of ethanol remained in the medium. At high ethanol concentration (60 g/l), almost no ethanol was consumed by the cells, although all of the glucose was consumed. Because ethanol is metabolized via the glyoxylate shunt, TCA cycle, and gluconeogenesis, this finding reflects changes in mitochondrial transport and, hence, an inability to use secondary carbon sources under stressful conditions. The transcriptional results indicated that at low ethanol concentrations, the pathways and cellular functions that use ethanol, such as the TCA cycle and transmembrane transport, together with other mitochondrion-related processes such as glutamine biosynthesis, cellular amide metabolism, and ion transport, were up-regulated (p < 0.001). All of these changes indicated a more active TCA cycle and respiratory chain. At the same time, oxidation-reduction reactions, lipid metabolism, cofactor binding, cellular alcohol metabolism, and glutathione metabolism were down-regulated. At the median ethanol condition, TCA cycle and glutamate metabolism were the only gene ontology (GO) groups showing significant up-regulation (p < 0.001), whereas down-regulation was observed among the same metabolic groups as for the low ethanol concentration. At the highest ethanol concentration, only a small number of transcripts from the TCA were up-regulated (CIT1, 2; MDH1; IDH1, 2), whereas down-regulation of ammonium transmembrane transport was detected (p < 0.001). This was consistent with strong down-regulation of the biosynthesis of mitochondrion-related amino acids, such as BCAA and arginine. As mentioned earlier, genes of the ergosterol biosynthetic pathway were down-regulated, and we measured almost twofold-higher ergosterol levels under conditions in which ethanol was consumed (Supplemental Figure S4). Higher ergosterol levels result in increased membrane fluidity (Ma and Liu, 2010), and this seems to be beneficial for coping with high-ethanol conditions. Increased membrane permeability is potentially the main cause of increased maintenance in the presence of ethanol. In addition to increased ergosterol levels, an increased ratio of monounsaturated to unsaturated fatty acids has been reported as a response to ethanol stress (Beaven et al., 1982; Sajbidor et al., 1995; You et al., 2003). We measured the total fatty acid composition under the ethanol stress conditions and determined a strong shift toward longer-chain fatty acids (C18), with the most abundant fatty acid under reference conditions, palmitoleic acid (C16:1 n-7), showing a gradual decrease under increasing ethanol concentrations and oleic acid (C18:1 n-9) concentration increasing under the same conditions (Figure 3 and Supplemental Table S4).
FIGURE 3:
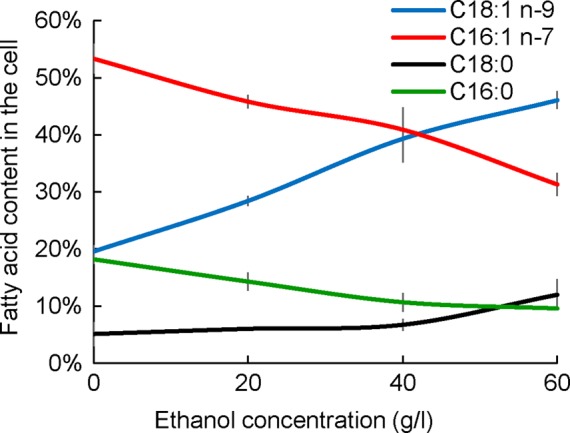
Total fatty acid content in cells cultivated in chemostats at constant specific growth rate of 0.1 h−1 on mineral medium supplemented with different ethanol concentrations. Four of the most abundant fatty acids are presented: C18:1, n-9 (blue line), C18:0 (black line), C16:1, n-7 (red line), and C16:0 (green line). Information about the profiles of other fatty acids is given in Supplemental Table S4.
High osmolarity induces reorganization in oxidation-reduction pathways
High osmolarity occurs in industrial processes due to high concentrations of salts, substrates, or other metabolites in industrial media. Although CEN.PK family–specific Na+ sensitivity has been reported (Daran-Lapujade et al., 2009), no long-term inhibition in a chemostat culture was observed. The first osmolarity-related effect on transcriptional level at the two lower NaCl concentrations (0.2 and 0.4 M NaCl) was related to the down-regulation of amino acid and ammonium transport, BCAA biosynthesis, and arginine, proline, and ornithine metabolism. Under the same conditions, up-regulation of oxidation-reduction pathways, glycerolipid metabolism, starch and sucrose metabolism, glycolysis/gluconeogenesis, and alcohol metabolism was observed. At the highest NaCl concentration (0.6 M), additional GO groups, such as cofactor binding, glutathione metabolism, and autophagy regulation, were down-regulated, whereas transmembrane transport and the TCA cycle showed enrichment among the up-regulated genes. In addition to the aforementioned enriched GO groups, genes typically related to osmotic stress (STL1, SUC2, GPD1, GPP1, and CTT1) were activated already at the lowest osmotic condition tested. Osmotic stress showed the highest distance compared with the other stress conditions studied, clustering separately on hierarchical clustering plots. Although oxidation-reduction pathways were down-regulated under all of the studied stress conditions (including biosynthesis pathways for steroids, glutathione, and secondary metabolites), the two highest osmotic stress conditions showed the largest perturbations in this pathway, having significant changes in both directions (Supplemental Figure S6 and Supplemental Table S5). When clustering genes from the GO group oxidoreductase activity (GO:0016491) for previously published osmotic stress experiments in batch and our osmotic stress experiments, we observed higher similarities with the stress adaptation period of sorbitol stress described by Gasch et al. (2000; later time points in batch osmostress; Supplemental Figure S8). When all of the stress experiments from the present study were used for oxidoreductase activity gene clustering, similar patterns for temperature and ethanol stress genes were detected (Supplemental Figure S9). However, osmotic stress response seemed to be regulated differently from the other stresses. When only osmotic stress response genes from the oxidoreductase activity group were clustered, significant enrichments were detected in oxidative phosphorylation, TCA, pyruvate, and methionine metabolism for the up-regulated genes and in glutathione metabolism and glycine catabolic processes for down-regulated genes (p < e-4; Supplemental Figure S10). In addition, groups such as cellular oxidant detoxification (represented by genes PRX1, MPR1, TRR1, TSA2, SOD2, DOT5, RSM26, SRX1, CCP1, CCS1, GLR1, and CTT1) and response to oxidative stress showed significant enrichment in up-regulated clusters (p < e-5). This implies the presence of oxidative stress in NaCl- treated cells, as previously seen in primary human corneal epithelia cells (Deng et al., 2015). Oxidative stress hinders the activity of proteins and therefore contributes to increased maintenance.
