

Abstract
The proteasome is a ubiquitous and highly plastic multi-subunit protease with multi-catalytic activity that is conserved in all eukaryotes. The most widely known function of the proteasome is protein degradation through the 26S ubiquitin-proteasome system, responsible for the vast majority of protein degradation during homeostasis. However, the proteasome also plays an important role in adaptive immune responses and adaptation to oxidative stress. The unbound 20S proteasome, the core common to all proteasome conformations, is the main protease responsible for degrading oxidized proteins. During periods of acute stress, the 19S regulatory cap of the 26S proteasome disassociates from the proteolytic core, allowing for immediate ATP/ubiquitin-independent protein degradation by the 20S proteasome. Despite the abundance of unbound 20S proteasome compared to other proteasomal conformations, many publications fail to distinguish between the two proteolytic systems and often regard the 26S proteasome as the dominant protease. Further confounding the issue are the differential roles these two proteolytic systems have in adaptation and aging. In this review, we will summarize the increasing evidence that the 20S core proteasome constitutes the major conformation of the proteasome system and that it is far from a latent protease requiring activation by binding regulators.
Keywords: Proteolysis, Proteasome, Immunoproteasome, Oxidative Stress, Adaptation, Nrf2
1. Introduction
For any organism, cell function is fundamentally dependent on protein molecules, which provide structure, allow movement, carry out enzymatic reactions, and conduct cell signaling to manage biological pathways. The concentration of proteins within a cell at any given time is determined by the rate of protein synthesis (translation) minus the rate of protein degradation (proteolysis); a process usually described as protein turnover. The half-lives of proteins vary widely from minutes to days. These differential rates of protein degradation represent a critical aspect of cellular regulation. For instance, many transcription factors have fast proteolysis rates, which are necessary for the rapid modulation of gene expression (Kallio et al., 1999; Maniatis et al., 1987). In addition to regulating cell signaling, protein degradation is crucial to remove incorrectly synthesized nascent proteins and oxidatively, or otherwise, damaged proteins.
A major pathway for protein degradation in the cell is the proteasome system. Most enzymatic protein break-down was initially assumed to take place in the relatively harsh acidic environment of the lysosome (Lüllmann-Rauch, 2005). However, in the 1970s Goldberg et al. (Etlinger and Goldberg, 1977; Goldberg et al., 1980) demonstrated that cells lacking lysosomes were still capable of protein turn-over via an autonomous ATP-stimulated proteolytic pathway. Both a 20S and a 26S proteasome was discovered, named according to their Svedberg sedimentation coefficients (a reflection of their relative molecular mass), and were thought to represent two separate entities within the cell until it was demonstrated that the 26S proteasome consisted of two 19S regulators bound to a 20S core (Coux et al., 1996; Davies, 2001; Driscoll and Goldberg, 1990; Eytan et al., 1989; Hershko and Ciechanover, 1992).
Since its initial discovery, the proteasome has been under intense investigation in order to understand its mechanism of action, regulation, substrate preferences, and its potential contributions to, or roles in, disease. Degradation by the proteasome falls into two major categories: ubiquitin-dependent degradation by the 26S proteasome, and ubiquitin-independent proteolysis by the 20S core proteasome; although there is also evidence that the 26S proteasome can conduct limited ATP-independent proteolysis (Coux et al., 1996; Davies, 2001; Driscoll and Goldberg, 1990; Eytan et al., 1989; Hershko and Ciechanover, 1992). Despite the fact that the 20S core is the primary form and dominant configuration of the proteasomal proteolytic complex, the overwhelming interest in the ubiquitin-proteasomal system has led to an underlying suggestion in the literature that the 20S core is latent unless bound to two 19S regulators, so as to form an active 26S proteasome. In this review, we have highlighted research demonstrating that the 20S core is the stoichiometrically dominant form of the proteasome and the conformation responsible for degrading oxidized proteins. We have also evaluated roles for the 20S proteasome during stress-adaptation and aging, and outlined common methodological deficiencies in distinguishing between 20S and 26S proteasomal proteolytic activities.
2. The 20S core proteasome
The 20S proteasome is the core unit of all proteasomal systems and (in mammals) constitutes approximately 1% of total cellular protein. The 20S proteasome is most abundant throughout the cytoplasm and the nucleus (Baumeister et al., 1998). In addition, it has been found to be directly attached to various organelle membranes, particularly the endoplasmic reticulum, where it degrades membrane-bound substrates (Hirsch and Ploegh, 2000). The 20S proteasome is essentially a hollow, cylindrical, 700 kDa structure made up of four, stacked, heptameric rings (Fig. 1A). The two identical outermost rings (one on each end) are composed of seven unique α-subunits. The two identical innermost rings are composed of seven unique β-subunits. The two inner β-rings form a catalytic chamber that comprises the entire proteolytic activity of the proteasome (Groll et al., 1999). Specifically, the β1, β2, and β5 subunits, in the two juxtaposed β-rings, combine to form six proteolytic sites comprised of trypsin-like, chymotrypsin-like, and peptidyl-glutamyl peptide hydrolyzing sites that can be differentially inhibited by distinct pharmacological agents (Borissenko and Groll, 2007).
Fig. 1.

The 20S proteasome and immunoproteasome. (A) The 20S proteasome consists of four stacked rings: two rings composed of α-subunits and two rings composed of β-subunits. (B) The plasticity of the 20S proteasome allows for the synthesis of a subtly different form of the core 20S proteasome, called the immunoproteasome.
The two flanking outer α-rings allow for the association of the 20S proteasome with several regulatory complexes (Sharon et al., 2006). The α-rings also bind substrates and act as antechambers that control the translocation of substrates into the catalytic cavity (Sharon et al., 2006). When a substrate binds to either α-ring, it induces a conformational change, which remains even after the substrate is translocated. A potential outcome of the substrate binding and its subsequent progression into the proteolytic core may have the complimentary effect of releasing the previously degraded product (Sharon et al., 2006). X-ray crystallographic analysis of the three-dimensional structure of the 20S proteasome from the archaebacterium, Thermoplasma acidophilum, indicates that the orifice of the α-rings is bordered by disordered residues and has an opening with a ~13 Å diameter (Lowe et al., 1995). However, in the 20S proteasome found in Saccharomyces cerevisiae, the N-termini of the α-subunits project and fill the opening of the orifice with interdigitating side chains, thus limiting accessibility (Groll et al., 1997). Substrate entry into mammalian 20S proteasomes is also thought to be similarly limited by stearic hindrance. In order for protein degradation to occur, the orifice to the α-rings must be opened to allow entry into the proteolytic β-ring core. In addition to immediate protein degradation, in vitro studies using tandem mass spectrometry have demonstrated that the α-ring antechambers are capable of substrate storage prior to degradation in circumstances where translocation rates are slower than proteolytic capacity (Sharon et al., 2006). The orifice of the α-ring opens when the extensions lift out of the cylinder upon interacting with hydrophobic residues. For instance, association with regulatory complexes allows the α-ring extensions to rise, resulting in the opening of the orifice. Similarly, binding to hydrophobic substrates (such as oxidatively damaged proteins) appears to also open the α-ring channel, and increase the rate of degradation of subsequent substrates (Coux et al., 1996; Davies, 2001; Driscoll and Goldberg, 1990; Eytan et al., 1989; Hershko and Ciechanover, 1992). For this reason, proteasomes are highly plastic and can adopt several different molecular forms that coexist within a cell. However, the most prevalent conformation of the proteasome is the free 20S core unit. While the 20S proteasome has largely been considered a dormant protease that is only activated upon binding to regulatory complexes, significant and increasing evidence has clearly demonstrated that the 20S core has a major a role in overall protein degradation, independent of ubiquitin and ATP.
3. Alternative subunits and binding regulators of the 20S proteasome
The simplest evidence of proteasome plasticity is the formation of the immunoproteasome (Fig. 1B). Proteasome assembly is an irreversible process in which two ~300 kDa 15S preproteasome complexes, composed of a complete α-ring in association with certain unprocessed β-subunits, will come together at the β-ring interface (Groll et al., 1997). One pathway of 15S preproteasome assembly includes the incorporation of presequence β1, β2, or β6 subunits (Frentzel et al., 1994). An adaptive immune response stimulated by γ-interferon induces an alternative pathway for preproteasome assembly, favoring the IFN-gamma-inducible proteasome form, which requires the upregulation of three alternative catalytic subunits (β1i, β2i, and β5i) and their incorporation into preproteasome complexes (Griffin et al., 1998). Interestingly, assembly of immunoproteasomes is cooperative and preproteasomes with incorporated β1i or β2i will not mature until β5i has been integrated (Frentzel et al., 1994; Griffin et al., 1998; Yang et al., 1995). The immunoproteasome has altered proteolytic affinity that enables the production of peptides for antigen presentation. Specifically, immunoproteasomes are able to generate peptides with a hydrophobic C terminus that fit into the groove of MHC class I molecules (Kloetzel and Ossendorp, 2004; Teoh and Davies, 2004). When antigenic peptides are displayed on the cell surface, the adaptive immune response is triggered, allowing CD8 T cells to reject pathogen-infected cells (Chen et al., 2001). New studies have also indicated that the immunoproteasome is involved in the pathogenesis of inflammatory diseases (Kimura et al., 2015), in addition to having non-immune functions such as regulating skeletal muscle differentiation (Cui et al., 2014). Our laboratory has demonstrated that the immunoproteasome (±PA28/11S) is also responsible for degrading oxidatively damaged proteins, and that synthesis of the immunoproteasome is strongly induced during adaptation to oxidative stress (Pickering and Davies, 2012b). The highly inducible nature of the immunoproteasome, and the fact that it is clearly involved in functions other than MHC class I antigen processing, may suggest that it would be better termed the ‘Inducible-Proteasome’, but we leave it to others (and the test of time) to determine if such a nomenclature change would be appropriate.
The proteolytic activity of the 20S proteasome can also be modulated by the reversible binding of various regulatory complexes. Three regulators have so far been identified to bind to the 20S proteasome: PA200, 11S/PA28, and 19S (Fig. 2). It is interesting to note that regulators may bind to the 20S core asymmetrically (i.e., different regulators at each end), resulting in hybrid proteasomes. Alternatively, only one regulator may bind to the 20S core, leaving the other end unbound. Frequently, when reference is made to the proteasome in publications, authors do not differentiate between the 20S or the 20S bound to regulatory proteins.
Fig. 2.
