

Abstract
Multicellular systems develop from single cells through distinct lineages. However, current lineage tracing approaches scale poorly to whole, complex organisms. Here we use genome editing to progressively introduce and accumulate diverse mutations in a DNA barcode over multiple rounds of cell division. The barcode, an array of CRISPR/Cas9 target sites, marks cells and enables the elucidation of lineage relationships via the patterns of mutations shared between cells. In cell culture and zebrafish, we show that rates and patterns of editing are tunable, and that thousands of lineage-informative barcode alleles can be generated. By sampling hundreds of thousands of cells from individual zebrafish, we find that most cells in adult organs derive from relatively few embryonic progenitors. In future analyses, genome editing of synthetic target arrays for lineage tracing (GESTALT) can be used to generate large-scale maps of cell lineage in multicellular systems for normal development and disease.
Introduction
The tracing of cell lineages was pioneered in nematodes by Charles Whitman in the 1870s, at a time of controversy surrounding Ernst Haeckel’s theory of recapitulation, which argued that embryological development paralleled evolutionary history (1). This line of work culminated a century later in the complete description of mitotic divisions in the roundworm C. elegans - a tour de force facilitated by its visual transparency as well as the modest size and invariant nature of this nematode’s cell lineage (2).
Over the past century, a variety of creative methods have been developed for tracing cell lineage in developmentally complex organisms (3). In general, subsets of cells are marked and their descendants followed as development progresses. The ways in which cell marking has been achieved include dyes and enzymes (4–6), cross-species transplantation (7), recombinase-mediated activation of reporter gene expression (8, 9), insertion of foreign DNA (10–12), and naturally occurring somatic mutations (13–15). However, despite many powerful applications, these methods have limitations for the large-scale reconstruction of cell lineages in multicellular systems. For example, dye and reporter gene-based cell marking are uninformative with respect to the lineage relationships between descendent cells. Furthermore, when two or more cells are independently but equivalently marked, the resulting multitude of clades cannot be readily distinguished from one another. Although these limitations can be overcome in part with combinatorial labeling systems (16, 17) or through the introduction of diverse DNA barcodes (10–12), these strategies fall short of a system for inferring lineage relationships throughout an organism and across developmental time. In contrast, methods based on somatic mutations have this potential, as they can identify lineages and sub-lineages within single organisms (13, 18). However, somatic mutations are distributed throughout the genome, necessitating whole genome sequencing (14, 15), which is expensive to scale beyond small numbers of cells and not readily compatible with in situ readouts (19, 20).
What are the requirements for a system for comprehensively tracing cell lineages in a complex multicellular system? First, it must uniquely and incrementally mark cells and their descendants over many divisions and in a way that does not interfere with normal development. Second, these unique marks must accumulate irreversibly over time, allowing the reconstruction of lineage trees. Finally, the full set of marks must be easily read out in each of many single cells.
We hypothesized that genome editing, which introduces diverse, irreversible edits in a highly programmable fashion (21), could be repurposed for cell lineage tracing in a way that realizes these requirements. To this end, we developed genome editing of synthetic target arrays for lineage tracing (GESTALT), a method that uses CRISPR/Cas9 genome editing to accumulate combinatorial sequence diversity to a compact, multi-target, densely informative barcode. Edited barcodes can be efficiently queried by a single sequencing read from each of many single cells (Fig. 1A). In both cell culture and in the zebrafish Danio rerio, we demonstrate the generation of thousands of uniquely edited barcodes that can be related to one another to reconstruct cell lineage relationships. In adult zebrafish, we observe that the majority of cells of each organ are derived from a small number of progenitor cells. Furthermore, ancestral progenitors, inferred on the basis of shared edits amongst subsets of derived alleles, make highly non-uniform contributions to germ layers and organ systems.
Figure 1. Genome editing of synthetic target arrays for lineage tracing (GESTALT).
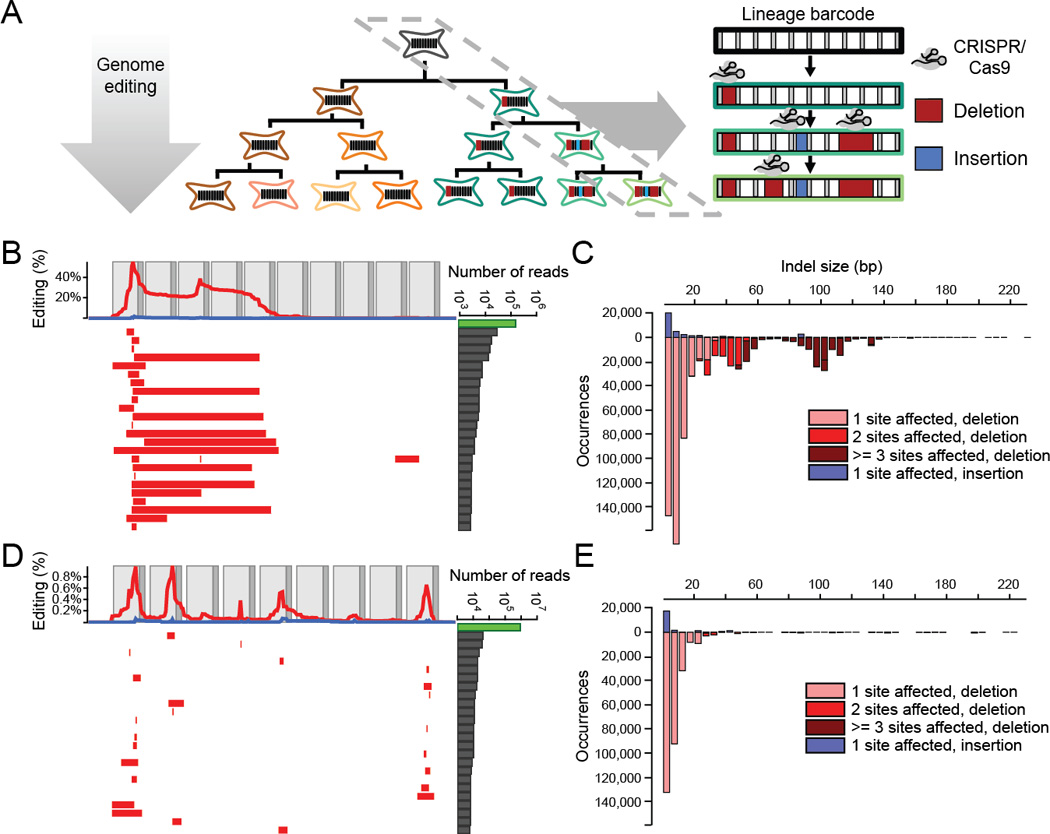
(A) An unmodified array of CRISPR/Cas9 target sites (i.e., a barcode) is engineered into a genome (gray cell). Editing reagents are introduced during expansion of cell culture or in vivo development of an organism, resulting in a unique pattern of insertions and deletions (right), and are stably accumulated in specific lineages (green cell lineage). The lineage relationships of alleles that differ in sequence can often be inferred on the basis of these accumulated edits. (B) The 25 most frequent alleles from the edited v1 barcode are shown. Each row corresponds to a unique sequence, with red bars indicating deleted regions and blue bars indicating insertion positions. Blue bars begin at the insertion site, with their width proportional to the size of the insertion, which will rarely obscure immediately adjacent deletions. The number of reads observed for each allele is plotted at the right (log10 scale; the green bar corresponds to the unedited allele). The frequency at which each base is deleted (red) or flanks an insertion (blue) is plotted at the top. Light gray boxes indicate the location of CRISPR protospacers while dark gray boxes indicate PAM sites. For the v1 array, inter-target deletions involving sites 1, 3 and 5, or focal (single target) edits of sites 1 and 3 were observed predominantly. (C) A histogram of the size distribution of insertion (top) and deletion (bottom) edits to the v1 array is shown. The colors indicate the number of target sites impacted. Although most edits are short and impact a single target, a substantial proportion of edits are inter-target deletions. (D) We tested three array designs in addition to v1, each comprising nine to ten weaker off-target sites for the same sgRNA (v2-v4) (22). Editing of the v2 array is shown with layout as described in panel (B). Editing of the v3 and v4 array are shown in fig. S3A and B. The weaker sites within these alternative designs exhibit lower rates of editing than the v1 array, but also a much lower proportion of inter-target deletions. (E) A histogram of the size distribution of insertion (top) and deletion (bottom) edits to the v2 array is shown. In contrast with the v1 array, almost all edits impact only a single target.