High temperature affects protein turnover
Higher tolerance toward increased temperatures is beneficial in industrial processes, as one can reduce costs for cooling bioreactors and reduce risks for contamination (Caspeta et al., 2014). From a physiological point of view, the two lower-temperature values tested resulted in relatively small transcriptional changes compared with the reference conditions and therefore also clustered closely together in the transcriptional analysis. In low-temperature conditions, the only enriched pathways at the transcriptome level were ergosterol and arginine biosynthesis, which showed partial down-regulation. With the increase in temperature to 36°C, significant and even more pronounced down-regulation was detected among the genes in the same pathways. The highest number of transcriptional changes took place at the highest temperature condition studied (38°C), at which, in addition to a sevenfold decrease in biomass yield (compared with the reference conditions), ethanol production was observed due to the change from respiratory to respirofermentative metabolism. Owing to the significant amount of ethanol present in the environment (3.94 ± 0.16 g/l), the highest ethanol condition and high-temperature conditions shared more than 60% of the changes compared with the reference condition at optimal environmental conditions. Although the present concentration itself cannot produce a significant stress to the cells (as also indicated by much less pronounced changes in total fatty acid composition), we assume that ethanol present in the environment already initiates changes in metabolism that in our transcriptome-based PC analysis were described on the first PC. High ethanol and temperature shared changes in steroid (ergosterol), amino acid (Cys, Met, His, Phe, Tyr, Trp, Val, Ile, Leu), mitochondrial ribosome and peroxisome biosynthesis, and propanoate and pyruvate metabolism. The only changes specific to high temperature (p < 0.001) took place in oxidative phosphorylation, translation (ribosomes), threonine-type peptidase activity, proteasome activity, and apoptosis (controlled cell death). The last two were indications of increased protein turnover in high-temperature conditions, which may be the main cause of the highest maintenance energy costs.
Regulation
To understand the underlying stress regulation mechanisms and control the responses to increased stress tolerance in industrial processes, it is important to determine the regulation level of relevant fluxes and the transcription factors controlling metabolism under relevant conditions. To quantify metabolic fluxes under the different conditions, we used condition-dependent near-genome scale models for flux balance analysis and compared these data with the transcriptome. Fluxes were calculated separately for each condition and replicate experiment, and the models were constrained by the measured uptake and secretion fluxes. The calculated median flux variability was <20%. Transcriptional control of fluxes can be identified by comparing the directions and z scores of two different conditions at the transcriptional and flux levels (Bordel et al., 2010). Because our data set contained 10 different conditions, we used all conditions to identify potential global transcriptional regulation. The RNA levels and calculated fluxes from 10 different conditions were plotted separately for each gene, and p values for the Pearson correlations were calculated. Of 758 fluxes with a nonzero value, 216 showed significant changes in flux profiles under at least two environmental conditions (p < 0.01; Supplemental Figure S11). Of these 216 fluxes, 55 showed R2 > 0.6 with p < 0.001 when correlated against their corresponding RNA levels. Enrichment analysis of those 55 potentially transcriptionally regulated genes indicated that the majority belonged to the oxidative phosphorylation pathway or were in some way related to mitochondria (Supplemental Table S6). Thus ∼25% of the metabolic fluxes that changed in response to the long-term stress studied here are transcriptionally regulated, and most of these changes drive an altered energy metabolism by mitochondria.
To gain further insight into the drivers of transcriptional reprogramming in response to stress, we searched for transcription factors (TFs) that play an important role in stressful conditions. We used gene set analysis to determine significantly enriched transcription factors using gene–TF relationship information from the Yeastract database (www.yeastract.com; as of 2 December 2015). With a p-value threshold of 0.001, we were able to detect 3, 16, and 8 TFs responsible for the highest level of stress toward ethanol, osmosis, and temperature, respectively (Supplemental Figure S12). Only one TF, GCN4, was represented in all three stress conditions studied. GCN4 is a transcriptional activator of amino acid biosynthetic genes, and its activity could be related to an increase in amino acid synthesis under conditions in which the translation rate is increased (Canelas et al., 2010). Our study showed strong down-regulation of the genes that GCN4 regulates, and we detected a twofold transcriptional down-regulation of GCN4 itself under conditions of high stress. For osmotic and temperature stress conditions, TFs regulating protein turnover, such as HAP4 and HSF1, were significantly affected. Surprisingly, the well-known general stress regulator MSN2 was significantly enriched only under high osmotic stress.
Initiation of overflow metabolism
Overflow metabolism refers to the seemingly wasteful metabolic strategy by which cells incompletely oxidize their growth substrate into by-products instead of using the more energetically efficient respiratory pathway, even in the presence of oxygen. In our experiments, high ethanol and high temperature were the two environmental conditions that showed the highest maintenance cost. These two conditions also clustered together during hierarchical clustering and on a transcriptome-based PC analysis. However, these two conditions represent very different physiological states. Under high ethanol conditions, cells were respiring (respiration quotient [RQ] < 1), whereas under high-temperature conditions, cells were mainly fermenting (RQ >> 1). This allowed us to look into the main differences between those conditions and detect the main changes that triggered the overflow metabolism under high-temperature conditions. Under both high ethanol and temperature conditions, we detected significant decrease in fluxes and down-regulation among genes in the glyoxylate shunt (MLS1, ICL1), gluconeogenesis (PCK1), and biosynthesis of mitochondrion-related amino acids (BCAAs, Arg), which indicates reduced transport into the mitochondria. These changes seem to be responsible for hindering the consumption of alternative carbon sources (in the present case, ethanol). On the other hand, up-regulation of mitochondrial ribosomes, down-regulation of the TCA genes CIT1, MDH1, KGD1, and FUM1, and, of most importance for explaining the overflow metabolism, down-regulation of many catabolic genes from the oxidative phosphorylation pathway (SDH, ATP, and COX genes; Figure 2) only under high-temperature conditions signal the genes likely responsible for the initiation of overflow metabolism. Because genes from the oxidative phosphorylation pathway are regulated on the transcriptional level (see earlier discussion), the initiation of overflow metabolism seems to be precisely controlled by metabolic regulation.