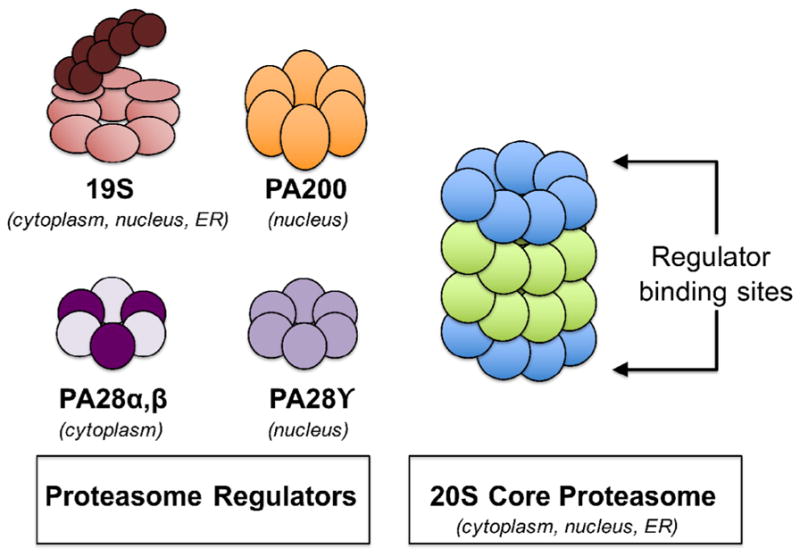
The binding regulators of the 20S proteasome. The 20S core proteasome and the immunoproteasome are able to bind to regulator proteins which alter their proteolytic capacities and/or peptide product profiles. Quantitatively, however, the major configuration of proteasomes found within mammalian cells is the unbound 20S core proteasome.
The PA200 regulator is a 200 kDa nuclear polypeptide and is the least investigated of the three 20S activators. PA200 enhances 20S hydrolysis of peptide substrates, but actually decreases the activity of the 20S proteasome against the intact proteins so far studied (Pickering and Davies, 2012b). PA200 activates the peptidase activity of the 20S proteasome by binding to one or both of the outer α-rings and increasing the opening into the proteolytic chamber (Ortega et al., 2005). In addition, PA200 was shown to interact with the 26S proteasome, presumably to form a 19S-20S-PA200 hybrid proteasome (Ortega et al., 2005). PA200 is a nuclear phosphoprotein and forms intranuclear foci following γ-irradiation, indicating that it may be involved in DNA repair (Ustrell et al., 2002). This is particularly interesting because poly-ADP ribose polymerase has been shown to polyadenylate and activate nuclear proteasomes to degrade oxidatively damaged histones as an early step in the DNA repair process (Ullrich et al., 1999).
Another regulator is alternately known as 11S or PA28; both names are widely used but we will use the PA28 terminology in this review. PA28 regulators exist in two major forms, PA28α,β and PA28γ. PA28α,β is composed of two 28 kDa α- and β-subunits and is expressed in the cytoplasm. PA28α,β is induced by γ-interferon and enhances the ability of the 20S to degrade short peptide substrates (Dubiel et al., 1992; Ma et al., 1992) as well as oxidized substrates (Pickering and Davies, 2012b). Binding of the PA28α,β regulator, however, significantly increases both the selectivity for, and the proteolytic activity against, oxidatively modified protein substrates (Pickering and Davies, 2012b). As well as being able to bind to both ends of the 20S proteasome, PA28 can bind to just one end while allowing the 19S regulator to bind to the other end, to form the asymmetric PA28α,β-20S-19S hybrid proteasome (Cascio et al., 2002; Hendil et al., 1998; Kopp et al., 2001; Tanahashi et al., 2000). This PA28α,β-20S-19S hybrid proteasome is able to hydrolyze tri- and tetra-peptides more efficiently than the 26S proteasome (Cascio et al., 2002; Kopp et al., 2001). Several lines of evidence indicate that PA28α,β plays a role in MHC class I antigen presentation, but the exact molecular mechanism by which this occurs is not yet understood (Rechsteiner et al., 2000). PA28γ is expressed in the nucleus and has been shown to promote MDM2-mediated p53 ubiquitinylation, thus facilitating protein turnover of the tumor suppressor p53 (Zhang and Zhang, 2008). Not surprisingly, PA28γ has been shown to be dysregulated in multiple cancers (Okamura et al., 2003; Roessler et al., 2006).
The third, but most well-known, binding activator of the 20S core proteasome is the 19S regulator. Binding of 19S regulators to both ends of the 20S core forms the symmetrical 26S proteasome which is responsible for ATP-ubiquitin-dependent degradation of proteins. The 19S regulator is composed of 19 different subunits, six of which contain AAA ATPase activity (Rpt 1–6) and thirteen which do not contribute to ATPase activity (Rpn 1–3 and Rpn 5–13) (Lasker et al., 2012). The cytosolic facing portion of the 19S regulatory region forms a symmetric ring comprised of three dimmers, known as the N-ring. The six-fold ring formed from the AAA ATPase domain is adjacent to the α-ring of the 20S core. The Rpn6 and Rpn13 subunits recognize and bind to ubiquitin-tagged proteins, while Rpn11 removes ubiquitin from the tagged protein substrate in the presence of ATP (Lasker et al., 2012). Thus the N-ring binds, unfolds, and detags ubiquitinylated substrates and feeds them into the ATPase domain. Subsequently, ATP hydrolysis is used to actively transport the substrate into the proteolytic 20S core. In addition to recognizing ubiquitinylated substrates and shuttling them into the 20S, the ATPase ring also displays chaperone-like activity and uses ATP hydrolysis for the final unfolding of substrates during their translocation into the 20S proteolytic chamber (Braun et al., 1999; Glickman and Raveh, 2005; Rechsteiner and Hill, 2005).
4. The kinetics of 20S proteasome catalytic activity
The rate of substrate degradation by the 20S proteasome is dependent on the orifice of the outer ring of the proteasome and the length of the peptide substrate. The α-rings act as gates that determine the rate at which substrates can enter or exit the proteolytic core, as well as the mean length of the peptide products (Groll et al., 2000; Köhler et al., 2001). Opening of the α-rings allows substrates to enter, whereas the duration of gate constriction promotes substrate degradation into peptides ranging in length from 3 to 24 amino acids (Kisselev et al., 1999). Size exclusion chromatography has uncovered three main subpopulations of peptides that are generated after proteolysis: 2–3 amino acid peptides, 8–10 amino acid peptides, and 20–30 amino acid peptides (Köhler et al., 2001). In certain instances, short periods of α-ring closure can result in an average increase in length of the peptides generated (Kisselev et al., 1999; Köhler et al., 2001). This was demonstrated by studying proteasome mutants incapable of constricting the α-rings (Groll et al., 2000). These mutants were able to degrade substrates faster, but the average length of the peptide products generated increased by approximately 20% (Groll et al., 2000; Köhler et al., 2001). Rapid opening of the α-ring has been implicated during the immune response. By reducing the degradation rate of foreign material, such as viruses or bacteria, the proteasome may expedite the ability to signal a microbial attack, as well as ensure that large fragments of foreign material are incorporated for antigen presentation (Kisselev et al., 1999).
To better understand the rate of proteolysis by the 20S core, a novel mathematical approach was generated that determines degradation rates based entirely upon the amino acid length of the protein (Luciani et al., 2005). Previously, mathematical modeling of the rate of 20S proteasome proteolysis was based on specific proteins with known cleavage sites (Holzhütter and Kloetzel, 2000; Peters et al., 2002). To create a generalized mathematical model for the rate of degradation by the proteolytic core, two key assumptions were incorporated: 1) preferential cleavage occurs after nine amino acids from the terminal end of the substrate (Groll and Huber, 2003), and 2) the rate at which degraded fragments exit the proteasome is inversely proportional to the substrate length (Luciani et al., 2005). This model follows Michaelis–Menten kinetics in that, at least initially, there is a rapid spike in proteolytic activity as the proteasome fills and begins to break down substrates, regardless of substrate length and the influx of substrate. The rate of degradation continually increases as substrate concentration increases, until it reaches a plateau of maximum capacity at high substrate concentrations. When both the cleavage and export rates are similar, the three subpopulations of amino acid fragments are generated, with the 8–10 amino acid peptides being the most favored due to preferred cleavage occurring every 9 amino acids (Köhler et al., 2001). However, increased length of the substrate coupled with high substrate concentration results in a reduction of the overall maximum degradation velocity that can be achieved by the proteolytic core (Luciani et al., 2005). The small peptides that exit the proteasome are subsequently degraded by the plethora of other peptidases and proteases found in the cytosol, and it is generally held that intracellular peptidase activity is never rate limiting to overall rates of protein turnover (Reits et al., 2004). However, when substrates are more than 80% degraded, some re-entry of small peptides (i.e., 2–4 amino acids long) may occur, leading to an overall reduction in the rate of proteolytic activity (Köhler et al., 2001; Luciani et al., 2005). Ultimately, the rate-limiting component for proteolytic capacity may be the ratio of substrate to peptide products. As more substrate is degraded and the peptide product pool increases, the likelihood of peptide re-entry into the proteasome also increases, resulting in a decline in the rate of proteolysis.
5. Types of proteolysis by the proteasomal system
5.1. ATP/ubiquitin-dependent proteolysis
Degradation of proteins by the proteasomal system is either ubiquitin-dependent or ubiquitin-independent. Ubiquitin is an 8.5 kDa protein that has seven lysine residues (e.g., K6, K11, K27, K29, K33, K48, and K63) and an N-terminus that serves as points of ubiquitinylation. The best characterized chains are K48, which is involved in ubiquitin-dependent proteolysis, and K63, which is involved in other processes like translation and DNA repair, while other chains, including mixed and branched chains, are less understood (Komander, 2009). The majority of substrates destined for degradation by the 26S proteasome pathway are shortlived regulatory proteins with an imbedded intrinsic signal for degradation called a degron (Shang and Taylor, 2011). Recently, it has been shown that HSP90 and HSP70 play a decisive role in whether a protein can be salvaged, or is subsequently shuttled for ubiquitinylation and degradation, by distinguishing between properly folded proteins and those requiring either repair or destruction.
Ubiquitin-dependent proteolysis begins with the initiation of an enzymatic cascade in which ubiquitin (Ub) is activated by the ubiquitin-activating enzyme (E1) through the formation of a thioester in an ATP-dependent process. Activated Ub is then transferred to a ubiquitin-conjugating enzyme (E2), also through the formation of a thioester bond. While some E2s are capable of transferring Ub directly to the respective protein substrate, most require a ubiquitin ligase enzyme (E3) (Ciechanover, 1994; Glickman and Ciechanover, 2002). Thus, through processive addition of ubiquitin monomers, a polyubiquitin chain is formed. This polyubiquitin chain is then bound to the substrate protein at epsilon amino groups of lysine residues (Finley et al., 1994). Many polyubiquitin-tagged proteins are targeted to the 19S regulator of 26S proteasomes, where they are deubiquitinylated and unfolded in ATP-requiring steps, and then degraded within the 20S core of the 26S complex.
5.2. Ubiquitin-targeting to lysosomes and ubiquitin-mediated signaling
It should be noted that ubiquitin tagging can also be involved in autophagy, where it can signal entrance into the lysosomal degradation pathway (Chen and Mallampalli, 2009). Furthermore, ubiquitinylated proteins are not always degraded, and the ubiquitin ‘tag’ can also have nonproteolytic functions, such as determining subcellular localization. For instance, while monoubiquitinylation is a precursor to polyubiquitinylation, it is also an important post-translational modification that has been shown to regulate transcription, histone modification, and DNA repair (Friedberg, 2006; Pemberton and Paschal, 2005). In addition, the process of ubiquitinylation can be reversed by deubiquitinase enzymes (DUB). In mammals, there are approximately 79 deubiquitinylases, which are capable of opposing E3 ligase activity (Nijman et al., 2005). Traditionally, an increase in the ubiquitin levels measured in total cell lysates has been used as an indicator of declining proteasome function. Given that ubiquitinylation plays roles in cell signaling independent of proteasomal degradation, assessing changes in ubiquitin levels may be a deceptive indicator of proteasome activity.
