

Abstract
Idiopathic basal ganglia calcification is a brain calcification disorder that has been genetically linked to autosomal dominant mutations in the sodium‐dependent phosphate co‐transporter, SLC20A2. The mechanisms whereby deficiency of Slc20a2 leads to basal ganglion calcification are unknown. In the mouse brain, we found that Slc20a2 was expressed in tissues that produce and/or regulate cerebrospinal fluid, including choroid plexus, ependyma and arteriolar smooth muscle cells. Haploinsufficient Slc20a2 +/− mice developed age‐dependent basal ganglia calcification that formed in glymphatic pathway‐associated arterioles. Slc20a2 deficiency uncovered phosphate homeostasis dysregulation characterized by abnormally high cerebrospinal fluid phosphate levels and hydrocephalus, in addition to basal ganglia calcification. Slc20a2 siRNA knockdown in smooth muscle cells revealed increased susceptibility to high phosphate‐induced calcification. These data suggested that loss of Slc20a2 led to dysregulated phosphate homeostasis and enhanced susceptibility of arteriolar smooth muscle cells to elevated phosphate‐induced calcification. Together, dysregulated cerebrospinal fluid phosphate and enhanced smooth muscle cell susceptibility may predispose to glymphatic pathway‐associated arteriolar calcification.
Keywords: cerebral vascular calcification, cerebrospinal fluid, glymphatic spaces, idiopathic basal ganglia calcification, phosphate, Slc20a2
Abbreviations
- BBB
blood brain barrier
- BGC
basal ganglia calcification
- CSF
cerebrospinal fluid
- EBSS
Earle's buffered salt solution
- IBGC
idiopathic basal ganglia calcification
- KO
knockout
- MRI
magnetic resonance imaging
- NT
no treatment
- OC
osteochondrogenic
- PAS
Periodic acid Schiff
- RARE
rapid acquisition with relaxation enhancement
- SANTA
Small ANimal Tomographic Analysis
- SMA
α‐smooth muscle actin
- SMC
smooth muscle cell
- TR
repetition time
- vWF
von Willebrand factor.
Introduction
Idiopathic basal ganglia calcification (IBGC) is a heritable disorder of bisymmetrical calcification in the basal ganglia, thalamus and cerebellum of the brain; it is a rare disease with unknown prevalence due to variable clinical features and inconsistent nomenclature 1. People with IBGC display varied degrees of both motor control and neuropsychiatric problems that worsen with age, including ataxia and dementia 23. In contrast to most calcification disorders, serum levels of both calcium and phosphorus are normal in patients with IBGC 1, 8. Currently, an animal model recapitulating the autosomal dominant feature of IBGC is lacking. There are few treatments available for symptomatic IBGC, which underscores the need for mechanistic studies in animal models that mimic the human condition 1.
Recently, human genome sequencing identified mutations in SLC20A2, PDGFB, PDGFRB and most recently XPR1 as causes of IBGC 2, 14, 16, 17, 18, 20, 21, 22, 28, 29, 30. However, the majority of genetically linked IBGC cases are attributed to mutations in SLC20A2 (also called PiT‐2, Ram1, MLVAR, IBGC1 and IBGC3), a member of the type III sodium‐dependent phosphate transporter family 2, 18, 26, 28, 29, 30. SLC20A2‐mutation linked IBGC cases have been termed IBGC3 28. In several cases the SLC20A2 mutation sites fall within putative phosphate transport motifs, however, in many cases haploinsufficiency of SLC20A2 is expected 2, 17, 28, 29, 30. Thus, in the present study we sought to determine whether Slc20a2 haploinsufficiency could cause BGC in mice, and to shed light on potential underlying mechanisms of IBGC3. This study presents a new hypothesis: that lack of Slc20a2 reduces phosphate clearance via glymphatic spaces, a newly identified metabolite clearance compartment of the central nervous system including perivascular and Virchow‐Robins spaces.
Materials and Methods
Animal studies
All mouse work was performed with IACUC approval from the University of Washington, Seattle. C57BL/6NTac‐Slc20a2<tm1a(EUCOMM)Wtsi>/Ieg (Slc20a2 +/−) mice were purchased from the European Mouse Mutant Archive (EMMA). Wildtype (Slc20a2 +/+) C57Bl/6 mice were purchased from Jackson labs (Sacramento, CA) and from Taconic labs (Hudson, NY). At least 3 animals/genotype were analyzed for each experiment; specific animal numbers are noted throughout. Saphenous blood draws were performed and RNA was immediately extracted with Trizol according to the manufacturer's instructions (Life Technologies, 15596‐018). Serum calcium and phosphorus were monitored at regular intervals between 2.5 months and 10 months, and remained in normal ranges. Mice were euthanized by IP injection of 40mg/kg body weight Beuthanasia‐D. Tissues were isolated immediately and fixed in Clark's Fixative (3:1 Methanol:Acetic Acid). For Evans Blue experiments mice were injected with 4 mL/kg of body weight or a minimum of 150 μL of 2% Evans Blue in filtered PBS (Sigma, E2129) and dye was allowed to circulate for 3 h. Mice were euthanized with Beuthanasia‐D and perfused with PBS, and then brains were immediately extracted, fixed in 10% formalin and imaged. After embedding and sectioning, retention of Evans Blue was assessed through surveying sections for blue color and red fluorescence.
Histology
Brains were isolated from mice between ages 2.5 months and 1.5 years old. Brain tissue was fixed in Clark's Fixative (3:1 Methanol: Acetic Acid) for 2 h at RT with shaking. The tissue was dehydrated into 100% EtOH, treated with Xylenes and embedded in paraffin wax at 60°C. Sections were cut at 7μm, dried, and baked at 60°C for 1 h prior to further processing.
Alizarin Red was performed as in Speer et al. 2009 with the exception of a 1 h 15 min Alizarin Red treatment time 25. Von Kossa was performed as in Speer et al. 2009 with the inclusion of an optimally reduced 22 min Von Kossa treatment to permit subsequent fluorescent von Willebrand factor (vWF) staining 25. Periodic acid Schiff (PAS) staining was performed with Periodic Acid Solution (Sigma, P7875, 5 min), Schiff's Reagent (Sigma, 3952016, 20 min) and Harris Hematoxylin counterstain (Sigma, HHS32, 1 min). Trichrome staining was performed with Bouin's fixative at 56°C (1 h), Weigerts Iron Hematoxylin (10 min), Beibrich Scarlet‐Acid Fuchsin (15 min), Phosphomolybdic‐phosphotungstic Acid (10‐15 min) and Aniline Blue (10‐20 min) (UW Pathology Services). Hematoxylin and Eosin staining was performed with Harris Hematoxylin (Sigma HHS32) and counterstained with Eosin Y (Sigma E4382). Alkaline phosphatase activity was detected with the ImmPACT Vector Red kit according to the manufacturer's instructions and a 30 minute AP detection system incubation (Vector, SK‐5105). X‐Gal staining was performed on brain slices. Briefly, brains were isolated and fixed for 1 h in X‐Gal fix containing 1% formaldehyde and 0.2% gluteraldehyde, sliced uniformly with a 1mm coronal brain matrix (EMS 69022), postfixed for 1 h in X‐Gal fix, washed in X‐Gal buffer and incubated in X‐Gal stain containing 5 mM of K3Fe and 5 mM K4Fe overnight at 37°C. X‐Gal stained brain slices were fixed in 4% paraformaldehyde, imaged with a Nikon D5100 DSLR and Nikon SMZ1500 Stereo Microscope, paraffin embedded, and sectioned.
Immunohistochemistry was performed using 4% serum block and antibody diluted in 2% serum, visualized with DAB (Sigma, D4293‐50SET) or DAB+nickel (Vector, SK‐4100), and counterstained with methyl green nuclei stain (Sigma, 323829, 5 min). Sections that underwent immunohistochemistry and/or the above histological stains were mounted with Permount (Fisher, SP15‐500). Immunofluorescence was followed by DAPI dilactate for nuclear counterstain (Life Technologies, D3571) and mounted with ProLong Gold Antifade Media (Life Technologies, P36930).