We lack a good explanation for why an onset of ethanol overflow appears in response to stress at the relatively low specific growth rate used in our study. To gain further insight into this phenomenon, we plotted glycolytic flux through the TCA cycle versus the specific glucose uptake rate. The flux through the TCA cycle was quantified as CO2 excretion not related to ethanol production, and this should represent the activity of respiration. Increase in the specific glucose uptake rate under constant specific growth rate conditions was observed in our studies due to decreased biomass yield. Of interest, we discovered a linear relationship between respiratory flux and the specific glucose uptake rate, but only up to a specific glucose uptake rate of ∼3.4 mmol/g (dry weight [DW]) h, at which respiration achieved a maximal value. With an additional increase in the specific glucose uptake rate, respiration began to decrease (Figure 4). At this time, RQ increased to >1, and overflow metabolism resulting in ethanol production was established. This profile is similar to that observed for respiratory metabolism in chemostat cultures operated at different dilution rates; therefore we overlaid data from such experiments on ours and found remarkable consistency (Figure 4; van Hoek et al., 1998). For specific growth rate–dependent experiments, glucose uptake rate increases due to higher specific growth rate. In our stress experiments, which were carried out at constant specific growth rate, increase in glucose uptake rate was observed due to decreasing biomass yield. These two approaches represent different concepts in metabolism but do show similar outcomes concerning the onset of overflow metabolism. This indicates that the Crabtree effect is a consequence of a limitation of energy supply, and allocation of proteome takes place to support translation at higher growth rates. This is consistent with a recent study showing that the Crabtree effect is due to a limitation in ATP formation by the F1F0-ATP synthase (Nilsson and Nielsen, 2016). To further determine the consistency of the relationship between respiration and glucose uptake rate under conditions in which yeast is exposed to different types and levels of stress, we performed four additional chemostat experiments in three biological replicates at the same dilution rate (0.1 h−1) but with different stress factors. A few additional data points were generated around the critical glucose uptake rate value and were found to fit very well with the overall profile (Figure 4).
FIGURE 4:
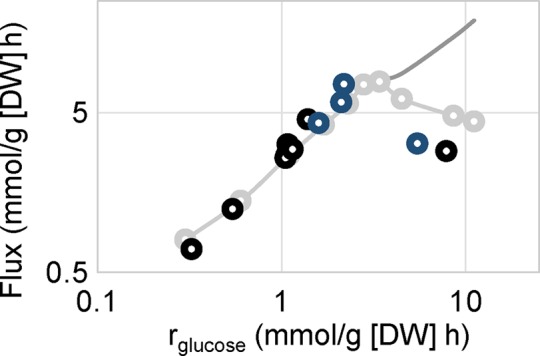
Specific glucose-uptake rate dependence of respiration in the yeast S. cerevisiae. A unique curve for respiration is presented for stress-related and specific growth rate–dependent data, which indicates a protein production limitation, where a trade-off between translational activity and respiration emerges. Specific growth rate-dependent data are presented in gray; the present stress experiments and additional stress experiments are shown in black and dark blue, respectively.
DISCUSSION
There are evident differences between a sudden effect of stress and an adapted stress response (Gibney et al., 2013). Our study focused on the adapted stress response to observe how the transcriptome of yeast cells adapt to different stressful conditions and determine the effect of a general stress response. Previously it was observed that general environmental stress response genes have a high overlap with expression changes occurring when a specific growth rate is reduced at optimal conditions in substrate-limited continuous cultivations (Gasch et al., 2000; Regenberg et al., 2006). This overlap exists because a significant decrease in the specific growth rate, which is associated with nutrient limitations, is evidently accompanied by the application of stressful conditions. Owing to this change in the specific growth rate, cell cycle genes are often described as one of the main stress responses (Gasch et al., 2000; Kanshin et al., 2015). Using a traditional experimental design, it has been difficult to identify the true stress response. Our experimental design, however, for the first time provides data giving us insight into the transcriptional reprogramming that is specifically due to an adapted response to three different types of stress.
One highly evident characteristic for adapted stress compared with a sudden stress response is the behavior of HSPs. Because an increase in HSP levels is a common response to sudden stress, it is surprising that there was significant down-regulation in the majority of cytosolic HSPs and HSP subunits under our adapted stress conditions (Supplemental Table S2). Because HSP transcript levels increase with decreasing specific growth rate of yeast, the observed phenomena in our experiments can be explained by already high HSP levels at relatively low specific growth rate used as the reference condition, providing a different background for data comparison (Regenberg et al., 2006). In addition, HSPs need to quickly rearrange the proteome or address dysfunctional proteins. Under adapted stress conditions, this function could no longer be as critical. There is increasing evidence that protein turnover is higher under conditions of high glucose-uptake rates (Boender et al., 2011; Lahtvee et al., 2014), resulting in lower need for additional HSPs in the adapted steady-state conditions. In contrast to cytosolic HSPs, however, HSPs mainly related to mitochondrial activities, such as HSP10 and HSP60, showed significant up-regulation at the transcriptional level under stressful conditions (Böettinger et al., 2015; Chong et al., 2015), indicating a requirement for proper mitochondrial functions under stress conditions. Transcriptome data comparison with previously published stress experiments using batch cultures with nutrient excess showed significant differences, as ESR genes were largely unchanged under stress conditions applied in chemostats (Supplemental Figure S7). One can argue that differences are due to the different reference conditions and that cells grown in a chemostat culture with carbon limitation are already experiencing stressful conditions. With the present experimental setup, we are not able to reject this hypothesis. We can claim, however, that typical stress-related physiological changes observed in our experiments (decrease in biomass yield, initiation of glycerol or ethanol production, changes in the membrane composition, etc.) are not fully covered by the previously described ESR genes but instead rely on other mechanisms, which are also addressed in this study.