5.3. ATP/ubiquitin-independent proteolysis by the proteasome
The primary substrates for ATP/ubiquitin-independent protein degradation are unfolded proteins. Several types of denaturing stressors are known to lead to protein unfolding including heat, cold, heavy metals, multiple toxins, hydrophobic agents, and various forms of oxidative stress. Specifically, oxidation leads to numerous chemical modifications to proteins that result in conformational changes and the subsequent exposure of hydrophobic residues (Carrard et al., 2002). These hydrophobic patches on the surface of a substrate protein are recognized by the unbound 20S proteasome and can promote the opening of the α-ring (Kisselev et al., 2002). In vitro studies have indicated that increased hydrophobicity facilitates recognition by the proteasome resulting in accelerated protein degradation (Giulivi et al., 1994; Pacifici et al., 1993). This method for 20S proteasome substrate recognition, and subsequent channeling into the 20S core, may take the place of the unfolding chaperone-like activity conducted by the 19S regulator during ATP/ubiquitin-dependent proteolysis by the 26S proteasome (Braun et al., 1999).
6. Distinguishing between 20S and 26S proteolysis
Assessment of proteolysis has traditionally relied on in vitro assays that measure the degradation of a fluorogenic or radioactively labeled peptide or protein substrate (Grune et al., 1996; Pacifici et al., 1988; Reinheckel et al., 2000a; Shringarpure et al., 2003). A common semi-quantitative technique that can distinguish between the 20S and 26S is to perform a proteasome activity gel by running cell lysate on a native SDS-PAGE gel and probing with Suc-LLVY-AMC, a small peptide substrate covalently linked to a 7-amino-4-methylcoumarin fluorophore that reflects the chymotrypsin-like activity of proteasomes. Liberated AMC molecules fluoresce upon exposure to UV light, and the 20S and 26S proteasomes are identified by the different molecular weight bands on the gel. Other fluorogenic peptide substrates are also available to test the trypsin-like and peptidyl-glutamyl-peptide hydrolyzing activities of the proteasome.
Furthermore, protein degradation can be measured by incubating intact cells with a mixture of 35S-cysteine/methionine to allow incorporation of the radiolabel into newly synthesized proteins, in a pulse-chase technique (Davies, 2001). By varying the length of incubation, different protein pools can be preferentially labeled (Grune et al., 1995). Incorporation and reutilization of the 35S-cysteine/methionine is then prevented by addition of excess cold cysteine/methionine. Various biochemical, HPLC, GC-MS, or gel-based techniques can then be used to follow the fate of the labeled proteins.
A quantitative approach to measuring proteolysis in cell extracts (or further purified cell fractions) involves incubating cell lysates with fluorogenic substrates, like the Suc-LLVY-AMC peptide, and assessing release of AMC by measuring fluorescence at 360 nm excitation/485 nm emission. Alternatively, the degradation of intact radiolabeled or fluorescent-labeled whole proteins can be measured by release of the label. In the widely-used radioactive method, epsilon amino groups of lysine residues of a purified protein substrate are first radiolabeled (typically with 3H or 14C) at trace levels. As the protein is degraded, during subsequent incubation with cell extracts or further purified cellular components, radiolabeled amino acids are progressively released into small peptides (<5 kDa) that are soluble in strong acids (such as trichloroacetic acid), whereas remaining intact proteins are precipitated by acid-treatment. Therefore, increasing acid-soluble radioactivity can be quantified by liquid scintillation as a simple measure of proteolysis (Stadtman, 1993). Recently, a simpler non-radioactive method for labeling intact whole protein substrates and measuring their degradation has been developed that offers several advantages, including: shorter assay times, real-time monitoring of proteolysis, and the elimination of radioactivity exposure and waste (Pickering and Davies, 2012a).
The above methods can also be combined with addition of various proteasome inhibitors, in an attempt to distinguish between the 20S and 26S proteasomes. Since most proteasome inhibitors are selective rather than specific, and because most target the protease activities of the 20S core (which is common to both the 20S and 26S proteasome forms), this approach is often unsuccessful. Some of the most widely used inhibitors primarily target the chymotrypsin-like sites of the catalytic β-subunits. However, at higher concentrations these inhibitors have also been shown to block the trypsin and caspase-like activity of the proteolytic core, rendering all proteolytic components non-functional (Kisselev et al., 2012). This is important as it was shown that merely blocking only the chymotrypsin activity of the proteolytic core is not sufficient to block proteolysis (Kisselev et al., 2006). The most common, relatively site-specific inhibitors used in the field are epoxomicin, MG132, and lactacystin, all of which inhibit β-subunits of the 20S core. Lactacystin was shown to permanently inhibit all three catalytic β-subunits (Craiu et al., 1997; Fenteany et al., 1995), while epoxomicin has been shown to have varying rates of inhibition for all three β-subunits (Meng et al., 1999). In addition, bortezomib is one of the first proteasome-specific inhibitors to receive FDA approval in the treatment of multiple myeloma due to its ability to selectively cause apoptosis in cancer-derived cell lines (Adams and Kauffman, 2004; Kisselev et al., 2012). Allosteric proteasome inhibitors include PR-39, which has been shown to both physically block the α7 subunits of the 20S proteasome and inhibit the interaction between the 20S and the 19S regulator (Gaczynska et al., 2003). It is important to note that these inhibitors act against the 20S proteasome and do not distinguish between the 20S and the 26S during in vitro assays. However, both the radiolabeling and fluorescence labeling methods described above, whether used in intact cells, cell extracts, or purified cell components, can be performed under conditions that inhibit ATP/ubiquitin-stimulated proteolysis. Studies with a conditional ubiquitin E1 mutant have demonstrated the utility of this technique for distinguishing between ubiquitin-dependent (26S) and ubiquitinin-dependent (20S) proteolysis by the proteasome (Shringarpure et al., 2003). Similarly, depleting cell extracts of ubiquitin and/or ATP, or inhibiting ubiquitinylation, is a relatively simple way with which to differentiate ubiquitin-dependent and ubiquitin-independent proteolytic capacity in vitro (Shringarpure et al., 2003).
Alternative techniques to distinguish the proteolytic activity of the 26S proteasome from the 20S proteasome include a novel approach that measures the degradation rate of ubiquitin-luciferase proteins in vivo (Luker et al., 2003). The concept behind this approach is that the 26S proteasome should only degrade proteins that have been ubiquitin-tagged, whereas the 20S proteasome should degrade oxidized proteins lacking a ubiquitin tag (Voges et al., 1999). The luciferase reporter was used for bioluminescence measurements with real-time imaging in both cell culture and mice. Under normal conditions, proteins that incorporated the ubiquitin-luciferase tag were rapidly degraded, but in the presence of an inhibitor, fluorescence increased, indicating inhibition of 26S activity (Luker et al., 2003). This approach may provide a more accurate method to measure 26S proteolysis specifically as it relies on the substrate preference differences between the 20S and the 26S proteasome. In addition, this method allows for real-time in vivo measurements, whereas the standard approach for measuring proteolytic activity relies on in vitro methods that may result in partial loss or disassociation of the 19S regulator from the 20S complex (Voges et al., 1999).
7. The degradation of oxidized proteins
Multiple studies from a number of laboratories have independently confirmed that the main pathway for the degradation of oxidatively damaged proteins in cells is via the 20S proteasome (Davies, 1993; Grune et al., 1996; Pacifici et al., 1989; Reinheckel et al., 1998; Salo et al., 1990). Recently, we have shown that the Pa28 (11S) regulator can actually improve the ability of the 20S proteasome to selectively degrade oxidized proteins and that the immunoproteasome (±PA28/11S) is at least as effective, or perhaps more effective (Pickering and Davies, 2012b). In contrast, direct side-by-side comparisons have found the 26S proteasome to be unable to selectively degrade oxidized proteins, whether in the presence or absence of ATP (Shringarpure et al., 2003). As discussed earlier, oxidation causes chemical modifications to proteins that result in conformational changes and subsequent exposure of hydrophobic residues (Carrard et al., 2002). These hydrophobic patches on the surface of oxidized proteins are then recognized by 20S proteasomes and can promote the opening of the α-rings (Kisselev et al., 2002) and enhance selective degradation within the β-rings (Giulivi et al., 1994; Pacifici et al., 1993). Essentially, the hydrophobic patches of oxidized proteins allow for substrate recognition by the 20S proteasome, and subsequent channeling into the 20S core, obviating the need for ubiquitin targeting and unfolding chaperone-like activity conducted by the 19S regulator as observed in ATP/ubiquitin-dependent proteolysis of non-oxidized proteins by the 26S proteasome (Braun et al., 1999).
While it is now abundantly clear that the 20S proteasome is able to degrade oxidized proteins, it is important to test for the possible role of other potential factors, such as ubiquitin. Interestingly, a few studies have found an increase in protein ubiquitinylation, or in the accumulation of poly-ubiquitinylated proteins, following oxidative stress. Pigs fed a hypercholesterolemic diet for 12 weeks exhibited increased ubiquitin conjugates in their coronary arteries, as assessed by immunoblotting, when compared with pigs fed normal chow (Hermann et al., 2003). Moreover, exposure of lens epithelial cells to H2O2 induced a transient increase in ubiquitin conjugation and ubiquitin-activating enzyme (E1) activity (Shang et al., 1997). It is, nevertheless, important to recognize that an increase in the steady-state cellular concentration of polyubiquitin-protein conjugates does not, in and of itself, provide evidence for increased proteolysis by the ATP/ubiquitin-dependent 26S proteasome pathway. In fact, significant increases in the cellular content of polyubiquitinylated proteins may actually be taken as de facto evidence for decreased 26S proteasome degradation, such that its normal ubiquitinylated substrates back-up or accumulate.
In order to determine if oxidized proteins are actually being ubiquitinylated, a study by Kastle et al. used MALDI tandem mass spectrometry to identify ubiquitinylated proteins isolated from WM-451-Lu melanoma cells that had been treated with 5 mM H2O2 (Kastle et al., 2012). A total of 24 different proteins were obtained upon UbiQapture-Q fractionation; however, oxidized proteins, as measured by carbonyl formation, were mainly isolated in the unbound fraction and not the ubiquitinylated pool of proteins. The proteins identified to undergo ubiquitinylation upon oxidative stress were categorized into functional groups including chaperones, energy metabolism, cytoskeleton/intermediate filaments, and protein translation/ribosome biogenesis (Kastle et al., 2012). Interestingly, the robust increase of chaperones in response to oxidative stress is tightly regulated by molecular mechanisms that include ubiquitinylation. Notably, the proportion of chaperones increased by one-third upon exposure to oxidative stress and, of the identified proteins, heat shock proteins were preferentially ubiquitinylated while oxidized proteins were not (Kastle et al., 2012).