Results
Combinatorial and cumulative editing of a compact genomic barcode in cultured cells
To test whether genome editing can be used to generate a combinatorial diversity of mutations within a compact region, we synthesized a contiguous array of ten CRISPR/Cas9 targets separated by 3 base-pair (bp) linkers (total length of 257 bp). The first target perfectly matched one single guide RNA (sgRNA), whereas the remainder were off-target sites for the same sgRNA, ordered from highest to lowest activity (22). This array of targets (‘v1 barcode’) was cloned downstream of an EGFP reporter in a lentiviral construct (23). We then transduced HEK293T cells with lentivirus and used FACS to purify an EGFP-v1 positive population. To edit the barcode, we co-transfected these cells with a plasmid expressing Cas9 and the sgRNA and a vector expressing Discosoma red fluorescent protein (DsRed). Cells were sorted three days post-transfection for high DsRed expression, and genomic DNA (gDNA) was harvested on day 7. The v1 barcode was PCR amplified and the resulting amplicons subjected to deep sequencing.
To minimize confounding sequencing errors, which are primarily substitutions, we analyzed edited barcodes for only insertion-deletion changes relative to the ‘wild-type’ v1 barcode. In this first experiment, we observed 1,650 uniquely edited barcodes (each observed in ≥25 reads) with diverse edits concentrated at the expected Cas9 cleavage sites, predominantly inter-target deletions involving sites 1, 3 and 5, or focal edits of sites 1 and 3 (Fig. 1B and C, and table S1). These results show that combinatorial editing of the barcode can give rise to a large number of unique sequences, i.e. “, alleles”.
To evaluate reproducibility, we transfected the same editing reagents to cultures expanded from three independent EGFP-v1 positive clones. Targeted RT-PCR and sequencing of EGFP-v1 RNA showed similar distributions of edits to the v1 barcode in the transcript pool, between replicates as well as in comparison to the previous experiment (fig. S1). These results show that the observed editing patterns are largely independent of the site of integration and that edited barcodes can be queried from either RNA or DNA.
To evaluate how editing outcomes vary as a function of Cas9 expression, we co-transfected EGFP-v1 positive cells with a plasmid expressing Cas9 and the sgRNA as well as an DsRed vector, and after four days sorted cells into low, medium, and high DsRed bins and harvested gDNA. Overall editing rates matched DsRed expression (frequency of non-wild-type barcodes: low DsRed = 40%; medium DsRed = 69%; high DsRed = 91%). The profile of edits observed remained similar, but there were fewer inter-target deletions in the lower DsRed bins (fig. S2). These results show that adjusting expression levels of editing reagents can be used to modify the rates and patterns of barcode editing.
We also synthesized and tested three barcodes (v2-v4) with nine or ten weaker off-target sites for the same sgRNA as used for v1 (22). Genome editing resulted in derivative barcodes with substantially fewer edits than seen with the v1 barcode, but a much greater proportion of these edits were to a single target site, i.e. fewer inter-target deletions were observed (Fig. 1D and E, and fig. S3A and B). As only a few targets were substantially edited in designs v1-v4, we combined the most highly active targets to a new, twelve target barcode (v5). This barcode exhibited more uniform usage of constituent targets, but with relative activities still ranging over two orders of magnitude (fig. S3C and table S1). These results illustrate the potential value of iterative barcode design.
To determine whether the means of editing reagent delivery influences patterns of barcode editing, we introduced a lentiviral vector expressing Cas9 and the same sgRNA to cells containing the v5 barcode (24). After two weeks of culturing a population bottlenecked to 200 cells by FACS, we observed diverse barcode alleles but with substantially fewer inter-target deletions than with episomal delivery of editing reagents (fig. S3D). This finding demonstrates that the allelic spectrum can also be modulated by the delivery mode of editing reagents.
Taken together, these results show that editing multiple target sites within a compact barcode can generate a combinatorial diversity of alleles, and also that these alleles can be read out by single sequencing reads derived from either DNA or RNA. Rates and patterns of barcode editing are tunable by using targets with different activities and/or off-target sequences, by iteratively recombining targets to new barcode designs, and by modulating the concentration and means of delivery of editing reagents.
Reconstruction of lineage relationships in cultured cells
To determine whether GESTALT could be used to reconstruct lineage relationships, we applied it to a designed lineage in cell culture (Fig. 2). A monoclonal population of EGFP-v1 positive cells was transfected with editing reagents to induce a first round of mutations in the v1 barcode. Clones derived from single cells were expanded, sampled, split, and re-transfected with editing reagents to induce a second round of mutations of the v1 barcode. For each clonal population, two 100-cell samples of the re-edited populations were expanded and harvested for gDNA. In these experiments, we began incorporating unique molecular identifiers (UMIs; 10 bp) during amplification of barcodes by a single round of polymerase extension (fig. S4A). Each UMI tags the single barcode present within each single cell, thereby allowing for correction of subsequent PCR amplification bias and enabling each UMI-barcode combination to be interpreted as deriving from a single cell (25).
Figure 2. Reconstruction of a synthetic lineage based on genome editing and targeted sequencing of edited barcodes.
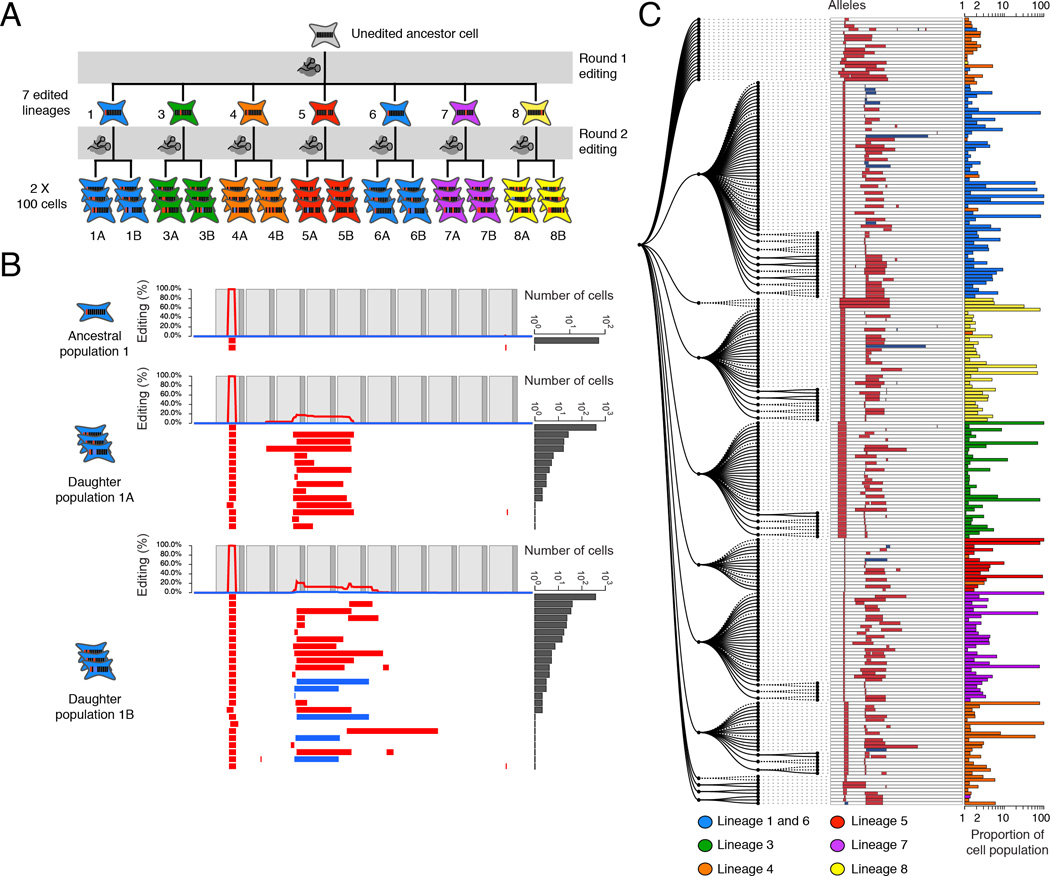
(A) A monoclonal population of cells was subjected to editing of the v1 array. Single cells were expanded, sampled (#1 to #12), re-transfected to induce a second round of barcode editing, and then expanded and sampled from 100-cell subpopulations (#1a, 1b to #12a, 12b). For clarity, the five clones where the original population was unedited are not shown. (B) Alleles observed in the synthetic lineage experiment are shown, with layout as described in the Fig. 1B legend. Cell population #1 represents sampling of cells that had been subjected to only the first round of editing; virtually all cells contain a shared edit to the first target. Populations #1a and #1b are derived from #1 but subjected to a second round of editing prior to sampling. These retain the edit to the first target, but subpopulations bear additional edits to other targets. (C) Maximum parsimony reconstruction using PHYLIP Mix (see Materials and Methods and fig. S4B) from alleles seen two or more times in the seven cell lineages represented in panel (A). Lineage membership and abundance of each allele are shown on the right. Progenitor cell lineage #4 (orange) appears to be derived from two cells, one edited and the other wild-type: only 62% of lineage #4 falls into a single clade, consistent with the proportion (64%) of the lineage edited after the first round. We assume that cells unedited in the first round either accrued edits matching other lineages (thus causing mixing), or accrued different edits (thus remaining outside the major clades).
