

Summary
The rapid development of mass spectrometry (MS) technologies has solidified shotgun proteomics as the most powerful analytical platform for large-scale proteome interrogation. The ability to map and determine differential expression profiles of the entire proteome is the ultimate goal of shotgun proteomics. Label-free quantitation has proven to be a valid approach for discovery shotgun proteomics, especially when sample is limited. Label-free spectral count quantitation is an approach analogous to RNA sequencing whereby count data is used to determine differential expression. Here we show that statistical approaches developed to evaluate differential expression in RNA sequencing experiments can be applied to detect differential protein expression in label-free discovery proteomics. This approach, termed MultiSpec, utilizes open-source statistical platforms; namely edgeR, DESeq and baySeq, to statistically select protein candidates for further investigation. Furthermore, to remove bias associated with a single statistical approach a single ranked list of differentially expressed proteins is assembled by comparing edgeR and DESeq q-values directly with the false discovery rate (FDR) calculated by baySeq. This statistical approach is then extended when applied to spectral count data derived from multiple proteomic pipelines. The individual statistical results from multiple proteomic pipelines are integrated and cross-validated by means of collapsing protein groups.
Graphical abstract
Introduction
Mass spectrometry based proteomics is the most diverse platform for protein identification and characterization, in part due to advances in protein isolation techniques, chromatographic separation options and a diverse array of ionization, fragmentation and data acquisition techniques [1]. In shotgun proteomics mere protein identification is usually not sufficient to understand the complexity of biological phenomena. Label-free spectral counting is a robust semi-quantitative technique directly applicable and widely used in shotgun proteomics [2–7]. Spectral count data from shotgun proteomics experiments are heavily influenced by chromatographic separations and sample complexity, as well as the choice of analytical instrumentation and the implementation of dynamic exclusion parameters, and therefore should be considered semi-quantitative and semi-random [8–11]. Regardless of bias associated with collection of mass spectrometry peptide data, ultimately protein identification must be inferred from the generated peptide spectra with the use of database search engines [12–19]. The next challenge is determining differential protein expression between cohorts of complex proteomes and prioritizing these protein candidates for validation.
In discovery based proteomic experiments the number of samples collected is often small (<10). In these instances it is not possible to prove that the counts fit a Gaussian (normal) distribution. Powerful statistical alternatives have been routinely applied in the proteomics community to perform differential expression analysis of spectral count data [20–25]. Models based on the Poisson distribution have historically been applied to model count data. The main limitation to the Poisson distribution is that it has only one model parameter and can not effectively model under- or over-dispersed data. When there is not sufficient data to confirm that the sample variances are equal, quasi-likelihood or generalized linear mixed effects modeling approaches can be used [20, 22]. Another alternative to the Poisson distribution is the negative binomial distribution. The approaches described herein leverage the negative binomial distribution to model over-dispersed count data through determining the unique mean-variance relationship [26–31]. Very similar to spectral count data from shotgun proteomics, RNA sequencing data is over-dispersed, multivariate in nature, and often limited by few biological replicates. This has encouraged the development of so-called tag-count based statistical approaches to determine differential expression. These tag-count based statistical approaches are powerful alternatives to traditional parametric and non-parametric tests when analyzing RNA sequencing data [32–36].
The approach described here, termed MultiSpec, employs a multi-model tag-count based statistical approach. Individual statistical results are combined and re-ranked using a median q-value/FDR approach. This holistic representation of differential expression can be extended to the analysis of spectral count data obtained from multiple proteomic pipelines. MultiSpec is built upon open-source statistical platforms, namely edgeR, DESeq and baySeq and is executable in the R programming language (v 3.0) [37]. The highlighted analysis (EAE/Sham) is a product of the multi-model statistical analyses of spectral count results derived from three proteomic pipelines (MassMatrix, MyriMatch and Proteome Discoverer). The three independent statistical analyses and integration of the results across proteomic pipelines utilized a maximum of 242 MB of real memory and was complete in 317 seconds. Files containing the raw spectral counts from each proteomic pipeline, detailed descriptions of the figures generated by MultiSpec and corresponding result tables are available in the supplemental material. The authors anticipate continuous advancement of this modular R script. Therefore, the most current version is available from the authors upon request. The version of the R script used in this study is available in Supplemental Material 13.
Materials And Methods
Experimental Data Sets
Two publicly available and previously described datasets were used to illustrate the utility of label-free spectral counting and highlight the statistical capabilities of MultiSpec.
First the dataset described by Chen et al. was used to validate that spectral counts generated by each proteomic pipeline is an acceptable approach to estimate fold changes [38]. In addition, this dataset was also utilized to evaluate the potential influence of TMM normalization on estimating fold changes. This dataset consisted of 36 human proteins spiked into a Pyrococcus furiosus (Pfu) lysate. Each cassette consisted of six human proteins from the Universal Proteome Standard from Sigma Aldrich (UPS). Each cassette was spiked into a Pfu lysate at different ratios: 1:1, 4:3, 3:5, 2:1, 4:1 and 1:8 as described in Supplemental Table I. For the purpose of this study, these spike ratios were assumed to be correct. Five technical replicates were produced for each condition (UPSA or UPSB) using a 95-minute gradient and data collected with an LTQ-Orbitrap Velos. This dataset consisting of 10 RAW files was analyzed by three separate label-free proteomic pipelines (MassMatrix, MyriMatch and Proteome Discoverer) as described below. Data were searched against a custom FASTA database containing 2,152 protein sequences: 36 Universal Proteome Standard (UPS) protein sequences, 71 common contaminant proteins and the Pfu UniProt database (08/20/2012) [38]. This custom database was concatenated to a reverse decoy database to estimate peptide and protein false discovery rates (FDR). The common constraints applied to database searches included: (1) limiting the search to b/y ions, (2) in-silico sequence digestion after Lys and Arg except those proceeding a Pro, (3) fixed modification due to carbamidomethylation of Cys (+57.0215 Da), (4) variable modifications for the formation of Glu to pyro-Glu (−18.011) and for oxidation of Met (+15.9949) and (5) precursor mass and fragment mass tolerances were set at 10 ppm and 0.6 Da, respectively. The full set of search parameters is provided in Supplemental Table 2.
Second, to highlight the ability for MultiSpec to identify unknown proteomic changes a dataset from a murine model of multiple sclerosis (EAE/Sham) was obtained from the PRoteomics IDEntifications (PRIDE) data repository [39]. The EAE/Sham dataset consisted of 18 RAW files from six biological replicates (three EAE and three Sham surgery) each analyzed in technical triplicate. The overall workflow of the data analysis is outlined in Figure 2. The EAE/Sham data were analyzed by three separate label-free proteomic pipelines (MassMatrix, MyriMatch and Proteome Discoverer), against the complete, reviewed, forward/reverse UniProt murine database (May 2014), containing 16,677 forward sequences. Like the Pfu dataset, the search parameters were harmonized across search engines (Supplemental Table 2).
Figure 2. Schematic representation of spectral count data analysis performed on EAE/Sham lysates.

