

Abstract
Carnitine is essential for the transfer of long-chain fatty acids across the inner mitochondrial membrane for subsequent β-oxidation. It can be synthesized by the body or assumed with the diet from meat and dairy products. Defects in carnitine biosynthesis do not routinely result in low plasma carnitine levels. Carnitine is accumulated by the cells and retained by kidneys using OCTN2, a high affinity organic cation transporter specific for carnitine. Defects in the OCTN2 carnitine transporter results in autosomal recessive primary carnitine deficiency characterized by decreased intracellular carnitine accumulation, increased losses of carnitine in the urine, and low serum carnitine levels. Patients can present early in life with hypoketotic hypoglycemia and hepatic encephalopathy, or later in life with skeletal and cardiac myopathy or sudden death from cardiac arrhythmia, usually triggered by fasting or catabolic state. This disease responds to oral carnitine that, in pharmacological doses, enters cells using the amino acid transporter B0,+. Primary carnitine deficiency can be suspected from the clinical presentation or identified by low levels of free carnitine (C0) in the newborn screening. Some adult patients have been diagnosed following the birth of an unaffected child with very low carnitine levels in the newborn screening. The diagnosis is confirmed by measuring low carnitine uptake in the patients’ fibroblasts or by DNA sequencing of the SLC22A5 gene encoding the OCTN2 carnitine transporter. Some mutations are specific for certain ethnic backgrounds, but the majority are private and identified only in individual families. Although the genotype usually does not correlate with metabolic or cardiac involvement in primary carnitine deficiency, patients presenting as adults tend to have at least one missense mutation retaining residual activity.
Keywords: carnitine, arrhythmia, SLC22A5, OCTN2, newborn screening, autism
INTRODUCTION
Carnitine (β-hydroxy-γ-trimethylammonium butyrate) is a hydrophilic quaternary amine that plays an essential role in energy metabolism. The main function of carnitine is the transfer of long-chain fatty acids to mitochondria for subsequent β-oxidation [1]. Carnitine also binds acyl residues deriving from the intermediary metabolism of amino acids and help in their elimination functioning as a scavenger [2]. This mechanism is essential in binding/removing abnormal organic acids in several organic acidemias and explains the secondary carnitine deficiency that can result from them [3]. Carnitine conjugation decreases the number of acyl residues attached to coenzyme A (CoA) and increases the ratio between free and acyl-CoAs [4]. This mechanism is especially important in disorders of mitochondrial fatty acids oxidation, when fatty acyl-CoAs accumulate in organs such as the heart inducing apoptosis and inflammation [5]. The conjugation of different acyl residues with carnitine produces acylcarnitine species that can be used as markers for newborn screening and for the diagnosis of inborn errors of metabolism using tandem mass spectrometry [3, 6].
Carnitine homeostasis reflects the balance among absorption from the diet, endogenous biosynthesis, and efficient renal reabsorption [7]. The average adult diet provides about 75% of daily carnitine requirements, mostly from meat and dairy products [3, 8–10]. Animal tissues contain relatively high amount of carnitine (between 0.2–6 μmol/g), with the highest concentration in heart and skeletal muscle [11]. Carnitine is transported from the intestinal lumen into the enterocyte, with the fraction not absorbed in the small intestine being almost completely degraded by bacteria in the large intestine [7]. The balance of carnitine requirement (about 25% in adults) is synthesized endogenously from the amino acids lysine and methionine, at an approximate rate of 1 to 2 μmol/kg body weight/day [7, 11, 12].
Even though carnitine is present primarily in animal products, strict vegetarians (vegans) and lacto-ovo-vegetarians maintain normal carnitine levels, indicating that humans not only synthesize carnitine, but also effectively conserve it through renal tubular reabsorption [7]. At normal circulating concentrations (25–50 μM), carnitine is efficiently reabsorbed by active transport through the high affinity carnitine transporter called organic cation transporter novel 2 (OCTN2), localized in the renal brush border membrane [3, 7, 13–15]. As dietary intake of carnitine decreases, the efficiency of carnitine reabsorption increases; likewise, the rate of carnitine excretion increases rapidly and the efficiency of carnitine reabsorption decreases as the plasma concentration and filtered load of carnitine increases [15]. This homeostatic mechanism serves to maintain circulating carnitine concentrations in a relatively narrow range, allowing the excretion of excess carnitine, for example following ingestion of oral or intravenous carnitine supplements [7].
The high-affinity OCTN2 (SLC22A5 gene) carnitine transporter plays a key role in carnitine homeostasis. OCTN2 was first identified in 1998 for its homology to the organic cation transporter novel 1 (OCTN1, SLC22A4 gene) [14]. It is highly expressed in the heart, muscle, and kidney and operates a sodium-dependent transport of carnitine and a sodium-independent organic cation transport [3]. A defect in the OCTN2 carnitine transporter causes primary carnitine deficiency and results in urinary carnitine wasting, low serum carnitine levels (0–8 μM, normal 25–50 μM), and decreased intracellular carnitine accumulation [3, 16].
THE CARNITINE CYCLE IN FATTY ACID OXIDATION
Fatty acids oxidation is an important source of energy production in mammals. During periods of fasting, fatty acids turn into the predominant substrate for energy production via oxidation in the liver, cardiac muscle, and skeletal muscle (Fig. 1) [2, 3]. The brain does not directly uses fatty acids for oxidative metabolism, but utilizes ketone bodies derived from acetyl-CoA and acetoacetyl-CoA produced by β-oxidation of fatty acids in the liver [3]. Fatty acids originate from three primary sources: exogenous fatty acids that enter cells from the blood or from the gut lumen, fatty acids that arise via de novo synthesis from acetyl-CoA, and fatty acids that are released within the cell by the hydrolysis of acylated proteins, phospholipids, and triglycerides [17]. Endogenous fatty acids are mobilized from adipose tissue stores and transported in the circulation primarily bound to albumin (Fig. 2) [3]. The main proteins involved in the transfer of fatty acids across the plasma membrane are: fatty acid transport proteins (FATPs), fatty acid translocase (FAT/CD36), caveolins, and plasma membrane fatty acid binding proteins (FABPpm) [18–20].
FIGURE 1. Fatty acids oxidation during fasting.
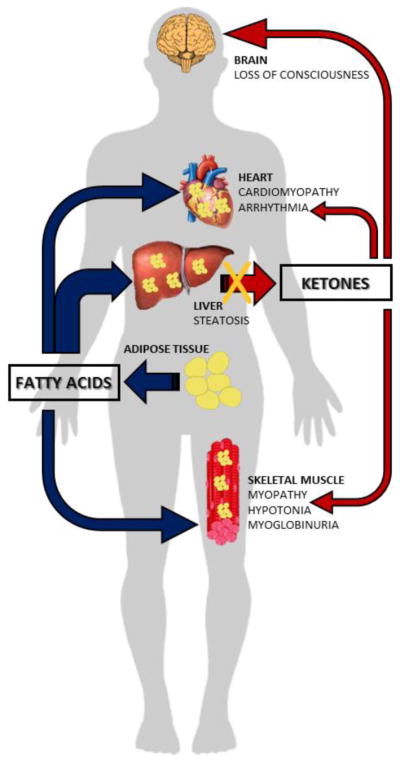
During periods of fasting, fatty acids released from the adipose tissues are oxidized in the liver, skeletal muscle, and cardiac muscle for energy production. The brain does not directly utilize fatty acids, but oxidizes ketone bodies derived from β-oxidation of fatty acids in the liver. When fatty acid oxidation is defective, fats released from the adipose tissue cannot be oxidized, and accumulate in organs such as the skeletal and cardiac muscles, impairing their function. Moreover, the liver is unable to produce ketones bodies resulting in energy deficiency.
FIGURE 2. The carnitine cycle in fatty acid oxidation.