Shringarpure et al. (2003) used cells with a conditionally mutated ubiquitinylating system to test for the possible importance of ubiquitinylation in the overall selective degradation of oxidized proteins. They found no evidence for preferential ubiquitinylation of oxidized proteins versus non-oxidized proteins, and inactivation of ubiquitinylation had no measurable effect on the selective degradation of oxidized proteins within intact cells. Additionally, purified oxidized proteins were degraded at the same rate by cell extracts, whether the extracts had a functioning polyubiquitinylating system or not (Shringarpure et al., 2003). The findings that oxidatively modified proteins are not preferentially ubiquitinylated, and that ubiquitinylation is not required for their selective degradation, supports the idea that the degradation of oxidized proteins is ubiquitin-independent (Fig. 3).
Fig. 3.

Mechanism of action for the degradation of oxidized proteins. Oxidative stress results in damage to proteins that cause them to unfold, thus revealing hydrophobic residues that are prone to aggregation. The 20S proteasome recognizes these hydrophobic patches on the surface of a substrate protein, which can promote the opening of the outer α-ring. In addition, both the immunoproteasome and the 20S proteasome bound to Pa28 regulators have a preference for degrading oxidatively damaged proteins.
Further supporting the ATP/ubiquitin-independence of oxidized protein degradation are in vitro and cell culture studies which indicate that the 20S proteasome is relatively stable upon H2O2 exposure, while the 26S proteasome becomes inactivated (Reinheckel et al., 1998, 2000b). Additionally, key sulfhydryl groups in the proteins responsible for the polyubiquitinylation cascade (E1,E2, and E3) are highly sensitive to transient inactivation during oxidative stress (Grune et al., 2011). Furthermore, in cultured bovine lens epithelial cells (BLECs), the formation of endogenous high molecular weight ubiquitin conjugates decreased by 73% upon exposure to H2O2 (Shang and Taylor, 1995). These studies indicate the ability of oxidative stress to block the ubiquitin linkage system through inhibition of E1/E2/E3 complexes. As a result, the ATP/ubiquitin-dependent pathway of protein degradation is also transiently blocked during stress; this is likely the reason that polyubiquitinylated proteins may accumulate in cells during certain stress conditions.
8. Modulation of the 26S proteasome in response to oxidative stress
In addition to transient inactivation of the E1, E2, and E3 ubiquitinylating enzymes during oxidative stress, it has recently been found that the 26S proteasome itself is tightly regulated, and is actually disassembled, and therefore transiently inactivated, immediately following an oxidative insult. Thus, ATP/ubiquitin-dependent protein degradation temporarily ceases for a period of about 3–5 hours following oxidative stress. In a study by Wang et al., it was found in yeast that the oxidative stress of H2O2 treatment triggered the disassociation of the 19S regulator from the 20S core proteasome, resulting in a loss of chymotrypsin-like proteolytic activity and increased intracellular accumulation of non-degraded, ubiquitinylated proteins (Wang et al., 2010). Furthermore, H2O2 induced the association of the proteasome scaffolding protein, Ecm29 with the 19S regulator, and (in separate experiments) deletion of ECM29 prevented the disassembly of the 26S proteasome (Wang et al., 2010). In addition, ecm29Δ mutants were more susceptible to oxidative stress than were wild-type yeast, indicating that disassociation of the 26S proteasome by Ecm29 is important for the successful degradation of oxidized proteins (Wang et al., 2010).
Simultaneously, an independent work by Grune et al. (2011), using human erythroid K562 cells, found that ATP-stimulated proteasome activity was mitigated immediately after treatment with H2O2, but recovered several hours later. Conversely, exposure of cells to mild oxidative stress correlated with an almost immediate (0–3 hours) increase in the capacity to degrade oxidized protein substrates, along with a subsequent (5–24 hours) increase in the synthesis of 20S proteasome, Pa28αβ (11S) regulator, and immunoproteasome (Grune et al., 2011). The immediate increases in ability to degrade oxidized proteins did not require transcription or translation, whereas the subsequent increases could be completely inhibited by transcriptional and translational inhibitors. In the same experiments, immunoprecipitation indicated that the 19S regulator lost affinity for the 20S proteasome initially following an oxidative insult in favor of increased affinity for HSP70, and that HSP70 synthesis increased significantly during the same period (Grune et al., 2011). Importantly, It is well established that HSP70 and other chaperones frequently act as holdases to coordinate transcriptional activity (Ali et al., 1998; Katschinski et al., 2004). Likewise, HSP70 siRNA did not impact disassembly of the 19S regulator, but did abrogate the reassembly of the 26S proteasome after recovery from oxidative stress (Grune et al., 2011). We conclude that Ecm29 and HSP70 act together as pivotal regulators of 26S/20S proteasome modulation during oxidative stress; by disassembling the 26S proteasome, Ecm29 immediately provides more 20S proteasome to degrade oxidized proteins while HSP70 binds and preserves the dissociated 19S regulators for 3–5 hours, allowing subsequent reassembly of 26S proteasomes which are vital to the protein processing machinery to allow successful stress adaptation (Fig. 4).
Fig. 4.

Disassembly of the 26S proteasome during oxidative stress. The components of the ubiquitin-26S proteasome system are highly sensitive to oxidative stress-induced inactivation. In response to oxidative stress, ECM29 facilitates the disassembly of the 26S proteasome, allowing for the unbound 20S proteasome to preferentially degrade oxidized proteins. 20S proteasomes may also bind to PA28 (11S) regulators to increase the efficiency of oxidized protein degradation. HSP70 acts as a holdase by binding the 19S regulatory unit until recovery from stress and is necessary for the reassembly of the 26S proteasome some 3–5 hours after an oxidative insult.
9. The stoichiometry of the 20S proteasome and binding regulators
While it is clear that the 20S proteasome core plays a critical role in ubiquitin-independent proteolysis, many publications refer to the 26S proteasome as the major protease in the cell. Through a series of labor-intensive glycerol gradient fractionations, immunoprecipitations, and immunoblotting experiments performed using HeLa cell extracts, Tanahashi et al. found that the sum total of 26S proteasomes and hybrid proteasomes were only equal to the content of free 20S proteasomes alone (Tanahashi et al., 2000). In support of this initial work, a recent study by Fabre et al. has used a quantitative proteomics approach to meticulously analyze the stoichiometry of the various proteins that make up the pool of proteasomes in the cell (Fabre et al., 2014). Proteasomes were precipitated from several different types of human cell culture lines, analyzed by nano-LC—MS/MS, and quantified in order to calculate the Protein Abundance Index (PAI), which is defined as the average for the ion chromatogram (XIC) area values for three of the most intense reference tryptic peptides identified for a given protein (Fabre et al., 2014). On average, across the many different cell types, just 36% of proteasomes were bound to a regulator (including the 19S, Pa28/11S, and Pa200 regulators) while 64% were unbound 20S core proteasomes (Fabre et al., 2014). This means that nearly two-thirds of the proteasomes available in the cell are in the free 20S form, and able to preferentially degrade oxidized proteins.
It has also been demonstrated that the 20S proteasome is responsible for the degradation of disordered proteins (Baugh et al., 2009) which, like oxidized proteins, have regions of increased hydrophobicity, and include substrates such as p53 (Asher et al., 2005), tau (David et al., 2002), α-synuclein (Tofaris et al., 2001), p21 (Li et al., 2007), and p27 (Tambyrajah et al., 2007). Therefore, in addition to managing protein homeostasis during oxidative stress, the 20S proteasome also degrades many proteins involved in signaling and regulation of cellular processes (Dyson and Wright, 2005). Due to the importance of ubiquitin-dependent proteolysis, it is not surprising that the 19S regulator was found to be the most abundant activator bound to the 20S proteasome, constituting 21–35% of the remaining cellular fraction of proteasomes, dependent on the cell type (Fabre et al., 2014). The abundance of 11S/PA28 and PA200 regulators was more variable across the cell lines analyzed, composing 1–14% of the proteasome pool (Fabre et al., 2014), but both have been shown to significantly increase under stressful conditions such as adaptation to oxidative stress (Grune et al., 2011), irradiation (Blickwedehl et al., 2008), and cancer progression (Ali et al., 2013). This work elegantly confirms that the major conformation of the proteasome is the 20S core responsible for ubiquitin-independent proteolysis and not the 26S proteasome responsible for ATP/ubiquitin-dependent degradation (Fabre et al., 2014).
10. The role of the 20S proteasome in adaptation to oxidative stress
Various terms, such as adaptive response, adaptive homeostasis, dose-response, and hormesis are used to describe the ability of cells and whole organisms to increase their tolerance to stress on a transient basis. Hormesis specifically refers to the adaptive response to low-dose toxin exposure, while dose-response is considered a pharmacological term. We favor the terms adaptive response and adaptive homeostasis because they are more physiological, and allow for inclusion of agents or conditions which may not be stressors in themselves, but which control signal transduction pathways that regulate stress resistance.
Transient adaptive stress-responses result in the transcription of protective genes that promote cell survival. A number of cellular stress-response pathways have evolved to manage dysfunction in protein homeostasis including the heat shock response, oxidative stress response, and proteasome-dependent protein degradation. The first studies to demonstrate adaption to oxidative stress in the 1980s used Escherichia coli, Salmonella typhimurium, and Saccharomyces cerevisiae to show that after recovery from a mild dose of H2O2, the organisms transiently became more tolerant of a subsequent, normally toxic or lethal exposure (Christman et al., 1985; Davies et al., 1995; Davies, 1995; Demple and Halbrook, 1983). Since then, this phenomenon has also been demonstrated in Caenorhabditis elegans (Pickering et al., 2013), Drosophila melanogaster (Pickering et al., 2013), and mammalian cell culture (Wiese et al., 1995). Recently, it has become apparent that a great degree of crosstalk exists between these mechanisms. For instance, adaptation to oxidative-stress provides some heat-stress tolerance, and results in an upregulation of heat shock proteins including HSP70, which is crucial for the reassembly of the 26S proteasome following an oxidative insult (Grune et al., 2011; Kastle et al., 2012).
The responses to oxidative stress also include the transactivation of Nrf2 and the subsequent induction of cytoprotective genes, such as glutathione peroxidases and glutathione transferases. Under normal homeostatic conditions, Nrf2 is bound to the Kelch-like ECH-associated protein 1 (KEAP1)-Cullin 3 (CUL3) E3 ligase complex and undergoes constitutive polyubiquitinylation and subsequent degradation by the 26S proteasome (Taguchi et al., 2011). KEAP1 acts as a sensor of oxidative stress and upon oxidation by an electrophile, or alkylation, is modified and can no longer bind to Nrf2. Nrf2 is then stabilized, phosphorylated by Akt, and translocated to the nucleus where it binds to Electrophile Response Elements or EPRE’s (also called Antioxidant Response Elements or AREs) (Taguchi et al., 2011). Interestingly, Nrf2 also targets several 20S proteasomal subunits for transcriptional activation, as well as the PA28αβ (11S) proteasome regulator (Pickering and Davies, 2012b). The promoter regions of many 20S α- and β-subunits, and PA28αβ subunits, possess EPRE/ARE domains consisting of the core motif RTGACnnnGC or the extended motif TMAnnRTGAYnnnGCAwwww (Nerland, 2007; Pickering et al., 2012). In cell culture, it was shown that adaptive doses of H2O2 lead to increased cellular levels of Nrf2, translocation of Nrf2 to the nucleus, and an increase in recruitment to the β5 subunit promoter (Pickering et al., 2012). Inhibition of Nrf2 with siRNA and retinoic acid abrogated the adaptive increase in 20S proteasomes, PA28αβ, proteolytic capacity, and stress resistance (Pickering et al., 2012). Interestingly, this pathway is conserved in the C. elegans homolog, SKN-1, and the D. melanogaster homologs, CnC and dKeap1 (Pickering et al., 2013; Sykiotis and Bohmann, 2008; Tsakiri et al., 2013). Both knockdown using RNAi, and a deletion mutant for skn-1, negatively impact oxidative stress-induced 20S proteasome expression and cytoprotection (Pickering et al., 2013). While adaptation applies to a number of cytoprotective pathways in response to a variety of stressors, the response to oxidative damage is of particular interest because of the association for oxidative damage accumulation during aging.