Seven of twelve clonal populations we isolated contained mutations in the v1 barcode that were unambiguously introduced during the first round of editing (Fig. 2A). Additional edits accumulated in re-edited cells but generally did not disrupt the early edits (Fig. 2B and fig. S5). We next sought to reconstruct the lineage relationships between all alleles observed in the experiment using a maximum parsimony approach (fig. S4B)(26). The resultant tree contained major clades that were defined by the early edits present in each lineage (Fig. 2C). Four clonal populations (#3, #5, #7 and #8) were cleanly separated upon lineage reconstruction, with >99.7% of cells accurately placed into each lineage’s major clade. Two lineages (#1 and #6) were mixed because they shared identical mutations from the first round of editing. These most likely represent the recurrence of the same editing event across multiple lineages, but could also have been daughter cells subsequent to a single, early editing event prior to isolating clones. Consequently, 99.9% of cells of these two lineages were assigned to a single clade (Fig. 2C, blue). One clonal population (#4) appears to have derived from two independent cells, one of which harbored an unedited barcode. Later editing of these barcodes confounded the assignment of this lineage on the tree. Overall, however, these results demonstrate that GESTALT can be used to capture and reconstruct cell lineage relationships in cultured cells.
Combinatorial and cumulative editing of a compact genomic barcode in zebrafish
To test the potential of GESTALT for in vivo lineage tracing in a complex multicellular organism, we turned to the zebrafish Danio rerio. We designed two new barcodes, v6 and v7, each with ten sgRNA target sites that are absent from the zebrafish genome and predicted to be highly editable (Materials and Methods). In contrast to v1-v5, in which the target sites are variably editable by one sgRNA, the targets within v6 or v7 are designed to be edited by distinct sgRNAs. We generated transgenic zebrafish that harbor each barcode in the 3’ UTR of DsRed driven by the ubiquitin promoter (27, 28) and a GFP marker that is expressed in the cardiomyocytes of the heart (fig. S6) (29). To evaluate whether diverse alleles could be generated by in vivo genome editing, we injected Cas9 and ten different sgRNAs with perfect complementarity to the barcode target sites into single-cell v6 embryos (Fig. 3A). Editing of integrated barcodes had no noticeable effects on development (fig. S7). To characterize barcode editing in vivo, we extracted gDNA from a series of single 30 hours post fertilization (hpf) embryos, and UMI-tagged, amplified and sequenced the v6 barcode. In control embryos (Cas9-; n = 2), all 4,488 captured barcodes were unedited. In contrast, in edited embryos (Cas9+; n = 8), fewer than 1% of captured barcodes were unedited. We recovered barcodes from hundreds of cells per embryo (median 943; range 257–2,832) and identified dozens to hundreds of alleles per embryo (median 225; range 86–1,323). 41% +/− 10% of alleles were observed recurrently within single embryos, most likely reflecting alleles that were generated in a progenitor of two or more cells. Fewer than 0.01% of alleles were shared in pairwise comparisons of embryos, revealing the highly stochastic nature of editing in different embryos. These results demonstrate that GESTALT can generate very high allelic diversity in vivo.
Figure 3. Generating combinatorial barcode diversity in transgenic zebrafish.
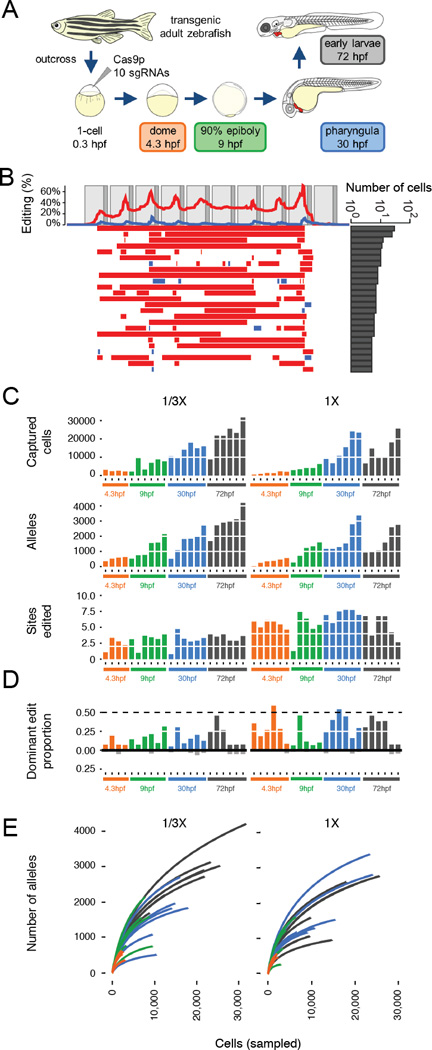
(A) One-cell zebrafish embryos were injected with complexed Cas9 ribonucleoproteins (RNPs) containing sgRNAs that matched each of the 10 targets in the array (v6 or v7). Embryos were collected at time points indicated. UMI-tagged barcodes were amplified and sequenced from genomic DNA. (B) Patterns of editing in alleles recovered from a 30 hpf v6 embryo, with layout as described in the Fig. 1B legend. (C) Bar plots show the number of cells sampled (top), unique alleles observed (middle) and proportion of sites edited (bottom) for 45 v7 embryos collected at four developmental time-points and two levels of Cas9 RNP (1/3x, 1x). Colors correspond to stages shown in panel (A). Although more alleles are observed with sampling of larger numbers of cells at later time points, the proportion of target sites edited remains relatively constant. (D) Bar plots show the proportion of edited barcodes containing the most common editing event in a given embryo. Six of 45 embryos had the most common edit in approximately 50% of cells (dashed line), consistent with this edit having occurred at the two-cell stage (see fig. S8A for example). Colors correspond to stages shown in panel (A). These same edits are rarer or absent in other embryos (black bars below). (E) For each of the 45 v7 embryos, all barcodes observed were sampled without replacement. The cumulative number of unique alleles observed as a function of the number of cells sampled is shown (average of the 500 iterations shown per embryo; two levels of Cas9 RNP: 1/3x on left, 1x on right). The number of unique alleles observed, even in later developmental stages where we are sampling much larger numbers of cells, appears to saturate, and there is no consistent pattern supporting substantially greater diversity in later time-points, consistent with the bottom row of panel (C) in supporting the conclusion that the majority of editing occurs before dome stage.
Reconstruction of lineage relationships in embryos
To evaluate whether lineage relationships can be reconstructed using edited barcodes, we focused on the v6 embryo with the lowest rates of inter-target deletions and edited target sites (Fig. 3B; avg. 58% +/− 27% of target sites no longer a perfect match to the unedited target, compared to 87% +/− 21% for all other 30 hpf v6 embryos). Application of our parsimony approach (fig. S4B) to the 1,961 cells in which we observed 1,323 distinct alleles generated the large tree shown in Fig. 4. 1,307 of the 1,323 (98%) alleles could be related to at least one other allele by one or more shared edits, 85% by two or more shared edits, and 56% by three or more shared edits. These results illustrate the principle of using patterns of shared edits between distinct barcode alleles to reconstruct their lineage relationships in vivo.