This dataset consisted of 18 raw files. Three technical replicates were collected for each of the six biological replicates. Prior to MultiSpec analysis, the technical replicates were merged into one file. The number depicted at the top of the data funnel represents the total number of homologous protein groups identified by each search engine and protein grouping approach. The specifics for each approach are highlighted to the right of each respective graphic. The boxed number at the bottom of the data funnel depicts the protein groups remaining after a ten total count filter is applied. The MultiSpec statistical approach was applied to identify differential protein expression. The median q-value/FDR was chosen as a representation of statistical significance. The number of differentially expressed proteins is highlighted in the lower pie shaped region. Protein identifications are then collapsed and differential expression is validated across search engines. A minimum q-value is chosen to rank candidates across proteomic pipelines. A final result of 192 proteins were differentially expressed by the MultiSpec analysis of the EAE/Sham dataset.
Label-free Proteomic Pipelines
Leveraging multiple search engines has been shown to increase peptide and protein identifications [40–43]. In this approach three proteomic pipelines (MassMatrix, MyriMatch and Proteome Discoverer) each consisting of unique peptide spectrum match (PSM) filtering criteria, search engine and protein grouping mechanism were used to cross validate differential protein expression. The complete set of parameters for each proteomic pipeline is outlined Supplemental Table 2.
MassMatrix [12, 44–46]
RAW data was converted to mzXML data format using MSConvert in ProteoWizard (v 3.0.7494) [47, 48]. In the case of the EAE/Sham dataset, the mzXML files for the three technical replicates were merged to represent a single biological replicate. An MS spectra was mapped to not more than one peptide sequence. Peptides with a p-value less than 0.05 were retained and mapped to either forward or reverse protein sequences. Decoy and non-decoy protein identifications and their associated spectral counts were parsed and recombined using an in-house Python script as described below (Supplemental Material 3). In cases where a homologous protein group was identified, it was represented by a comma separated list of unique identifiers (UniProtIDs). The maximum protein score of the proteins in a homologous protein group was used to represent the protein group. The final harmonized protein list was ranked by protein score and filtered where each valid protein ID contained at least two unique peptides. The FDR was estimated using a target-decoy strategy and all proteins were retained until the incorporation of protein decoy exceeded the 5% protein FDR [49]. To account for the missing value problem across multiple samples, the homologous protein groups were split across search results based on their protein ranking and grouping in the database search. The regrouping used a bipartite approach similar to that described by Zhang et al. but at the protein group level rather than the peptide level [16]. A table of the database search results and protein groupings for each sample was supplied as an input. The final output was a combined spreadsheet of spectral counts with a harmonized grouping of proteins across all samples. If a homologous protein group was identified, it was represented by a comma-separated list of unique identifiers (UniProtIDs).
MyriMatch/IDPicker3 [13, 50]
MyriMatch (v 2.2.140) and IDPicker3 (v 3.1.593) are open source software suites and together constitute a proteomic analysis pipeline. For both the Pfu and EAE/Sham datasets, RAW data files were searched directly with the MyriMatch search engine and pepXML output files passed to IDPicker3. Peptides were regrouped and reassigned to protein groups adhering to peptide parsimony rules [16]. The peptide FDR was manually adjusted to produce an overall protein FDR no greater than 5%. Again, if a homologous protein group was identified, it was represented by a comma-separated list of unique identifiers (UniProtIDs).
Proteome Discoverer
Proteome Discoverer (v 1.4, ThermoFisher Scientific) and Scaffold (v 4.3.2, Proteome Software) were used to identify PSMs, perform protein inference and final protein grouping. RAW data files were loaded directly into Proteome Discoverer which uses the SEQUEST search engine [14, 51, 52]. For a given spectrum, candidate peptide matches were filtered based on precursor mass tolerance and a maximum of 500 candidates peptides were accepted for further scrutiny. Candidate peptides with fewer than 10% of the theoretical fragment ions present were considered poor candidate peptides and rejected. The remaining candidates were subjected to cross-correlation scoring analysis (Xcorr) [53]. A maximum of ten PSMs was accepted with each having an Xcorr value greater than 0.4. The ΔCn parameter or the Xcorr relative difference was set less than 0.05 to select for the best possible PSM(s). The Percolator algorithm was applied to limit PSM FDR to ≤1% [54]. As part of the Scaffold software suite, the ProteinProphet scoring algorithm was employed to regroup PSMs into homologous protein groups [19]. A harmonized protein list was produced with the Shared Peptide Grouping algorithm and filtered to include all protein group identifications with ≤5% FDR [55]. Homologous protein groups were treated the same as they were in the MassMatrix and MyriMatch/IDPicker3 proteomic pipelines. That is, a comma-separated list of unique identifiers (UniProtIDs) was constructed to represent a homologous protein group.
Spectral Count List Filtering
Regardless of which proteomic pipeline was used it is common to apply a final filtration in order to remove protein identifications that were identified in only a few biological replicates, usually represented by sparse and low count data. In the case of the EAE/Sham dataset the threshold of ten total spectral counts across the six biological replicates was deemed acceptable. It is very difficult to accurately calculate a p-value for a protein that is represented by sparse low-count data. In these tag-count approaches the calculation of p-values from sparse low-count data creates a bias of p-values near 1.0. Since an assumption when calculating a q-value is that p-values are uniformly distributed, a large proportion of large p-values may be overrepresented and bias the q-value calculation. However, it is imperative to critically evaluate data quality and filter criteria as they pertain to the individual experiment in order to avoid removing protein identifications which represent true biological differences between proteomes. It is the responsibility of the researcher to select an appropriate filter that balances both the retention of biologically relevant data and the removal of data that may bias the statistical analysis. To assist in this endeavor, MultiSpec produces a directional p-value histogram which serves as a data diagnostic plot but can also be used to identify if a given filtering threshold is not appropriate (Supplemental Material 12).
Description of the MultiSpec Statistical Platform
Serial Analysis of Gene Expression is one of the first biological techniques where the negative binomial distribution was found useful to model multivariate count data [56]. Traditional maximum-likelihood techniques based on the negative binomial distribution have been used to model count data from transcriptomic experiments but it has been suggested that they can routinely be applied to proteomic spectral count data [57]. However, current approaches used to model RNA sequencing data, namely, edgeR [26], DESeq [27] and baySeq [28] have not been routinely applied to spectral count proteomics experiments, with few exceptions [58–61]. Each of these statistical approaches is uniquely valuable when determining differential expression and can be applied to small scale discovery proteomics experiments with as few as three biological replicates. Furthermore, by leveraging multiple statistical approaches bias of one approach can be highlighted and corrected. In the MultiSpec approach, p-values calculated in edgeR and DESeq are converted into FDR estimates by calculating a q-value [62, 63]. These q-values are then directly contrasted with the Bayesian FDR calculated by BaySeq.