The following antibodies and antibody concentrations were used: CD13 (R&D Systems, AF2335, 5 μg/mL), Collagen IV (Millipore, AB756P, 20 μg/mL), RUNX2 (R&D Systems, MAB2006, 2.5 μg/mL), SMA (Sigma, A2547, 60 μg/mL), SLC20A1 (gift of Dr. M. Levi, University of Colorado, 1:100), SLC20A2 (gift of Dr. M. Levi, University of Colorado, 8 μg/mL), SOX9 (Santa Cruz, SC20095, 1 μg/mL) and vWF (Dako, A0082, 15.5 μg/mL). Secondary antibodies included: DyLight 488‐Conjugated AffiniPure Donkey Anti‐Goat IgG (Jackson ImmunoResearch, 705‐485‐147, 7.5 μg/mL), DyLight 549‐Conjugated AffiniPure Donkey Anti‐Rabbit IgG (Jackson ImmunoResearch, 711‐505‐152, 7.5 μg/mL), Biotin‐SP‐Conjugated AffiniPure Goat Anti‐Rabbit IgG (Jackson ImmunoResearch, 111‐065‐144, 2.4 μg/mL), Rabbit Anti‐Goat‐Biotinylated IgG (Vector Labs, BA‐5000, 3 μg/mL), Alexa Fluor 488‐Conjugated AffiniPure Donkey Anti‐Rabbit IgG (Jackson ImmunoResearch, 711‐545‐152, 15 μg/mL).
Quantification of histological features
All stained and mounted sections were imaged on a Nikon E800 Upright Microscope. All histology quantifications were performed blind. For quantification of calcification, 18 matched sections were taken at ∼40 μm increments throughout the thalamus for each animal, n = 3 animals/genotype. Lesion sites were imaged at 40× magnification and file names were coded. Calcified lesion number and area were determined by outlining contiguous Alizarin Red positive sites with ImageJ.
For CD13 and SLC20A1 cell counts, 6 matched sections were analyzed per animal, n=3 animals/genotype. The thalamus was imaged at 10× in the same location for each hemisphere (a total of 12 images per animal). Anatomical brain structures including the hippocampus and brain ventricles were used to reliably image the same region. Image names were coded and cell numbers were determined blindly by counting DAPI stained nuclei surrounded by the immunofluorescence signal of the protein target of interest and the ImageJ Cell Counter plugin was used to assist with cell counting.
siRNA‐mediated knockdown of SLC20A2
WT mouse SMCs were seeded at a density of 2.5 × 104 cells/well in a 6‐well plate with growth media (DMEM with 10% FBS and 1× antibiotic‐antimycotic). After 24 h, growth media was replaced with transfection media (DMEM with 10% FBS) before 2nM of mouse Slc20a2 siRNA (Ambion, 4390771) or Silencer Select Negative Control No. 1 (scramble, SCR) siRNA (Ambion, 4390843), and 1.5 μL of Lipofectamine RNAiMAX (Life Technologies, 13778150) were added to each well. SMCs without siRNA treatment (no treatment, NT) were also used as a negative control. Transfection media was replaced with growth media after 24 h. SMCs were redosed with the respective siRNAs after 5 days. Sense: 5'‐GGCGUGCUGUUCAUACUAA‐3'; Antisense: 5'‐UUAGUAUGAACAGCACGCC‐3'.
In vitro calcification of mouse SMCs
SMC calcification was induced by treatment with calcification media (DMEM with 3% FBS and 1× antibiotic‐antimycotic) supplemented with NaH2PO4/Na2HPO4 to a final concentration of 2.8 mM phosphate. SMCs were decalcified with 0.6N HCl overnight at 4°C, and the calcium content was determined colorimetrically by the o‐cresolphtalein complexone method (Calcium Reagent Set, Teco Diagnostics, C503‐480). After decalcification, SMCs were washed 3 times with PBS and solubilized with 0.2N NaOH. The calcium content was normalized to protein content, measured with a BCA Protein Assay (Thermo Scientific, 23235).
Cisternal CSF collection and phosphate detection
CSF was collected from the cisterna magna of one year old Slc20a2 +/− and +/+ mice as in Liu et al. 2008 19. Briefly, glass capillary tubes were purchased from World Precision Instruments, TW100F4 and pulled on a Sutter P‐87 flaming micropipette puller, with the heat index set at 100 and the pressure index set at 330. Pipette tips were cut with scissors such that the tip has an inner diameter of ∼0.5mm. CSF collection took less than five minutes per animal and yielded an average volume of 4.5 μL. CSF was spun down for 10 min at 2000rpm at 4°C, transferred to a new tube and stored at 4°C until analysis of phosphate levels. Phosphate levels were determined with the QuantiChrom Phosphate Assay Kit (Bioassays Systems, DIPI500).
RNA isolation and TaqMan quantitative real‐time PCR (QPCR)
Choroid plexus was isolated and choroid plexus RNA was extracted using the RNeasy Mini Kit according to the manufacturers directions (Qiagen, 74106). cDNA was made with 300ng of total RNA per sample using the Omniscript Reverse Transcriptase kit (Qiagen, 205113). TaqMan probes are conjugated with a fluorochrome reporter (FAM) tag at the 5'‐end and an MGB quencher at the 3'‐end were used to assess expression levels. Amplification and detection was carried out in 96‐well optical plates on an ABI Prism 7000 Sequence Detection System (Applied Biosystems), with TaqMan Universal PCR 2× master mix (Life Technologies, 4305719) in a final volume of 25 μL per reaction. Each reaction (in triplicates) was carried out at 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15sec, and 60°C for 1 min. Results were analyzed with the manufacturer's software, SDS 1.1 (Applied Biosystems). Gene mRNA expression was normalized to the housekeeping gene 18S, and normalized to the NT control using the quantitative method ( , where ΔΔC T = [ − ]treated − [ − ]control). The following Taqman Assays were used: Slc20a2 (Life Technologies, Mm00660204_mH, FAM‐MGB) and 18S Ribosomal RNA Control Reagents (Life Technologies, Cat. No. 4308329).
Phosphate uptake studies
Phosphate uptake studies were performed with mouse SMCs, as previously described 13. Briefly, SMCs were incubated in Earle's buffered salt solution (EBSS) containing 0.1, 0.25, 0.5 mM phosphate and 5μCi/mL PO4 (PerkinElmer Life Science, Inc.). Radioactivity was measured by liquid scintillation. Sodium‐dependent phosphate uptake was determined by subtracting uptake levels measured in EBSS containing choline chloride from uptake levels measured in EBSS containing sodium chloride. Uptake values were normalized to cellular protein content.
Assessment of ocular abnormalities
Mouse eyes were photographed, coded, and scored blindly. Each eye was scored as positive or negative for microphthalmia identified by a small eye, as well as positive or negative for cataracts identified by the presence of visible lens opacity.
Micro‐computed tomography
Micro‐computed tomography (micro CT) scans were performed at the Small ANimal Tomographic Analysis (SANTA) facility located at the Seattle Children's Research Institute using a SkyScan 1076 instrument (Skyscan, Belgium). Two independent scans were performed on each animal. Skull scans were done at an isotropic resolution of 8.60µm using the following settings: 55kV, 180µA, 1.0mm Aluminum filter, 1450ms exposure, rotation step of 0.7°, 180° scan, and 3 frame averaging. Full body scans were performed at an isotropic resolution of 34.42µm (60kV, 170µA, 0.5mm Aluminum filter, 120ms exposure, rotation step of 0.6°, 180° scan, and three frame averaging). Raw data were reconstructed using the software NRecon V1.6.9 (SkyScan) and 3D rendered images of each dataset were generated using the Drishti V2 Volume Exploration software (http://sf.anu.edu.au/Vizlab/drishti).