Figure 4. Lineage reconstruction of an edited zebrafish embryo.
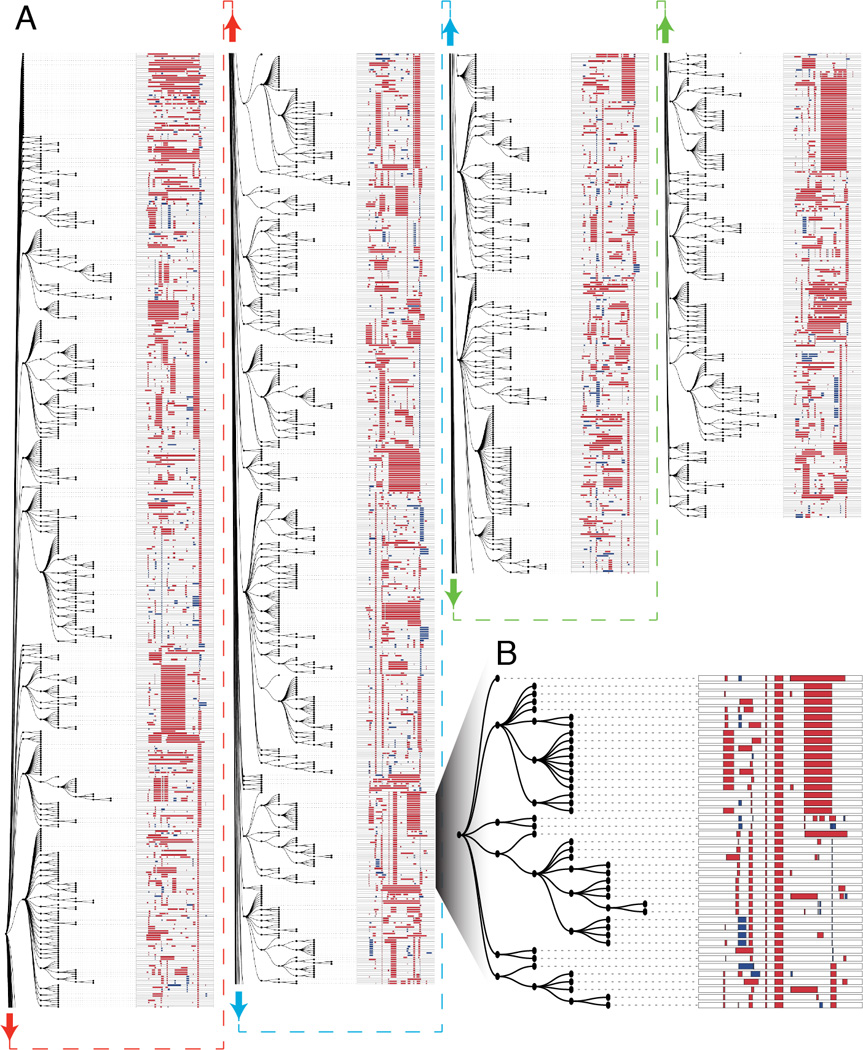
(A) A lineage reconstruction of 1,323 alleles recovered from the v6 embryo also represented in Fig. 3B, generated by a maximum parsimony approach implemented in the PHYLIP Mix package (see Materials and Methods and fig. S4B). A dendrogram to the left of each column represents the lineage relationships, and the alleles are represented on the right. Each row represents a unique allele. Matched colored arrows and dashed lines connect subsections of the tree together. There are many large clades of alleles sharing specific edits, as well as sub-clades defined by ‘dependent’ edits. These dependent edits occur within a clade defined by a more frequent edit but are rare or absent elsewhere in the tree. (B) A portion of the tree is shown at higher resolution. Two edits are shared by all alleles in this clade. Six independent edits define descendent sub-clades within this clade, and further edits define additional sub-sub-clades within the clade.
Developmental timing of barcode editing
To determine the developmental timing of barcode editing, we injected Cas9 and ten sgRNAs into one-cell stage v7 transgenic embryos and harvested genomic DNA before gastrulation (dome stage, 4.3 hpf; n = 10 animals), after gastrulation (90% epiboly / bud stage, 9 hpf; n = 11 animals), at pharyngula stage (30 hpf; n = 12 animals), and from early larvae (72 hpf; n = 12 animals) (Fig. 3A). We recovered barcode sequences from a median of 8,785 cells per embryo (range 461–31,640; total of 45 embryos), comprising a median of 1,223 alleles per embryo (range 15–4,195) (Fig. 3C). Within single embryos, 65% +/− 6% of alleles were observed recurrently, whereas in pairwise comparisons of embryos only 2% +/− 5% of alleles were observed recurrently. The abundances of alleles were well-correlated between technical replicates for each of two 72 hpf embryos (fig. S8A and B), and alleles containing many edits were more likely to be unique to an embryo than those with few edits (fig. S8C). To assess when editing begins, we analyzed the proportions of the most common editing events across all barcodes sequenced in a given embryo, reasoning that the earliest edits would be the most frequent. Across eight v6 and 45 v7 embryos, we never observed an edit that was present in 100% of cells. This observation indicates that no permanent edits were introduced at the one-cell stage. In nearly all embryos, we observe that the most common edit is present in >10% of cells, and in some cases in ~50% of cells (Fig. 3D and fig. S9). This observation also holds in ~4,000-cell dome stage embryos, which result from approximately 12 rounds of largely synchronous division unaccompanied by cell death. Most of these edits are rare or absent in other embryos, suggesting they are unlikely to have arisen recurrently within each lineage. These results suggest that the edits present in ~50% of cells were introduced at the two-cell stage and that the edits present in >10% of cells were introduced before the 16-cell stage.
How long does barcode editing persist? Two aspects of the data suggest that it tapers relatively early in development. First, in dome stage embryos (4.3 hpf), we captured barcodes from a median of 2,086 cells, in which a median of 4.8 targets were edited. Although the number of cells and alleles that we were able to sample increased at the later developmental stages, the proportion of edited sites appeared relatively stable (Fig. 3C). If editing were occurring throughout this time course, we would instead expect the proportion of edited sites to increase substantially. Second, the number of unique alleles appears to saturate early, never exceeding 4,200 (Fig. 3E). For example, only 4,195 alleles were observed in a 72 hpf embryo in which we sampled the highest number of cells (n = 31,639). These results suggest that the majority of editing events occurred before dome stage.
Editing diversity in adult organs
To evaluate whether barcodes edited during embryogenesis can be recovered in adults, we dissected two edited 4-month old v7 transgenic zebrafish (ADR1 and ADR2) (Fig. 5A). We collected organs representing all germ layers - the brain and both eyes (ectodermal), the intestinal bulb and posterior intestine (endodermal), the heart and blood (mesodermal), and the gills (neural crest, with contributions from other germ layers). We further divided the heart into four samples – a piece of heart tissue, dissociated unsorted cells (DHCs), FACS-sorted GFP+ cardiomyocytes, and non-cardiomyocyte heart cells (NCs) (fig. S10). We isolated genomic DNA from each sample, amplified and sequenced edited barcodes with high technical reproducibility (fig. S11), and observed barcode editing rates akin to those in embryos (fig. S12). For zebrafish ADR1, we captured barcodes from between 776 and 44,239 cells from each tissue sample (median 17,335), corresponding to a total of 197,461 cells and 1,138 alleles. For zebrafish ADR2, we captured barcodes from between 84 and 52,984 cells from each tissue sample (median 20,973), corresponding to a total of 217,763 cells and 2,016 alleles. These results show that edits introduced to the barcode during embryogenesis are inherited through development and tissue homeostasis and can be detected in adult organs.
Figure 5. Organ-specific progenitor cell dominance.