Fatty acids bound to albumin are transferred across the plasma membrane by the action of fatty acid transport proteins (FATP), fatty acid translocase (FAT/CD36), caveolins and plasma membrane fatty acid binding proteins (FABPpm). Inside the cell, fatty acids undergo vectorial acylation, a process catalyzed by acyl-CoA synthases (ACS), that traps them in the cytoplasm as acyl-CoA thioesters. The acyl-CoA thioesters are then conveyed through different metabolic pathways in mitochondria, peroxisomes and microsomes based on the cell energy status. ACS: acyl-CoA synthases; CACT: carnitine acyl carnitine translocase; CPT-1: carnitine palmitoyl transferase-1; CPT-2: carnitine palmitoyl transferase-2; FA: fatty acid; FABPpm: plasma membrane fatty acid binding proteins; FAT/CD36: fatty acid translocase; FATP: fatty acid transport proteins; OCTN2: organic cation transporter novel 2.
FATPs are transmembrane proteins that enhance the uptake of long-chain (16–20 carbon atoms) fatty acids into cells [2, 19, 20]. In humans, FATPs comprise a family of six highly homologous proteins, FATP1–FATP6 (SLC27A1-6 gene) [20, 21]. FATPs have acyl-CoA synthase (ACS) activity [20], converting fatty acids to acyl-CoA thioesters in order to activate them for metabolic processes [2, 20, 22]. The intrinsic ACS activity of FATPs maintains a constant gradient of free fatty acids from the extracellular to the intracellular milieu, with trapping of the long-chain fatty acyl-CoA thioesters inside the cell (vectorial acylation) [17, 20, 23–26], a process conserved from Saccharomyces cerevisiae [27]. FATP1 is highly expressed in the skeletal muscle, heart, white adipose tissue and brown adipose tissue, with an increase in expression upon differentiation of 3T3-L1 cells from pre-adipocytes to adipocytes [20]. Increased expression of FATP1 has been associated with insulin resistance, with a negative correlation between SLC27A1 (the gene encoding FATP1) expression and body mass index in humans [20, 28]. FATP2 is expressed primarily in the liver and kidney and has two splice variants in humans (FATP2a and FATP2b): both variants mediate fatty acids uptake, but only FATP2a has ACS activity, with a preference for very long-chain fatty acids [20, 29]. Knockdown of FATP2 in mice suggests a major role in peroxisomal ACS activity [20]. Very little is known about the physiological role of FATP3 and its subcellular localization is still unclear [20]. FATP4 is primarily expressed on the apical side of enterocytes and in the skin where it plays a major role in epidermal development [20, 30]. Mutations in SLC27A4 (the gene encoding FATP4) cause autosomal recessive congenital ichthyosis prematurity syndrome (OMIM #608649). This disease is characterized by thickened epidermis and respiratory complications, and displays symptoms that are similar to the phenotype seen in Slc27a4 knockout mice [20, 31–33]. FATP5 is expressed exclusively in the basal membrane of hepatocytes. Hepatocytes from mice lacking FATP5 have reduced long-chain fatty acids uptake, are resistant to hepatic steatosis, and are essential for bile acid re-conjugation [20, 21, 34]. In mice, FATP5 (SLC27A5 gene) is involved in body weight regulation, while in humans SLC27A5 expression is increased in patients with early-stage non-alcoholic fatty liver disease [20, 21, 35]. FATP6 is expressed primarily in the heart, specifically in the sarcolemma of cardiomyocytes and plasma membranes juxtaposed to the blood vessels of the heart, where it co-localizes with another fatty acid transport protein, FAT/CD36 [20]. FATP6 (SLC27A6 gene) functions as a fatty acid transporter in the heart and variations in SLC27A6 are associated with lower triglyceride levels , blood pressure, and left ventricular hypertrophy [20, 36].
FAT/CD36 (CD36 gene) is expressed in a wide variety of cells including macrophages, adipocytes, myocytes, enterocytes, and hepatocytes and functions as a receptor for collagen, thrombospondin, Plasmodium falciparum infected red blood cells [37], apoptotic neutrophils, oxidized low density lipoproteins, and as a transporter for long chain fatty acids [18]. This transmembrane protein facilitates the uptake and intracellular trafficking of free fatty acids, as well as esterification into triglycerides in heart and skeletal muscle cells [2, 18, 38, 39]. Patients with FAT/CD36 deficiency have postprandial hypertriglyceridemia, insulin resistance, and hypertension, with increased atherosclerotic risk [40]. FAT/CD36 deficiency has also been associated with malaria susceptibility in humans [41].
Caveolins (caveolins 1, 2, and 3) are integral membrane proteins that serve as core components of caveolae. Caveolae are 60–80 nm flask-shaped, non-clathrin-coated plasma membrane invaginations that project into the cytoplasm, particularly abundant in cholesterol- and sphingolipid-rich lipid rafts in adipocytes, where they are increased during adipocyte differentiation [42]. Caveolin-1 (CAV1 gene) is ubiquitously expressed, but shows high expression in adipocytes, fibroblasts, pneumocytes, neurons, glia, endothelial and epithelial cells [42]. Caveolin-1 directly binds cholesterol and long-chain free fatty acids facilitating their transport inside the cell and upregulates the expression of FAT/CD36 on the plasma membrane [18]. Loss of function of CAV1 cause Berardinelli-Seip congenital lipodystrophy and congenital generalized lipodystrophy type 3 (OMIM #612526), characterized by marked paucity of adipose tissue, extreme insulin resistance, hypertriglyceridemia, hepatic steatosis, and early onset of diabetes [43]. Similar features are seen in Cav1 knockout mice [44]. Caveolin-2 (CAV2 gene) localizes with caveolin-1 [45]. No clear phenotype has been associated with CAV2 deficiency in humans, while Cav2 knockout mice demonstrate severe pulmonary dysfunction along with little or no change in caveolin-1 expression and caveolae formation [46]. Caveolin-3 (M-caveolin, CAV3 gene) is muscle-specific and plays a key role in muscle development and physiology, being critical for the electrical transmission of the contractile impulse, and for the muscle cell membrane stability [42, 47, 48]. Mutations in CAV3 can cause a clinical continuum of skeletal muscle phenotypes: limb-girdle muscular dystrophy type 1C (OMIM #607801), rippling muscle disease-2 (OMIM #606072), isolated hyperCKemia (OMIM #123320), and distal myopathy (OMIM #614321) [48]. The same mutation can cause heterogeneous phenotypes, with intra-familial variability and no genotype/phenotype correlation [48].
The FABPs are a family of 14–15 kDa proteins that bind with high affinity to hydrophobic ligands such as free saturated and unsaturated long-chain fatty acids, their CoA derivatives, bilirubin, organic anions, and other small molecules [18]. FABP isoforms have tissue-specific expression and differ in stoichiometry, affinity, and specificity toward their ligands [49, 50]. They are evolutionarily conserved and similar genes are present in Drosophila melanogaster, Caenorhabditis elegans, mice and humans [49]. FABPs enhance free fatty acids solubility and facilitate transfer to specific cellular compartments (mitochondria and peroxisomes for oxidation; endoplasmic reticulum for re-esterification; lipid droplets for storage; or to the nucleus for gene expression regulation) [18]. There are no reported human phenotypes and knock-out mice show inconsistent or no phenotypes [50–60].
Fatty acid activation inside the cells
Independently from their source (exogenous, de novo synthesis or intracellular hydrolysis), intracellular fatty acids undergo thio-esterification to CoA [61]. This process is catalyzed by acyl-CoA synthases (ACSs) and results in the formation of acyl-CoA products, the activated form of intracellular fatty acids [61].
There are at least 26 proven or likely human ACS genes/proteins, all containing nucleotide-[adenosine monophosphate/adenosine triphosphate (AMP/ATP)] and fatty acid-binding motifs [50, 61]. The crystal structure of bacterial and yeast ACS enzymes suggest that ATP binding induces a conformational change that opens a “gate” to the fatty acids binding site [50]. The enzyme-bound fatty acid is converted to a fatty acid-AMP intermediate, then CoA is attached to the fatty acid-AMP, and AMP is removed [50]. Finally, the acyl-CoA and AMP are released, and the enzyme reverts to its original form [50]. The fatty acid-binding site determines substrate specificity toward defined carbon-chain length and saturation [50, 62]. Bile acids and cholesterol derivatives are also substrates of ACS enzymes [50].