Magnetic resonance imaging
The magnetic resonance imaging (MRI) data were acquired on a Bruker Avance III, 14 Tesla (600 MHz), Ultrashield, high resolution, 89mm, vertical bore magnet running ParaVision version 5.1 software. A 3‐D, T2‐weighted, rapid acquisition with relaxation enhancement (RARE) pulse sequence was used. The sequence parameters were repetition time (TR); 3 sec, echo time (TE); 6 msec, RARE factor; 16, field of view; 2.56 × 1.75 × 1.37 mm, spatial resolution; 0.1 × 0.1 × 0.1 mm. The total acquisition time was 1 h 15 min.
Statistics
At least three independent samples per genotype were analyzed for all histological stains. Three biological replicates per condition were generated for all cell culture experiments. The following statistical tests were used to analyze quantitative data: Wilcoxon signed ranks test was used to determine whether calcified brain lesions were bilateral. For comparison of two groups a P‐value was determined by a two‐tailed Student's T‐test with unequal variance. For comparison of three or more groups, One‐way ANOVA was used and followed by Tukey's Post Hoc Test when appropriate to compare means between groups.
Results
Slc20a2 +/− mice developed age‐dependent basal ganglion calcification
Mice heterozygous for the Slc20a2 KO‐first allele were obtained from the European Mutant Mouse Archive (EMMA), and used to generate control wildtype (WT) Slc20a2 +/+, heterozygous (Het) Slc20a2 +/− and knockout (KO) Slc20a2 −/− mice. Slc20a2 expression across genotypes was determined at the RNA level and we found an average reduction of 75% in Slc20a2 +/− mice and 99% in Slc20a2 −/− mice compared to Slc20a2 +/+ mice. We then used the calcium stain Alizarin Red to confirm the previously reported finding that Slc20a2 −/− mice develop BGC [Figure 1A, Supporting Information Figure 1E–F, and Jensen et al. 10], and tested the novel hypothesis that Slc20a2 haploinsufficient mice develop BGC. We found that Slc20a2 +/− mice do indeed develop calcified lesions that stained positively with Alizarin Red (Figure 1B), as well as the phosphate stain, Von Kossa (Figure 1C). The severity of calcification assessed by lesion number and size was significantly higher in Slc20a2 +/− brains compared to wildtype Slc20a2 +/+ brains at 1 year (Figure 1D–F). As with human BGC, calcification was bilateral in Slc20a2 +/− brains (Figure 1G). Calcification was absent at 2.5 months (mo), minimal at 6mo, bilateral at 1 year (yr) and prominent at 1.5yr (Supporting Information Figure 1) in heterozygous mice.
Figure 1.
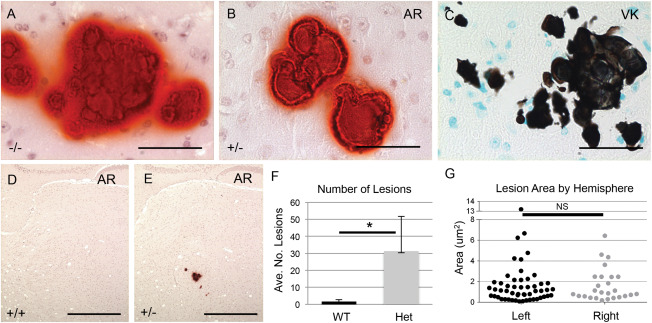
Slc20a2 deficient mice develop BGC. BGC presence was detected in Slc20a2 −/− mice by Alizarin Red (AR) (A). Brain sections from Slc20a2 +/− mice revealed mineral deposits positive for both AR (B) and von Kossa (VK) (C). Abundant calcified lesions were observed by AR staining in Slc20a2 +/− mice but not Slc20a2 +/+ controls (D, E). The average number of lesions was significantly higher in the Slc20a2 +/− mice compared to the Slc20a2 +/+ controls as determined by a two‐tailed Student's T‐test with unequal variance; N = 6; P‐value=0.018 (F). As seen in human BGC patients, the calcified lesions were located bilaterally as determined by a two‐tailed Wilcoxon signed ranks test; N = 6; P‐value=0.7265 (G). Scale bars: 50 μM (A‐C), 500 μM (D,E).
Slc20a2‐deficiency associated BGC was localized to cerebral arterioles
Calcified deposits were identified specifically in arterioles, and were characterized by the accumulation of basement membrane proteins. Co‐localization of Von Kossa with the endothelial cell marker vWF and α‐smooth muscle actin (SMA) confirmed that calcified mineral deposits associated with arterioles (Figure 2A–D). Alizarin Red stained sections compared with Periodic acid‐Schiff (PAS) stained sections revealed the presence of basement membrane components in the calcified lesion (Figure 2E–G), and Masson's Trichrome staining confirmed that the calcified lesions were rich in collagen and/or mucin (Figure 2H). Finally, immunohistochemical staining identified the basement membrane protein collagen type IV, as a major component of the calcified lesions (Figure 2I).
Figure 2.
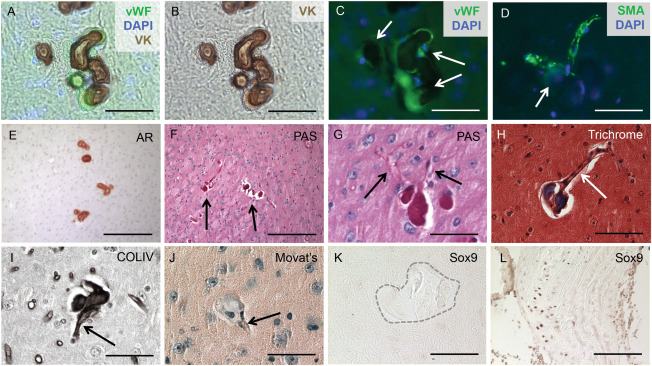
BGC is localized to cerebral arterioles. The calcified lesions (arrows in C) were localized to arterioles, characterized by the co‐localization of vWF and von Kossa (VK) staining and counterstaining with the nuclear marker DAPI (A‐C). Furthermore, the lesions (arrow in D) were also present in SMA‐positive vessels counterstained with DAPI (D). Alizarin Red (AR) staining compared with Periodic acid‐Schiff (PAS)‐positive staining of consecutive sections confirmed that the calcified lesions (arrows in F) contained basement membrane components (E‐G) and localized to blood vessels (arrows in G). Positive Masson's Trichrome staining indicated that the lesions were rich in collagen and/or mucin (H) and formed in blood vessels (arrow in Figure H). Immunohistochemical staining identified collagen type IV as the major basement membrane protein present in the calcified lesions (I) and further confirmed localization to blood vessels (arrow in I). The SMC in the calcified regions do not undergo an osteochondrogenic phenotype change, as the lesions (arrow in J) lacked proteoglycan‐rich or cartilage‐like structures, confirmed by negative Movat Pentachrome (J) and SOX9 (K) staining. Calcified aortic tissue from a diabetes mouse model was used as a positive control (L). Scale bars: 50μM (A‐D,G‐L), 200μM (E,F).
BGC lesions were void of osteochondrogenic cells
SMC can undergo an osteochondrogenic (OC) phenotype change and this mechanism has been implicated in several types of vascular calcification 3, 12, 24. Therefore, we tested the hypothesis that Slc20a2 loss might result in an arteriolar SMC to OC phenotype change. OC phenotype change has been observed in several disorders involving phosphate dysregulation, such as aortic medial calcification in chronic kidney disease rat models. However, brain sections stained with Movat Pentachrome lacked proteoglycan‐rich or cartilage‐like structures (Figure 2J) and were negative for alkaline phosphatase activity (data not shown). We then examined expression of sex‐determining region Y‐box 9 (SOX9) and Runt‐related transcription factor 2 (RUNX2), key transcription factors required for OC differentiation. In accordance with the Pentachrome stain, we determined that the BGC lesion sites were SOX9 and RUNX2 negative (Figure 2K and data not shown). Calcified aortic tissue from a diabetes mouse model was used as a positive control (Figure 2L).