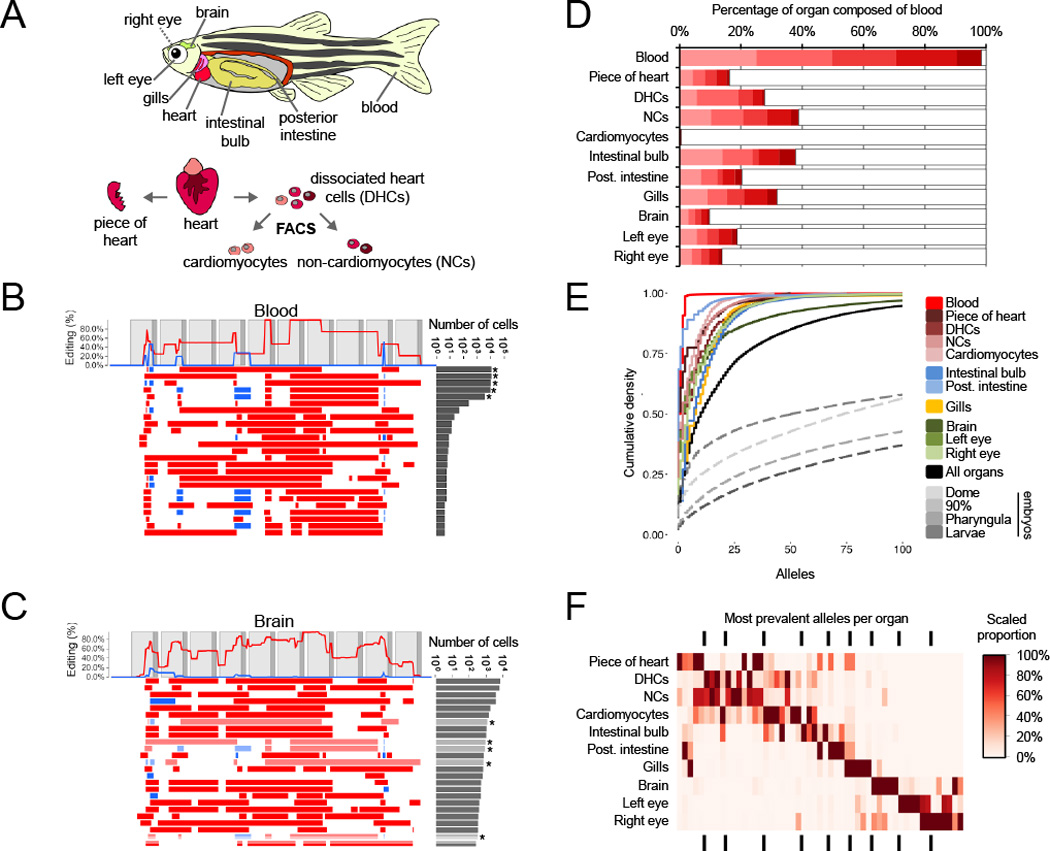
(A) The indicated organs were dissected from a single adult v7 transgenic edited zebrafish (ADR1). A blood sample was collected as described in the Methods. The heart was further split into the four samples shown (fig. S10). (B) Patterns of editing in the most prevalent 25 alleles (out of 135 total) recovered from the blood sample. Layout as described in the Fig. 1B legend. The most prevalent 5 alleles (indicated by asterisks) comprise >98% of observed cells. (C) Patterns of editing in the most prevalent 25 alleles (out of 399 total) recovered from brain. Layout as described in the Fig. 1B legend. Alleles that have identical editing patterns compared to the most prevalent blood alleles are indicated by asterisks and light shading. (D) The five dominant blood alleles (shades of red) are present in varying proportions (10–40%) in all intact organs except the FACS-sorted cardiomyocyte population (0.5%). All other alleles are summed in grey. (E) The cumulative proportion of cells (y-axis) represented by the most frequent alleles (x-axis) for each adult organ of ADR1 is shown, as well as the adult organs in aggregate. In all adult organs except blood, the five dominant blood alleles are excluded. All organs exhibit dominance of sampled cells by a small number of progenitors, with fewer than 7 alleles comprising the majority of cells. For comparison, a similar plot for the median embryo (dashed) from each time-point of the developmental time course experiment is also shown. (F) The distribution of the most prevalent alleles for each organ, after removal of the five dominant blood alleles, across all organs. The most prevalent alleles were defined as being at >5% abundance in a given organ (median 5 alleles, range 4–7). Organ proportions were normalized by column and colored as shown in legend. Underlying data presented in table S2.
Differential contribution of embryonic progenitors to adult organs
To analyze the contribution of diverse alleles to different organs, we compared the frequency of edited barcodes within and between organs. We first examined blood (of note, zebrafish erythrocytes are nucleated (30)). Only 5 alleles defined over 98% of cells in the ADR1 blood sample (Fig. 5B), suggesting highly clonal origins of the adult zebrafish blood system from a few embryonic progenitors. Consistent with the presence of blood in all dissected organs, these common blood alleles were also observed in all organs (10–40%; Fig. 5C) but largely absent from cardiomyocytes isolated by flow sorting (0.5%). Furthermore, the relative proportions of these five alleles remained constant in all dissected organs, suggesting that they primarily mark the blood and do not substantially contribute to non-blood lineages (Fig. 5D). In performing similar analyses of clonality across all organs (while excluding the five most common blood alleles), we observed that a small subset of alleles dominates each organ (Fig. 5E). Indeed, for all dissected organs, fewer than 7 alleles comprised >50% of cells (median 4, range 2–6), and, with the exception of the brain, fewer than 25 alleles comprised >90% of cells (median 19, range 4–38). Most of these dominant alleles were organ-specific, i.e. although they were found rarely in other organs, they tended to be dominant in only one organ (Fig. 5F). For example, the most frequent allele observed in the intestinal bulb comprised 13.6% of captured non-blood cells observed in that organ, but <0.01% of cells observed in any other organ. There are exceptions, however. For example, one allele is observed in 24.7% of sorted cardiomyocytes, 13.4% of the intestinal bulb, and at lower abundances in all other organs. Similar results were observed in ADR2 (fig. S13). These results indicate that the majority of cells in diverse adult organs are descended from a few differentially edited embryonic precursors.
Reconstructing lineage relationships in adult organs
To reconstruct the lineage relationships between cells both within and across organs on the basis of shared edits, we again relied on maximum parsimony methods (fig. S4B). The resulting trees for ADR1 and ADR2 are shown in Fig. 6 and fig. S14, respectively. We observed clades of alleles that shared specific edits. For example, ADR1 had 8 major clades, each defined by ‘ancestral’ edits that are shared by all captured cells assigned to that clade (Fig. 7A; also indicated by colors in the tree shown in Fig. 6). Collectively, these clades comprised 49% of alleles and 90% of the 197,461 cells sampled from ADR1 (Fig. 7A). Blood was contributed to by 3 major clades (#3, #6, #7) (Fig. 7B). After re-allocating the 5 dominant blood alleles from the composition of individual organs back to blood (Fig. 5B and fig. S15), we observed that all major clades made highly non-uniform contributions across organs. For example, clade #3 contributed almost exclusively to mesodermal and endodermal organs, while clade #5 contributed almost exclusively to ectodermal organs. These results reveal that GESTALT can be used to infer the contributions of inferred ancestral progenitors to adult organs.
Figure 6. Lineage reconstruction for adult zebrafish ADR1.