11. The role of the 20S proteasome in aging and in aging-related disease
A hallmark of aging is the decline in the quality control mechanisms of the cellular proteome (Taylor and Dillin, 2011). Loss of protein homeostasis results in an increase in protein oxidation, protein misfolding, and protein aggregation that can lead to aging-related diseases, including cardiovascular disease (CVD) (Wang and Robbins, 2006), ischemic stroke (Chen et al., 2011), and neurodegenerative disorders (Morimoto, 2008). Proteasome activity has long been described to decline with age, thus contributing to the overall loss in protein homeostasis during aging. However, this assertion appears to be in opposition to the aging-related increase in muscle wasting that is observed in C. elegans (Herndon et al., 2002), rats (Medina et al., 1995), and humans (Suetta et al., 2012). The confusion regarding proteasome activity during aging is probably largely due to the failure to distinguish between 20S and 26S proteasome activity. For instance, in the spinal cord of rats ranging from 3 weeks to 28 months, chymotrypsin-like activity measured by the liberation of AMC from a Suc-LLVY-AMC substrate steadily decreases with age (Keller et al., 2000). However, when this same assay is used to assess proteasome activity in the triceps of aging rats, it was found that proteolytic activity actually increased, in addition to elevated levels of the ubiquitin ligases and proteasome β-subunits (Altun et al., 2010). In support of these data, a recent human trial found that the ubiquitin-proteasome pathway was activated in both young and old individuals upon immobilization, thereby triggering muscular atrophy (Suetta et al., 2012). Thus, it seems that increased ubiquitin-dependent proteolysis is a major determinant of sarcopenia and muscle wasting during aging.
Interestingly, proteasome inhibitors have been used in dy(3K)/dy(3K) mice to partially alleviate detrimental phenotypes for muscle wasting diseases such as congenital muscular dystrophy, thus improving body weight, locomotion, and survival (Korner et al., 2014). Specifically, bortezomib reduced proteasome activity in congenital muscular dystrophy type 1A muscle tissue (Korner et al., 2014). Furthermore, patients diagnosed with Duchenne muscular dystrophy (DMD) exhibit dystrophin deficiency and a severe decrease in the expression and membrane localization of the DGC glycoprotein complex (Deconinck and Dan, 2007), in addition to increased activation of ubiquitin-dependent proteolysis (Kumamoto et al., 2000). For DMD patients, treatment with Velcade® (bortezomib) resulted in upregulation of dystrophin, α-sarcoglycan, and β-dystroglycan, as well as increased expression of the DGC glycoprotein complex (Gazzerro et al., 2010).
While activity of the 26S proteasome and ubiquitin-dependent proteolysis does appear to increase in a tissue-specific manner during aging, the ability to degrade oxidized proteins by the 20S proteasome does indeed decrease with age, thereby contributing to the overall decline in protein homeostasis. Oxidation is capable of damaging all cellular components, including DNA, lipids, and proteins, and several lines of evidence have indicated that oxidative stress is central to aging and the development of many aging-related diseases (Halliwell and Gutteridge, 1999; Jacob et al., 2013). Heart failure is the number one cause of hospitalization for people aged 65 and older, and nearly all patients with, or at risk of, heart disease have enhanced levels of oxidative stress (Hall et al., 2012; Madamanchi et al., 2005). Oxidative stress has also been demonstrated to have a major role in the initiation and progression of atherosclerosis, which is an underlying cause for a number of cardiovascular complications (Bonomini et al., 2008). Moreover, in hypertensive rats, oxidative stress was found to mediate progressive cardiac fibrosis by enhancing TGF-β1 expression (Zhao et al., 2008).
As oxidative stress increases with age, the capacity to degrade misfolded and oxidized proteins within the cell declines. A study using human lung fibroblast WI-38 cells as a model for replicative senescence found that both 20S expression and activity were mitigated in senescent cells, correlating with an increase in carbonylated and ubiquitinylated proteins (Chondrogianni et al., 2003). In addition, blocking the proteolytic activity of the 20S core in low passage WI-38 cells induced an aged-phenotype (Chondrogianni et al., 2003). In an in vivo study using rat liver tissue, 20S proteasome expression did not change for old animals compared to young animals, but peptidylglutamyl peptide hydrolyzing activity showed a 60% reduction in aged rats (Hayashi and Goto, 1998).
A number of studies, of both divisionally competent cells and post-mitotic cells (Grune et al., 2001, 2004, 2005; Sitte et al., 2000a, 2000b, 2000c), as well as model organisms (David et al., 2010; Demontis and Perrimon, 2010), have found an accumulation of oxidized, aggregated, and cross-linked proteins with senescence and aging. Inherently, protein oxidation leads to structural rearrangement, resulting in exposure of hydrophobic patches that can cause binding to 20S proteasomes and ensure degradation (as discussed earlier). Alternatively, progressive oxidative damage to proteins may lead to aggregation. Such oxidized protein aggregates often have volatile amino acid residues, such as tyrosyl or tryptophanyl radicals, which can react to form permanent covalent cross-links between proteins, thus further stabilizing the damaged aggregates (Leeuwenburgh et al., 1997). Importantly, protein aggregates of too large a size cannot enter the limited aperture of the 20S proteasome and, therefore, are not readily degraded. These cross-linked protein aggregates do, however, attract 20S proteasomes whose α-subunits can bind to their hydrophobic regions, thus trapping the proteasomes and removing them from the active pool of proteases able to degrade damaged proteins (Shringarpure and Davies, 2002; Stenoien et al., 1999; Verhoef et al., 2002). Essentially, this is a special case of reversible non-competitive proteasome inhibition. While some studies have indicated an increase in 20S proteasome synthesis and activity in cell lysates during aging, this may not produce an accurate reflection of functional proteasomes in vivo. Proteasomes that are sequestered at aggregation sites within the cell cannot degrade oxidized proteins, but lysates from these cells will reflect the increased number of proteasomes and demonstrate their activity in vitro once liberated from protein aggregates, thus giving a false indication of overall proteolytic capacity during aging. These studies clearly show that oxidatively-damaged protein aggregates, such as observed in aging and senescent cells, can decrease the functional pool of 20S proteasomes, and therefore the overall capacity of aged cells to degrade oxidatively damaged proteins.
12. Final remarks
The proteasome plays a central role in protein homeostasis, whether through ATP/ubiquitin-dependent or ubiquitin-independent proteolysis. Due to the plasticity and dynamic regulation of the proteasome in response to stress and aging, it is critical that every effort is made to distinguish between the two systems. Just as important is future research aimed to identify the mechanism for the loss of adaptive capacity of the 20S proteasome during aging.
Protein oxidation is a factor in multiple diseases of aging, including Alzheimer’s and Parkinson’s disease (Cutler et al., 2004; Lin and Beal, 2006; Yan et al., 2013), atherosclerosis and arteriosclerosis (Li et al., 2014), stroke (Zhang et al., 2011), cancer (Reuter et al., 2010), arthritis (Wruck et al., 2011), cataract (Sawada et al., 2009; Spector, 1995), macular degeneration (Beatty et al., 2000; Blasiak et al., 2014), and frailty (Ingles et al., 2014; Serviddio et al., 2009). Loss of protein homeostasis and the adaptive capacity to respond to oxidative stress during aging may have a staggering impact on the world economy (Yang et al., 2013). An epidemiological study of the progression of cognitive impairment in subjects over the age of 85, and initially in optimal cognitive health, revealed that 23% developed Alzheimer’s disease within 10 years (Gonzales Mc Neal et al., 2001). Furthermore, inpatient census data for the USA indicate that the incidence of stroke increases logarithmically with age, and escalates rapidly for individuals over the age of 60 (Williams, 2001). These studies highlight the need to elucidate age-dependent factors and mechanisms, such as a decline in 20S proteasome adaptation, that contribute to disease in order to promote health span, alleviate the economic problems of the growing aging population, and the potential burden on society. Because the ability to mount homeostatic adaptive responses declines with age, resulting in the decline of overall protein homeostasis (Calderwood et al., 2009; Rattan, 2008; Texel and Mattson, 2011), measuring the adaptive response to oxidative stress and the elimination of damaged proteins by the 20S proteasome may potentially provide useful biomarkers and prognostic data to streamline therapeutics for aging patients.
Acknowledgments
This work was supported by R01 grant no. ES003598 from the National Institute of Environmental Health Sciences of the US National Institutes of Health, to K.J.A. Davies.