Normalization
Slight differences in sample type, preparation and quality can drastically affect overall count abundance, commonly referred to as the library size. In many cases differential expression of proteins between biological conditions is the primary goal. In this case, a comparison between spectral counts of two different proteins within the same sample is not a primary concern. The most simplistic approach to equalize library sizes is to redistribute counts with the use of a normalization factor. A total count approach takes the total number of spectral counts in a given run, the library size, and divides this by the mean of all library sizes. A total count normalization approach is effective in many cases. However, when proteins between biological conditions are disproportionally represented, this type of normalization can bias the results toward one of these conditions. Assuming that the majority proteins between sample conditions are not differentially expressed, an upper quartile (UQ) or weighted trimmed mean of M-values (TMM) normalization method provides a more robust approach to calculate a library size normalization factor [64, 65]. UQ normalization calculates a library-scaling factor from the 75% quantile of the counts from each library after the removal of all non-positive count values. The quantile can be adjusted as seen fit based on the experiment. TMM normalization chooses one replicate as a reference and subsequently computes a TMM factor for the remaining replicates. The TMM factor is determined after ‘trimming’ the bottom 30% log2-fold-changes and removal of the top 5% high-count data. In the case of UQ or TMM normalization, the scaling factor equalizes the library size from which pseudo-counts can be calculated. A similar normalization approach, termed relative log expression (RLE), is determined by taking the median of an RLE scaling factor. The RLE scaling factor is determined by dividing the observed counts by its geometric mean on a tagwise basis [27, 35]. The UQ, RLE and TMM approaches have been thoroughly vetted and are effective in removing bias in count data due to unequal library sizes [35]. The normalization approaches available to the tag-count based approach used in MultiSpec are as follows: edgeR = (TMM, RLE and UQ); DESeq = RLE; baySeq = TMM.
edgeR
Using a negative binomial model, edgeR addresses the issue of few biological replicates through the implementation of a ‘common’ dispersion parameter that is estimated using a quantile-adjusted conditional maximum likelihood approach [56, 57]. A common dispersion parameter is appropriate if the frequency of observed spectral counts is consistent for all proteins in a given sample, or if the number of biological replicates in the dataset is small and determining a dispersion estimate for each individual protein is not appropriate or possible. However, when three or more biological replicates are available in each condition, edgeR can calculate a tagwise dispersion estimate using an empirical Bayesian approach. In this approach, the common dispersion estimate is calculated then used as a prior and a weighted conditional likelihood estimator is used to squeeze the observed tagwise dispersion toward the common dispersion estimate [57]. Differential expression is ultimately determined with an ‘exact test’ [56]. This ‘exact test’ is similar to the conditional Fisher’s exact test but rather than determining p-values based on the hypergeometric distribution, it utilizes a negative binomial distribution [56].
DESeq
DESeq leverages a link between the sample mean and sample dispersion by assuming a local linear regression approach or by fitting a general linear model to the mean-dispersion relationship [27]. Biological replicates can be pooled within or between conditions prior to the estimation of the dispersion parameter. In other words, DESeq addresses the issue of few biological replicates effectively by borrowing data between biological replicates as well as across multiple proteins that have similar count values. Protein identifications that have these common characteristics are expected to have similar dispersion. Then, like edgeR, DESeq uses the ‘exact test’ to determine differential expression but utilizes its unique pseudo-counts to estimate the mean and dispersion to calculate the variance [27].
baySeq
The baySeq statistical algorithm assumes the data can be fit to a negative binomial distribution [28]. In classical Bayesian analyses, model parameters are considered to be random variables. A prior distribution is assumed or calculated. If the model that fits the observed data is similar to the prior distribution, then the probability that the data come from different distributions is low. This will result in a high FDR. Conversely, if the observed data cannot be modeled well by the prior distribution parameters, then it is possible that they originate from two different distributions. In this case, the probability that the data come from different distributions is large and will result in a low FDR.
Consider the case of a pair-wise study that encompasses two distinct conditions, each condition with biological replicates. On a protein-by-protein basis, a posterior distribution is calculated by bootstrapping data from all conditions assuming a negative binomial prior distribution. This model represents all sample data, both differentially expressed and non-differential data. This prior distribution, based on the negative binomial model, has a set of quasi-likelihood parameter estimates that describe the sample mean and variance. In a pairwise analysis, a second model is fit to the data where each condition is subsetted based on the experimental design. Each biological replicate is nested within the appropriate condition (subset). A posterior distribution is determined in a similar manner as they were for the entire dataset but the parameter estimates are not recalculated, rather those from the whole dataset are retained. Proteins that are not differentially expressed should fall under the same distribution but those proteins that are differentially expressed should not. The posterior probabilities where the data have different distribution means are calculated, as well as the FDR.
FDR Estimation and Cross Algorithm Equivalence Testing
There are many ways to control family-wise error rate (FWER) when performing multiple hypothesis tests simultaneously. The Bonferroni multi-test correction is a traditional approach to control FWER in the strong sense but with a substantial loss in statistical power. In a multivariate setting, strict control of FWER is only needed when a strong conclusion will be drawn based off the differential expression of one hypothesis test. In the case of many shotgun proteomics experiments, differential expression of a set of proteins centralized in a pathway, serving a similar biological function or the result of an external stimulus, is more likely to be of interest. In such a case, the control of FWER in the strong sense is unnecessarily restrictive and the use of FDR is more appropriate. The p-value is a common statistical measurement that describes the rate in which truly non-significant results are considered significant. In contrast, the FDR is the rate that a significant test is truly not significant [66]. The Benjamini-Hochberg FDR approximation is a common approach to control FWER in the rapidly developing ‘omics’ era [67]. Although this approach is valid, it is argued that it over controls in terms of FDR resulting in a loss of statistical power [68]. In the proteomics field specifically, the q-value approach has been applied to control PSM false discovery rate [62] so it was applied in the MultiSpec approach to control the protein FWER. The q-value obtained from MultiSpec can be defined as a percentage of false discoveries acceptable from corresponding p-values at a given threshold. The q-value has also been described as a posterior Bayesian p-value [69]. In this multi-model statistical approach, the q-values calculated from the edgeR and DESeq p-values are directly compared to the FDR calculations from baySeq.
Ranking Differential Expression Candidates
By comparing q-values or FDR across statistical platforms statistically divergent results can be criticized. These results are those that are found to be significant in one statistical approach but not found to be significant by an alternative, yet appropriate, approach. Although this information is very valuable, it may be difficult to interpret. While there are a variety of methods to combine p-values, no such method exists for combining q-values and FDR [70]. Therefore, in the MultiSpec approach a median q-value/FDR was used to rank final protein candidates. For simplicity, this median value is referred to as a MultiSpec q-value.
Collapsing MultiSpec Differential Expression Candidates
Using multiple database search engines (or proteomic pipelines) is a simple way to increase protein identifications [40, 41]. While a truly differentially expressed protein should be identified as differentially expressed regardless of the proteomic pipeline used, every proteomic pipeline has a unique manner to identify PSMs and infer protein identification. Leveraging multiple proteomic pipelines provides valuable information to weigh differentially expressed candidates that can assist in understanding the underlying biological differences.