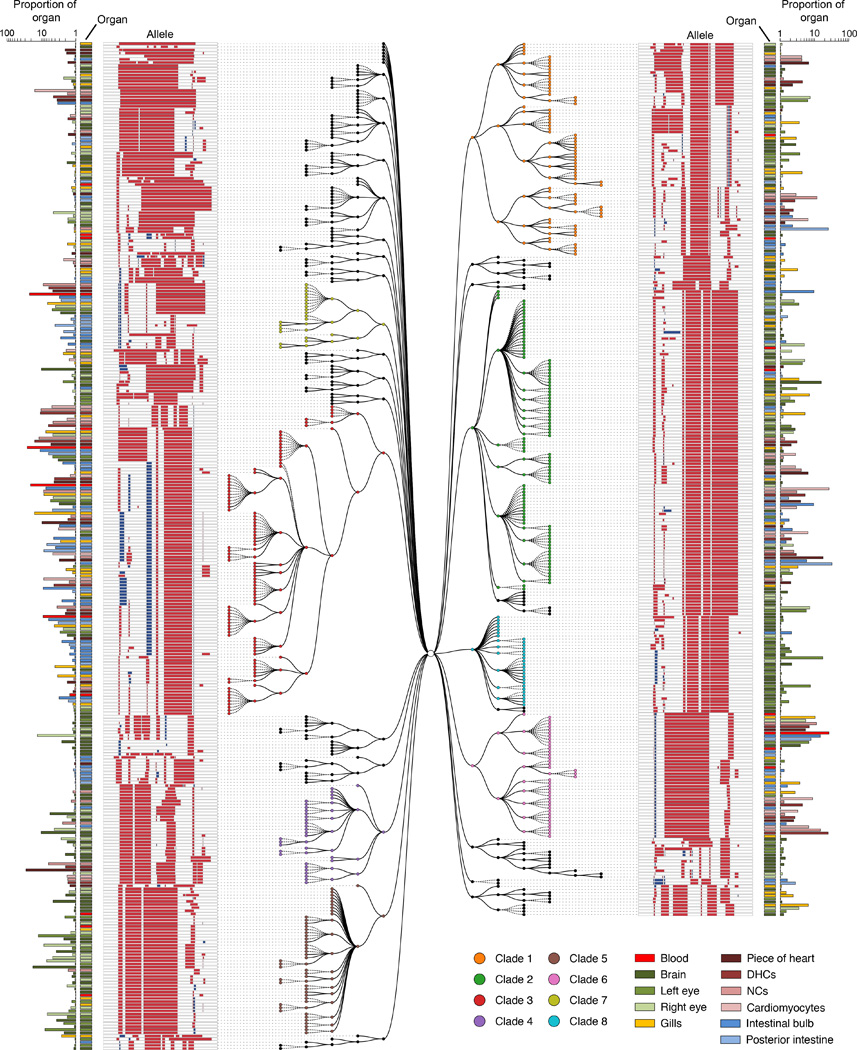
Unique alleles sequenced from adult zebrafish organs can be related to one another using a maximum parsimony approach implemented in the PHYLIP Mix package (see Materials and Methods and fig. S4B). For reasons of space, we show a tree reconstructed from the 601 ADR1 alleles observed at least five times in individual organs. Eight major clades are displayed with colored nodes, each defined by ‘ancestral’ edits that are shared by all alleles assigned to that clade (shown in Fig. 7A). Editing patterns in individual alleles are represented as shown previously. Alleles observed in multiple organs are plotted on separate lines per organ and are connected with stippled branches. Two sets of bars outside the alleles identify the organ in which the allele was observed and the proportion of cells in that organ represented by that allele (log scale).
Figure 7. Clades and subclades corresponding to inferred progenitors exhibit increasing levels of organ restriction.
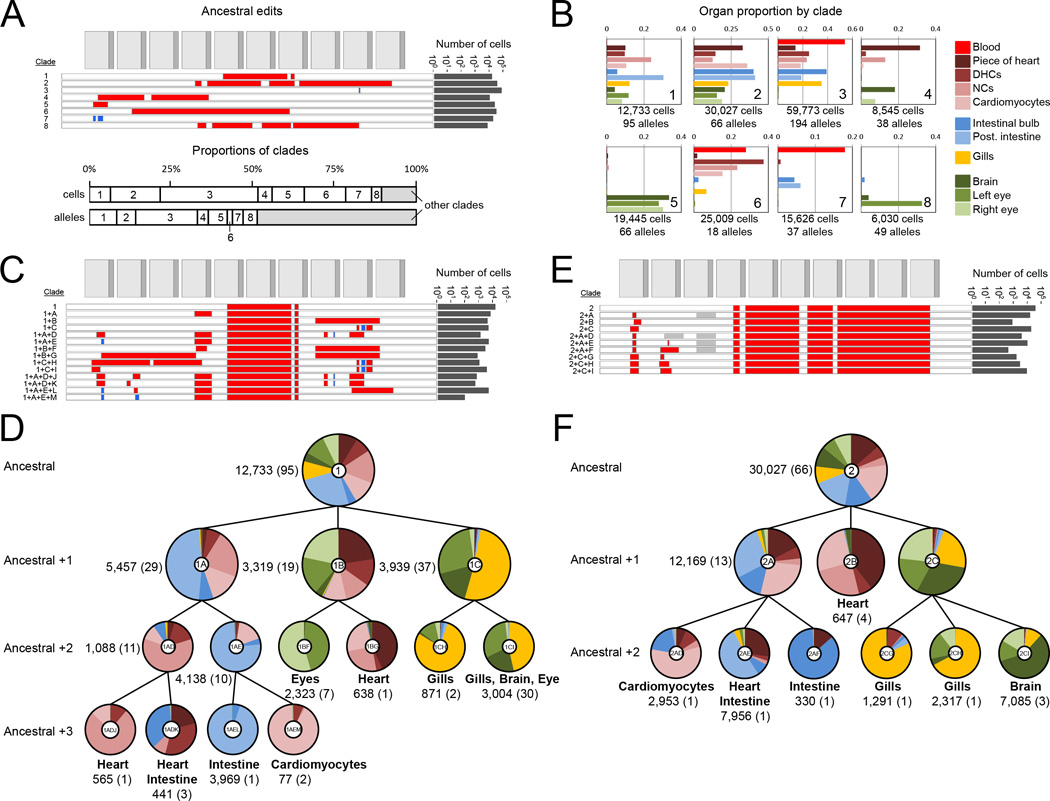
(A) Top panel: The parsimony inferred ancestral edits that define eight major clades of ADR1 are shown, with the total number of cells in which these are observed indicated on the right. Bottom panel: Contributions of the eight major clades to all cells or all alleles. 19 alleles (out of 1,138 total) that contained ancestral edits from more than one clade were excluded from assignment to any clade, and any further lineage analysis. (B) Contributions of each of the eight major clades to each organ, displayed as a proportion of each organ. To accurately display the contributions of the eight major clades to each organ, we first re-assigned the five dominant blood alleles from other organs back to the blood. The total number of cells and alleles within a given major clade are listed below. The clade contributions of all clades and subclades are presented in table S3. For heart subsamples, ‘piece of heart’ = a piece of heart tissue, ‘DHCs’ = dissociated unsorted cells; ‘cardiomyocytes’ = FACS-sorted GFP+ cardiomyocytes; and ‘NCs’ = non-cardiomyocyte heart cells. (C) and (E) Edits that define subclades of clade #1 (C) and clade #2 (E), with the total number of cells in which these are observed indicated on the right. A grey box indicates an unedited site or sites, distinguishing it from related alleles that contain an edit at this location. (D) and (F) Lineage trees corresponding to subclades of clade #1 (D) and clade #2 (F) that show how dependent edits are associated with increasing lineage restriction. The pie chart at each node indicates the organ distribution within a clade or subclade. Ratios of cell proportions are plotted, a normalization that accounts for differential depth of sampling between organs. Labels in the center of each pie chart correspond to the subclade labels in (C) and (E). Alleles present in a clade but not assigned to a descendent subclade (either they have no additional lineage restriction or are at low abundance) are not plotted for clarity. The number of cells (and the number of unique alleles) are also listed, and terminal nodes also list major organ restriction(s), i.e. those comprising >25% of a subclade by proportion.
Although some ancestral clades appear to contribute to all germ layers, we find that subclades, defined by additional shared edits within a clade, exhibit greater specificity. For example, although clade #1 contributes substantially to all organs except blood, additional edits divide clade #1 into three subclades with greater tissue restriction (Fig. 7C and D). The #1+A subclade primarily contributes to mesendodermal organs (heart, both gastrointestinal organs) whereas the #1+C subclade primarily contributes to neuroectodermal organs (brain, left eye, and gills). Similar patterns are observed for clade #2 (Fig. 7E and F), where the #2+A subclade contributes primarily to mesendodermal organs, the #2+B subclade to the heart, and the #2+C clade to neuroectodermal organs. Additional edits divide these subclades into further tissue-specific sub-subclades. For example, whereas the #2+A subclade is predominantly mesendoderm, additional edits define #2+A+D (heart, primarily cardiomyocytes), #2+A+E (heart and posterior intestine), and #2+A+F (intestinal bulb). All of the major clades exhibit similar patterns of increasing restriction with additional edits (Fig. 7C-F and fig. S16). Similar observations were made in fish ADR2 (fig. S17). These results indicate that GESTALT can record lineage relationships across many cell divisions and capture information both before and during tissue restriction.