Although the general stress response showed consequential effects in increased maintenance, decreased biomass yield, and inhibited respiration, different stress types contributed differently to the effects (Figure 5). Increased membrane permeability has been reported as the main stress response in the case of high ethanol concentrations (Cartwright et al., 1987; Rosa and Sá-Correia, 1996). In accordance with the literature, we detected significant changes in the membrane composition, both in fatty acid composition and total ergosterol content. However, in contrast to the changes in reported unsaturated fatty acids, our data demonstrated that under adapted environmental conditions, the levels of C18 saturated and unsaturated fatty acids increased, whereas C16 fatty acid content decreased with the increase of ethanol concentration in the medium. In bacteria, the presence of ethanol increases errors in translation (Haft et al., 2014). Although we lack this information for yeast, we observed decreased gene expression profiles for several amino acid biosynthesis pathways, which could be caused by inefficient translation and therefore higher intracellular amino acid pools, which feed back to inhibit biosynthesis. Regarding osmotic stress, the major physiological response is related to glycerol excretion, in which glycerol is known to serve as an osmolyte of proliferating yeast cells (Hohmann, 2002). Although we failed to find connections between osmotic and oxidative stress in the literature, our analysis demonstrated that many oxidoreductase genes that were up-regulated under osmotic stress belong to the oxidative stress category (p < e-7). Oxidative stress hinders the activity of proteins and therefore contributes to increased maintenance. Increased temperature has a direct effect on protein folding, and we found indirect evidence of increased protein turnover in our experiments. Faster protein turnover under high- temperature conditions was also reported in early studies of yeast stress response (McAlister and Finkelstein, 1980; Peter and Loomis, 1982). Increased protein turnover also increases the maintenance costs of the cell (Figure 5). We observed that increased flux rates have a similar effect on cells as reflected by increased specific growth rate at optimal conditions, and a similar tradeoff between production of translational and respirational proteins can be observed as the synthesis of total protein is limited. With a limitation in translational capacity, respiration is hindered, which in turn accelerates increases in fermentative capacity and is less demanding in terms of the production of the total proteome but also less energy efficient (Nilsson and Nielsen, 2016). In addition, respirofermentative metabolism in yeast is accompanied by the production of ethanol, which affects the permeability of membranes at high concentrations.
FIGURE 5:

Overview of key differences among the three studied stress conditions. High ethanol concentrations affect the cell membrane and increase maintenance due to impaired membrane permeability. Osmotic stress causes oxidative stress, which results in increased maintenance. High temperature increases protein turnover, which increases maintenance. High maintenance energy causes an increase in specific flux rates and an increased demand for ATP, which eventually results in the onset of fermentation and a reduction in respiration, which is consistent with what occurs at high specific growth rates. Upward- and downward-facing arrows represent up- and down-regulation, respectively.
A confounding metabolic response for all three stress conditions is increased energy expenditure for maintenance, which results in decreased biomass yield (Figure 5). This imposes a requirement for increased glucose uptake rate, which causes a similar effect on the cells as observed for increased specific growth rates under optimal environmental conditions. We therefore observe a similar tradeoff between the production of translational and respirational proteins for these two conditions (Figure 4). This results in replacement of respiratory with respirofermentative growth and subsequent production of ethanol (van Hoek et al., 1998; Vemuri et al., 2007), indicating more efficient allocation of the proteome (Molenaar et al., 2009; Basan et al., 2015; Schmidt et al., 2015; Nilsson and Nielsen, 2016). In conclusion, we showed how three different types of stress initiate different cellular adaptation mechanisms, which converge on altered mitochondrial metabolism. As cells seek to retain the global ATP balance, they allocate their proteome to address increased requirements for maintenance energy.
Materials and Methods
Yeast strain, cultivation conditions, and sampling
S. cerevisiae CEN.PK113-7D (MATa, MAL2-8c, SUC2) was used throughout the study. To screen maximal stress conditions possible to apply in chemostat experiments, micro titer-plate batch experiments were carried out for ethanol and osmotic stress (NaCl) experiments. For temperature experiments, turbidostat experiments with slowly changing temperature were carried out in triplicate (Supplemental Figure S13). Maximal concentrations that still supported growth at 0.1 h−1 were selected and divided roughly into three equal categories to represent the dynamic dose-dependent profile. The stress conditions were as follows: ethanol, 20, 40, and 60 g/l; osmolarity, 0.2, 0.4, and 0.6 M NaCl; temperature, 33, 36, and 38°C. Cultivations were carried out in 1-l bioreactors (DasGip GmbH, Jülich, Germany; working volume, 0.5 l) equipped with in situ pH, dO2, turbidity, and external O2 and CO2 sensors (BlueSens GmbH, Herten, Germany). The dO2 levels were kept at >30% throughout the experiments. Base medium used contained 10 g of glucose, 5 g of (NH4)2SO4, 3 g of KH2PO4, and 0.5 g of MgSO4 per liter, in addition to 1 ml of trace elements solution and 1 ml of vitamin solution. The trace element solution contained, per liter (pH = 4), EDTA (sodium salt), 15.0 g; ZnSO4⋅7H2O, 4.5 g; MnCl2⋅2H2O, 0.84 g; CoCl2⋅6H2O, 0.3 g; CuSO4⋅5H2O, 0.3 g; Na2MoO4⋅2H2O, 0.4 g; CaCl2⋅2H2O, 4.5 g; FeSO4⋅7H2O, 3.0 g; H3BO3, 1.0 g; and KI, 0.10 g. The vitamin solution contained, per liter (pH = 6.5), biotin, 0.05 g; p-amino benzoic acid, 0.2 g; nicotinic acid, 1 g; Ca pantothenate, 1 g; pyridoxine-HCl, 1 g; thiamine-HCl, 1 g; and myoinositol, 25 g. Batch phase on base medium was always started at reference conditions (pH 5.5, 600 rpm, 30°C, 1 vvm of air) from 1% of overnight culture inoculum. After the end of the ethanol phase in batch, the chemostat mode was started at stress conditions. Sampling was carried out in steady-state conditions after passage through at least six culture volumes of fresh medium. For extracellular metabolome measurements, culture broth was withdrawn from bioreactors and filtered into vials that were stored at −20°C. For all other samples, biomass was withdrawn from bioreactors into precooled Eppendorf tubes and centrifuged for 22 s at 4°C and 14,000 rpm, and biomass was snap-frozen after decanting of the supernatant. The whole sampling process took <45 s. Samples were stored at −80°C. For biomass concentration measurements, ∼40 ml of culture broth was collected from the outflow into Falcon tubes placed in ice and measured gravimetrically using preweighed filter plates.