An obvious limitation with simply overlapping identifications from multiple proteomic pipelines is the impact of homologous protein groups. This MultiSpec approach collapses protein identification across proteomic pipelines as highlighted in Supplemental Data 4. For example the protein groups (A,B,C), (D,E,F) and (G) were identified in ‘Pipeline 1’. In ‘Pipeline 2’, protein groups (A,B), (E,F,G) and (H) were identified. In ‘Pipeline 3’, the identified protein groups were (B,A), (D) and (G,F). Regardless of the number of proteins encompassed in a given protein group or the number of proteomic pipeline chosen, each protein group are broken into their representative parts. For instance, (D,E,F) would be broken into (D), (E) and (F). Then original protein groupings as defined by the proteomic pipelines were reapplied and the union of all the proteins taken as a representation of the protein group. So in this case (D) would be represented as (D,E,F). Likewise, (E) would be represented as (D,E,F,G). This process is repeated until the number of protein groups can no longer be reduced. In the example highlighted in Supplemental Data 4, the protein groups for pipeline 3 were originally (B,A), (D) and (G,F) are now (A,B,C) and (D,E,F,G) after two iterations of protein group collapsing. For this instance, the protein groups (D) and (G,F) were collapsed into protein group (D,E,F,G). So there are two corresponding MultiSpec q-values for the protein group (D,E,F,G) in ‘Pipeline 3’. The minimum of the two is chosen to represent the statistical significance of that protein group from ‘Pipeline 3’. Finally, the minimum MultiSpec q-values across ‘Pipelines’ is chosen as a ‘representative collapsed MultiSpec q-value’ and this value was used to produce a final ranked list of differentially expressed protein candidates.
Welch’s t-test
To evaluate a typical analysis of count data, the spectral count data were quantile normalized and then logarithm transformed after the addition of a single count to attempt to fit the data to a normal distribution [71]. Under the assumptions that the transformed data follow a normal distribution, a two-tailed Welch’s t-test is an appropriate hypothesis-based test to evaluate if two means are the same when the associated variances may be unequal. The assumption of unequal variances is appropriate because there is a known mean-variance relationship with spectral count data derived from shotgun proteomics experiments. The sample degrees of freedom are estimated using the Welch-Satterhwaite equation. This process was completed by calling t.test() in base R. To evaluate if the data follow a normal distribution, the Shapiro-Wilk test statistic can be completed based on biological condition in a tagwise fashion, assuming independence [72]. To allow direct comparison to MultiSpec platforms the FDR was determined using the same q-value approach.
Likelihood Ratio Test (G-test)
Spectral count data was TMM normalized prior to conducting a likelihood ratio test, also known as a G-test. The multivariate data was collapsed into a [2 x k] matrix to evaluate if the observed spectral counts of a given protein in k biological conditions are independent. The p-value was then calculated, assuming that the data can be fit well to a χ2 distribution with k−1 degrees of freedom using the likelihood.test() function in base R. Again to maintain consistency, q-values were calculated.
Results and Discussion
MultiSpec is multi-model statistical approach that evaluates edgeR, DESeq and baySeq statistical analyses in order to arrive at a holistic statistical representation for differential expression of proteomic count data. The approach is well suited for pairwise hypothesis testing but can be extended to analyze more sophisticated experimental designs through the use of generalized log-linear models. Statistical significance can be drawn from each statistical component individually, but by leveraging the orthogonally of each approach, the bias of individual statistical approaches can be highlighted. In order to control for multiple comparisons in multivariate data, the p-values derived by edgeR and DESeq are used to calculate q-values. The q-values are then directly compared to the FDR that is calculated by baySeq. As a holistic representation of the MultiSpec statistical approach, the median q-value/FDR from edgeR, DESeq and baySeq was calculated. This value termed as a MultiSpec q-value is used to rank differentially expressed candidate proteins.
The MultiSpec statistical analysis can be completed on the spectral counts obtained from a single proteomic pipeline. However, as illustrated below, it was intended to be used for the statistical analysis of spectral count results obtained from multiple proteomic pipelines. Using multiple proteomic pipelines is a simple way to increase protein identifications and may prove to be valuable when attempting to understand biological phenomenon. Furthermore, differentially expressed proteins that are cross-validated by multiple proteomic pipelines are strong candidates for follow-up experiments. MultiSpec uses a protein group collapsing mechanism to reduce discrepancies inherited during the assembly of homologous protein groups inferred from peptide identifications. This approach used here can easily be applied to both spectral counting and precursor intensity label-free shotgun proteomics experiments.
MultiSpec is written in the R statistical computing language (≥ v.3.0) and executable by calling the program with a set of command line arguments. The input for analysis is a table of raw spectral counts and unique protein identifiers (UniProtIDs or FASTA headers). In the case of a homologous protein group unique identifiers should be comma separated values (e.g. P10275, P19091). Spectral counts are filtered based on user supplied criteria (e.g. total counts across all biological samples, minimum counts of a given protein, etc.). Each statistical component embraces its unique normalization and statistical approaches described above. Many parameters are defaulted to traditional values but can be adjusted based on caveats within the count data. The output from the analysis is a comprehensive list of differentially expressed candidates along with the raw output from each model analysis. The script further outputs several diagnostic plots to rapidly identify departure from specific model assumptions. For example, edgeR and DESeq assume a sample mean and variance relationship and a uniform p-value distribution. As customary in proteomics data analysis, an arbitrary cut-off of 5% FDR was chosen for the analysis described herein but can be relaxed.
Database search produces robust spectral count data for label-free relative protein quantitation
Before evaluating the ability of MultiSpec to determine differential protein expression, the robustness of label-free spectral counting and the effect of TMM normalization across three proteomic database search pipelines, each with their unique software search engine, was reexamined. A benchmark dataset consisting of a Pyrococcus furiosus (Pfu) lysate spiked with 36 human proteins at six different ratios was used to evaluate the underlying accuracy and precision of spectral counting across these three different shotgun proteomics analysis applications. Five technical replicates were collected for each of the Universal Proteome Standard A (UPSA) and Universal Proteome Standard B (UPSB) samples. Under the assumption that each technical replicate represents an injection of an equivalent amount of each peptide mixture, raw spectral counts were normalized using TMM library size normalization. A strong linear relationship between the normalized counts, ‘pseudo counts’, for each database search pipeline was observed. This observation supports spectral counting as a robust approach to estimate relative protein abundance differences (Supplemental Data 5). The pseudo counts and calculated log2-fold-changes from each analysis further highlight the capability of each proteomic pipeline to independently determine accurate differences in protein abundance (Figure 1 and Supplemental Data 6). Furthermore, TMM normalization had a uniform but minimal impact on the log2-fold-changes (Supplemental Data 7). The MassMatrix and MyriMatch pipelines identified peptides from all UPS proteins. However, the Proteome Discoverer pipeline was unable to identify peptides in at least one technical replicate for five UPS proteins (P62988 from cassette 1, P01133 from cassette 2, P00709 from cassette 3 and P02741/P10145 from cassette 6). As a measure of the robustness of spectral counting, Spearman rank correlations were calculated for the log2-fold-changes of TMM normalized spectral counts. The Spearman rank correlations for MassMatrix and MyriMatch pipelines were 0.955 and 0.938, respectively. After removal of proteins with missing log2-fold-change values from the Proteome Discoverer results, the Spearman rank correlation was 0.959 (Supplemental Data 8). These data support that each proteomic software pipeline is capable of estimating log2-fold-change values from pseudo counts after TMM normalization.
Figure 1. Label-free spectral counts derived from MassMatrix, MyriMatch and Proteome Discoverer proteomic pipelines robustly estimate true fold changes.