Acyl-CoAs, which are generally bound to proteins and membranes [17, 50], are channeled toward or away from specific metabolic pathways based on the cell energy status. Their intracellular flux and destination are coordinated by multiple proteins such as FABP1 (or L-FABP), sterol carrier protein 2 (SCP2) and acyl-CoA binding domain proteins (ACBD1-7), that channel them for storage or energy production [63].
Carnitine in fatty acid oxidation
When cells require energy, acyl-CoAs can be conveyed to mitochondria and peroxisomes, which work together to maintain lipid homeostasis [19, 64]. Mitochondrial and peroxisomal β-oxidation differs in the transport of substrates, substrate specificities, end products, and energy production [19].
The mitochondrial membrane is impermeable to acyl-CoAs and fatty acids must be conjugated to carnitine to enter mitochondria (Fig. 2). Carnitine is accumulated inside the cell by the high-affinity OCTN2 carnitine transporter in the heart, muscle, and kidney. Hepatocytes have a different low-affinity, high-capacity carnitine transporter [15]. Carnitine forms a high-energy ester bond with long chain fatty acids by the action of carnitine palmitoyl transferase 1 (CPT-1), located in the inner aspect of the outer mitochondrial membrane, with the formation of acylcarnitines [3]. There are three isoforms of CPT-1: CPT-1A, CPT-1B, and CPT-1C [19]. CPT-1A, also called liver CPT-1, is expressed in the liver, brain, kidney, lung, spleen, intestine, pancreas, ovary and fibroblasts [2]. CPT-1B is the muscle isoform and is highly expressed in heart, skeletal muscle and testis [2]. CPT1-C is the neuron specific isoform, but its function in neural metabolism remains controversial [65]. CPT-1 is sensitive to inhibition by malonyl-CoA [2]. Acylcarnitines are then translocated across the inner mitochondrial membrane by the carnitine acylcarnitine translocase (CACT, SLC25A20 gene) [3]. Once inside mitochondria, carnitine palmitoyl transferase 2 (CPT-2), located in the inner mitochondrial membrane, removes carnitine from acylcarnitines and re-generates acyl-CoAs [2]. Carnitine then returns to the cytoplasm for another cycle (using CACT), while the acyl-CoAs can enter (in aerobic conditions and in the presence of low levels of ATP) β-oxidation with final production of acetyl-CoA for oxidative phosphorylation or production of ketone bodies in the liver [3]. Both CPT-1 and CPT-2 are primarily involved in the import of long-chain acyl-CoAs, such as palmitoyl-CoA, oleoyl-CoA, and linoleoyl-CoA [2]. The oxidation of medium- (C6–C10) and short-chain (C4–C6) fatty acids seems largely independent from the carnitine shuttle [19, 66].
Under physiologic conditions, oxidation of long- and medium-chain fatty acids is primarily handled by the mitochondrial β-oxidation system, with only minimal contribution from the peroxisomal system [67]. Preferential substrates of peroxisomal β-oxidation are very long-chain fatty acids (which are not substrate of CPT-1 and thus cannot enter mitochondria), pristanic acid (2,6,10,14-tetramethylpentadecanoic acid), di- and trihydroxycholestanoic acid, the tetracosaenoic acid (C24:6n-3), and long-chain dicarboxylic acids [19, 66–68]. These acyl-CoAs enter peroxisomes by interacting with ATP-binding cassette (ABC) transporters (ABCD1, ABCD2, and ABCD3) on the peroxisomal membrane [19, 66]. All three are half transporters and act as dimers that remove CoA to allow entry of free fatty acids within peroxisomes [19, 69–72]. ABCD1 exhibits highest affinities for very hydrophobic saturated very long-chain fatty acids (C24:0–C26:0); ABCD2 to slightly more hydrophilic very long-chain to long chain-fatty acids (C22:0–C24:0, C22:6); ABCD3 to the most hydrophilic fatty acid species (C20:5) as well as dicarboxylic acids (C16:0DCA) [69]. ABCD1 is defective in X-linked adrenoleukodystrophy, the most frequent disorder of peroxisomal β-oxidation causing childhood cerebral adrenoleukodystrophy and adrenomyeloneuropathy [67]. The peroxisomal β-oxidation system is comparable to that of mitochondria and consists of subsequent steps of dehydrogenation, hydration, dehydrogenation again and thiolytic cleavage [67, 68]. Although peroxisomes contain the full enzymatic machinery to β-oxidize fatty acids, oxidation does not go to completion, and the final products of the peroxisomal β-oxidation can be shuttled to mitochondria for complete oxidation to CO2 and H2O and thus energy production [67, 68, 73]. The final products of peroxisomal β-oxidation can reach mitochondria after conjugation with carnitine trough carnitine octanoyltransferase (CrOT) (whose substrates are medium and long-chain fatty acyl-groups) or peroxisomal carnitine acetyltransferase (CrAT), which is more active toward short-chain acyl-CoAs [18, 66]. The resulting acylcarnitines can be then transported from peroxisomes to mitochondria. Overexpression of CrOT in HepG2 cells decreases intracellular medium-chain and very long-chain fatty acids, with no effects on long chain fatty acids. Opposite effects are seen with CrOT knockdown [73]. Alternatively, the final product of peroxisomal β-oxidation can be used by other metabolic pathways [67, 68, 73]. For example, peroxisomal β-oxidation of di- and trihydroxycholestanoic acid leads to the formation of choloyl-CoA and chenodeoxycholoyl-CoA, then converted by the action of bile acil-CoA: amino acid N-acyltransferase into the taurine and glycine conjugates, which are exported out of the peroxisome into the cytosol and then excreted into the biliary ducts by the bile salt export pump [67].
Both peroxisomes and mitochondria are required for the α-oxidation of branched-chain fatty acids, among which phytanic acid [67, 74]. 3-Methyl branched-chain fatty acids cannot be β-oxidized directly because of the presence of the methyl-group at the 3-position, and they require removal of the last carbon atom by α-oxidation to generate 2-methyl fatty acids that can then be β-oxidized [67, 68].
Fatty acids can also undergo ω-oxidation by action of a microsomal oxidase that uses molecular oxygen, and both an alcohol and aldehyde dehydrogenase to produce dicarboxylic acids, molecules with a carboxyl group at each end [75, 76]. These dicarboxylic acids can be further degraded by peroxisomal β-oxidation to succinate and acetyl-CoA or completely oxidized after transport into the mitochondrial β-oxidation system [75]. Under physiologic conditions, ω−oxidation is a minor pathway of fatty acid metabolism, but a failure of β-oxidation can result in increased ω−oxidation activity, with production of excess dicarboxylic acids that are non-specific markers of fatty acid oxidation defects [76, 77].
DISORDERS OF FATTY ACID OXIDATION
Inherited defects of fatty acids oxidation are transmitted as autosomal recessive traits in humans [3]. When the oxidation of fatty acids is defective, fats are released from the adipose tissue during fasting and reach the liver, skeletal muscle and heart where they can accumulate (Fig. 1). In the liver, a defect in the carnitine cycle or in mitochondrial β-oxidation will result in steatosis and decreased production of ketones. Ketones can be used as an alternate energy source by the heart, skeletal muscle, and brain, sparing glucose. In the liver, acetyl-CoA allosterically activates pyruvate carboxylase to favor gluconeogenesis. The net result of ketone bodies production and of gluconeogenesis is glucose sparing and production (although this latter not from fat itself). If fatty acid oxidation is defective, fat cannot be utilized, glucose is consumed without regeneration via gluconeogenesis (due to inhibition of pyruvate carboxylase), ketones cannot be produced and there is a drop in glucose levels (hypoglycemia). The lack of usable supplies of energy will impair brain function with loss of consciousness [3, 78]. Fats can go directly to the heart and skeletal muscle, where they can accumulate and impair organ/tissue function (cardiomyopathy/myopathy). Free fatty acids and long-chain acylcarnitines can alter the electrical activity of cardiac cells resulting in arrhythmia [3, 78–80]. In certain diseases, the muscle fibers can also break down during sustained exercise resulting in myoglobinuria [3, 81].