Slc20a2 was expressed in choroid plexus and ependyma, and was required for regulation of CSF phosphate concentration
We took advantage of the LacZ expression construct in the Slc20a2 KO‐first allele, and determined localization of activity in mutant Slc20a2 mice by staining for b‐galactosidase. Both X‐Gal staining and immunohistochemical staining of SLC20A2 in heterozygous animals revealed a notable consensus expression pattern in neurovascular tissues and tissues related to cerebrospinal fluid (CSF) production and regulation, including the ependyma (Ep) (Figure 3A–C) and the choroid plexus epithelium (CP Ep) (Figure 3D–G). Contrary to SLC20A2, SLC20A1 was localized to the choroid plexus endothelium (CP En) and largely absent from choroid plexus epithelium (Figure 3H). Of note, SLC20A2 was recently identified in shark choroid plexus with a similar tissue specific expression pattern; furthermore, the kinetics of phosphate transport across the shark choroid plexus were identical to SLC20A2 phosphate transport parameters 7.
Figure 3.
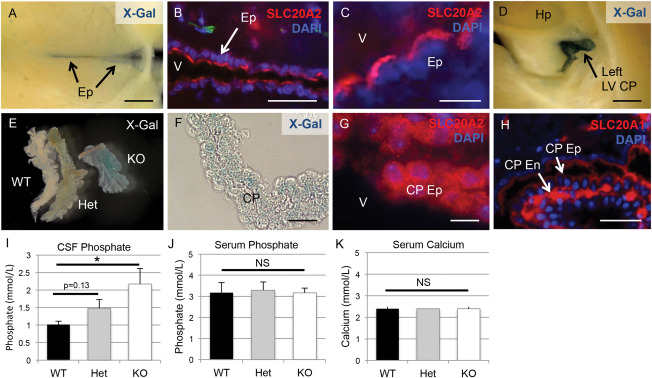
Slc20a2 is expressed in CSF‐associated tissues and regulates CSF phosphate levels. X‐Gal staining and/or immunofluorescence counterstain with the nuclear marker DAPI were used to determine tissue‐specific expression patterns of Slc20a2. Both X‐Gal staining and antibody mediated detection of revealed localization in neurovascular tissues involved in cerebrospinal fluid (CSF) regulation, including the ependyma (Ep) (A‐C) and the choroid plexus (CP) epithelium (Ep) (D‐G). Furthermore, immunohistochemical staining localized SLC20A1 to the choroid plexus endothelium (CP En) but not the CP Ep (H). CSF phosphate levels were trending in Slc20a2 +/− mice and significantly increased in the Slc20a2 −/− mice compared to the WT mice as determined by a One‐way ANOVA followed by Tukey's Post Hoc Test; N = 11; KO P‐value is < 0.05 when tested against WT samples (I). Data shown is representative of three independent experiments. Compared to Slc20a2 +/+ mice, serum phosphate and calcium levels in both the Slc20a2 +/− and Slc20a2 −/− mice were not significantly different, as determined by a One‐way ANOVA; N = 23 (J, K). Scale bars: 2 mm (A,D,E), 50μM (B), 10μM (C,F,G), 20μM (H). Abbreviations: CP En=choroid plexus endothelium; CP Ep=choroid plexus epithelium; Ep=ependyma; LV=lateral ventricle; V=ventricle.
We then tested the hypothesis that SLC20A2 regulates CSF phosphate levels. As shown in Figure 3I, phosphate levels in CSF trended higher in Slc20a2 +/− compared to WT mice, but this did not reach statistical significance (P‐value=0.13). On the other hand, severe deficiency of Slc20a2 in the global Slc20a2 −/− mice allowed us to observe a statistically significant increase in CSF phosphate concentration (Figure 3I). This condition of high phosphate in the CSF occurred in the absence of changes in serum phosphate and calcium levels in Slc20a2 +/− or Slc20a2 −/− mice compared to WT mice (Figure 3J,K).
Slc20a2 loss and high CSF phosphate levels were associated with multiple neural manifestations
One third of weaned Slc20a2 KO mice died prematurely between 4 weeks and 21 weeks of age (N=33), likely caused by severe hydrocephalus. Severe lethal hydrocephalus cases were diagnosed by brain morphology (Figure 4A) and histology of H&E stained coronal sections (Figure 4B). Both Slc20a2 Het and KO mice developed nonpresenting hydrocephalus in accordance with gene dosage. MRI suggests moderate to severe hydrocephalus in KO mice (Figure 4C) and mild hydrocephalus in Het mice (Figure 4D). Nonpresenting mice did not exhibit major cranial defects, indicating that parietal plate development was completed before gross disruption of CSF flow in these animals. Both male and female Slc20a2 KO mice had reduced body weight (Figure 4E,F) and abnormal skeletal system structure was frequently observed. Finally, Slc20a2 null mice had reduced survival rates between 4 weeks and 21 weeks of age (Figure 4G).
Figure 4.
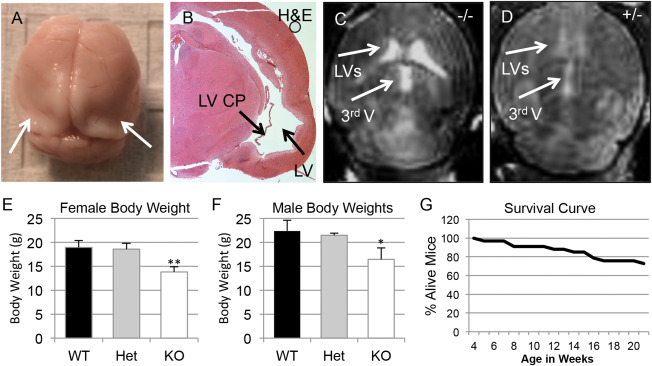
Complete Slc20a2 loss results in hydrocephalus and premature death. Severe lethal hydrocephalus cases and premature death were seen in our weaned Slc20a2 −/− mice, as diagnosed by brain morphology (A) and by histology of H&E stained coronal sections (B). Moderate to severe hydrocephalus was confirmed in Slc20a2 −/− mice by MRI, n = 2 (C) and detection of ventricles in Slc20a2 +/− mice by MRI indicates mild hydrocephalus (n = 1) in Slc20a2 +/− mice (D). Reduced body weights were observed in both Slc20a2 −/− female (N = 25; P‐value<0.01) and Slc20a2 −/− male (N = 21; P‐value<0.05) mice when compared to age and sex matched Slc20a2 +/+ controls at six weeks of age, determined by a One‐way ANOVA followed by Tukey's Post Hoc Test (E,F). Poor survival was observed in Slc20a2 −/− mice between 4 and 21 weeks of age (G).
Ocular calcification was detected in Slc20a2 KO mice
Both microphthalmia and cataracts were observed in Slc20a2 KO mice (Figure 5A,B). Since high CSF phosphate was identified in Slc20a2 null mice and glymphatic fluid contacts the optic nerve head, we hypothesized that the eye and optic nerve might be susceptible to calcification. Indeed, analysis of 9 μm CT scans detected calcification in both cataracts and in the proximity of the optic nerve head (Figure 5C–E).
Figure 5.