Discussion
We describe a new method, GESTALT, which uses combinatorial and cumulative genome editing to record cell lineage information in a highly multiplexed fashion. We successfully applied this method to both artificial lineages (cell culture) as well as to a whole organism (zebrafish). Full tree reconstructions for cell culture, zebrafish embryo, and zebrafish adult experiments are provided at http://gestalt.gs.washington.edu/.
The strengths of GESTALT include: 1) the combinatorial diversity of mutations that can be generated within a dense array of CRISPR/Cas9 target sites; 2) the potential for informative mutations to accumulate across many cell divisions and throughout an organism’s developmental history; 3) the ability to scalably query lineage information from at least hundreds of thousands of cells and with a single sequencing read per single cell; 4) the likely applicability of GESTALT to any organism, from bacteria and plants to vertebrates, that allows genome editing, as well as human cells (e.g. tumor xenografts). Even in organisms in which transgenesis is not established, lineage tracing by genome editing may be feasible by expressing editing reagents to densely mutate an endogenous, non-essential genomic sequence.
Our experiments also highlight several remaining technical challenges. Chief amongst these are: 1) the chance recurrence of identical edits or similar patterns of edits in distantly related cells can confound lineage inference; 2) non-uniform editing efficiencies and inter-target deletions within the barcode contribute to suboptimal sequence diversity and loss of information, respectively; 3) the transient means by which Cas9 and sgRNAs are introduced likely restrict editing to early embryogenesis; 4) the computational challenge of precisely defining the multiple editing events that give rise to different alleles complicates the unequivocal reconstruction of lineage trees; and 5) the difficulty of isolating tissues without contamination by blood and other cells can hinder the assignment of alleles to specific organs. A broader set of challenges includes the lack of information about the precise anatomical location and exact cell type of each queried cell, the fact that genome editing events are not directly coupled to the cell cycle, and the failure to recover all cells. These challenges currently hinder the reconstruction of a lineage tree as complete and precise as the one that Sulston and colleagues described for C. elegans. Despite these limitations, our proof-of-principle study shows that GESTALT can inform developmental biology by richly defining lineage relationships among vast numbers of cells recovered from an organism.
The current challenges highlight the need for further optimization of the design of targets and arrays, as well as the delivery of editing reagents. For example, an array containing twice as many targets as used here could fit within a single read on contemporary sequencing platforms, thus yielding more lineage information per cell without sacrificing throughput. Also, as we have shown, adjustments to the target sequences and dosages of editing reagents can be used to fine-tune mutation rates and to minimize undesirable inter-target deletions. Finally, sgRNA sequences and lengths (31), Cas9 cleavage activity and target preferences (32, 33), and the means by which Cas9 and sgRNA(s) are expressed (e.g. transient, constitutive (34), or induced (35, 36)), can be altered to control the pace, temporal window and tissue(s) at which the barcodes are mutated. For example, coupling editing to cell cycle progression might enable higher resolution reconstruction of lineage relationships throughout development.
Our application of GESTALT to a vertebrate model organism, zebrafish, demonstrates its potential to yield insights into developmental biology. First, our results suggest that relatively few embryonic progenitor cells give rise to the majority of cells of many adult zebrafish organs, reminiscent of clonal dominance (37, 38). For example, only 5 of the 1,138 alleles observed in ADR1 gave rise to >98% of blood cells, and for all dissected organs, fewer than 7 alleles comprised >50% of cells. There are several mechanisms by which such dominance can emerge, e.g. by uneven starting populations in the embryo, drift, competition, interference, unequal cell proliferation or death, or a combination of these mechanisms (39–42). Controlling the temporal and spatial induction of edits and isolating defined cell types from diverse organs should help resolve the mechanisms by which different embryonic progenitors come to dominate different adult organs.
Second, we show that GESTALT can inform the lineage relationships amongst thousands of differentiated cells. For example, following the accumulation of edits from ancestral to more complex reveals the progressive restriction of progenitors to germ layers and then organs. Cells within an organ can both share and differ in their alleles, revealing additional information about organ development. Future studies will need to determine whether such lineages reflect distinct cell fates (e.g., blood sub-lineages or neuronal subpopulations), because the anatomical resolution at which we queried alleles was restricted to grossly dissected organs and tissues. Because edited barcodes are expressed as RNA, we envision that combining our system with other platforms will permit much greater levels of anatomical resolution without sacrificing throughput. For example, in situ RNA sequencing of barcodes would provide explicit spatial and histological context to lineage reconstructions (19, 20). Also, capturing richly informative lineage markers in single cell RNA-seq or ATAC-seq datasets may inform the interpretation of those molecular phenotypes, while also adding cell type resolution to studies of lineage (43, 44). Such integration may be particularly relevant to efforts to build comprehensive atlases of cell types. Because these single cell methods generate many reads per single cell, this would also facilitate using multiple, unlinked target arrays. In principle, the combined diversity of the barcodes queried from single cells could be engineered to uniquely identify every cell in a complex organism. In addition, orthogonal imaging-based lineage tracing approaches in fixed and live samples (e.g., Brainbow and related methods (16, 29)) and longitudinal whole animal imaging approaches (45, 46) might be leveraged in parallel to validate and complement lineages resolved by GESTALT.
Although further work is required to optimize GESTALT towards enabling spatiotemporally complete maps of cell lineage, our proof-of-principle experiments show that using multiplex in vivo genome editing to record lineage information to a compact barcode at an organism-wide scale will be a powerful tool for developmental biology. This approach is not limited to normal development but can also be applied to animal models of developmental disorders, as well as to investigate the origins and progression of cancer. Our study also supports the notion that whereas its most widespread application has been to modify endogenous biological circuits, genome editing can also be used to stably record biological information (47), analogous to recombinase-based memories but with considerably greater flexibility and scalability. For example, coupling editing activity to external stimuli or physiological changes could record the history of exposure to intrinsic or extrinsic signals. In the long term, we envision that rich, systematically generated maps of organismal development, wherein lineage, epigenetic, transcriptional and positional information are concurrently captured at single cell resolution, will advance our understanding of normal development, inherited diseases, and cancer.
Supplementary Material
Acknowledgments
For discussion and advice, we thank J. Felsenstein, M. Kuhner, M. Rossmann, E. Fast, C. Burns, R. Ben-Yair, S. Salipante; members of the Shendure Lab, particularly M. Snyder; members of the Schier Lab, particularly J. Farrell, B. Raj, M. Norris, and N. Lord; and members of the Horwitz lab, particularly D. Anderson. We thank G. Church for work on predecessors of this concept with JS in 2000. We thank M. Desai, R. Losick, A. Murray and L. Zon for comments on the manuscript. We also thank the Bauer Core FACS Facility, the staff of the zebrafish facility, and L. Pieper for technical support. Data associated with this paper are archived at Dryad and can be download from doi:10.5061/dryad.478t9. This work was supported by grants from the Paul G. Allen Family Foundation (JS & MSH), an NIH Director’s Pioneer Award (JS; DP1HG007811), NIH/NIGMS (AFS; GM056211), NIH/NICHD (AFS; HD085905), and NIH/NIMH (AFS; MH105960). JAG was supported by a fellowship from the American Cancer Society. AHM was supported by a fellowship from the NIH/NHLBI (T32HL007312). JS is an investigator of the Howard Hughes Medical Institute. JS, AM, GF, JG, and AS have filed a provisional patent application (62/332,896) that relates to genome editing of synthetic target arrays for lineage tracing.
Footnotes
Author contributions
GF, AM and JS developed the initial concept. GF led the cell culture experiments and developed the UMI protocol, with assistance from AM. JG led the fish experiments, with assistance from AM and GF. AM led development of the analysis pipeline. AM, GF, and JG processed and analyzed the data. AM, GF, JG, AS and JS designed experiments and interpreted the data. MH provided critical early insight. AM, GF, JG, AS, and JS wrote the manuscript.
Supplementary Materials
Materials and Methods
Figs. S1 to S17
References (48–56)
Table S1–S4
NOTE: Tables S1 to S4 are included with the manuscript as a separate single file.