(a) Plot of the average log2-fold-changes (UPSB:UPSA) derived from TMM normalized spectral counts. The vertical dashed lines indicate the true spike ratios for each cassette. The UniProtID is aligned and color-coded by cassette. If the protein was not identified in one condition the corresponding log2-fold-change was not calculated. (b) Plot of TMM normalized counts from cassette 5. The expected spike ratio for cassette 5 is 4:1 (UPSA:UPSB). UPSA is the baseline, so the expected log2-fold-change (UPSB:UPSA) for cassette 5 is −2.0.
While the Pfu experiment allows one to test the robustness of spectral count quantitation and the influence that TMM normalization has on the determining log2-fold-changes, a limitation is that only technical variability was introduced. Both edgeR and DESeq platforms are intended to be applied to samples with biological variability. In the absence of biological variability the variance between UPSA and UPSB conditions is constant. In this instance of constant variance, log2-fold-changes can be calculated and the effects of normalization can be investigated but a dispersion parameter can not be estimated. Without an accurate dispersion parameter estimate, it is not possible to calculate a p-value and hence not possible to perform a true differential expression analysis. The EAE/Sham dataset was used to illustrate the full capabilities of MultiSpec.
MultiSpec statistical analysis identifies differential expression in a murine experimental autoimmune encephalomyelitis (EAE/Sham) model
The utility of the MultiSpec multi-model statistical analysis was evaluated using a public dataset of 18 RAW files for spinal chord lysates collected from a murine model of experimental autoimmune encephalomyelitis (EAE) and a corresponding sham surgery control (Sham) were downloaded from the PRoteomics IDEntifications (PRIDE) data repository [39]. The flow a typical MultiSpec analysis is outlined in Figure 2.
The first step to analyze a shotgun proteomics dataset with the MultiSpec analytical workflow is to choose a set of proteomic pipelines. In this example, proteomic pipelines were assembled for MassMatrix, MyriMatch and Proteome Discoverer which identified 1156, 1228 and 1118 proteins, respectively. After requiring a ten total count filter across the six biological replicates 1,064, 1,026, and 849 protein groups remained. MultiSpec differential expression analysis was completed with the protein count data obtained from each proteomic pipeline independent of the other pipelines. The data from the retained protein groups were then normalized (see above for details about normalization).
Again, MultiSpec differential expression analysis combines three separate yet orthogonal statistical approaches: edgeR, DESeq and baySeq. For edgeR and baySeq analyses, TMM normalization was conducted while RLE normalization was conducted in DESeq. Using normalized counts (pseudo-counts) tag-wise and local-linear regression approaches were used to estimate the dispersion in edgeR and DESeq respectively. BaySeq posterior distributions were calculated in a tagwise fashion, under both models, by bootstrapping 1,000 iterations and assuming a negative binomial prior distribution. The output of each statistical approach can be used separately to rank protein candidates. However, in order to control for false discoveries p-values as determined by edgeR and DESeq were converted to q-values [62, 66, 68, 69]. In MultiSpec, q-values from edgeR and DESeq are directly contrasted with baySeq FDR and the median of these three values is chosen to represent statistical significance. The median q-value, or ‘MultiSpec q-value’, is then used to rank candidates. In general, a median value is more robust to outliers than is the mean [70]. Furthermore the use of the median value is advantageous when three orthogonal statistical approaches are used because a protein must be determined as significant in at least two of the approaches. So in this case, the MultiSpec q-value is a holistic representation of each individual statistical approach.
In order to evaluate the origin from which the MultiSpec q-value is chosen, the program script produces a Venn diagram and origin/rank plot (Figure 3A and 3B). For the MultiSpec analysis from count data derived by the MassMatrix proteomic pipeline, the median q-value is often selected from DESeq. At the MultiSpec q-value cut-off of 0.05, edgeR tends to produce lower q-values and baySeq tends to produce higher q-values. However, at higher MultiSpec q-value, the influence of baySeq appears to be stronger but the trend of edgeR producing lower q-values is consistent (Supplemental Data 9). The overall effect of employing a median q-value approach is best supported by evaluating q-values from the entire dataset. The program script produces a Spearman rank correlation matrix (Supplemental Data 10). From this matrix, it is clear that overall edgeR tends to produce lower q-values while DESeq produces higher q-values. Also, in general the baySeq FDR is chosen to represent the MultiSpec q-value but not at lower q-values. This is likely an artifact introduced by the selection of these statistical platforms. While edgeR and DESeq both leverage the negative-binomial distribution to perform hypothesis tests, the most significant results are represented by very small q-values. However, baySeq FDR calculations are capped at 1 × 10−8 by default. In this example, using the MassMatrix count data, there are 76 proteins that were determined to be significant only by edgeR at a q-value ≤ 0.05. The MultiSpec edgeR Orphan Plot compares the edgeR q-value against the DESeq q-value or baySeq FDR (Figure 3C). This enables a quick evaluation of how edgeR deviates from DESeq and baySeq and why these proteins were chosen to be excluded in the MultiSpec approach. This result is not unexpected. One criticism of edgeR is that it is anti-conservative for differences especially in regards to low counts [27]. Furthermore, as seen in Supplemental Data 11, a large proportion of edgeR significant tests are derived from low count data (average counts <10). This is a prime example of how edgeR statistical analyses can benefit greatly by corroboration with other statistical approaches and why this multi-model statistical approach to analyze spectral count data is advantageous.
Figure 3. MultiSpec Differential Expression Analysis of spectral counts derived from the MassMatrix analysis of EAE/Sham lysates.

(a) The Origin/Rank Plot highlights the relationships between the tag-count approaches used by MultiSpec. This two part figure describes the q-value/FDR as a function of the final MultiSpec q-value/rank. The top portion indicates which statistical platform (edgeR, DESeq or baySeq) the MultiSpec q-value is derived from while the bottom portion tracks the q-values from each individual statistical approach. (b) Venn diagram of the results derived from MultiSpec. The proteins identified as statistically significant by the MultiSpec approach must have a significant q-value/FDR in at least two of the three statistical approaches. The proteins that met this criteria are highlighted in the gray shaded oval. Proteins that were identified solely by one statistical approach are considered ‘orphans’ and are not significant in the MultiSpec approach. (c) The Orphan Plot allows a quick evaluation of the statistical outputs of orphan candidates. In the edgeR Orphan plot the edgeR q-value is plotted on the x-axis while the DESeq q-values (red) and the baySeq FDR (blue) are plotted on the y-axes.