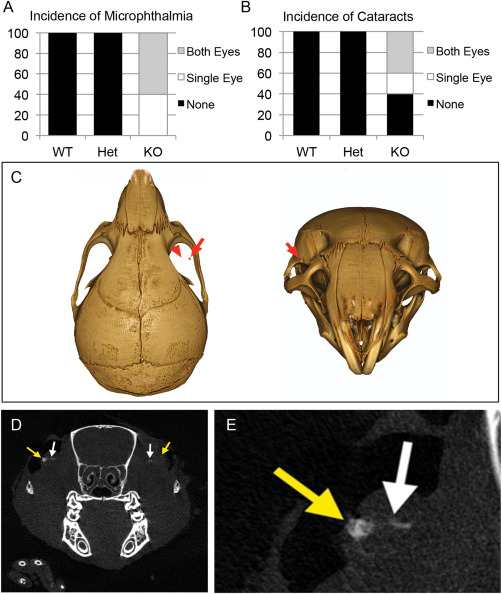
Ocular abnormalities associated with Slc20a2 deficiency. Both microphthalmia and cataracts were observed in six week old Slc20a2 KO mice (WT n = 8; Het n = 4; KO n = 5) (A,B). Furthermore, 9 μM CT scans detected calcification (red arrows in C) in both cataracts (yellow arrow in D,E) and in close proximity to the optic nerve head (white arrows in D,E).
Slc20a2 was highly expressed in cerebral SMC
In addition to tissues associated with CSF, analysis of Slc20a2 KO brain slices revealed X‐Gal positive cells in neurovascular tissues, dispersed throughout the brain parenchyma, and in the cerebellum. X‐Gal activity was detected in arteries within the dura mater (Figure 6A), as well as the cortex (Figure 6B), the cerebellum (Figure 6C), in the region of the cervical lymphatics (Figure 6D), and in bilateral arterioles throughout the thalami (Figure 6E). Wildtype Slc20a2 +/+ brain slices from littermates were stained as a negative control, and were X‐Gal negative as expected. We then determined whether vascular X‐Gal staining co‐localized with SMCs or astrocytes.
Figure 6.
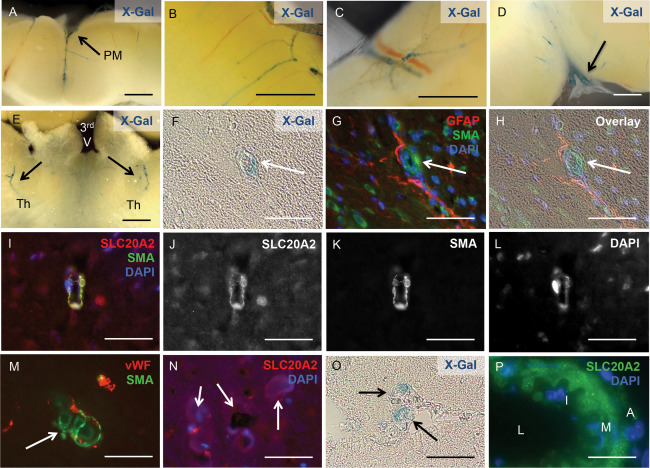
Slc20a2 is highly expressed in cerebral SMC. X‐Gal activity was detected in arteries within the pia mater (PM) (A), the cortex (B), the cerebellum (C), in the region of the cervical lymphatics (D), and in the thalamus of both hemispheres (Th); location of the third ventricle (3rd V) is indicated for spatial orientation (E). X‐Gal staining and anti‐SLC20A2 antibody signal co‐localized with SMA‐positive cells and GFAP‐positive cell projections (F‐L). Calcified lesions (arrows in M, N) also co‐localized with vWF and SMA positive cells (M), and revealed the presence of SLC20A2 positive cells in low numbers (N). Slc20a2 was also expressed in media of vessels that connect to the CP (O) and in large cerebral arteries (L: lumen, I: intima, M: media, A: adventitia) (P). Scale bars: 2mm (A‐E), 50μM (F‐P).
We found clear localization of X‐Gal staining in cells of the neurovascular unit in Slc20a2 +/− mice with BGC; X‐Gal positive cells co‐localized with the SMC marker SMA and were juxtaposed to GFAP positive astrocytes (Figure 6F–H). We then confirmed that SLC20A2 protein was localized to arteriolar SMCs by co‐localization with SMA (Figure 6I–L). As expected, BGC lesions were present in SMA positive vessels (Figure 6M). SLC20A2 positive cells were present in low numbers in highly calcified lesions (Figure 6N). X‐Gal activity was also detected in the media of vessels that connect to the choroid plexus (Figure 6O) and large cerebral arteries (Figure 6P). Together these data implicate SMC as a strong candidate for a causative cell type in Slc20a2 deficient cerebrovascular calcification.
Loss of Slc20a2 in SMC enhanced susceptibility to high phosphate‐induced calcification
It has been suggested that BGC may develop in response to high extracellular phosphate levels that trigger an active cell‐mineralization process 26, 28. In order to test the hypothesis that Slc20a2 loss leads to enhanced susceptibility to calcification in SMCs, we developed an in vitro system for mechanistic studies by treating SMC with Slc20a2 siRNA and pro‐calcific medium, and assessing calcification levels. Dosing SMCs with Slc20a2 specific siRNA yielded an 88% knockdown (KD), compared to both NT and scramble siRNA (SCR) treated cells (Figure 7A). Decreased phosphate uptake was observed in KD cells as compared to SCR controls using radiolabelled Phosphorus‐33 (Figure 7B). In order to produce a more accurate model of cerebral vessels and glymphatic spaces, in which a dysfunctional phosphate transporter may result in local accumulation of high extracellular phosphate concentrations, we treated cultured SMCs with normal or high phosphate calcification media. Indeed, Slc20a2 deficient SMCs showed greater calcification in response to calcification media starting at 4 days, and this attained statistical significance at 6 days (Figure 7C).
Figure 7.
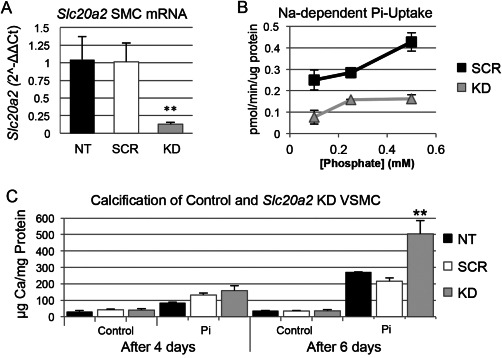
Loss of Slc20a2 in SMC results in a susceptibility to high phosphate‐induced calcification. Knockdown (KD) of Slc20a2 in SMCs was induced by short interfering RNA (siRNA). The level of Slc20a2 in KD SMCs was 90% on average (P‐value<0.0001) compared to the NT and scramble siRNA treated (SCR) controls (n = 3 per condition), determined by a One‐way ANOVA with Tukey's Post Hoc Test (A). Furthermore, the KD SMCs (n = 3 per concentration) had a lower phosphate uptake at all phosphate concentrations compared to SCR controls (n = 3 per concentration): 0.1 mM phosphate (P‐value=0.0085); 0.25 mM phosphate (P‐value=0.011); and 0.5 mM phosphate (P‐value=0.0027) determined by a two‐tailed Student's T‐test with unequal variance (B). Cultured SMCs (n = 3 per condition) were also treated with normal or high phosphate media calcification media. Calcification was time‐dependent; after 6 days, the KD SMCs had an increased level of calcification (P‐value=0.006) compared to the NT and SCR controls, using a One‐way ANOVA with Tukey's Post Hoc Test (C).