Primary carnitine deficiency (Carnitine uptake defect, CUD)
Primary carnitine deficiency (OMIM #212140) is an autosomal recessive disorder of the carnitine cycle that results in defective fatty acid oxidation [3, 12, 82]. The incidence of primary carnitine deficiency varies with a frequency of approximately 1:40,000 newborns in Japan [83], 1:37,000-1:100,000 newborns in Australia [84], and 1:142,000 in the USA [3, 85]. The highest incidence of primary carnitine deficiency (1:300) is in the Faroe Islands, an archipelago in the North Atlantic that remained geographically isolated for many centuries [82, 86–88].
The gene for primary carnitine deficiency, SLC22A5 (MIM# 603377), spans about 30 kb on chromosome 5q31 (chr5:132,369,752–132,395,614, hg38) [3, 14, 82, 89, 90] (Fig. 3). It is composed of 10 exons and the resulting mRNA has an open reading frame of 1,674 nucleotides. The SLC22A5 gene encodes for the organic cation transporter novel 2 (OCTN2), which is composed of 557 amino acids with 12 predicted transmembrane spanning domains with both the amino- and carboxyl-terminus facing the cytoplasm as in other organic cation transporters [14, 91]. There is a long extracellular loop consisting of 107 amino acids between the first two transmembrane domains, which is highly conserved among organic cation transporters encoded by members of the solute carrier (SLC) 22 family, suggesting an essential role in transporter function [92]. This loop contains 3 putative glycosylation sites, N57, N64, and N91 that are normally glycosylated, and is important for substrate and sodium recognition (Fig. 3) [14, 91, 92]. Substitution of asparagine 57, 64, and 91 with glutamine reduces OCTN2 glycosylation and results in cytoplasmic retention of carnitine transporters [93]. Natural mutations affecting this extracellular loop in patients with primary carnitine deficiency can cause a defect in the glycosylation and affect carnitine recognition by the OCTN2 transporter [3, 93, 94]. OCTN2 also contains five potential sites for protein kinase C-dependent (Ser-164, Ser-225, Ser-280, Ser-322 and Ser-323) and one potential site for protein kinase A-dependent phosphorylation (Ser-402) in putative intracellular domains [91]. Natural mutations in serine 225 and 280 [94] markedly reduce carnitine transport, although it is unknown whether it is related to abnormal phosphorylation. OCTN2 contains a glucose-transporter signature motif between transmembrane domains 2 and 3 in which substitution of arginine 169 with glutamine, proline or tryptophan (all identified in patients with primary carnitine deficiency) impairs transporter function without impairing maturation to the plasma membrane [95]. Of two natural variations identified in the nucleotide binding motif (GTEILGKS) in the intracellular loop between the fourth and fifth transmembrane domains (T219K and S225L) only one (S225L) impairs significantly carnitine transport activity ([91] and unpublished results). The predicted size of OCTN2 without glycosylation is 62,751 [91].
FIGURE 3. The human carnitine transporter.

The SLC22A5 gene composed of 10 exons on the long arm of chromosome 5 encodes for the high-affinity carnitine transporter OCTN2 mRNA that has an open reading frame (ORF) of 1,674 nt. The transporter has 12 predicted transmembrane domains, 3 extracellular glycosylation sites, a glucose-transporter signature motif between transmembrane domains 2 and 3, a nucleotide binding motif in the intracellular loop between transmembrane domains 4 and 5, and several intracellular motifs that might be involved in regulatory mechanisms through the action of protein kinase A (PK-A) and C (PK-C).
OCTN2 operates as a Na+-independent organic cation transporter as well as a high affinity (Km = 2.9±0.7 μM) Na+-dependent carnitine transporter [14, 89, 98–100]) (Fig. 4). At low concentrations, sodium lowers the Km of OCTN2 toward carnitine [99], suggesting binding to a site close to the carnitine binding site, while higher sodium concentrations provide the electrochemical gradient to transfer the carnitine-sodium complex inside the cell [99]. Half-maximal stimulation of carnitine transport was obtained at a sodium concentration of 13 ± 2.7 mM in CHO cells expressing the normal OCTN2 cDNA [98, 99], as well as in human fibroblast that express the same transporter (11.4 ± 2.1 mM, [15]).
FIGURE 4. Confocal imaging of the OCTN2 transporter tagged with the green fluorescent protein expressed in Chinese Hamster Ovary cells.

The OCTN2 transporter localizes to the plasma membrane and operates a sodium/carnitine co-transport. The cytoplasm (with the Golgi apparatus stained in red by Bodipy-ceramide) has a low concentration of sodium (about 20 mM) and membrane potential is about -65mV [96]. The resulting sodium electrochemical gradient energizes a more than 80-fold intracellular carnitine accumulation [97].
The study of natural mutations [3] and chimeric transporters obtained by fusing specific domains of the OCTN2 transporter with the organic cation transporter novel 1 (OCTN1, SLC22A4, that does not transport carnitine) [98] has identified multiple domains of OCTN2 essential for function. The amino-terminus and residues in transmembrane domains 7, 9 and 10 have important roles in carnitine recognition [98]. The C-terminus of OCTN2 is required for an efficient transmembrane transfer of the Na+/carnitine complex (Fig. 4), with an essential role played by tyrosine residues in the intracellular loop between transmembrane domains 10 and 11 [98, 101].
Clinical presentation
Defective carnitine transport activity of OCTN2 results in urinary carnitine wasting, low serum carnitine levels (0–8 μM, normal 25–50 μM), and decreased intracellular carnitine accumulation [3, 82]. Patients with primary carnitine deficiency lose most (10–95%) of the filtered carnitine in urine, and their heterozygous parents lose 2 to 3 times the normal amount, explaining their mildly reduced plasma carnitine levels [3, 16, 82]. The decreased intracellular carnitine accumulation results in impaired fatty acid oxidation.
If carnitine supplements are not promptly started, patients with primary carnitine deficiency can present with an acute metabolic decompensation early in life, or later in life with skeletal and cardiac myopathy or sudden death from arrhythmia [82, 100, 102]. Other children, diagnosed because of an affected sibling, had only mild developmental delays or were asymptomatic [3, 103, 104].
The metabolic presentation is more frequent before two years of age [3]. Typically, these children stop eating and become irritable following an upper respiratory tract infection or an acute illness (e.g. gastroenteritis) [47]. Subsequently, they become lethargic and minimally responsive [86]. In most cases, they have hepatomegaly in addition to signs and symptoms of the triggering condition [48]. Laboratory evaluation usually reveals hypoglycemia, with minimal or no ketones in urine, and hyperammonemia, with variably elevated liver function tests. Hyperammonemia is likely caused by decreased expression of urea cycle enzymes in the liver and is usually observed only during acute decompensation [105]. Creatine kinase can also be mildly elevated [13]. If children are not treated promptly with intravenous glucose, they progress to coma and death [21].
The heart’s constant need for energy expenditure and its heavy dependence on fatty acids (the cardiac muscle obtains 50–70% of its energy from fatty acids) explain why patients with primary carnitine deficiency can develop cardiomyopathy, usually between 1 and 4 years of age, that responds poorly to standard therapy [3, 90, 106–108]. If the condition is not correctly diagnosed and no carnitine is supplemented, progressive heart failure eventually leads to transplantation or death. Cardiomyopathy is not commonly seen in adults with primary carnitine deficiency [87, 88, 109]. OCTN2 mutations are not more frequent in adult patients with cardiomyopathy than in the general population [90, 109].
Cardiac arrhythmia has been reported in children and in many adult patients with primary carnitine deficiency [109–113]. Long QT syndrome leading to ventricular tachycardia and cardiac arrest has been reported in primary carnitine deficiency and disappeared after initiation of carnitine supplementation [109]. Adult patients with primary carnitine deficiency, even if asymptomatic, are at risk of sudden death from cardiac arrhythmia as demonstrated by the decreased prevalence of primary carnitine deficiency with age in the Faroe Islands, from 4–5 cases/1,000 to 0.2–0.4 cases/1,000 (Ulrike Steuerwald, personal communication).