Extracellular metabolome, total fatty acid, and ergosterol measurements
Levels of glucose, acetate, glycerol, and ethanol in the culture medium were measured with liquid chromatography (ultimate 3000 HPLC system; Thermo Fisher Scientific, Waltham, MA), using a Bio-Rad (Hercules, CA) HPX-87H column with isocratic elution of 5 mM H2SO4 at a flow rate of 0.6 ml/min and at 45°C. A refractive index detector was used for detection and quantification of substances. Because evaporation of ethanol from the environment was detected during the cultivation, blank (not inoculated) cultivation experiments at similar environmental conditions were run to detect average ethanol evaporation rate and taken into account in the final reported concentrations for ethanol. Total fatty acid composition was measured according to Khoomrung et al. (2012), which is a modification of a method described in Abdulkadir and Tsuchiya (2008). Briefly, the biomass sample (∼10 mg) was mixed with 4 ml of hexane, 2 ml of 14% BF3 in MeOH, and 25 μl of internal standard (17:0; 4000 μg/ml). The sample was flushed into the tube with nitrogen gas for 30 s, and the tube was closed tightly. The tube was heated using a microwave digestion system equipped with a PRO-24 medium-pressure high-throughput rotor (Milestone Start D; Sorisole, Bergamo, Italy). After cooling of the sample to room temperature, 2 ml of Milli-Q water was added and shaken vigorously and centrifuged. The upper phase (hexane phase, which contained the fatty acid methyl esters [FAMEs]) was analyzed by gas chromatography (GC)–mass spectrometry (MS). The FAMEs were separated and quantified using a Focus GC ISQ single-quadrupole GC mass spectrometer (Thermo Fisher Scientific). The separation of FAMEs was performed on a Zebron (ZB-WAX; Phenomenex, Torrance, CA) GC column (30 m × 0.25 mm inside diameter, 0.25-μm film thickness). The sample was injected in splitless injection mode (1 μl at 240°C), and helium was used a carrier gas (1 ml/min). The identification of unknown FAMEs from yeast cells was achieved by comparing their retention times and mass spectrum profiles with known standards. The quantification of FAMEs was performed based on the five-point external calibration curve in the range of 0.1–25 μg/ml.
Ergosterol was measured as described in Khoomrung et al. (2013). Briefly, dry biomass was extracted in anaerobic conditions in CHCl3:MeOH (2:1) solution and microwaved for 10 min at 60°C and 1000 W. After addition of 0.73% NaCl solution, the organic layer was transferred to a new tube, concentrated, and analyzed using high-performance liquid chromatography (ultimate 3000 HPLC system; Dionex) equipped with a charged aerosol detector (Corona; ESA, Chelmsford, MA).
RNA sequencing
RNA from the biomass samples was extracted and purified using Qiagen RNeasy Mini Kit extraction and DNA degradation according to the user’s manual (Qiagen, Hilden, Germany). The integrity of the product was verified using a 2100 Bioanalyzer according to the user’s manual (Agilent Technologies, Santa Clara, CA). RNA concentration was determined by a NanoDrop 2000 (Thermo Scientific, Wilmington, DE).
The Illumina TruSeq sample preparation kit, version 2 (Illumina, San Diego, CA), with poly-A selection, was used to prepare RNA samples for sequencing. Fragments were clustered on cBot and sequenced on two lanes on an Illumina HiSeq 2500 with paired ends (2× 100 base pairs), according to the manufacturer’s instructions.
The short reads were mapped to the CEN.PK 113-7D reference genome (cenpk.tudelft.nl) using TopHat version 2.0.10 (Kim et al., 2013). Each sample had between 8.3 and 16.2 million mappable reads, with an average map rate of 88%. Read counts were determined using the featureCounts software from the subread package, version 1.4.0-p1 (Liao et al., 2014). The number of expected fragments per kilobase of transcript per million fragments mapped was calculated using Cufflinks version 2.1.1 (Trapnell et al., 2010).
Read counts were used in the differential expression analysis, with the software DESeq (Anders and Huber, 2010). The p values were adjusted for multiple testing using the Benjamini–Hochberg procedure (Benjamini and Hochberg, 1994) as implemented in DESeq. All conditions were compared with the reference samples. Raw data from the experiments were deposited in ArrayExpress and assigned the identifier E-MTAB-4044.