Slc20a2 deficiency did not alter blood brain barrier permeability or Slc20a1 levels
In addition to OC phenotype change discussed above, pericyte deficiency, blood brain barrier (BBB) disruption, and Slc20a1 compensation have been proposed as potential mechanisms for BGC. To test these possible mechanisms, several experiments were performed. Pericytes were quantified by counting CD13 positive cells in the thalamus of 1yr old Slc20a2 +/− and +/+ mice. No difference in the CD13 positive cell numbers or the association of CD13 positive cells with vessels was observed between Slc20a2 +/− and Slc20a2 +/+ brains (Supporting Information Figure 2A). Furthermore, BBB function was maintained as determined by lack of Evans Blue permeability at 1yr (Supporting Information Figure 2B). Together these data indicate that BBB integrity is maintained in Slc20a2 haploinsufficient mice with BGC. Furthermore, this functional data complements the protein localization patterns and in vitro studies, and suggests that neither pericytes nor endothelial cells are the causative cell type, again implicating a role for SMC. Finally, we examined the potential for Slc20a1 compensation in neurovascular cells that might predispose to vascular calcification 4, 22. There was no difference in the number of SLC20A1 positive cells or overall protein expression in the thalamus of Slc20a2 +/+ vs Slc20a2 +/− mice (Supporting Information Figure 2C–E). Although we did not find an upregulation of SLC20A1 protein levels or the number of SLC20A1 positive cells in Slc20a2 +/− mice with BGC, we did find that SLC20A1 was expressed by endothelial cells associated with the calcified lesions (Supporting Information Figure 2F–H).
Discussion
We found for the first time that Slc20a2 +/− mice developed age‐dependent BGC that formed in cerebral arterioles, similar to what has been observed in humans with autosomal dominant BGC. Slc20a2 displayed a consensus gene expression pattern in tissues that produced or regulated CSF, and Slc20a2 depletion resulted in elevated CSF phosphate levels. Slc20a2 −/− mice were subviable prior to weaning and had poor survival rates between 4 and 21 weeks of age, with deaths likely caused by severe hydrocephalus. In vessels, SLC20A2 protein was abundant in neurovascular SMCs within the glymphatic passages. SMC knockdown by Slc20a2 specific siRNA resulted in increased susceptibility to calcification in vitro. In contrast, Slc20a2 haploinsufficient brains showed no difference in pericyte numbers and maintained BBB integrity, with normal levels of Slc20a1 and numbers of SLC20A1 positive cells. These data strongly suggest that BGC caused by Slc20a2 deficiency was caused by abnormal CSF phosphate homeostasis combined with increased susceptibility of neurovascular SMC to calcification.
Similar to previous findings, our data confirmed that Slc20a2 −/− mice develop BGC 10. In contrast to the previous findings, however, we are the first to describe several additional central and peripheral phenotypes in the Slc20a2 −/− mice. Centrally, Slc20a2 −/− mice displayed significantly elevated phosphate in CSF, calcified optic nerve tissue, and moderate to severe hydrocephalus. Peripherally, the mice displayed poor skeletal development, low body weight, and significantly decreased post‐weaning survival rates. Of note, optic nerve sheath calcification has been associated with human BGC in rare cases 27. Slc20a2 +/− mice for the most part lacked these additional phenotypes and thus are the first animal model to accurately recapitulate the autosomal dominant feature of IBGC.
We identified two major roles of Slc20a2 that most likely contribute to Slc20a2 haploinsufficiency‐associated BGC. First, we found that SLC20A2 was required to maintain normal CSF phosphate levels. Several tissues are involved in CSF production and maintenance. Slc20a2 displayed a consensus expression pattern in all of these tissues, including the choroid plexus, ependyma, and SMC within glymphatic pathways (indicated in red in Figure 8A–C). The choroid plexus is a well‐accepted site of CSF production. CSF is produced from the choroid plexus as an ultrafiltrate from blood that flows through the choroid plexus capillaries. The choroid plexus epithelium that surrounds the choroid plexus capillaries is the primary interface between the ultrafiltrate and the brain ventricles, and actively regulates CSF composition 11. Slc20a2 was abundantly expressed by the choroid plexus epithelium (illustrated in red in Figure 8A). Phosphate transport properties across the experimentally tractable spiny dogfish choroid plexus are consistent with SLC20A2 activity 7. Ventricular CSF clearance and passage into the brain interstitium is mediated by the ependymal cells that line the ventricles. SLC20A2 protein lines the apical (ventricular lumen) side of the ependymal cells (illustrated in red in Figure 8A), and thus was exquisitely poised to participate in ventricular phosphate re‐uptake from the ventricles to the interstitium. Analogous to the role of type II transporters in controlling urinary phosphate levels, SLC20A2 appears to be a major determinant of CSF phosphate levels 15.
Figure 8.
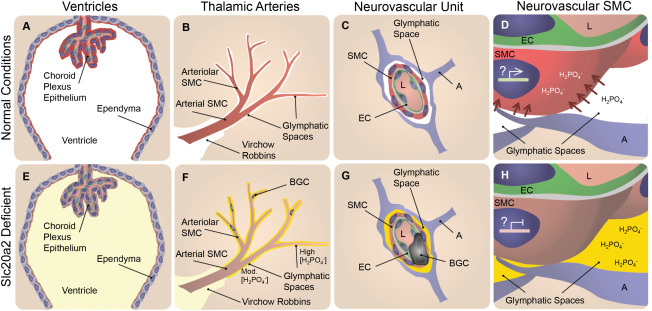
Slc20a2 deficiency‐associated high CSF phosphate and SMC susceptibility to calcification may result in BGC in a 2‐hit mechanism. In normal conditions phosphate CSF concentrations are maintained at homeostatic levels as indicated in white in A‐D. SLC20A2 is poised to regulate CSF phosphate and is localized to the choroid plexus and ependyma (red in A), and the neurovascular SMCs (red in B‐D) adjacent to CSF‐containing glymphatic spaces. In Slc20a2 deficient conditions, these tissues contain reduced levels of SLC20A2 indicated in brown (E‐H). Slc20a2 deficiency leads to higher CSF phosphate concentrations in glymphatic spaces as indicated in yellow (E‐H). The glymphatic spaces are delineated by astrocytes and permeated by astrocyte endfeet, creating pockets. Fluid flow through this porous and segmented structure (G and H), is assumed to increase retention parameters caused by turbulent flow and increased surface‐ion interactions mediated by Brownian Forces. In this case a higher concentration of phosphate likely accumulates in glymphatic spaces that surround the exterior of smaller vessels, indicated by the yellow gradient and segmentation by astrocyte endfeet (F‐H). In normal conditions, SLC20A2 may act to promote SMC phosphate clearance from arteriolar glymphatic spaces (red arrows in D), and SLC20A2 plays a currently unidentified protective role against SMC calcification, illustrated by active gene expression in a SMC nucleus (D). In disease conditions, SLC20A2 deficiency results in high CSF phosphate concentrations compounded by the loss of protective roles against SMC calcification (H). We pose the hypothesis that these abnormalities lead to BGC in a 2‐hit mechanism. Abbreviations: BGC=basal ganglia calcification; SMC = smooth muscle cell; EC = endothelial cell; A = astrocyte; L = lumen.
In addition to CSF production from the choroid plexus, it is now recognized that a significant portion of CSF is produced by vessels within the glymphatic pathway 11. Although much less is known about this mechanism, CSF is likely produced from the blood via the vessels in the glymphatic spaces (perivascular and Virchow Robbins spaces). CSF accumulates in the glymphatic spaces that surround vascular tissue and is intimately associated with AQP4 positive astrocyte endfeet 9. Thalamic glymphatic spaces in the region of the vessels that calcify in Slc20a2 deficient mice are continuous with CSF flow from the cisterna magna 7. In addition to the choroid plexus epithelium and the apical ependymal staining, we found abundant SLC20A2 protein in SMCs that lined glymphatic pathways (illustrated in red in Figure 8B,C). Lastly, we found functional data that supported a role for Slc20a2 in maintaining normal levels of CSF phosphate, as Slc20a2 deficient mice developed elevated phosphate levels in the CSF evident by sampling of cisternal CSF (illustrated in yellow in Figure 8E–H). Flow within and around the thalamic arterioles is believed to be slower than it is at more superficial cerebral vessels of larger diameter, although the mechanisms that regulate this are poorly understood. We suspect that the astrocyte/SMC contact sites play a role in restricting the rate of flow, and this is supported by the observed accumulation of fluorescent markers 9. As such, it is likely that an increasing gradient of phosphate persists in these afferent vessels in Slc20a2 deficient conditions caused by decreased levels of phosphate clearance (illustrated by a yellow gradient in Figure 8F–H). Thus, SLC20A2 regulates CSF phosphate levels, and high phosphate CSF that fills the arteriolar glymphatic spaces/pockets may play a role in the development of BGC (Figure 8F).