There is no correlation between genotype and cardiac or metabolic (hypoketotic hypoglycemia, hepatic encephalopathy) phenotype in primary carnitine deficiency, since different types of presentation have been observed within an individual family and other factors, intrinsic or environmental (fasting, recurrent infections, and others), are responsible for the different phenotypes [82, 103, 114, 115]. Nonsense and frameshift mutations, however, are typically associated with lower carnitine transport and are more prevalent in symptomatic individuals, whereas missense mutations and in-frame deletions (with some residual carnitine transport activity) are more prevalent in asymptomatic mothers identified by newborn screening of their unaffected infants [82].
Newborn screening and diagnosis
Primary carnitine deficiency can be identified clinically following a symptomatic presentation or it can be identified by newborn screening using tandem mass spectrometry [3]. Primary carnitine deficiency is usually associated with low levels of free carnitine (C0) and other acylcarnitines, including C3-, C16-, C18-, and C18:1-carnitine in the newborn screening blood spots since the lack of carnitine prevents their formation [116] (Fig. 5). Apart from C0, low cutoff values have not been defined for the acylcarnitine species, relying heavily on a single metabolite (C0) for the identification of patients at risk for primary carnitine deficiency.
FIGURE 5. Carnitine and acylcarnitine levels in infants with primary carnitine deficiency (patients) and in infants whose mothers are affected by primary carnitine deficiency (mothers).

Carnitine and acylcarnitines were measured in newborn screening blood spots by tandem mass spectrometry using standard methods. The first newborn screening (NBS1) was collected between 12 and 36 hours of life; the second between 7 and 21 days of life (NBS2). Data are averages of values from 4 patients and 4 mothers and from 150,000 unaffected controls, with the standard deviations indicated. Values for carnitine (C0), other acylcarnitine species (C3-, C16-, C18-, C18:1-carnitine) in patients and mothers were significantly (p<0.001 using t-test) below those measured in controls. Levels of C0 and C16-carnitine were significantly different in patients as compared to mothers in the first newborn screening, while levels of C3 were significantly different in patients as compared to mothers in the second screening (*, p<0.05 using t-test). The levels of C3-, C16-, C18-, C18:1-carnitine in patients and of C-16 carnitine in mothers were significantly different between the first and second screening, (°, p<0.05 using t-test).
Carnitine is transferred by the placenta to the growing fetus during intrauterine life and shortly after birth the infants’ carnitine level reflects the mothers’ carnitine levels [12, 84]. As a consequence, if the screening is done too early in life, plasma carnitine levels can falsely be above the cut-off in an infant with primary carnitine deficiency and drop below the cut-off over time (States mandating 2 screens where the second screening is usually collected between 7 and 21 days of age might better identify infants with primary carnitine deficiency, although no data in this respect have been published). Extremely low carnitine (C0) levels have been detected on the first newborn screening of infants of mothers with primary carnitine deficiency (with the infant being a carrier for this condition) [110, 112, 113] (Fig. 5). In these infants, carnitine levels might remain low if the infant is breastfed, since the milk from mothers with primary carnitine deficiency does not provide sufficient amounts of carnitine. By contrast, standard infant formulas and breastmilk of women without carnitine deficiency contain about 60 nmol/ml of carnitine, providing about 1.5 mg/kg per day of carnitine.
The mothers identified through their baby’s newborn screen are in most cases asymptomatic, although there is anecdotal evidence that some feel better (increased stamina) after initiation of carnitine therapy [113]. These mothers, as other adults with primary carnitine deficiency, are at high risk for cardiac arrhythmia and sudden death even if asymptomatic [87, 109, 113, 117–119]. Therefore, low carnitine levels in infants might unmask primary carnitine deficiency in the mother [104, 109–113, 120, 121]. Additional maternal conditions that can cause low carnitine levels in newborns include maternal glutaric acidemia type I [122], medium chain acyl-CoA dehydrogenase deficiency [123], and 3-methyl-crotonyl-CoA carboxylase deficiency [124]. The diagnosis of primary carnitine deficiency needs to be biochemically confirmed by demonstration of low free carnitine levels in plasma (<8 μM, normal 25–50 μM) with reduced renal reabsorption (less than 90%) and normal renal function with no abnormalities in the urine organic acids (although a non-specific dicarboxylic aciduria has been reported [16]). Given the possibility of a maternal disorder causing primary carnitine deficiency, plasma and urine carnitine, plasma acylcarnitine profile, and urine organic acids should be evaluated in the mother as well. The diagnosis is then definitively confirmed by molecular testing of the SLC22A5 gene or demonstrating reduced carnitine transport in fibroblasts (<20% of normal controls). The functional assay remains very useful to confirm or exclude the condition in patients with genetic variations of unknown clinical significance or who continue to have low carnitine levels despite negative molecular studies [3]. Heterozygous parents of affected children have half-normal carnitine transport in their fibroblasts and might have borderline low levels of plasma carnitine [16]. In our experience, functional studies in fibroblasts are the most definitive test, since up to 16% of mutant alleles causing primary carnitine deficiency cannot be identified by sequencing and deletion/duplication analysis of all 10 exons of the SLC22A5 gene and flanking regions [125].
Sequencing of the coding and flanking regions of the SLC22A5 gene encoding the OCTN2 carnitine transporter in patients with primary carnitine deficiency has identified 138 mutations (Table I, Fig. 6), among which 97 are missense, 29 cause the premature insertion of a stop codon as a result of a direct mutational event (14) or as a result of a frameshift (15), 7 changes are predicted to result in abnormal splicing affecting canonical splice-donor or-acceptor sites, 2 are in-frame deletions of single amino acids, and 3 are large gene deletions, one of which affecting the whole SLC22A5 gene.
Table 1.
Mutations in the SLC22A5 gene encoding for the OCTN2 carnitine transporter in patients with primary carnitine deficiency.