References
- Adams J, Kauffman M. Development of the proteasome inhibitor Velcade™ (Bortezomib) Cancer Invest. 2004;22:304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- Ali A, Bharadwaj S, O’Carroll R, Ovsenek N. HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol Cell Biol. 1998;18:4949–4960. doi: 10.1128/mcb.18.9.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A, Wang Z, Fu J, Ji L, Liu J, Li L, et al. Differential regulation of the REGgamma-proteasome pathway by p53/TGF-beta signalling and mutant p53 in cancer cells. Nat Commun. 2013;4:2667. doi: 10.1038/ncomms3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun M, Besche HC, Overkleeft HS, Piccirillo R, Edelmann MJ, Kessler BM, et al. Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. J Biol Chem. 2010;285:39597–39608. doi: 10.1074/jbc.M110.129718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, Tsvetkov P, Kahana C, Shaul Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005;19:316–321. doi: 10.1101/gad.319905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh JM, Viktorova EG, Pilipenko EV. Proteasomes can degrade a significant proportion of cellular proteins independent of ubiquitination. J Mol Biol. 2009;386:814–827. doi: 10.1016/j.jmb.2008.12.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister W, Walz J, Zuhl F, Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- Beatty S, Koh HH, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- Blasiak J, Petrovski G, Vereb Z, Facsko A, Kaarniranta K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. Biomed Res Int. 2014;2014:768026. doi: 10.1155/2014/768026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blickwedehl J, Agarwal M, Seong C, Pandita RK, Melendy T, Sung P, et al. Role for proteasome activator PA200 and postglutamyl proteasome activity in genomic stability. Proc Natl Acad Sci USA. 2008;105:16165–16170. doi: 10.1073/pnas.0803145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomini F, Tengattini S, Fabiano A, Bianchi R, Rezzani R. Atherosclerosis and oxidative stress. Histol Histopathol. 2008;23:381–390. doi: 10.14670/HH-23.381. [DOI] [PubMed] [Google Scholar]
- Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- Braun BC, Glickman M, Kraft R, Dahlmann B, Kloetzel PM, Finley D, et al. The base of the proteasome regulatory particle exhibits chaperone-like activity. Nat Cell Biol. 1999;1:221–226. doi: 10.1038/12043. [DOI] [PubMed] [Google Scholar]
- Calderwood SK, Murshid A, Prince T. The shock of aging: molecular chaperones and the heat shock response in longevity and aging – a mini-review. Gerontology. 2009;55:550–558. doi: 10.1159/000225957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrard G, Bulteau AL, Petropoulos I, Friguet B. Impairment of proteasome structure and function in aging. Int J Biochem Cell Biol. 2002;34:1461–1474. doi: 10.1016/s1357-2725(02)00085-7. [DOI] [PubMed] [Google Scholar]
- Cascio P, Call M, Petre BM, Walz T, Goldberg AL. Properties of the hybrid form of the 26S proteasome containing both 19S and PA28 complexes. EMBO J. 2002;21:2636–2645. doi: 10.1093/emboj/21.11.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BB, Mallampalli RK. Masking of a nuclear signal motif by monoubiquitination leads to mislocalization and degradation of the regulatory enzyme cytidylyltransferase. Mol Cell Biol. 2009;29:3062–3075. doi: 10.1128/MCB.01824-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, et al. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid Redox Signal. 2011;14:1505–1517. doi: 10.1089/ars.2010.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Norbury CC, Cho Y, Yewdell JW, Bennink JR. Immunoproteasomes shape immunodominance hierarchies of antiviral CD8(+) T cells at the levels of T cell repertoire and presentation of viral antigens. J Exp Med. 2001;193:1319–1326. doi: 10.1084/jem.193.11.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chondrogianni N, Stratford FL, Trougakos IP, Friguet B, Rivett AJ, Gonos ES. Central role of the proteasome in senescence and survival of human fibroblasts: induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J Biol Chem. 2003;278:28026–28037. doi: 10.1074/jbc.M301048200. [DOI] [PubMed] [Google Scholar]
- Christman MF, Morgan RW, Jacobson FS, Ames BN. Positive control of a regulon for defenses against oxidative stress and some heat-shock proteins in Salmonella typhimurium. Cell. 1985;41:753–762. doi: 10.1016/s0092-8674(85)80056-8. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- Craiu A, Gaczynska M, Akopian T, Gramm CF, Fenteany G, Goldberg AL, et al. Lactacystin and clasto-lactacystin β-lactone modify multiple proteasome β-subunits and inhibit intracellular protein degradation and major histocompatibility complex class I antigen presentation. J Biol Chem. 1997;272:13437–13445. doi: 10.1074/jbc.272.20.13437. [DOI] [PubMed] [Google Scholar]
- Cui Z, Hwang SM, Gomes AV. Identification of the immunoproteasome as a novel regulator of skeletal muscle differentiation. Mol Cell Biol. 2014;34:96–109. doi: 10.1128/MCB.00622-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DC, Layfield R, Serpell L, Narain Y, Goedert M, Spillantini MG. Proteasomal degradation of tau protein. J Neurochem. 2002;83:176–185. doi: 10.1046/j.1471-4159.2002.01137.x. [DOI] [PubMed] [Google Scholar]
- David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010;8:e1000450. doi: 10.1371/journal.pbio.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JM, Lowry CV, Davies KJ. Transient adaptation to oxidative stress in yeast. Arch Biochem Biophys. 1995;317:1–6. doi: 10.1006/abbi.1995.1128. [DOI] [PubMed] [Google Scholar]
- Davies KJ. Protein modification by oxidants and the role of proteolytic enzymes. Biochem Soc Trans. 1993;21:346–353. doi: 10.1042/bst0210346. [DOI] [PubMed] [Google Scholar]
- Davies KJ. Biochemical Society Symposia. Portland Press; London: 1995. Oxidative stress: the paradox of aerobic life; pp. 1–32. [DOI] [PubMed] [Google Scholar]
- Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83:301–310. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- Deconinck N, Dan B. Pathophysiology of Duchenne muscular dystrophy: current hypotheses. Pediatr Neurol. 2007;36:1–7. doi: 10.1016/j.pediatrneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Demontis F, Perrimon N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 2010;143:813–825. doi: 10.1016/j.cell.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demple B, Halbrook J. Inducible repair of oxidative DNA damage in Escherichia coli. Nature. 1983;304:466–468. doi: 10.1038/304466a0. [DOI] [PubMed] [Google Scholar]
- Driscoll J, Goldberg AL. The proteasome (multicatalytic protease) is a component of the 1500-kDa proteolytic complex which degrades ubiquitin-conjugated proteins. J Biol Chem. 1990;265:4789–4792. [PubMed] [Google Scholar]
- Dubiel W, Pratt G, Ferrell K, Rechsteiner M. Purification of an 11 S regulator of the multicatalytic protease. J Biol Chem. 1992;267:22369–22377. [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Etlinger JD, Goldberg AL. A soluble ATP-dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes. Proc Natl Acad Sci USA. 1977;74:54–58. doi: 10.1073/pnas.74.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eytan E, Ganoth D, Armon T, Hershko A. ATP-dependent incorporation of 20S protease into the 26S complex that degrades proteins conjugated to ubiquitin. Proc Natl Acad Sci USA. 1989;86:7751–7755. doi: 10.1073/pnas.86.20.7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre B, Lambour T, Garrigues L, Ducoux-Petit M, Amalric F, Monsarrat B, et al. Label-free quantitative proteomics reveals the dynamics of proteasome complexes composition and stoichiometry in a wide range of human cell lines. J Proteome Res. 2014;13:3027–3037. doi: 10.1021/pr500193k. [DOI] [PubMed] [Google Scholar]
- Fenteany G, Standaert RF, Lane WS, Choi S, Corey E, Schreiber SL. Inhibition of proteasome activities and subunit-specific aminoterminal threonine modification by lactacystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- Finley D, Sadis S, Monia BP, Boucher P, Ecker DJ, Crooke ST, et al. Inhibition of proteolysis and cell cycle progression in a multiubiquitination-deficient yeast mutant. Mol Cell Biol. 1994;14:5501–5509. doi: 10.1128/mcb.14.8.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frentzel S, Pesold-Hurt B, Seelig A, Kloetzel PM. 20S proteasomes are assembled via distinct precursor complexes. Processing of LMP2 and LMP7 proproteins takes place in 13–16 S preproteasome complexes. J Mol Biol. 1994;236:975–981. doi: 10.1016/0022-2836(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Friedberg EC. Reversible monoubiquitination of PCNA: a novel slant on regulating translesion DNA synthesis. Mol Cell. 2006;22:150–152. doi: 10.1016/j.molcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Gaczynska M, Osmulski PA, Gao Y, Post MJ, Simons M. Prolineand arginine-rich peptides constitute a novel class of allosteric inhibitors of proteasome activity. Biochemistry. 2003;42:8663–8670. doi: 10.1021/bi034784f. [DOI] [PubMed] [Google Scholar]
- Gazzerro E, Assereto S, Bonetto A, Sotgia F, Scarfi S, Pistorio A, et al. Therapeutic potential of proteasome inhibition in Duchenne and Becker muscular dystrophies. Am J Pathol. 2010;176:1863–1877. doi: 10.2353/ajpath.2010.090468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulivi C, Pacifici RE, Davies KJ. Exposure of hydrophobic moieties promotes the selective degradation of hydrogen peroxide-modified hemoglobin by the multicatalytic proteinase complex, proteasome. Arch Biochem Biophys. 1994;311:329–341. doi: 10.1006/abbi.1994.1245. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Raveh D. Proteasome plasticity. FEBS Lett. 2005;579:3214–3223. doi: 10.1016/j.febslet.2005.04.048. [DOI] [PubMed] [Google Scholar]
- Goldberg AL, Strnad NP, Swamy KS. Studies of the ATP dependence of protein degradation in cells and cell extracts. Protein Degradation in Health and Disease, Ciba Foundation Symposium; 1980. pp. 227–251. [DOI] [PubMed] [Google Scholar]
- Gonzales McNeal M, Zareparsi S, Camicioli R, Dame A, Howieson D, Quinn J, et al. Predictors of healthy brain aging. J Gerontol A Biol Sci Med Sci. 2001;56:B294–B301. doi: 10.1093/gerona/56.7.b294. [DOI] [PubMed] [Google Scholar]
- Griffin TA, Nandi D, Cruz M, Fehling HJ, Kaer LV, Monaco JJ, et al. Immunoproteasome assembly: cooperative incorporation of interferon gamma (IFN-gamma)-inducible subunits. J Exp Med. 1998;187:97–104. doi: 10.1084/jem.187.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groll M, Huber R. Substrate access and processing by the 20S proteasome core particle. Int J Biochem Cell Biol. 2003;35:606–616. doi: 10.1016/s1357-2725(02)00390-4. [DOI] [PubMed] [Google Scholar]
- Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- Groll M, Heinemeyer W, Jäger S, Ullrich T, Bochtler M, Wolf DH, et al. The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study. Proc Natl Acad Sci USA. 1999;96:10976–10983. doi: 10.1073/pnas.96.20.10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groll M, Bajorek M, Köhler A, Moroder L, Rubin DM, Huber R, et al. A gated channel into the proteasome core particle. Nat Struct Mol Biol. 2000;7:1062–1067. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- Grune T, Reinheckel T, Joshi M, Davies KJ. Proteolysis in cultured liver epithelial cells during oxidative stress. Role of the multicatalytic proteinase complex, proteasome. J Biol Chem. 1995;270:2344–2351. doi: 10.1074/jbc.270.5.2344. [DOI] [PubMed] [Google Scholar]