References
- 1.Stent GS. Developmental cell lineage. Int. J. Dev. Biol. 1998;42:237–241. [PubMed] [Google Scholar]
- 2.Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Developmental Biology. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 3.Kretzschmar K, Watt FM. Lineage Tracing. Cell. 2012;148:33–45. doi: 10.1016/j.cell.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Kimmel CB, Law RD. Cell lineage of zebrafish blastomeres. III. Clonal analyses of the blastula and gastrula stages. Developmental Biology. 1985;108:94–101. doi: 10.1016/0012-1606(85)90012-0. [DOI] [PubMed] [Google Scholar]
- 5.Keller RE. Vital dye mapping of the gastrula and neurula of Xenopus laevis. I. Prospective areas and morphogenetic movements of the superficial layer. Developmental Biology. 1975;42:222–241. doi: 10.1016/0012-1606(75)90331-0. [DOI] [PubMed] [Google Scholar]
- 6.Weisblat DA, Sawyer RT, Stent GS. Cell lineage analysis by intracellular injection of a tracer enzyme. Science. 1978;202:1295–1298. doi: 10.1126/science.725606. [DOI] [PubMed] [Google Scholar]
- 7.Le Douarin NM, Teillet MA. Experimental analysis of the migration and differentiation of neuroblasts of the autonomic nervous system and of neurectodermal mesenchymal derivatives, using a biological cell marking technique. Developmental Biology. 1974;41:162–184. doi: 10.1016/0012-1606(74)90291-7. [DOI] [PubMed] [Google Scholar]
- 8.Dymecki SM, Tomasiewicz H. Using Flp-recombinase to characterize expansion of Wnt1-expressing neural progenitors in the mouse. Developmental Biology. 1998;201:57–65. doi: 10.1006/dbio.1998.8971. [DOI] [PubMed] [Google Scholar]
- 9.Zinyk DL, Mercer EH, Harris E, Anderson DJ, Joyner AL. Fate mapping of the mouse midbrain-hindbrain constriction using a site-specific recombination system. Current Biology. 1998;8:665–668. doi: 10.1016/s0960-9822(98)70255-6. [DOI] [PubMed] [Google Scholar]
- 10.Walsh C, Cepko CL. Widespread dispersion of neuronal clones across functional regions of the cerebral cortex. Science. 1992;255:434–440. doi: 10.1126/science.1734520. [DOI] [PubMed] [Google Scholar]
- 11.Lu R, Neff NF, Quake SR, Weissman IL. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nature Biotechnology. 2011;29:928–933. doi: 10.1038/nbt.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porter SN, Baker LC, Mittelman D, Porteus MH. Lentiviral and targeted cellular barcoding reveals ongoing clonal dynamics of cell lines in vitro and in vivo. 2014;15:1–14. doi: 10.1186/gb-2014-15-5-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salipante SJ, Horwitz MS. Phylogenetic fate mapping. Proc Natl Acad Sci USA. 2006;103:5448–5453. doi: 10.1073/pnas.0601265103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Behjati S, et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature. 2014;513:422–425. doi: 10.1038/nature13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lodato MA, et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 2015;350:94–98. doi: 10.1126/science.aab1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livet J, et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- 17.Church GM, Shendure J President And Fellows Of Harvard College, assignee. Nucleic acid memory device. 20030228611. Patent US. N.d. Print.
- 18.Carlson CA, et al. Decoding cell lineage from acquired mutations using arbitrary deep sequencing. Nat Meth. 2011;9:78–80. doi: 10.1038/nmeth.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J-H, et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 2014;343:1360–1363. doi: 10.1126/science.1250212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ke R, et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat Meth. 2013;10:857–860. doi: 10.1038/nmeth.2563. [DOI] [PubMed] [Google Scholar]
- 21.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096–1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 22.Tsai SQ, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 2014;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sancak Y, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Meth. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miner BE, Stöger RJ, Burden AF, Laird CD, Hansen RS. Molecular barcodes detect redundancy and contamination in hairpin-bisulfite PCR. Nucleic Acids Research. 2004;32:e135–e135. doi: 10.1093/nar/gnh132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Felsenstein J. PHYLIP - Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 27.Kawakami K. Tol2: a versatile gene transfer vector in vertebrates. Genome Biol. 2007;8(Suppl 1):S7. doi: 10.1186/gb-2007-8-s1-s7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porter SN, Baker LC, Mittelman D, Porteus MH. Lentiviral and targeted cellular barcoding reveals ongoing clonal dynamics of cell lines in vitro and in vivo. Genome Biol. 2014;15:1–14. doi: 10.1186/gb-2014-15-5-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan YA, et al. Zebrabow: multispectral cell labeling for cell tracing and lineage analysis in zebrafish. Development. 2013;140:2835–2846. doi: 10.1242/dev.094631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thisse C, Zon LI. Organogenesis--heart and blood formation from the zebrafish point of view. Science. 2002;295:457–462. doi: 10.1126/science.1063654. [DOI] [PubMed] [Google Scholar]
- 31.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nature Biotechnology. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kleinstiver BP, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slaymaker IM, et al. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Platt RJ, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ablain J, Durand EM, Yang S, Zhou Y, Zon LI. A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Developmental Cell. 2015;32:756–764. doi: 10.1016/j.devcel.2015.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin L, et al. Multiplex Conditional Mutagenesis Using Transgenic Expression of Cas9 and sgRNAs. Genetics. 2015;200:431–441. doi: 10.1534/genetics.115.176917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gupta V, Poss KD. Clonally dominant cardiomyocytes direct heart morphogenesis. Nature. 2012;484:479–484. doi: 10.1038/nature11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snippert HJ, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 39.Klein AM, Simons BD. Universal patterns of stem cell fate in cycling adult tissues. Development. 2011;138:3103–3111. doi: 10.1242/dev.060103. [DOI] [PubMed] [Google Scholar]
- 40.Blanpain C, Simons BD. Unravelling stem cell dynamics by lineage tracing. Nat Rev Mol Cell Biol. 2013;14:489–502. doi: 10.1038/nrm3625. [DOI] [PubMed] [Google Scholar]
- 41.Henson PM, Hume DA. Apoptotic cell removal in development and tissue homeostasis. Trends Immunol. 2006;27:244–250. doi: 10.1016/j.it.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 42.Pellettieri J, Sánchez Alvarado A. Cell turnover and adult tissue homeostasis: from humans to planarians. Annu. Rev. Genet. 2007;41:83–105. doi: 10.1146/annurev.genet.41.110306.130244. [DOI] [PubMed] [Google Scholar]
- 43.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nature Biotechnology. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cusanovich DA, et al. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348:910–914. doi: 10.1126/science.aab1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Megason SG, Fraser SE. Imaging in systems biology. Cell. 2007;130:784–795. doi: 10.1016/j.cell.2007.08.031. [DOI] [PubMed] [Google Scholar]
- 46.Liu Z, Keller PJ. Emerging Imaging and Genomic Tools for Developmental Systems Biology. Developmental Cell. 2016;36:597–610. doi: 10.1016/j.devcel.2016.02.016. [DOI] [PubMed] [Google Scholar]
- 47.Farzadfard F, Lu TK. Genomically encoded analog memory with precise in vivo DNA writing in living cell populations. Science. 2014;346:1256272–1256272. doi: 10.1126/science.1256272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends in Genetics. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 51.Mosimann C, et al. Ubiquitous transgene expression and Cre-based recombination driven by the ubiquitin promoter in zebrafish. Development. 2011;138:169–177. doi: 10.1242/dev.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang C-J, Tu C-T, Hsiao C-D, Hsieh F-J, Tsai H-J. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev. Dyn. 2003;228:30–40. doi: 10.1002/dvdy.10356. [DOI] [PubMed] [Google Scholar]
- 53.Meeker ND, Hutchinson SA, Ho L, Trede NS. Method for isolation of PCR-ready genomic DNA from zebrafish tissues. Biotech. 2007;43:610–612. 614. doi: 10.2144/000112619. [DOI] [PubMed] [Google Scholar]
- 54.Gagnon JA, et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE. 2014;9:e98186. doi: 10.1371/journal.pone.0098186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Babaei F, et al. Novel blood collection method allows plasma proteome analysis from single zebrafish. J. Proteome Res. 2013;12:1580–1590. doi: 10.1021/pr3009226. [DOI] [PubMed] [Google Scholar]
- 56.Gupta T, Mullins MC. Dissection of organs from the adult zebrafish. J Vis Exp. 2010:e1717–e1717. doi: 10.3791/1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.