| Exon | mRNA | Protein | Effect | Reference |
|---|---|---|---|---|
| OCTN2 deletion | Deletion | [112] | ||
| 5'-UTR | c.-91_22del113 | Deletion | [126] | |
| 1 | c.3G>T | p.M1L | Missense | [127] |
| 1 | c.4_5insC | p.R2Pfs*135 | Frameshift | [126] |
| 1 | c.12C>G | p.Y4* | Stop codon | [103] |
| 1 | c.34G>A | p.G12S | Missense | [112] |
| 1 | c.43G>T | p.G15W | Missense | [110] |
| 1 | c.47C>T | p.P16L | Missense | * |
| 1 | c.51C>G | p.F17L | Missense | [111, 128] |
| 1 | c.56G>C | p.R19P | Missense | [103] |
| 1 | c.59T>A | p.L20H | Missense | * |
| 1 | c.64_66delTTC | p.F23del | In-frame deletion | [115] |
| 1 | c.77G>A | p.S26N | Missense | [82] |
| 1 | c.83G>T | p.S28I | Missense | [129] |
| 1 | c.95A>G | p.N32S | Missense | [88, 115] |
| 1 | c.131C>T | p.A44V | Missense | * |
| 1 | c.136C>T | p.P46S | Missense | [113] |
| 1 | c.137C>T | p.P46L | Missense | * |
| 1 | c.149G>A | p.C50Y | Missense | * |
| 1 | c.196A>C | p.T66P | Missense | [112] |
| 1 | c.224G>C | p.R75P | Missense | [112] |
| 1 | c.232delC | p.H79Tfs*150 | Frameshift | [94] |
| 1 | c.248G>T | p.R83L | Missense | [130] |
| 1 | c.254_264dupGGCTCGCCACC | p.I89Gfs*44 | Frameshift | [103, 115] |
| 1 | c.278C>G | p.S93W | Missense | * |
| 1 | c.283C>G | p.L95V | Missense | * |
| 1 | c.287G>C | p.G96A | Missense | [112] |
| 1 | c.338G>A | p.C113Y | Missense | [131] |
| 1 | c.344A>G | p.D115G | Missense | * |
| 1 | c.350G>A | p.W117* | Stop codon | [112] |
| 1 | c.368T>G | p.V123G | Missense | [112] |
| 1 | c.393G>C | p.E131D | Missense | * |
| IVS1 | c.393+5 G>A | Splicing | [131] | |
| IVS1 | c.394-16T>A | Splicing | [82] | |
| 2 | c.396G>A | p.W132* | Stop codon | [83, 126, 132] |
| 2 | c.419G>A | p.W140* | Stop codon | * |
| 2 | c.424G>T | p.A142S | Missense | [94] |
| 2 | c.428C>T | p.P143L | Missense | [112] |
| 2 | c.433dupA | p. T145Nfs*50 | Frameshift | [131] |
| 2 | c.451G>A | p.V151M | Missense | * |
| 2 | c.454G>C | p.G152R | Missense | [133] |
| 2 | c.458_459delTG | p.V153fs*40 | Frameshift | [127] |
| IVS2 | c.497+1G>T | Splicing | [131] | |
| 3 | Exon 3 deletion | p.F167Dfs*61 | Deletion | * |
| 3 | c.505C>T | p.R169W | Missense | [114, 115] |
| 3 | c.506G>C | p.R169P | Missense | * |
| 3 | c.506G>A | p.R169Q | Missense | [134] |
| 3 | c.517delC | p.L173Cfs*2 | Frameshift | [131] |
| 3 | c.523G>A | p.V175M | Missense | * |
| 3 | c.529A>G | p.M177V | Missense | [112] |
| 3 | c.535A>T | p.M179L | Missense | [83] |
| 3 | c.557T>C | p.L186P | Missense | [112] |
| 3 | c.565_568delTTCT | p.F189Rfs*13 | Frameshift | * |
| 3 | c.573delG | p.N192Ifs*11 | Frameshift | [112] |
| 3 | c.614T>G | p.M205R | Missense | * |
| 3 | c.629A>G | p.N210S | Missense | * |
| 3 | c.632A>G | p.Y211C | Missense | [135] |
| 3 | c.641C>T | p.A214V | Missense | * |
| IVS3 | c.652+1G>A | Splicing | [115] | |
| IVS3 | c.653-2A>C | Splicing | * | |
| 4 | c.656C>A | p.T219K | Missense | * |
| 4 | c.674C>T | p.S225L | Missense | * |
| 4 | c.680G>A | p.R227H | Missense | [112] |
| 4 | c.688T>C | p.F230L | Missense | [112] |
| 4 | c.692C>T | p.S231F | Missense | * |
| 4 | c.695C>T | p.T232M | Missense | [127] |
| 4 | c.700G>C | p.G234R | Missense | [112, 131] |
| 4 | c.718G>A | p.A240T | Missense | [112] |
| 4 | c.725G>T | p.G242V | Missense | [114] |
| 4 | c.740C>G | p.P247R | Missense | * |
| 4 | c.745_748delTTTG | p. F249Lfs*13 | Frameshift | [131] |
| 4 | c.760C>T | p.R254* | Stop codon | [136] |
| 4 | c.761G>A | p.R254Q | Missense | * |
| 4 | c.768G>A | p.W256* | Stop codon | [94] |
| 4 | c.769C>T | p.R257W | Missense | [112] |
| 4 | c.791C>T | p.T264M | Missense | [90] |
| 4 | c.791C>G | p.T264R | Missense | [112] |
| 4 | c.797C>T | p.P266L | Missense | [131] |
| 4 | c.806delT | p.L269Rfs*26 | Frameshift | [137] |
| 4 | c.806T>C | p.L269P | Missense | * |
| 5 | c.825G>A | p.W275* | Stop codon | [127] |
| 5 | c.839delC | p.R282Dfs*13 | Frameshift | [115] |
| 5 | c.839C>T | p.S280F | Missense | [94] |
| 5 | c.844C>T | p.R282* | Stop codon | [102, 134, 135] |
| 5 | c.844delC | p.V295* | Stop codon | [138] |
| 5 | c.845G>A | p.R282Q | Missense | [94] |
| 5 | c.847T>C; T>A | p.W283R | Missense | [139] |
| 5 | c.849G>T | p.W283C | Missense | [83] |
| 5 | c.865C>T | p.R289X* | Stop codon | [127] |
| 5 | c.902C>A | p.A301D | Missense | [114] |
| 5 | c.934A>G | p.I312V | Missense | [90, 112] |
| 5 | c.949G>A | p.E317K | Missense | [90] |
| 5 | c.955C>T | p.Q319* | Stop codon | [112] |
| 6 | c.1008delA | p.T337Pfs*11 | Frameshift | [115] |
| 6 | c.1043T>C | p.I348T | Missense | * |
| 6 | c.1051T>C | p.W351R | Missense | [114] |
| 7 | c.1064C>T | p.S355L | Missense | [112] |
| 7 | c.1072T>A | p.Y358N | Missense | [112] |
| 7 | c.1085C>T | p.S362L | Missense | [111] |
| 7 | c.1088T>C | p.L363P | Missense | [140] |
| 7 | c.1161T>G | p.Y387* | Stop codon | [136] |
| 7 | c.1175_1177delTGC | p.L394del | In-frame deletion | * |
| 7 | c.1193C>T | p.P398L | Missense | [94] |
| 7 | c.1195C>T | p.R399W | Missense | [110] |
| 7 | c.1196G>A | p.R399Q | Missense | [103] |
| 7 | c.1202_1203insA | p.Y401* | Stop codon | [102, 115] |
| 7 | c.1232G>T | p.G411V | Missense | [133] |
| 7 | c.1234A>G | p.S412G | Missense | * |
| IVS7 | c.1267+3_1267+23del | Splicing | [127] | |
| 8 | c.1302delG | p.G435Afs*23 | Frameshift | [102] |
| 8 | c.1316T>G | p.V439G | Missense | * |
| 8 | c.1319C>T | p.T440M | Missense | [115] |
| 8 | c.1324_1325GC>AT | p.A442I | Missense | [110] |
| 8 | c.1327T>G | p.F443V | Missense | [112] |
| 8 | c.1336G>T | p.V446F | Missense | [139] |
| 8 | c.1340A>G | p.Y447C | Missense | [101, 129] |
| 8 | c.1342G>T | p.V448L | Missense | * |
| 8 | c.1345T>G | p.Y449D | Missense | [101] |
| 8 | c.1354G>A | p.E452K | Missense | [100] |
| 8 | c.1364C>G | p.P455R | Missense | [112] |
| 8 | c.1372delG | p. V458* | Stop codon | [131] |
| 8 | c.1385G>T | p.G462V | Missense | * |
| 8 | c.1392_1409delinsCA | p.V465Tfs*28 | Frameshift | * |
| 8 | c.1400C>G | p.S467C | Missense | [83] |
| 8 | c.1403C>G | p.T468R | Missense | [115] |
| 8 | c.1409C>T | p.S470F | Missense | [115] |
| 8 | c.1411C>T | p.R471C | Missense | [111] |
| 8 | c.1412G>C | p.R471P | Missense | [93] |
| 8 | c.1412G>A | p.R471H | Missense | [104] |
| 8 | c.1427T>G | p.L476R | Missense | [141] |
| 8 | c.1433C>T | p.P478L | Missense | [132] |
| IVS8 | c.1451-1G>A | Splicing | [126] | |
| 9 | c.1451G>T | p.G484V | Missense | * |
| 9 | c.1462C>T | p.R488C | Missense | [113] |
| 9 | c.1463G>A | p.R488H | Missense | [94] |
| 9 | c.1520T>C | p.L507S | Missense | [112] |
| 9 | c.1556_1559dupACAC | p.I521Hfs*2 | Frameshift | [113] |
| 10 | c.1645C>T | p.P549S | Missense | [112, 128] |
Novel mutations not previously reported.
FIGURE 6.

Missense mutations in the OCTN2 carnitine transporter in patients with primary carnitine deficiency.