- Grune T, Reinheckel T, Davies KJ. Degradation of oxidized proteins in K562 human hematopoietic cells by proteasome. J Biol Chem. 1996;271:15504–15509. doi: 10.1074/jbc.271.26.15504. [DOI] [PubMed] [Google Scholar]
- Grune T, Shringarpure R, Sitte N, Davies K. Age-related changes in protein oxidation and proteolysis in mammalian cells. J Gerontol A Biol Sci Med Sci. 2001;56:B459–B467. doi: 10.1093/gerona/56.11.b459. [DOI] [PubMed] [Google Scholar]
- Grune T, Jung T, Merker K, Davies KJ. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int J Biochem Cell Biol. 2004;36:2519–2530. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Grune T, Merker K, Jung T, Sitte N, Davies KJ. Protein oxidation and degradation during postmitotic senescence. Free Radic Biol Med. 2005;39:1208–1215. doi: 10.1016/j.freeradbiomed.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Grune T, Catalgol B, Licht A, Ermak G, Pickering AM, Ngo JK, et al. HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic Biol Med. 2011;51:1355–1364. doi: 10.1016/j.freeradbiomed.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MJ, Levant S, DeFrances CJ. Hospitalization for congestive heart failure: United States, 2000–2010. NCHS data brief. 2012:1–8. [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM. Free Radicals in Biology and Medicine. Vol. 3. Oxford University Press; Oxford: 1999. [Google Scholar]
- Hayashi T, Goto S. Age-related changes in the 20S and 26S proteasome activities in the liver of male F344 rats. Mech Ageing Dev. 1998;102:55–66. doi: 10.1016/s0047-6374(98)00011-6. [DOI] [PubMed] [Google Scholar]
- Hendil KB, Khan S, Tanaka K. Simultaneous binding of PA28 and PA700 activators to 20 S proteasomes. Biochem J. 1998;332(Pt 3):749–754. doi: 10.1042/bj3320749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann J, Gulati R, Napoli C, Woodrum JE, Lerman LO, Rodriguez-Porcel M, et al. Oxidative stress-related increase in ubiquitination in early coronary atherogenesis. FASEB J. 2003;17:1730–1732. doi: 10.1096/fj.02-0841fje. [DOI] [PubMed] [Google Scholar]
- Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, et al. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002;419:808–814. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system for protein degradation. Annu Rev Biochem. 1992;61:761–807. doi: 10.1146/annurev.bi.61.070192.003553. [DOI] [PubMed] [Google Scholar]
- Hirsch C, Ploegh HL. Intracellular targeting of the proteasome. Trends Cell Biol. 2000;10:268–272. doi: 10.1016/s0962-8924(00)01768-2. [DOI] [PubMed] [Google Scholar]
- Holzhütter HG, Kloetzel PM. A kinetic model of vertebrate 20S proteasome accounting for the generation of major proteolytic fragments from oligomeric peptide substrates. Biophys J. 2000;79:1196–1205. doi: 10.1016/S0006-3495(00)76374-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingles M, Gambini J, Carnicero JA, Garcia-Garcia FJ, Rodriguez-Manas L, Olaso-Gonzalez G, et al. Oxidative stress is related to frailty, not to age or sex, in a geriatric population: lipid and protein oxidation as biomarkers of frailty. J Am Geriatr Soc. 2014;62:1324–1328. doi: 10.1111/jgs.12876. [DOI] [PubMed] [Google Scholar]
- Jacob KD, Noren Hooten N, Trzeciak AR, Evans MK. Markers of oxidant stress that are clinically relevant in aging and age-related disease. Mech Ageing Dev. 2013;134:139–157. doi: 10.1016/j.mad.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio PJ, Wilson WJ, O’Brien S, Makino Y, Poellinger L. Regulation of the hypoxia-inducible transcription factor 1alpha by the ubiquitin-proteasome pathway. J Biol Chem. 1999;274:6519–6525. doi: 10.1074/jbc.274.10.6519. [DOI] [PubMed] [Google Scholar]
- Kastle M, Reeg S, Rogowska-Wrzesinska A, Grune T. Chaperones, but not oxidized proteins, are ubiquitinated after oxidative stress. Free Radic Biol Med. 2012;53:1468–1477. doi: 10.1016/j.freeradbiomed.2012.05.039. [DOI] [PubMed] [Google Scholar]
- Katschinski DM, Le L, Schindler SG, Thomas T, Voss AK, Wenger RH. Interaction of the PAS B domain with HSP90 accelerates hypoxia-inducible factor-1alpha stabilization. Cell Physiol Biochem. 2004;14:351–360. doi: 10.1159/000080345. [DOI] [PubMed] [Google Scholar]
- Keller JN, Huang FF, Markesbery WR. Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience. 2000;98:149–156. doi: 10.1016/s0306-4522(00)00067-1. [DOI] [PubMed] [Google Scholar]
- Kimura H, Caturegli P, Takahashi M, Suzuki K. New insights into the function of the immunoproteasome in immune and nonimmune cells. J Immunol Res. 2015;2015:541984. doi: 10.1155/2015/541984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselev AF, Akopian TN, Woo KM, Goldberg AL. The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes. Implications for understanding the degradative mechanism and antigen presentation. J Biol Chem. 1999;274:3363–3371. doi: 10.1074/jbc.274.6.3363. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, Kaganovich D, Goldberg AL. Binding of hydrophobic peptides to several non-catalytic sites promotes peptide hydrolysis by all active sites of 20 S proteasomes. Evidence for peptide-induced channel opening in the alpha-rings. J Biol Chem. 2002;277:22260–22270. doi: 10.1074/jbc.M112360200. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, Callard A, Goldberg AL. Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J Biol Chem. 2006;281:8582–8590. doi: 10.1074/jbc.M509043200. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol. 2012;19:99–115. doi: 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloetzel PM, Ossendorp F. Proteasome and peptidase function in MHC-class-I-mediated antigen presentation. Curr Opin Immunol. 2004;16:76–81. doi: 10.1016/j.coi.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Komander D. The emerging complexity of protein ubiquitination. Biochem Soc Trans. 2009;37:937–953. doi: 10.1042/BST0370937. [DOI] [PubMed] [Google Scholar]
- Kopp F, Dahlmann B, Kuehn L. Reconstitution of hybrid proteasomes from purified PA700-20 S complexes and PA28alphabeta activator: ultrastructure and peptidase activities. J Mol Biol. 2001;313:465–471. doi: 10.1006/jmbi.2001.5063. [DOI] [PubMed] [Google Scholar]
- Korner Z, Fontes-Oliveira CC, Holmberg J, Carmignac V, Durbeej M. Bortezomib partially improves laminin alpha2 chain-deficient muscular dystrophy. Am J Pathol. 2014;184:1518–1528. doi: 10.1016/j.ajpath.2014.01.019. [DOI] [PubMed] [Google Scholar]
- Köhler A, Cascio P, Leggett DS, Woo KM, Goldberg AL, Finley D. The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release. Mol Cell. 2001;7:1143–1152. doi: 10.1016/s1097-2765(01)00274-x. [DOI] [PubMed] [Google Scholar]
- Kumamoto T, Fujimoto S, Ito T, Horinouchi H, Ueyama H, Tsuda T. Proteasome expression in the skeletal muscles of patients with muscular dystrophy. Acta Neuropathol. 2000;100:595–602. doi: 10.1007/s004010000229. [DOI] [PubMed] [Google Scholar]
- Lasker K, Förster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, et al. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci USA. 2012;109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeuwenburgh C, Rasmussen JE, Hsu FF, Mueller DM, Pennathur S, Heinecke JW. Mass spectrometric quantification of markers for protein oxidation by tyrosyl radical, copper, and hydroxyl radical in low density lipoprotein isolated from human atherosclerotic plaques. J Biol Chem. 1997;272:3520–3526. doi: 10.1074/jbc.272.6.3520. [DOI] [PubMed] [Google Scholar]
- Li H, Horke S, Forstermann U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis. 2014;237:208–219. doi: 10.1016/j.atherosclerosis.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Li X, Amazit L, Long W, Lonard DM, Monaco JJ, O’Malley BW. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGgamma-proteasome pathway. Mol Cell. 2007;26:831–842. doi: 10.1016/j.molcel.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lowe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science. 1995;268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- Luciani F, Keşmir C, Mishto M, Or-Guil M, de Boer RJ. A Mathematical model of protein degradation by the proteasome. Biophys J. 2005;88:2422–2432. doi: 10.1529/biophysj.104.049221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker GD, Pica CM, Song J, Luker KE, Piwnica-Worms D. Imaging 26S proteasome activity and inhibition in living mice. Nat Med. 2003;9:969–973. doi: 10.1038/nm894. [DOI] [PubMed] [Google Scholar]
- Lüllmann-Rauch R. Lysosomes. Springer; 2005. History and morphology of the lysosome; pp. 1–16. [Google Scholar]
- Ma CP, Slaughter CA, DeMartino GN. Identification, purification, and characterization of a protein activator (PA28) of the 20 S proteasome (macropain) J Biol Chem. 1992;267:10515–10523. [PubMed] [Google Scholar]
- Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005;25:29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Goodbourn S, Fischer JA. Regulation of inducible and tissue-specific gene expression. Science. 1987;236:1237–1245. doi: 10.1126/science.3296191. [DOI] [PubMed] [Google Scholar]
- Medina R, Wing SS, Goldberg AL. Increase in levels of polyubiquitin and proteasome mRNA in skeletal muscle during starvation and denervation atrophy. Biochem J. 1995;307(Pt 3):631–637. doi: 10.1042/bj3070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci USA. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerland DE. The antioxidant/electrophile response element motif. Drug Metab Rev. 2007;39:235–248. doi: 10.1080/03602530601125000. [DOI] [PubMed] [Google Scholar]
- Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Okamura T, Taniguchi S, Ohkura T, Yoshida A, Shimizu H, Sakai M, et al. Abnormally high expression of proteasome activator-gamma in thyroid neoplasm. J Clin Endocrinol Metab. 2003;88:1374–1383. doi: 10.1210/jc.2002-021413. [DOI] [PubMed] [Google Scholar]
- Ortega J, Heymann JB, Kajava AV, Ustrell V, Rechsteiner M, Steven AC. The axial channel of the 20S proteasome opens upon binding of the PA200 activator. J Mol Biol. 2005;346:1221–1227. doi: 10.1016/j.jmb.2004.12.049. [DOI] [PubMed] [Google Scholar]
- Pacifici RE, Lin SW, Davies KJ. The measurement of protein degradation in response to oxidative stress. Basic Life Sci. 1988;49:531–535. doi: 10.1007/978-1-4684-5568-7_82. [DOI] [PubMed] [Google Scholar]
- Pacifici RE, Salo DC, Davies KJ. Macroxyproteinase (M.O.P.): a 670 kDa proteinase complex that degrades oxidatively denatured proteins in red blood cells. Free Radic Biol Med. 1989;7:521–536. doi: 10.1016/0891-5849(89)90028-2. [DOI] [PubMed] [Google Scholar]
- Pacifici RE, Kono Y, Davies KJ. Hydrophobicity as the signal for selective degradation of hydroxyl radical-modified hemoglobin by the multicatalytic proteinase complex, proteasome. J Biol Chem. 1993;268:15405–15411. [PubMed] [Google Scholar]
- Pemberton LF, Paschal BM. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic. 2005;6:187–198. doi: 10.1111/j.1600-0854.2005.00270.x. [DOI] [PubMed] [Google Scholar]
- Peters B, Janek K, Kuckelkorn U, Holzhütter HG. Assessment of proteasomal cleavage probabilities from kinetic analysis of time-dependent product formation. J Mol Biol. 2002;318:847–862. doi: 10.1016/S0022-2836(02)00167-5. [DOI] [PubMed] [Google Scholar]