Metabolic modeling and integrative data analysis
Transcriptome data were used to create condition and biological replicate–specific near genome–scale metabolic models of yeast based on the model iTO977 (Osterlund et al., 2013). The tINIT algorithm was used with the 30% cut-off, expecting that low-abundance mRNAs are not translated into active proteins (Agren et al., 2014). Models were tested to be able to produce biomass and constrained with the measured exchange fluxes. All models were deposited in BioModels (Chelliah et al., 2015) and assigned the identifiers from MODEL1511100000 to MODEL1511100030. Intracellular flux distribution was calculated using FBA and the RAVEN toolbox with Mosek solver on MATLAB R2012b (Agren et al., 2013), where ATP drain was used as an optimization function. FBA was used separately for every condition and biological replica, and SDs for flux values were calculated based on biological replicates. Additional data analysis included hierarchical clustering (R), gene set analysis (Platform for Integrative Analysis of Omics data [PIANO]; Väremo et al., 2013), enrichment analysis (Reimand et al., 2011), and TF analysis, with TF-gene relationships acquired from Yeastrack (on 12.02.2015; PIANO).
Supplementary Material
Acknowledgments
We acknowledge Francesco Gatto for creating condition-dependent genome scale models. This investigation was financially supported by the Knut and Alice Wallenberg Foundation and the Novo Nordisk Foundation.
Abbreviations used:
- BCAA
branched chain amino acids
- ESR
environmental stress response
- ETC
electron transport chain
- FAME
fatty acid methyl esters
- FBA
flux balance analysis
- GO
gene ontology
- GSA
gene set analysis
- GSH
glutathione
- HPLC
high-performance liquid chromatography
- HSP
heat shock protein
- PC
principal component
- RNAseq
RNA sequencing
- RQ
respiration quotient
- TCA
tricarboxylic acid cycle
- TF
transcription factor.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-03-0187) on June 15, 2016.
REFERENCES
- Abdulkadir S, Tsuchiya M. One-step method for quantitative and qualitative analysis of fatty acids in marine animal samples. J Exp Mar Bio Ecol. 2008;354:1–8. [Google Scholar]
- Agren R, Liu L, Shoaie S, Vongsangnak W, Nookaew I, Nielsen J. The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput Biol. 2013;9:e1002980. doi: 10.1371/journal.pcbi.1002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agren R, Mardinoglu A, Asplund A, Kampf C, Uhlen M, Nielsen J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol Syst Biol. 2014;10:721. doi: 10.1002/msb.145122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auesukaree C, Damnernsawad A, Kruatrachue M, Pokethitiyook P, Boonchird C, Kaneko Y. Genome-wide identification of genes involved in tolerance to various environmental stresses in Saccharomyces cerevisiae. J Appl Genet. 2009;50:301–310. doi: 10.1007/BF03195688. [DOI] [PubMed] [Google Scholar]
- Basan M, Hui S, Okano H, Zhang Z, Shen Y, Williamson JR, Hwa T. Overflow metabolism in bacteria Escherichia coli results from efficient proteome allocation. Nature. 2015;528:99–104. doi: 10.1038/nature15765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaven MJ, Charpentier C, Rose AH. Production and tolerance of ethanol in relation to phospholipid fatty-acyl composition in Saccharomyces cerevisiae NCYC 431. Microbiology. 1982;128:1447–1455. [Google Scholar]
- Belloch C, Orlic S, Barrio E, Querol A. Fermentative stress adaptation of hybrids within the Saccharomyces sensu stricto complex. Int J Food Microbiol. 2008;122:188–195. doi: 10.1016/j.ijfoodmicro.2007.11.083. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1994;57:289–300. [Google Scholar]
- Boender LGM, van Maris AJA, de Hulster EaF, Almering MJH, van der Klei IJ, Veenhuis M, de Winde JH, Pronk JT, Daran-Lapujade P. Cellular responses of Saccharomyces cerevisiae at near-zero growth rates: transcriptome analysis of anaerobic retentostat cultures. FEMS Yeast Res. 2011;11:603–620. doi: 10.1111/j.1567-1364.2011.00750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böettinger L, Oeljeklaus S, Guiard B, Rospert S, Warscheid B, Becker T. The mitochondrial heat shock protein 70 (Hsp70) and Hsp10 cooperate in the formation of Hsp60 complexes. J Biol Chem. 2015;290:11611–11622. doi: 10.1074/jbc.M115.642017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordel S, Agren R, Nielsen J. Sampling the solution space in genome-scale metabolic networks reveals transcriptional regulation in key enzymes. PLoS Comput Biol. 2010;6:e1000859. doi: 10.1371/journal.pcbi.1000859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, Botstein D. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol Biol Cell. 2008;19:352–367. doi: 10.1091/mbc.E07-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cakar ZP, Seker UOS, Tamerler C, Sonderegger M, Sauer U. Evolutionary engineering of multiple-stress resistant Saccharomyces cerevisiae. FEMS Yeast Res. 2005;5:569–578. doi: 10.1016/j.femsyr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Canelas AB, Harrison N, Fazio A, Zhang J, Pitkänen JP, van den Brink J, Bakker BM, Bogner L, Bouwman J, Castrillo JI, et al. Integrated multilaboratory systems biology reveals differences in protein metabolism between two reference yeast strains. Nat Commun. 2010;1:145. doi: 10.1038/ncomms1150. [DOI] [PubMed] [Google Scholar]
- Cartwright CP, Veazey FJ, Rose AH. Effect of ethanol on activity of the plasma-membrane ATPase in, and accumulation of glycine by, Saccharomyces cerevisiae. J Gen Microbiol. 1987;133:857–865. doi: 10.1099/00221287-133-4-857. [DOI] [PubMed] [Google Scholar]