Most families have private mutations and a few mutations, occurring at mutation-prone DNA sequences, have been reported more than once. Founder mutations have been reported in specific geographic areas, such as p.W132X and p.S467C in Japan [83], p.R254X in the Chinese population [136], and p.N32S in the Faroe Islands [88]. In the Faroe Islands, patients with primary carnitine deficiency are either homozygous or compound heterozygous for four different mutations (c.95A>G, p.N32S; c.136C>T, p.P46S; c.131C>T, p.A44V; c.825-52G>A) and a risk-haplotype, with p.N32S being the most prevalent [142]. Patients homozygous for p.N32S had lower mean plasma carnitine levels, the lowest mean residual OCTN2 carnitine transport activity in their fibroblasts, and had a high risk of having symptoms (hypoglycemia or cardiomyopathy in children, sudden death or fatigue in adults) [142], with plasma and tissue carnitine levels dropping rapidly after discontinuation of oral carnitine supplements [143]. All nonsense and frameshift mutations analyzed result in nonsense mediated RNA decay and completely abolish protein function [99, 144]. By contrast, some missense mutations retain residual carnitine transport activity when expressed in Chinese Hamster Ovary cells or measured in patient’s fibroblasts [100]. A specific mutation, p.P46S, has only been identified in asymptomatic or minimally symptomatic (easy fatigability, muscle pain with exercise, fasting intolerance) adult women [145]. This mutation reduces, but does not abolish carnitine transport and affects glycosylation and maturation to the plasma membrane of the OCTN2 carnitine transporter [146]. This and other similar missense mutations seem protective against early clinical manifestations of primary carnitine deficiency, but can still be associated with fatal cardiac arrhythmia leading to sudden death [147].
Many missense mutations affect maturation of the OCTN2 transporter to the plasma membrane [148], while others result in abnormal function of membrane-localized transporters [99, 149]. Pharmacological chaperones can increase stability and membrane maturation of selected mutant OCTN2 transporters [148].
Therapy: carnitine supplementation
Patients with primary carnitine deficiency respond to dietary carnitine supplementation (100–200 mg/kg/day), if started before irreversible organ damage occurs [3]. The dose of carnitine should be divided in at least 3 daily administrations and adapted to each individual patient by serial measurements of plasma carnitine levels. Carnitine has few side effects [3]. It can cause diarrhea and intestinal discomfort with high doses. This is usually self-limiting, and resolves by reducing carnitine dosage. Sometimes, bacterial metabolism in the intestine can result in carnitine degradation, with production of trimethylamine, a non-toxic chemical with a very unpleasant odor. This responds to oral therapy with metronidazole, an antibiotic active against anaerobic bacteria, and/or administration of probiotics, such as those present in certain yogurts. Suspension of therapy for a few days usually does not result in any complication, since the tissue carnitine content requires time to become depleted. The long-term prognosis is favorable as long as children and adults remain on carnitine supplements [3]. Repeated attacks of hypoglycemia or sudden death from arrhythmia without cardiomyopathy have been reported in patients discontinuing carnitine against medical advice [3].
In patients with primary carnitine deficiency, carnitine is absorbed by the gut through the amino acid transporter B0,+ (ATB0,+), that has lower affinity toward carnitine (Km=800 μM) than OCTN2 (Km 3–5 μM) [14, 98–100]), but exhibits much higher capacity than OCTN2 [150].
Differential diagnosis: secondary carnitine deficiency
Primary carnitine deficiency should be differentiated from other causes of carnitine deficiency. These include a number of organic acidemias, defects of fatty acid oxidation and of the carnitine cycle (such as very long chain acyl-CoA dehydrogenase (VLCAD), medium chain acyl-CoA dehydrogenase (MCAD), long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD), carnitine palmitoyltransferase 2 (CPT-2), and carnitine-acylcarnitine translocase (CACT) deficiencies). These metabolic disorders are associated with accumulation of acylcarnitines that can inhibit renal carnitine reabsorption and are lost in urine [151, 152]. In all these disorders, analysis of urine organic acids and plasma acylcarnitine profile, in conjunction with the clinical presentation, allows a definitive diagnosis [3].
Low carnitine levels can also be seen in patients with generalized renal tubular dysfunction, such as renal Fanconi syndrome. In this case, the urinary wasting of other compounds, such as bicarbonate, phosphorus and amino acids, allows a net differentiation, since patients with primary carnitine deficiency have selective carnitine losses [3].
Some pharmacological therapies, such as cyclosporine, pivampicillin, and valproate can bind carnitine forming compounds that are excreted in urine and resulting in carnitine depletion [13, 153–155]. Other medications can inhibit OCTN2 leading to secondary carnitine deficiency, for example anticancer drugs (etoposide, actinomycin D and vinblastine), omeprazole, β-lactam antibiotics (cephaloridine, cefepime, and cefluprenam), and quinolone antibiotic (levofloxacin and grepafloxacin) [13, 153–155].
As carnitine biosynthesis constitutes only 25% of the carnitine pool, defects in carnitine biosynthesis do not usually result in carnitine deficiency in adult individuals with normal renal absorptive function and regular diet. On the other hand, decreased dietary intake in vegetarians does not result in carnitine deficiency due to endogenous carnitine synthesis and the effective renal carnitine absorption [3, 7, 156]. Carnitine biosynthesis in infants is modest and carnitine deficiency was reported in infants fed early preparations of soy formulas that were deficient in carnitine as well as in neonates receiving carnitine-free total parenteral nutrition [157, 158]. We have also found an abnormally low carnitine in the second newborn screen (collected between 7 and 21 days of age) of infants receiving “organic” formulas without carnitine.
CARNITINE BIOSYNTHESIS
The majority of carnitine in the human body derives from the diet (75%), with biosynthesis accounting only for 25% of the total carnitine pool. Carnitine is synthesized by liver, kidney, and in smaller amounts by brain from methionine and protein bound lysine. Carnitine biosynthesis is performed in different organelles and requires several cofactors, such as pyridoxal phosphate, niacin, vitamin C, and iron (Fig. 7) [4, 12, 159]. Mammals cannot synthesize carnitine from free lysine, but lysine residues that compose certain proteins undergo post-translational N-methylation through the action of methyltransferases that use S-adenosyl methionine as methyl donor, with formation of 6-N-trimethyllysine in the cytoplasm [159, 160]. 6-N-trimethyllysine-proteins undergo hydrolysis in the lysosomes with release of ε-N-trimethyllysine, which is the first metabolite of carnitine biosynthesis. Then, ε-N-trimethyllysine enters mitochondria where it undergoes hydroxylation by the ε-N-trimethyllysine hydroxylase to produce β-hydroxy-N-ε-trimethyllysine, a process that requires vitamin C and iron as cofactors [12]. Subsequently, a pyridoxal phosphate dependent aldolase in the cytosol cleaves β-hydroxy-N-ε-trimethyllysine to glycine and 4-N-trimethylaminobutyraldehyde. Dehydrogenation of 4-N-trimethylaminobutyraldehyde by a NAD+-dependent dehydrogenase in the cytosol leads to the formation of 4-N-trimethylaminobutyrate (γ-butyrobetaine) [12]. In the last step, γ−butyrobetaine hydroxylase, a cytosolic dioxygenase, adds the hydroxyl group that carnitine uses to form esters with acyl moieties, with formation of 3-hydroxy-4-N-trimethylaminobutyrate (carnitine) [11, 12].
FIGURE 7. Carnitine biosynthesis in humans.

Carnitine is synthetized in mammals from lysine residues of certain proteins that are post-translationally N-methylated by S-adenosyl-methionine: ε-N-lysine methyltransferase (1), with formation of protein-6-N-trimethyllysine that directs the protein to the lysosome for further degradation. Hydrolysis of protein-6-N-trimethyllysine in the lysosomes by a peptidase (2) releases ε-N-trimethyllysine that is transferred to mitochondria and hydroxylated to β-hydroxy-N-ε-trimethyllysine by ε-N-trimethyllysine hydroxylase (3). In the cytosol, β-hydroxy-N-ε-trimethyllysine is cleaved by a β-hydroxy-N-ε-trimethyllysine aldolase (4) to produce glycine and 4-N-trimethylaminobutyraldehyde that is converted by 4-N-trimethylaminobutyraldehyde dehydrogenase (5) to γ-butyrobetaine. In the last step of carnitine endogenous biosynthesis, γ-butyrobetaine is hydrolyzed by a cytosolic γ−butyrobetaine hydroxylase (6) to carnitine. Brain, liver and kidney are capable of full carnitine biosynthesis, while other tissues, such as cardiac and skeletal muscle, can only synthetize γ−butyrobetaine, and obtain carnitine from the circulation using the OCTN2 carnitine transporter.