In summary, we predicted that lack of Slc20a2 would cause decreased phosphate uptake and clearance in all CSF generating tissues including the choroid plexus and blood vessels within the glymphatic pathway, and would lead to phosphate wasting into the CSF, which we readily observed in Slc20a2 −/− mice. We speculate that our inability to detect statistically significant derangements in CSF phosphate concentrations in Slc20a2 +/− mice may have been caused by variability arising from the detected differences in gradients of CSF phosphate levels using our bulk CSF collection method, and compensation by other phosphate transporters or enhanced water transport that led to increased CSF volume. The latter hypothesis is supported by our observation of mild hydrocephalus in the Slc20a2 +/− mice.
The second major role of Slc20a2 that we identified is anti‐calcific protection of SMC. Slc20a2 deficient SMC treated in vitro with a pro‐calcification high phosphate media displayed significantly greater levels of calcification than either NT or scramble siRNA treated controls. This finding supported the idea that Slc20a2 is protective against calcification, yet suggested that a high phosphate insult is required to reveal the susceptibility of Slc20a2 deficient SMC to calcification. Together, these findings suggest that global Slc20a2 deficiency leads to BGC in a “2 hit” etiology. The data support a hypothetical disease mechanism in which dysfunctional CSF phosphate clearance results in elevated CSF phosphate levels and accumulation of high phosphate in glymphatic spaces that surrounding thalamic arterioles. Subsequently, direct exposure of this high phosphate fluid to SMC that lacked Slc20a2‐dependent anti‐calcific properties would be expected to result in cerebral vascular calcification. Currently, the molecular mechanism delineating how Slc20a2 protects arteriolar SMCs from vascular calcification remains unknown. However, we have observed decreased phosphate uptake in Slc20a2 deficient SMC in vitro (Figure 7B); in vivo this may play a protective role against high phosphate in the glymphatic space.
The idea that deranged phosphate homeostasis accounts, at least in part, for BGC is further supported by recent findings that mutations in the phosphate effluxer, XPR1, are also causative for BGC in people 16. Indeed, the identified mutations in XPR1 are localized to the highly conserved phosphate efflux SPX domain and XPR1 RNAi results in decrease in phosphate efflux in HEK293T cells 16. Furthermore, phosphate efflux was restored when endogenous XPR1 was expressed 16. In the human brain XPR1 was expressed in several regions, including the thalamus and the choroid plexus epithelium. SLC20A2 and XPR1 may both be integral to regulation of phosphate homeostasis in glymphatic spaces and protection against BGC. Further analysis of the SLC20A2 and XPR1 genetic and molecular interactions may shed light onto how these two phosphate regulators play a role in the development of BGC.
In addition to SLC20A2 and XPR1, mice that lack normal PDGF signaling also developed BGC, and both PDGFB and PDGFRB mutations have been linked to IBGC. Though the mechanism of IBGC in these cases is still unproven, disrupted PDGF signaling led to a loss of normal pericyte recruitment into the brain and loss of BBB function in mice 1, 14. While it has been speculated that loss of pericytes and BBB dysfunction might underlie IBGC 1, 14, we found no evidence of pericyte deficiency in the thalamus or BBB dysfunction in Slc20a2 +/− mice compared to Slc20a2 +/+ mice. These data led us to speculate that BGC resulting from Slc20a2 deficiency may arise from a different mechanism than BGC associated with PDGFB signaling defects. Alternatively, PDGF signaling may be a critical upstream regulator of Slc20a2 expression, since PDGFB has been shown to induce expression of Slc20a2 in cultured mesenchymal cells 5. It will be important to determine whether PDGF deficiency leads to decreased Slc20a2 levels in the brain and subsequent elevations in CSF phosphate concentrations.
The Slc20a2 haploinsufficient mouse model will be an invaluable tool for testing efficacy of therapies and preventative treatments. Our work confirmed that the gene expression, histological phenotypes, and blood chemistry of Slc20a2 +/− mice are representative of human IBGC. The BGC that develops in Slc20a2 +/− can now be used as a reliable bioassay to test candidate IBGC treatments. It remains to be seen whether Slc20a2 +/− mice develop motor control and neuropsychiatric disorders. If these mechanisms are conserved, their assessment may provide a noninvasive bioassay that will greatly assist analyzing alleviation of disease progression over time. Now that an accurate mouse model has been established, the field can also move forward in determining the causative cell type(s) in BGC through the use of tissue specific KOs.
Finally, our studies are pertinent not only to understanding the disease mechanism of IBGC, but more broadly reveal for the first time the importance of the phosphate transporter, SLC20A2 in controlling CSF phosphate homeostasis and neural health. They also are among the first to describe a severe pathology associated with the newly identified glymphatic clearance pathway in the brain, and highlight the susceptibility of this pathway to damage. Finally, our studies may provide insights into other diseases, such as Alzheimer's. Functional choroid plexus studies in mouse support that deregulation of choroid plexus‐CSF clearance pathways may be a central disease event causative of Alzheimer's Disease, and abnormal build up of brain wastes are suspected of exacerbating sporadic Alzheimer's Disease 6. Indeed, a healthy brain‐CSF barrier would confer protective advantages on the brain, including protection against metabolic dysfunctions in diseased, aging central nervous systems, and promotion of an optimal environment for neurons 6.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Figure S1. Calcification is bilaterally localized and increases with age. Calcified lesions have bilateral localization and are present in the thalamus of Slc20a2 +/‐ mice as early as 6mo (A,B). The severity and lesion size of the calcified deposits increase with age, as seen in 1yr and 1.5yr old mice (C‐D, G‐H). Abundant calcification is observed throughout the brain of Slc20a2 ‐/‐ mice at ∼1 yr (E‐F). Scale bars: 500 μM (A,C,F), 250 μM (B,D,G).
Figure S2. Testing of alternate hypothetical disease mechanisms. Pericyte deficiency, blood brain barrier (BBB) disruption, Slc20a1 compensation were tested as potential hypotheses to explain the development of BGC. Pericyte numbers were quantified by counting CD13 positive cells in the thalami of 1yr old Slc20a2 +/−, n = 3 and +/+ mice n = 3 (10 sections per animal); no difference in CD13 positive cells were detected as determined by a two‐tailed Student's T‐test with unequal variance; P‐value = 0.744 (A). BBB permeability was tested by Evans Blue permeability in 1 yr old Slc20a2 +/− and +/+; both control and Slc20a2 +/− brains maintained BBB integrity (B). Furthermore, there was no difference in Slc20a1 expression or the number of SLC20A1‐positive cells in the thalami of +/+ (n = 3) and +/− mice (n = 3); as determined by a two‐tailed Student's T‐test with unequal variance; 10 sections per animal; P‐value=0.228 (C‐E, arrows indicate calcified lesions), suggesting that there is an absence of Slc20a1 upregulation following the loss of Slc20a2. However, endothelial cells associated with calcified lesions did contain SLC20A1 as confirmed by immunohistochemical staining (F‐H, arrows indicate calcified lesions). Scale bars: 1 cm (B), 100 μM (C,D), 50 μM (F‐H).