The MultiSpec approach leverages multiple proteomic pipelines to cross-validate differential expression profiles
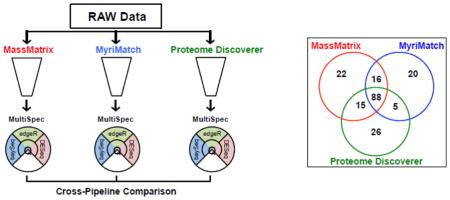
Using multiple database search engines is a simple approach to increase protein identifications [40, 41]. Once MultiSpec q-values are determined from several proteomic pipelines (MassMatrix, MyriMatch and Proteome Discoverer for the EAE/Sham lysate example), differentially expressed candidates are cross-validated. Prior to cross-pipeline validation homologous protein groups are collapsed as described above and in Supplemental Data 4. This approach effectively removes potential discontinuities related to protein group labeling, which is an inherited result of how each proteomic pipeline performs peptide-spectral matching/filtering and protein inference/filtering. For the MultiSpec analytical approach, the minimum MultiSpec q-value is chosen to represent a collapsed protein group within each proteomic pipeline. The results from multiple proteomic pipelines are then overlaid as shown in Figure 4D. At this point, the minimum MultiSpec q-value is chosen to represent the ‘Collapsed MultiSpec q-value’. This approach essentially takes the union of all differentially expressed proteins from every proteomic pipeline. Alternatively, the maximum or median MultiSpec q-value can be chosen as the ‘Collapsed MultiSpec q-value’ to assemble a final ranking system.
Figure 4. Cross-Pipeline Validation of MultiSpec Differential Expression Analyses.

(a) For each protein log2-fold-change depicted in panel (b) a gold point is drawn that represents if the protein was identified by the indicated proteomic pipeline. (b) MultiSpec fold-change plot of the median log2-fold-change of all proteins found to meet the MultiSpec q-value threshold (≤ 0.05) sorted from smallest to largest. Proteins identified by three, two or one pipeline(s) are green triangles, blue circles or red squares. (c) Venn diagram of significant protein groups across pipelines prior to cross-pipeline validation. A total of 1322 unique protein groups were identified. (d) Venn diagram of significant protein groups across pipelines after cross-pipeline validation. A total of 287 redundant protein groups were collapsed.
Overall the MassMatrix, MyriMatch and Proteome Discoverer pipelines identified a total of 1,322 protein groups prior to collapsing protein groups (Figure 4C). Collapsing protein groups removed 287 (~22%) redundant protein identifications. After implementing a significance threshold, (‘Collapsed MultiSpec q-value’ ≤ 0.05) 192 proteins were ranked (Figure 4D). These proteins are then ordered by the median log2-fold-change as calculated by edgeR. The ‘Collapsed Median Log2-Fold-Change Plot’ and ‘Pipeline Origin of Differential Expression’ is automatically produced by the MultiSpec script (Figures 4A and 4B). Each of the 192 proteins highlighted in the ‘Collapsed Median Log2-Fold-Change Plot’ is color coded by how many proteomic pipelines identified that protein, regardless of statistical significance. By relaxing the requirement that a protein must meet an arbitrary cutoff to be considered, it allows the entire dataset to be analyzed as a whole. The ‘Collapsed Median Log2-Fold-Change Plot’ indicates that the vast majority of significant proteins were identified by all three proteomic pipelines. After a detailed interrogation of the data table output 176/192 proteins were identified by three proteomic pipelines. Only 10/192 and 6/192 proteins were identified by two or only one proteomic pipelines. Although it is not possible to unequivocally determine that protein inference is correct, this cross validation approach provides additional support that the majority of significant protein identifications are at least uniformly identified and not an artifact of the applied bioinformatics.
Diagnostics Output and Visualization of Differential Expression
Each statistical package in MultiSpec provides a series of automated diagnostic and differential expression plots. The correct interpretation of diagnostic plots is vital because they are an indication of how well the data fits the model that is being used to perform hypothesis testing and ultimately arrive at statistical significance. Furthermore, several plots used to visualize data are automatically produced throughout the MultiSpec analysis including those shown in Figure 3 and Figure 4. MultiSpec utilizes the functionality and flexibility of R (v. 3.0) by calling edgeR [26, 56, 57, 73], DESeq [27], baySeq [28], limma [74], qvalue [63], optparse [75], caroline [76], MASS [77] and sm [78] packages. Detailed instructions on how to call MultiSpec from command line as well as an outline describing each plot that is automatically generated can be found in the ‘MultiSpec quiet tutorial’ (Supplemental Data 12). All of the data, including Raw spectral counts can be found in Supplemental Data 13.
Conclusion
Label-free shotgun proteomics has been an effective tool to profile protein expression between biological conditions. Although technological and methodological advancements continue to propel the proteomics field, there remains a need for hypothesis-test based statistical means to identify biological candidates for follow-up. The MultiSpec statistical approach enables hypothesis testing of proteomic spectral count data generated from any proteomic software suite. MultiSpec utilizes the tag-count analyses from edgeR, DESeq and baySeq. A median q-value/FDR (‘MultiSpec q-value) is chosen as a holistic representation of this multi-model approach providing an added level of data scrutiny. In the case at hand, the anti-conservative nature of edgeR is highlighted specifically in regards to proteins represented by few spectral counts. The utility of the MultiSpec statistical analysis is extended by integrating results from multiple proteomic pipelines. The protein group collapsing mechanism described here runs seamlessly with the MultiSpec statistical analysis but can be utilized to integrate results from label-free precursor intensities or label-based approaches. It is recommended that the MultiSpec approach be used to analyze spectral count data from multiple proteomic pipelines and implementation considered as a routine statistical approach.
Supplementary Material
Biological Significance.
Spectral count data from shotgun proteomics experiments is semi-quantitative and semi-random, yet a robust way to estimate protein concentration. Tag-count approaches are routinely used to analyze RNA sequencing data sets. This approach, termed MultiSpec, utilizes multiple tag-count based statistical tests to determine differential protein expression from spectral counts. The statistical results from these tag-count approaches are combined in order to reach a final MultiSpec q-value to re-rank protein candidates. This re-ranking procedure is completed to remove bias associated with a single approach in order to better understand the true proteomic differences driving the biology in question. The MultiSpec approach can be extended to multiple proteomic pipelines. In such an instance, MultiSpec statistical results are integrated by collapsing protein groups across proteomic pipelines to provide a single ranked list of differentially expressed proteins. This integration mechanism is seamlessly integrated with the statistical analysis and provides the means to cross-validate protein inferences from multiple proteomic pipelines.
Highlights.
Concise review of tag-count based approaches traditionally used in RNA sequencing data sets. Focus has been shifted to their use in determining protein differential expression from spectral count based shotgun proteomics experiments.
Mechanism to collapse protein groups which enables comparisons across proteomic pipelines.
Introduction of a q-value combinatorial approach for the comparison of statistical significance across multiple statistical approaches.
Acknowledgments
This work was supported by the Ohio State University Pelotonia Bridge grant (M.A.F.) and in part from NIH grant CA107106.