- Caspeta L, Chen Y, Ghiaci P, Feizi A, Buskov S, Hallstrom BM, Petranovic D, Nielsen J. Altered sterol composition renders yeast thermotolerant. Science. 2014;346:75–78. doi: 10.1126/science.1258137. [DOI] [PubMed] [Google Scholar]
- Caspeta L, Nielsen J. Economic and environmental impacts of microbial biodiesel. Nat Biotechnol. 2013;31:789–793. doi: 10.1038/nbt.2683. [DOI] [PubMed] [Google Scholar]
- Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA. Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell. 2001;12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelliah V, Juty N, Ajmera I, Ali R, Dumousseau M, Glont M, Hucka M, Jalowicki G, Keating S, Knight-Schrijver V, et al. BioModels: ten-year anniversary. Nucleic Acids Res. 2015;43:D542–D548. doi: 10.1093/nar/gku1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong YT, Koh JLY, Friesen H, Duffy K, Cox MJ, Moses A, Moffat J, Boone C, Andrews BJ. Yeast proteome dynamics from single cell imaging and automated analysis. Cell. 2015;161:1413–1424. doi: 10.1016/j.cell.2015.04.051. [DOI] [PubMed] [Google Scholar]
- Daran-Lapujade P, Daran JM, Luttik MAH, Almering MJH, Pronk JT, Kötter P. An atypical PMR2 locus is responsible for hypersensitivity to sodium and lithium cations in the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D. FEMS Yeast Res. 2009;9:789–792. doi: 10.1111/j.1567-1364.2009.00530.x. [DOI] [PubMed] [Google Scholar]
- Deng R, Hua X, Li J, Chi W, Zhang Z, Lu F, Zhang L, Pflugfelder SC, Li DQ. Oxidative stress markers induced by hyperosmolarity in primary human corneal epithelial cells. PLoS One. 2015;10:1–16. doi: 10.1371/journal.pone.0126561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar R, Sägesser R, Weikert C, Yuan J, Wagner a. Adaptation of Saccharomyces cerevisiae to saline stress through laboratory evolution. J Evol Biol. 2011;24:1135–1153. doi: 10.1111/j.1420-9101.2011.02249.x. [DOI] [PubMed] [Google Scholar]
- Ding MZ, Zhou X, Yuan YJ. Metabolome profiling reveals adaptive evolution of Saccharomyces cerevisiae during repeated vacuum fermentations. Metabolomics. 2010;6:42–55. [Google Scholar]
- Dong SJ, Yi CF, Li H. Changes of Saccharomyces cerevisiae cell membrane components and promotion to ethanol tolerance during the bioethanol fermentation. Int J Biochem Cell Biol. 2015;69:196–203. doi: 10.1016/j.biocel.2015.10.025. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerosa L, Kochanowski K, Heinemann M, Sauer U. Dissecting specific and global transcriptional regulation of bacterial gene expression. Mol Syst Biol. 2013;9:658. doi: 10.1038/msb.2013.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney PA, Lu C, Caudy AA, Hess DC, Botstein D. Yeast metabolic and signaling genes are required for heat-shock survival and have little overlap with the heat-induced genes. Proc Natl Acad Sci USA. 2013;110:E4393–E4402. doi: 10.1073/pnas.1318100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf A, Dragosits M, Gasser B, Mattanovich D. Yeast systems biotechnology for the production of heterologous proteins. FEMS Yeast Res. 2009;9:335–348. doi: 10.1111/j.1567-1364.2009.00507.x. [DOI] [PubMed] [Google Scholar]
- Haft RJF, Keating DH, Schwaegler T, Schwalbach MS, Vinokur J, Tremaine M, Peters JM, Kotlajich MV, Pohlmann EL, Ong IM, et al. Correcting direct effects of ethanol on translation and transcription machinery confers ethanol tolerance in bacteria. Proc Natl Acad Sci USA. 2014;111:E2576–E2585. doi: 10.1073/pnas.1401853111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann S. Osmotic stress signaling and osmoadaptation in yeasts. Microbiol Mol Biol Rev. 2002;66:300–372. doi: 10.1128/MMBR.66.2.300-372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullesson D, David F, Pfleger B, Nielsen J. Impact of synthetic biology and metabolic engineering on industrial production of fine chemicals. Biotechnol Adv. 2015;33:1395–1402. doi: 10.1016/j.biotechadv.2015.02.011. [DOI] [PubMed] [Google Scholar]
- Kanshin E, Kubiniok P, Thattikota Y, Amours DD, Thibault P. Phosphoproteome dynamics of Saccharomyces cerevisiae under heat shock and cold stress. Mol Syst Biol. 2015;11:813. doi: 10.15252/msb.20156170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoomrung S, Chumnanpuen P, Jansa-Ard S, Nookaew I, Nielsen J. Fast and accurate preparation fatty acid methyl esters by microwave-assisted derivatization in the yeast Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2012;94:1637–1646. doi: 10.1007/s00253-012-4125-x. [DOI] [PubMed] [Google Scholar]
- Khoomrung S, Chumnanpuen P, Jansa-Ard S, Ståhlman M, Nookaew I, Borén J, Nielsen J. Rapid quantification of yeast lipid using microwave-assisted total lipid extraction and HPLC-CAD. Anal Chem. 2013;85:4912–4919. doi: 10.1021/ac3032405. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahtvee P-J, Seiman A, Arike L, Adamberg K, Vilu R. Protein turnover forms one of the highest maintenance costs in Lactococcus lactis. Microbiology. 2014;160:1501–1512. doi: 10.1099/mic.0.078089-0. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Ma M, Liu ZL. Mechanisms of ethanol tolerance in Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2010;87:829–845. doi: 10.1007/s00253-010-2594-3. [DOI] [PubMed] [Google Scholar]
- McAlister L, Finkelstein DB. Alterations in translatable ribonucleic acid after heat shock of Saccharomyces cerevisiae. J Bacteriol. 1980;143:603–612. doi: 10.1128/jb.143.2.603-612.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar D, van Berlo R, de Ridder D, Teusink B. Shifts in growth strategies reflect tradeoffs in cellular economics. Mol Syst Biol. 2009;5:323. doi: 10.1038/msb.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J. Yeast cell factories on the horizon. Science. 2015;349:1050–1051. doi: 10.1126/science.aad2081. [DOI] [PubMed] [Google Scholar]