Acknowledgments
The Giachelli Lab is funded by NIH grants HL62329, HL081785, and HL114611, and a grant from the DOD PRORP #OR120074. Dr. Wallingford is funded by NHLBI Training Grant T32HL007828. The Speer Lab is supported by AHA grant 13GRNT14360045. The Cox Lab is supported by Laurel Foundation Endowment for Craniofacial Research (TCC). Ms. Chia is supported by a Howard Hughes Medical Institute's Med‐into‐Grad Program #56006778. Mr. Chavkin is funded by NIBIB Training Grant T32HL007828. Mr. Sawangmake was supported by the Fulbright Visiting Scholarship. The content of this project is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors thank the Small Animal Tomographic Analysis Facility (SANTA) in the Center for Developmental Biology and Regenerative Medicine at Seattle Children's Research Institute for assistance with micro‐CT scans, as well as Dr. Ken Marro at the University of Washington South Lake Union NMR Center for assistance with MRI.
References
- 1. Betsholtz C, Keller A (2014) PDGF, pericytes and the pathogenesis of idiopathic basal ganglia calcification (IBGC). Brain Pathol 24:387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen WJ et al (2013) Novel SLC20A2 mutations identified in southern Chinese patients with idiopathic basal ganglia calcification. Gene 529:159–162. [DOI] [PubMed] [Google Scholar]
- 3. Cheng SL, Shao JS, Charlton‐Kachigian N, Loewy AP, Towler DA (2003) MSX2 promotes osteogenesis and suppresses adipogenic differentiation of multipotent mesenchymal progenitors. J Biol Chem 278:45969–45977. [DOI] [PubMed] [Google Scholar]
- 4. Crouthamel MH et al (2013) Sodium‐dependent phosphate cotransporters and phosphate‐induced calcification of vascular smooth muscle cells: redundant roles for PiT‐1 and PiT‐2. Arteriosclerosis, Thrombosis, Vascular Biol 33:2625–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Demoulin JB et al (2004) Platelet‐derived growth factor stimulates membrane lipid synthesis through activation of phosphatidylinositol 3‐kinase and sterol regulatory element‐binding proteins. J Biol Chem 279:35392–35402. [DOI] [PubMed] [Google Scholar]
- 6. Gonzalez‐Marrero I et al (2015) Choroid plexus dysfunction impairs beta‐amyloid clearance in a triple transgenic mouse model of Alzheimer's disease. Front Cell Neurosci 9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guerreiro PM, Bataille AM, Parker SL, Renfro JL (2014) Active removal of inorganic phosphate from cerebrospinal fluid by the choroid plexus. Am J Physiol Renal Physiol 306:F1275–1284. [DOI] [PubMed] [Google Scholar]
- 8. Hsu SC et al (2013) Mutations in SLC20A2 are a major cause of familial idiopathic basal ganglia calcification. Neurogenetics 14:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Iliff JJ et al (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med 4(147):147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jensen N et al (2013) Loss of function of Slc20a2 associated with familial idiopathic Basal Ganglia calcification in humans causes brain calcifications in mice. J Mol Neurosci 51:994–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jessen NA, Munk AS, Lundgaard I, Nedergaard M (2015) The glymphatic system: A beginner's guide. Neurochem Res [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson K, Polewski M, van Etten D, Terkeltaub R (2005) Chondrogenesis mediated by PPi depletion promotes spontaneous aortic calcification in NPP1−/− mice. Arterioscler Thromb Vasc Biol 25:686–691. [DOI] [PubMed] [Google Scholar]
- 13. Jono S et al (2000) Phosphate regulation of vascular smooth muscle cell calcification. Circul Res 87:10–17. [DOI] [PubMed] [Google Scholar]
- 14. Keller A et al (2013) Mutations in the gene encoding PDGF‐B cause brain calcifications in humans and mice. Nat Genet 45:1077–1082. [DOI] [PubMed] [Google Scholar]
- 15. Lederer E (2014) Renal phosphate transporters. Curr Opin Nephrol Hypertens 23:502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Legati A et al (2015) Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat Genet 47(6):579–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lemos RR, Oliveira MF, Oliveira JR (2013) Reporting a new mutation at the SLC20A2 gene in familial idiopathic basal ganglia calcification. Eur J Neurol 20:43–44. [DOI] [PubMed] [Google Scholar]
- 18. Lemos RR et al (2015) Update and Mutational Analysis of SLC20A2: A Major Cause of Primary Familial Brain Calcification. Hum Mutat 36:489–495. [DOI] [PubMed] [Google Scholar]
- 19. Liu L, Duff K (2008) A technique for serial collection of cerebrospinal fluid from the cisterna magna in mouse. J Visual Exp JoVE 21:10–12, pii: 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nicolas G et al ( 2014. ) A de novo nonsense PDGFB mutation causing idiopathic basal ganglia calcification with laryngeal dystonia. Eur J Hum Genet 22:1236–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nicolas G et al (2014) PDGFB partial deletion: a new, rare mechanism causing brain calcification with leukoencephalopathy. J Mol Neurosci 53:171–175. [DOI] [PubMed] [Google Scholar]
- 22. Nicolas G et al (2013) Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology 80:181–187. [DOI] [PubMed] [Google Scholar]
- 23. Saiki M et al (2007) Neurological deficits are associated with increased brain calcinosis, hypoperfusion, and hypometabolism in idiopathic basal ganglia calcification. Movement Disord 22:1027–1030. [DOI] [PubMed] [Google Scholar]
- 24. Shroff RC et al (2010) Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 21:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Speer MY et al (2009/) Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res 104:733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taglia I, Bonifati V, Mignarri A, Dotti MT, Federico A (2015) Primary familial brain calcification: update on molecular genetics. Neurol Sci 36:787–794. [DOI] [PubMed] [Google Scholar]
- 27. Utman SAK, Atkinson PL, Smith RA, Baig HM (2011) Idiopathic dural optic nerve sheath calcification with intracranial parenchymal calcification. J Radiol Extra 78:49–51. [Google Scholar]
- 28. Wang C et al (2012) Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nature genetics 44:254–256. [DOI] [PubMed] [Google Scholar]
- 29. Yamada M et al (2014) Evaluation of SLC20A2 mutations that cause idiopathic basal ganglia calcification in Japan. Neurology 82:705–712. [DOI] [PubMed] [Google Scholar]
- 30. Zhang Y, Guo X, Wu A (2013) Association between a novel mutation in SLC20A2 and familial idiopathic basal ganglia calcification. PloS one 8(2):e57060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Figure S1. Calcification is bilaterally localized and increases with age. Calcified lesions have bilateral localization and are present in the thalamus of Slc20a2 +/‐ mice as early as 6mo (A,B). The severity and lesion size of the calcified deposits increase with age, as seen in 1yr and 1.5yr old mice (C‐D, G‐H). Abundant calcification is observed throughout the brain of Slc20a2 ‐/‐ mice at ∼1 yr (E‐F). Scale bars: 500 μM (A,C,F), 250 μM (B,D,G).
Figure S2. Testing of alternate hypothetical disease mechanisms. Pericyte deficiency, blood brain barrier (BBB) disruption, Slc20a1 compensation were tested as potential hypotheses to explain the development of BGC. Pericyte numbers were quantified by counting CD13 positive cells in the thalami of 1yr old Slc20a2 +/−, n = 3 and +/+ mice n = 3 (10 sections per animal); no difference in CD13 positive cells were detected as determined by a two‐tailed Student's T‐test with unequal variance; P‐value = 0.744 (A). BBB permeability was tested by Evans Blue permeability in 1 yr old Slc20a2 +/− and +/+; both control and Slc20a2 +/− brains maintained BBB integrity (B). Furthermore, there was no difference in Slc20a1 expression or the number of SLC20A1‐positive cells in the thalami of +/+ (n = 3) and +/− mice (n = 3); as determined by a two‐tailed Student's T‐test with unequal variance; 10 sections per animal; P‐value=0.228 (C‐E, arrows indicate calcified lesions), suggesting that there is an absence of Slc20a1 upregulation following the loss of Slc20a2. However, endothelial cells associated with calcified lesions did contain SLC20A1 as confirmed by immunohistochemical staining (F‐H, arrows indicate calcified lesions). Scale bars: 1 cm (B), 100 μM (C,D), 50 μM (F‐H).