Abbreviations
- PSM(s)
Peptide Spectrum Match(es)
- Pfu
Pyrococcus furiosus
- UPS
Universal Proteome Standard Mix
- PRIDE
Proteomics IDEntifications data repository
- EAE
Experimental Autoimmune Encephalomyelitis
- FDR
False Discovery Rate
- Xcorr
Cross-Correlation Scoring Analysis
- UQ
Upper Quartile
- TMM
Trimmed Mean of M-Values
- RLE
Relative Log Expression
- FWER
Family-Wise Error Rate
- MDS
Multidimensional Scaling
- CPM
Counts per Million Counts
Footnotes
Availability
The current version of the MultiSpec R script is included in Supplementary Data 13.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mallick P, Kuster B. Proteomics: a pragmatic perspective. Nature biotechnology. 2010;28:695–709. doi: 10.1038/nbt.1658. [DOI] [PubMed] [Google Scholar]
- 2.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Analytical chemistry. 2004;76:4193–201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 3.Lundgren DH, Hwang SI, Wu L, Han DK. Role of spectral counting in quantitative proteomics. Expert review of proteomics. 2010;7:39–53. doi: 10.1586/epr.09.69. [DOI] [PubMed] [Google Scholar]
- 4.Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, et al. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Molecular & cellular proteomics : MCP. 2005;4:1487–502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- 5.Patel VJ, Thalassinos K, Slade SE, Connolly JB, Crombie A, Murrell JC, et al. A comparison of labeling and label-free mass spectrometry-based proteomics approaches. J Proteome Res. 2009;8:3752–9. doi: 10.1021/pr900080y. [DOI] [PubMed] [Google Scholar]
- 6.Usaite R, Wohlschlegel J, Venable JD, Park SK, Nielsen J, Olsson L, et al. Characterization of global yeast quantitative proteome data generated from the wild-type and glucose repression saccharomyces cerevisiae strains: the comparison of two quantitative methods. J Proteome Res. 2008;7:266–75. doi: 10.1021/pr700580m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carvalho PC, Hewel J, Barbosa VC, Yates JR., 3rd Identifying differences in protein expression levels by spectral counting and feature selection. Genetics and molecular research : GMR. 2008;7:342–56. doi: 10.4238/vol7-2gmr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang B, VerBerkmoes NC, Langston MA, Uberbacher E, Hettich RL, Samatova NF. Detecting differential and correlated protein expression in label-free shotgun proteomics. J Proteome Res. 2006;5:2909–18. doi: 10.1021/pr0600273. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Wen Z, Washburn MP, Florens L. Effect of dynamic exclusion duration on spectral count based quantitative proteomics. Analytical chemistry. 2009;81:6317–26. doi: 10.1021/ac9004887. [DOI] [PubMed] [Google Scholar]
- 10.Florens L, Carozza MJ, Swanson SK, Fournier M, Coleman MK, Workman JL, et al. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods. 2006;40:303–11. doi: 10.1016/j.ymeth.2006.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin S, Daly DS, Springer DL, Miller JH. The effects of shared peptides on protein quantitation in label-free proteomics by LC/MS/MS. J Proteome Res. 2008;7:164–9. doi: 10.1021/pr0704175. [DOI] [PubMed] [Google Scholar]
- 12.Xu H, Freitas MA. A mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC bioinformatics. 2007;8:133. doi: 10.1186/1471-2105-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabb DL, Fernando CG, Chambers MC. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J Proteome Res. 2007;6:654–61. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994;5:976–89. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 15.Cooper B, Feng J, Garrett WM. Relative, label-free protein quantitation: spectral counting error statistics from nine replicate MudPIT samples. Journal of the American Society for Mass Spectrometry. 2010;21:1534–46. doi: 10.1016/j.jasms.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Zhang B, Chambers MC, Tabb DL. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J Proteome Res. 2007;6:3549–57. doi: 10.1021/pr070230d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Wen Z, Washburn MP, Florens L. Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Analytical chemistry. 2010;82:2272–81. doi: 10.1021/ac9023999. [DOI] [PubMed] [Google Scholar]
- 18.Zhou JY, Schepmoes AA, Zhang X, Moore RJ, Monroe ME, Lee JH, et al. Improved LC-MS/MS spectral counting statistics by recovering low-scoring spectra matched to confidently identified peptide sequences. J Proteome Res. 2010;9:5698–704. doi: 10.1021/pr100508p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Analytical chemistry. 2003;75:4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 20.Choi H, Fermin D, Nesvizhskii AI. Significance analysis of spectral count data in label-free shotgun proteomics. Molecular & cellular proteomics : MCP. 2008;7:2373–85. doi: 10.1074/mcp.M800203-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Booth JG, Eilertson KE, Olinares PD, Yu H. A bayesian mixture model for comparative spectral count data in shotgun proteomics. Molecular & cellular proteomics : MCP. 2011;10 doi: 10.1074/mcp.M110.007203. M110 007203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li M, Gray W, Zhang H, Chung CH, Billheimer D, Yarbrough WG, et al. Comparative shotgun proteomics using spectral count data and quasi-likelihood modeling. J Proteome Res. 2010;9:4295–305. doi: 10.1021/pr100527g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pham TV, Piersma SR, Warmoes M, Jimenez CR. On the beta-binomial model for analysis of spectral count data in label-free tandem mass spectrometry-based proteomics. Bioinformatics. 2010;26:363–9. doi: 10.1093/bioinformatics/btp677. [DOI] [PubMed] [Google Scholar]
- 24.Pavelka N, Fournier ML, Swanson SK, Pelizzola M, Ricciardi-Castagnoli P, Florens L, et al. Statistical similarities between transcriptomics and quantitative shotgun proteomics data. Molecular & cellular proteomics : MCP. 2008;7:631–44. doi: 10.1074/mcp.M700240-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Webb-Robertson BJ, Matzke MM, Datta S, Payne SH, Kang J, Bramer LM, et al. Bayesian Proteoform Modeling Improves Protein Quantification of Global Proteomic Measurements. Molecular & cellular proteomics : MCP. 2014 doi: 10.1074/mcp.O113.030932. [DOI] [PubMed] [Google Scholar]
- 26.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anders S, Huber W. Differential expression analysis for sequence count data. Genome biology. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hardcastle TJ, Kelly KA. baySeq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC bioinformatics. 2010;11:422. doi: 10.1186/1471-2105-11-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Auer PL, Doerge RW. A Two-Stage Poisson Model for Testing RNA-Seq Data. Statistical Applications in Genetics and Moledular Biology. 2011;10 [Google Scholar]
- 30.Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–8. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 31.Di Y, Shafer DW, Cumbie JS, Chang JH. The NBP Negative Binomial Model for Assessing Differential Gene Expression from RNA-Seq. Statistical Applications in Genetics and Moledular Biology. 2011;10 [Google Scholar]
- 32.Guo Y, Li CI, Ye F, Shyr Y. Evaluation of read count based RNAseq analysis methods. BMC genomics. 2013;14(Suppl 8):S2. doi: 10.1186/1471-2164-14-S8-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robles JA, Qureshi SE, Stephen SJ, Wilson SR, Burden CJ, Taylor JM. Efficient experimental design and analysis strategies for the detection of differential expression using RNA-Sequencing. BMC genomics. 2012;13:484. doi: 10.1186/1471-2164-13-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kvam VM, Liu P, Si Y. A comparison of statistical methods for detecting differentially expressed genes from RNA-seq data. American journal of botany. 2012;99:248–56. doi: 10.3732/ajb.1100340. [DOI] [PubMed] [Google Scholar]
- 35.Dillies MA, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Briefings in bioinformatics. 2013;14:671–83. doi: 10.1093/bib/bbs046. [DOI] [PubMed] [Google Scholar]