- Nilsson A, Nielsen J. Metabolic trade-offs in yeast are caused by F1F0-ATP synthase. Sci Rep. 2016;6:22264. doi: 10.1038/srep22264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Duibhir E, Lijnzaad P, Benschop JJ, Lenstra TL, van Leenen D, Groot Koerkamp MJ, Margaritis T, Brok MO, Kemmeren P, Holstege FC. Cell cycle population effects in perturbation studies. Mol Syst Biol. 2014;10:732. doi: 10.15252/msb.20145172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olz R, Larsson K, Adler L, Gustafsson L. Energy flux and osmoregulation of Saccharomyces cerevisiae grown in chemostats under NaCl stress. J Bacteriol. 1993;175:2205–2213. doi: 10.1128/jb.175.8.2205-2213.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterlund T, Nookaew I, Bordel S, Nielsen J. Mapping condition-dependent regulation of metabolism in yeast through genome-scale modeling. BMC Syst Biol. 2013;7:36. doi: 10.1186/1752-0509-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter E, Loomis WF. Analysis of the heat shock response of Saccharomyces cerevisiae. J Bacteriol. 1982;151:311–327. doi: 10.1128/jb.151.1.311-327.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper PW, Talreja K, Panaretou B, Byrne K, Praekelt UM, Meacock P. Induction of major heat-shock proteins of Saccharomyces cerevisiae including plasma membrane HspSO, by ethanol levels above a critical threshold. Microbiology. 1994;140:3031–3038. doi: 10.1099/13500872-140-11-3031. [DOI] [PubMed] [Google Scholar]
- Postmus J, Aardema R, de Koning LJ, de Koster CG, Brul S, Smits GJ. Isoenzyme expression changes in response to high temperature determine the metabolic regulation of increased glycolytic flux in yeast. FEMS Yeast Res. 2012;12:571–581. doi: 10.1111/j.1567-1364.2012.00807.x. [DOI] [PubMed] [Google Scholar]
- Postmus J, Canelas AB, Bouwman J, Bakker BM, van Gulik W, de Mattos MJT, Brul S, Smits GJ. Quantitative analysis of the high temperature-induced glycolytic flux increase in Saccharomyces cerevisiae reveals dominant metabolic regulation. J Biol Chem. 2008;283:23524–23532. doi: 10.1074/jbc.M802908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postmus J, Tuzun I, Bekker M, Müller WH, de Mattos MJT, Brul S, Smits GJ. Dynamic regulation of mitochondrial respiratory chain efficiency in Saccharomyces cerevisiae. Microbiology. 2011;157:3500–3511. doi: 10.1099/mic.0.050039-0. [DOI] [PubMed] [Google Scholar]
- Regenberg B, Grotkjaer T, Winther O, Fausbøll A, Akesson M, Bro C, Hansen LK, Brunak S, Nielsen J. Growth-rate regulated genes have profound impact on interpretation of transcriptome profiling in Saccharomyces cerevisiae. Genome Biol. 2006;7:R107. doi: 10.1186/gb-2006-7-11-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, Arak T, Vilo J. G:Profiler—a web server for functional interpretation of gene lists. Nucleic Acids Res. 2011;39:307–315. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa MF, Sá-Correia I. Intracellular acidification does not account for inhibition of Saccharomyces cerevisiae growth in the presence of ethanol. FEMS Microbiol Lett. 1996;135:271–274. doi: 10.1111/j.1574-6968.1996.tb08000.x. [DOI] [PubMed] [Google Scholar]
- Sajbidor J, Ciesarová Z, Smogrovicová D. Influence of ethanol on the lipid content and fatty acid composition of Saccharomyces cerevisiae. Folia Microbiol (Praha) 1995;40:508–510. doi: 10.1007/BF02814733. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Kochanowski K, Vedelaar S, Ahrne E, Volkmer B, Callipo L, Knoops K, Bauer M, Aebersold R, Heinemann M. The quantitative and condition-dependent Escherichia coli proteome. Nat Biotechnol. 2015;34:104–110. doi: 10.1038/nbt.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanegas JM, Contreras MF, Faller R, Longo ML. Role of unsaturated lipid and ergosterol in ethanol tolerance of model yeast biomembranes. Biophys J. 2012;102:507–516. doi: 10.1016/j.bpj.2011.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoek P, van Dijken JP, Pronk JT. Effect of specific growth rate on fermentative capacity of baker’s yeast. Appl Environ Microbiol. 1998;64:4226–4233. doi: 10.1128/aem.64.11.4226-4233.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väremo L, Nielsen J, Nookaew I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013;41:4378–4391. doi: 10.1093/nar/gkt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri GN, Eiteman MA, McEwen JE, Olsson L, Nielsen J. Increasing NADH oxidation reduces overflow metabolism in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2007;104:2402–2407. doi: 10.1073/pnas.0607469104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang S, Liu H, Zhang L, Yi C, Li H. Changes and roles of membrane compositions in the adaptation of Saccharomyces cerevisiae to ethanol. J Basic Microbiol. 2015;55:1417–1426. doi: 10.1002/jobm.201500300. [DOI] [PubMed] [Google Scholar]
- Yang H, Tong J, Lee CW, Ha S, Eom SH, Im YJ. Structural mechanism of ergosterol regulation by fungal sterol transcription factor Upc2. Nat Commun. 2015;6:6129. doi: 10.1038/ncomms7129. [DOI] [PubMed] [Google Scholar]
- You KM, Rosenfield C, Douglas C, Knipple DC. Ethanol tolerance in the yeast Saccharomyces cerevisiae is dependent on cellular oleic acid content. Appl Environ Microbiol. 2003;69:1499–1503. doi: 10.1128/AEM.69.3.1499-1503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X-Q, Bai F-W. Mechanisms of yeast stress tolerance and its manipulation for efficient fuel ethanol production. J Biotechnol. 2009;144:29–30. doi: 10.1016/j.jbiotec.2009.05.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.