- Pickering AM, Davies KJ. A simple fluorescence labeling method for studies of protein oxidation, protein modification, and proteolysis. Free Radic Biol Med. 2012a;52:239–246. doi: 10.1016/j.freeradbiomed.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering AM, Davies KJA. Differential roles of proteasome and immunoproteasome regulators Pa28 alpha beta, Pa28 gamma and Pa200 in the degradation of oxidized proteins. Arch Biochem Biophys. 2012b;523:181–190. doi: 10.1016/j.abb.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering AM, Linder RA, Zhang H, Forman HJ, Davies KJ. Nrf2-dependent induction of proteasome and Pa28alphabeta regulator are required for adaptation to oxidative stress. J Biol Chem. 2012;287:10021–10031. doi: 10.1074/jbc.M111.277145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering AM, Staab TA, Tower J, Sieburth D, Davies KJ. A conserved role for the 20S proteasome and Nrf2 transcription factor in oxidative stress adaptation in mammals, Caenorhabditis elegans and Drosophila melanogaster. J Exp Biol. 2013;216:543–553. doi: 10.1242/jeb.074757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattan SI. Hormesis in aging. Ageing Res Rev. 2008;7:63–78. doi: 10.1016/j.arr.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Rechsteiner M, Hill CP. Mobilizing the proteolytic machine: cell biological roles of proteasome activators and inhibitors. Trends Cell Biol. 2005;15:27–33. doi: 10.1016/j.tcb.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Rechsteiner M, Realini C, Ustrell V. The proteasome activator 11 S REG (PA28) and class I antigen presentation. Biochem J. 2000;345(Pt 1):1–15. [PMC free article] [PubMed] [Google Scholar]
- Reinheckel T, Sitte N, Ullrich O, Kuckelkorn U, Davies KJ, Grune T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem J. 1998;335(Pt 3):637–642. doi: 10.1042/bj3350637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinheckel T, Grune T, Davies KJ. The measurement of protein degradation in response to oxidative stress. Methods Mol Biol. 2000a;99:49–60. doi: 10.1385/1-59259-054-3:49. [DOI] [PubMed] [Google Scholar]
- Reinheckel T, Ullrich O, Sitte N, Grune T. Differential impairment of 20S and 26S proteasome activities in human hematopoietic K562 cells during oxidative stress. Arch Biochem Biophys. 2000b;377:65–68. doi: 10.1006/abbi.2000.1717. [DOI] [PubMed] [Google Scholar]
- Reits E, Neijssen J, Herberts C, Benckhuijsen W, Janssen L, Drijfhout JW, et al. A major role for TPPII in trimming proteasomal degradation products for MHC class I antigen presentation. Immunity. 2004;20:495–506. doi: 10.1016/s1074-7613(04)00074-3. [DOI] [PubMed] [Google Scholar]
- Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler M, Rollinger W, Mantovani-Endl L, Hagmann ML, Palme S, Berndt P, et al. Identification of PSME3 as a novel serum tumor marker for colorectal cancer by combining two-dimensional polyacrylamide gel electrophoresis with a strictly mass spectrometry-based approach for data analysis. Mol Cell Proteomics. 2006;5:2092–2101. doi: 10.1074/mcp.M600118-MCP200. [DOI] [PubMed] [Google Scholar]
- Salo DC, Pacifici RE, Lin SW, Giulivi C, Davies KJ. Superoxide dismutase undergoes proteolysis and fragmentation following oxidative modification and inactivation. J Biol Chem. 1990;265:11919–11927. [PubMed] [Google Scholar]
- Sawada H, Fukuchi T, Abe H. Oxidative stress markers in aqueous humor of patients with senile cataracts. Curr Eye Res. 2009;34:36–41. doi: 10.1080/02713680802500960. [DOI] [PubMed] [Google Scholar]
- Serviddio G, Romano AD, Greco A, Rollo T, Bellanti F, Altomare E, et al. Frailty syndrome is associated with altered circulating redox balance and increased markers of oxidative stress. Int J Immunopathol Pharmacol. 2009;22:819–827. doi: 10.1177/039463200902200328. [DOI] [PubMed] [Google Scholar]
- Shang F, Taylor A. Oxidative stress and recovery from oxidative stress are associated with altered ubiquitin conjugating and proteolytic activities in bovine lens epithelial cells. Biochem J. 1995;307:297–303. doi: 10.1042/bj3070297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang F, Taylor A. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic Biol Med. 2011;51:5–16. doi: 10.1016/j.freeradbiomed.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang F, Gong X, Taylor A. Activity of ubiquitin-dependent pathway in response to oxidative stress. Ubiquitin-activating enzyme is transiently up-regulated. J Biol Chem. 1997;272:23086–23093. doi: 10.1074/jbc.272.37.23086. [DOI] [PubMed] [Google Scholar]
- Sharon M, Witt S, Felderer K, Rockel B, Baumeister W, Robinson CV. 20S proteasomes have the potential to keep substrates in store for continual degradation. J Biol Chem. 2006;281:9569–9575. doi: 10.1074/jbc.M511951200. [DOI] [PubMed] [Google Scholar]
- Shringarpure R, Davies KJ. Protein turnover by the proteasome in aging and disease. Free Radic Biol Med. 2002;32:1084–1089. doi: 10.1016/s0891-5849(02)00824-9. [DOI] [PubMed] [Google Scholar]
- Shringarpure R, Grune T, Mehlhase J, Davies KJ. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J Biol Chem. 2003;278:311–318. doi: 10.1074/jbc.M206279200. [DOI] [PubMed] [Google Scholar]
- Sitte N, Huber M, Grune T, Ladhoff A, Doecke WD, Von Zglinicki T, et al. Proteasome inhibition by lipofuscin/ceroid during postmitotic aging of fibroblasts. FASEB J. 2000a;14:1490–1498. doi: 10.1096/fj.14.11.1490. [DOI] [PubMed] [Google Scholar]
- Sitte N, Merker K, Von Zglinicki T, Davies KJ, Grune T. Protein oxidation and degradation during cellular senescence of human BJ fibroblasts: part II – aging of nondividing cells. FASEB J. 2000b;14:2503–2510. doi: 10.1096/fj.00-0210com. [DOI] [PubMed] [Google Scholar]
- Sitte N, Merker K, Von Zglinicki T, Grune T, Davies KJ. Protein oxidation and degradation during cellular senescence of human BJ fibroblasts: part I – effects of proliferative senescence. FASEB J. 2000c;14:2495–2502. doi: 10.1096/fj.00-0209com. [DOI] [PubMed] [Google Scholar]
- Spector A. Oxidative stress-induced cataract: mechanism of action. FASEB J. 1995;9:1173–1182. [PubMed] [Google Scholar]
- Stadtman ER. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu Rev Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, DeMartino GN, et al. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet. 1999;8:731–741. doi: 10.1093/hmg/8.5.731. [DOI] [PubMed] [Google Scholar]
- Suetta C, Frandsen U, Jensen L, Jensen MM, Jespersen JG, Hvid LG, et al. Aging affects the transcriptional regulation of human skeletal muscle disuse atrophy. PLoS ONE. 2012;7:e51238. doi: 10.1371/journal.pone.0051238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykiotis GP, Bohmann D. Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev Cell. 2008;14:76–85. doi: 10.1016/j.devcel.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123–140. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- Tambyrajah WS, Bowler LD, Medina-Palazon C, Sinclair AJ. Cell cycle-dependent caspase-like activity that cleaves p27(KIP1) is the beta(1) subunit of the 20S proteasome. Arch Biochem Biophys. 2007;466:186–193. doi: 10.1016/j.abb.2007.07.019. [DOI] [PubMed] [Google Scholar]
- Tanahashi N, Murakami Y, Minami Y, Shimbara N, Hendil KB, Tanaka K. Hybrid proteasomes. Induction by interferon-gamma and contribution to ATP-dependent proteolysis. J Biol Chem. 2000;275:14336– 14345. doi: 10.1074/jbc.275.19.14336. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004440. pii: a004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teoh CY, Davies KJ. Potential roles of protein oxidation and the immunoproteasome in MHC class I antigen presentation: the ‘PrOxI’ hypothesis. Arch Biochem Biophys. 2004;423:88–96. doi: 10.1016/j.abb.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Texel SJ, Mattson MP. Impaired adaptive cellular responses to oxidative stress and the pathogenesis of Alzheimer’s disease. Antioxid Redox Signal. 2011;14:1519–1534. doi: 10.1089/ars.2010.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Layfield R, Spillantini MG. alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001;509:22–26. doi: 10.1016/s0014-5793(01)03115-5. [DOI] [PubMed] [Google Scholar]
- Tsakiri EN, Sykiotis GP, Papassideri IS, Terpos E, Dimopoulos MA, Gorgoulis VG, et al. Proteasome dysfunction in Drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell. 2013;12:802–813. doi: 10.1111/acel.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O, Reinheckel T, Sitte N, Hass R, Grune T, Davies KJ. Poly-ADP ribose polymerase activates nuclear proteasome to degrade oxidatively damaged histones. Proc Natl Acad Sci USA. 1999;96:6223–6228. doi: 10.1073/pnas.96.11.6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustrell V, Hoffman L, Pratt G, Rechsteiner M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 2002;21:3516–3525. doi: 10.1093/emboj/cdf333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoef LG, Lindsten K, Masucci MG, Dantuma NP. Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum Mol Genet. 2002;11:2689–2700. doi: 10.1093/hmg/11.22.2689. [DOI] [PubMed] [Google Scholar]
- Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99:1315–1328. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]
- Wang X, Yen J, Kaiser P, Huang L. Regulation of the 26S proteasome complex during oxidative stress. Sci Signal. 2010;3:ra88. doi: 10.1126/scisignal.2001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese AG, Pacifici RE, Davies KJA. Transient adaptation to oxidative stress in mammalian cells. Arch Biochem Biophys. 1995;318:231–240. doi: 10.1006/abbi.1995.1225. [DOI] [PubMed] [Google Scholar]
- Williams GR. Incidence and characteristics of total stroke in the United States. BMC Neurol. 2001;1:2. doi: 10.1186/1471-2377-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wruck CJ, Fragoulis A, Gurzynski A, Brandenburg LO, Kan YW, Chan K, et al. Role of oxidative stress in rheumatoid arthritis: insights from the Nrf2-knockout mice. Ann Rheum Dis. 2011;70:844–850. doi: 10.1136/ard.2010.132720. [DOI] [PubMed] [Google Scholar]
- Yan MH, Wang X, Zhu X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med. 2013;62:90–101. doi: 10.1016/j.freeradbiomed.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Fruh K, Ahn K, Peterson PA. In vivo assembly of the proteasomal complexes, implications for antigen processing. J Biol Chem. 1995;270:27687–27694. doi: 10.1074/jbc.270.46.27687. [DOI] [PubMed] [Google Scholar]
- Yang Z, Lin PJ, Levey A. Monetary costs of dementia in the United States. N Engl J Med. 2013;369:489. doi: 10.1056/NEJMc1305541. [DOI] [PubMed] [Google Scholar]
- Zhang XH, Lei H, Liu AJ, Zou YX, Shen FM, Su DF. Increased oxidative stress is responsible for severer cerebral infarction in stroke-prone spontaneously hypertensive rats. CNS Neurosci Ther. 2011;17:590–598. doi: 10.1111/j.1755-5949.2011.00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang R. Proteasome activator PA28 gamma regulates p53 by enhancing its MDM2-mediated degradation. EMBO J. 2008;27:852–864. doi: 10.1038/emboj.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Zhao T, Chen Y, Ahokas RA, Sun Y. Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Mol Cell Biochem. 2008;317:43–50. doi: 10.1007/s11010-008-9803-8. [DOI] [PubMed] [Google Scholar]