- 36.Guo Y, Zhao S, Ye F, Sheng Q, Shyr Y. MultiRankSeq: multiperspective approach for RNAseq differential expression analysis and quality control. BioMed research international. 2014;2014:248090. doi: 10.1155/2014/248090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome biology. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen YY, Chambers MC, Li M, Ham AJ, Turner JL, Zhang B, et al. IDPQuantify: combining precursor intensity with spectral counts for protein and peptide quantification. J Proteome Res. 2013;12:4111–21. doi: 10.1021/pr400438q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dagley LF, Croft NP, Isserlin R, Olsen JB, Fong V, Emili A, et al. Discovery of novel disease-specific and membrane-associated candidate markers in a mouse model of multiple sclerosis. Molecular & cellular proteomics : MCP. 2014;13:679–700. doi: 10.1074/mcp.M113.033340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shteynberg D, Deutsch EW, Lam H, Eng JK, Sun Z, Tasman N, et al. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Molecular & cellular proteomics : MCP. 2011;10 doi: 10.1074/mcp.M111.007690. M111 007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shteynberg D, Nesvizhskii AI, Moritz RL, Deutsch EW. Combining results of multiple search engines in proteomics. Molecular & cellular proteomics : MCP. 2013;12:2383–93. doi: 10.1074/mcp.R113.027797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones AR, Siepen JA, Hubbard SJ, Paton NW. Improving sensitivity in proteome studies by analysis of false discovery rates for multiple search engines. Proteomics. 2009;9:1220–9. doi: 10.1002/pmic.200800473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alves G, Wu WW, Wang G, Shen RF, Yu YK. Enhancing peptide identification confidence by combining search methods. J Proteome Res. 2008;7:3102–13. doi: 10.1021/pr700798h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu H, Yang L, Freitas MA. A robust linear regression based algorithm for automated evaluation of peptide identifications from shotgun proteomics by use of reversed-phase liquid chromatography retention time. BMC bioinformatics. 2008;9:347. doi: 10.1186/1471-2105-9-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu H, Freitas MA. MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics. 2009;9:1548–55. doi: 10.1002/pmic.200700322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu H, Freitas MA. Monte carlo simulation-based algorithms for analysis of shotgun proteomic data. J Proteome Res. 2008;7:2605–15. doi: 10.1021/pr800002u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–6. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, et al. A cross-platform toolkit for mass spectrometry and proteomics. Nature biotechnology. 2012;30:918–20. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods in molecular biology. 2010;604:55–71. doi: 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma ZQ, Dasari S, Chambers MC, Litton MD, Sobecki SM, Zimmerman LJ, et al. IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J Proteome Res. 2009;8:3872–81. doi: 10.1021/pr900360j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yates JR, 3rd, Eng JK, McCormack AL, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Analytical chemistry. 1995;67:1426–36. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- 52.Yates JR, 3rd, Eng JK, McCormack AL. Mining genomes: correlating tandem mass spectra of modified and unmodified peptides to sequences in nucleotide databases. Analytical chemistry. 1995;67:3202–10. doi: 10.1021/ac00114a016. [DOI] [PubMed] [Google Scholar]
- 53.MacCoss MJ, Wu CC, Yates JR., 3rd Probability-based validation of protein identifications using a modified SEQUEST algorithm. Analytical chemistry. 2002;74:5593–9. doi: 10.1021/ac025826t. [DOI] [PubMed] [Google Scholar]
- 54.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nature methods. 2007;4:923–5. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 55.Searle BC. Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics. 2010;10:1265–9. doi: 10.1002/pmic.200900437. [DOI] [PubMed] [Google Scholar]
- 56.Robinson MD, Smyth GK. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics. 2008;9:321–32. doi: 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- 57.Robinson MD, Smyth GK. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23:2881–7. doi: 10.1093/bioinformatics/btm453. [DOI] [PubMed] [Google Scholar]
- 58.Johnson EK, Zhang L, Adams ME, Phillips A, Freitas MA, Froehner SC, et al. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PloS one. 2012;7:e43515. doi: 10.1371/journal.pone.0043515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shapiro JP, Biswas S, Merchant AS, Satoskar A, Taslim C, Lin S, et al. A quantitative proteomic workflow for characterization of frozen clinical biopsies: laser capture microdissection coupled with label-free mass spectrometry. Journal of proteomics. 2012;77:433–40. doi: 10.1016/j.jprot.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harshman SW, Canella A, Ciarlariello PD, Rocci A, Agarwal K, Smith EM, et al. Characterization of multiple myeloma vesicles by label-free relative quantitation. Proteomics. 2013;13:3013–29. doi: 10.1002/pmic.201300142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fei SS, Wilmarth PA, Hitzemann RJ, McWeeney SK, Belknap JK, David LL. Protein database and quantitative analysis considerations when integrating genetics and proteomics to compare mouse strains. J Proteome Res. 2011;10:2905–12. doi: 10.1021/pr200133p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kall L, Storey JD, MacCoss MJ, Noble WS. Posterior error probabilities and false discovery rates: two sides of the same coin. J Proteome Res. 2008;7:40–4. doi: 10.1021/pr700739d. [DOI] [PubMed] [Google Scholar]
- 63.Dabney A, Storey JD, Warnes GR. R package version 1.24.20. 2010. qvalue: Q-value estimation for false discovery rate control. [Google Scholar]
- 64.Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome biology. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Muliple Testing. Journal of the Royal Statistical Society. 1995;57:289–300. [Google Scholar]
- 68.Storey JD. A direct approach to false discovery rates. Journal of the Royal Statistical Society. 2002;64:479–98. [Google Scholar]
- 69.Storey JD. The Positive False Discovery Rate: A Bayesian Interpretation and the q-Value. The Annals of Statistics. 2003;31:2013–35. [Google Scholar]
- 70.Chang LC, Lin HM, Sibille E, Tseng GC. Meta-analysis methods for combining multiple expression profiles: comparisons, statistical characterization and an application guideline. BMC bioinformatics. 2013;14:368. doi: 10.1186/1471-2105-14-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–93. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 72.Royston P. Remark AS R94: A Remark on Algorithm AS181: The W-test for Normality. Journal of the Royal Statistical Society Series C (Applied Statistics) 1995;44:547–51. [Google Scholar]
- 73.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic acids research. 2012;40:4288–97. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smyth G. Limma: linear models for microarray data. Bioinformatics and Computational Biology Solutions using R and Bioconductor. 2005:397–420. [Google Scholar]
- 75.Davis TL. optparse: Command line option parser. R package version 1.2.0. 2014 Avaliable online at: http://CRAN.R-project.org/package=optparse.
- 76.Schruth DM. caroline: A Collection of Database, Data Structure, Visualization and Utility Functions for R. R package version 0.7.6. 2013 Avaliable online at: http://CRAN.R-project.org/package=caroline.
- 77.Venables WN, Ripley BD. Modern applied statistics with S. 4. New York: Springer; 2002. [Google Scholar]
- 78.Bowman AWaAA. R package ‘sm’: nonparametric smoothing methods (version 2.2–5.4) 2014 Avaliable online at: http://www.stats.gla.ac.uk/~adrian/sm and http://azzalini.stat.unipd.it/Book_sm.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.