The first three enzymes of carnitine endogenous biosynthesis, ε-N-trimethyllysine hydroxylase, β-hydroxy-N-ε-trimethyllysine aldolase, and 4-N-trimethylaminobutyraldehyde dehydrogenase are widely distributed, while γ−butyrobetaine hydroxylase is only present in kidney (highest enzyme activity), liver and brain (lowest measurable enzyme activity). Therefore, the heart and the skeletal muscle cannot synthesize carnitine and fully rely on carnitine transport for long chain fatty acid oxidation [12, 161]. In newborns, γ−butyrobetaine hydroxylase activity in the liver is about 12% of the adult enzyme activity [162]. The reduced capacity that neonates have to synthesize carnitine might explain the low carnitine levels observed in infants receiving formulas or intravenous nutrition without carnitine.
Disorders of carnitine biosynthesis
Two defects in carnitine biosynthesis have been recently identified in humans. Deletions in the TMLHE gene on Xq28 (GRCh38: X:155,489,010–155,612,960) encoding for ε-N-trimethyllysine hydroxylase (Enzyme 3 in Fig. 7) have been associated with non-syndromic autism-spectrum disorders in males [163–166]. The TMLHE exon 2 deletion is relatively common even in healthy males with an estimated frequency of 1:350 males, but is more frequent in patients with autism, suggesting that ε-N-trimethyllysine hydroxylase deficiency is a risk factor for autism (meta-analysis Z-score = 2.90 and p = 0.00037), although with low penetrance (2 to 4%) [164]. In some families, mutations or point deletions in the TMLHE gene segregate with autism and therapy with carnitine can be helpful [165, 167]. In cultured lymphoblastoid cell lines, ε-N-trimethyllysine hydroxylase activity is undetectable or severely reduced in patients with mutations or deletions in the TMLHE gene. Biochemically, affected individuals have up to a three-fold increase in ε-N-trimethyllysine and reduced levels of β-hydroxy-N-ε-trimethyllysine and γ-butyrobetaine in both plasma and urine, with a high ε-N-trimethyllysine/γ-butyrobetaine ratio [164, 165, 167]. Carnitine levels are in the low normal range, reflecting the predominant contribution of dietary intake to plasma carnitine levels. Relative carnitine deficiency has also been shown in patients with autism [168]. Potential disease mechanisms of brain toxicity include toxic effects of excess or deficiency of intermediates in the carnitine biosynthetic pathway or carnitine deficiency in early stages of life during brain development in which the availability of carnitine/acetylcarnitine might affect gene transcription or neurotransmitters balance [169].
A defect in γ−butyrobetaine hydroxylase, which is the last enzyme in the biosynthetic pathway of L-carnitine and catalyzes the formation of L-carnitine from γ−butyrobetaine (Enzyme 6 in Fig. 7) was reported in a girl with microcephaly, speech delay, growth retardation, and minor facial anomalies, although her plasma carnitine levels were within normal limits [170]. The patient was homozygous for a microdeletion in 11p14.2, which contains the BBOX1 gene (GRCh38: 11:27,040,704–27,127,806) encoding for the γ−butyrobetaine hydroxylase enzyme, as well as the Fibin (Fin Bud Initiation Factor Homolog) gene encoding for fibin, a secreted protein identified in zebrafish, mice and humans potentially acting downstream of retinoic acid and wnt signaling, essential for pectoral fin bud initiation and tbx5 expression in zebrafish [170, 171]. The BBOX1 gene spans 86.9 kb and contains 9 exons, including alternatively spliced exons 1A, 1B, and 1C [172]. With only a single case report, it is however difficult to correlate this phenotype with the loss of function of any of these genes [170].
CONCLUSIONS
Carnitine is essential for mitochondrial fatty acid oxidation and can be synthesized endogenously or taken with the diet. The OCTN2 carnitine transporter is essential for retaining carnitine in the body and allowing adequate supplies to the heart and the skeletal muscle, which derive most of their energy from fat. Mutations impairing the function of the OCTN2 transporter result in carnitine deficiency that can present early in life with hypoketotic hypoglycemia, or later in life with cardiomyopathy and sudden death from cardiac arrhythmia. Newborn screening can identify infants with primary carnitine deficiency as well as mothers with this condition because their infants have low levels of carnitine at birth. Missense mutations retaining residual carnitine transport activity are identified with increased frequency in these mothers, who still remain at risk of sudden death from cardiac arrhythmia. The study of natural mutations has identified domains of the OCTN2 transporter important for substrate and co-substrate recognition and their transfer inside the cell. The association of disorders of carnitine biosynthesis with autism raises the possibility that carnitine could have some functions during critical periods of brain development.
Highlights.
The OCTN2 carnitine transporter mediates high-affinity carnitine transport
Defective OCTN2 activity reduces carnitine levels and impairs fatty acid oxidation
Missense mutations in OCTN2 define the function of selected transporter domains
Defects in carnitine biosynthesis do not reduce carnitine levels
Acknowledgments
This work was supported in part by NIH grant DK 53824.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roe C, Ding J. Mitochondrial fatty acid oxidation disorders. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 2297–2326. [Google Scholar]
- 2.Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 2010;33:469–477. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142C:77–85. doi: 10.1002/ajmg.c.30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bieber LL. Carnitine. Annu Rev Biochem. 1988;57:261–283. doi: 10.1146/annurev.bi.57.070188.001401. [DOI] [PubMed] [Google Scholar]
- 5.Drosatos K, Schulze PC. Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr Heart Fail Rep. 2013;10:109–121. doi: 10.1007/s11897-013-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasquali M, Monsen G, Richardson L, Alston M, Longo N. Biochemical findings in common inborn errors of metabolism. Am J Med Genet C Semin Med Genet. 2006;142C:64–76. doi: 10.1002/ajmg.c.30086. [DOI] [PubMed] [Google Scholar]
- 7.Rebouche CJ. Kinetics, pharmacokinetics, and regulation of L-carnitine and acetyl-L-carnitine metabolism. Ann N Y Acad Sci. 2004;1033:30–41. doi: 10.1196/annals.1320.003. [DOI] [PubMed] [Google Scholar]
- 8.Borum PR. Carnitine in neonatal nutrition. J Child Neurol. 1995;10(Suppl 2):S25–31. [PubMed] [Google Scholar]
- 9.Rebouche CJ, Engel AG. Kinetic compartmental analysis of carnitine metabolism in the human carnitine deficiency syndromes. Evidence for alterations in tissue carnitine transport. J Clin Invest. 1984;73:857–867. doi: 10.1172/JCI111281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stanley CA. Carnitine deficiency disorders in children. Ann N Y Acad Sci. 2004;1033:42–51. doi: 10.1196/annals.1320.004. [DOI] [PubMed] [Google Scholar]
- 11.Vaz FM, Wanders RJ. Carnitine biosynthesis in mammals. Biochem J. 2002;361:417–429. doi: 10.1042/0264-6021:3610417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scaglia F, Longo N. Primary and secondary alterations of neonatal carnitine metabolism. Semin Perinatol. 1999;23:152–161. doi: 10.1016/s0146-0005(99)80047-0. [DOI] [PubMed] [Google Scholar]
- 13.El-Hattab AW, Scaglia F. Disorders of carnitine biosynthesis and transport. Mol Genet Metab. 2015 doi: 10.1016/j.ymgme.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 14.Tamai I, Ohashi R, Nezu J, Yabuuchi H, Oku A, Shimane M, Sai Y, Tsuji A. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J Biol Chem. 1998;273:20378–20382. doi: 10.1074/jbc.273.32.20378. [DOI] [PubMed] [Google Scholar]
- 15.Scaglia F, Wang Y, Longo N. Functional characterization of the carnitine transporter defective in primary carnitine deficiency. Arch Biochem Biophys. 1999;364:99–106. doi: 10.1006/abbi.1999.1118. [DOI] [PubMed] [Google Scholar]
- 16.Scaglia F, Wang Y, Singh RH, Dembure PP, Pasquali M, Fernhoff PM, Longo N. Defective urinary carnitine transport in heterozygotes for primary carnitine deficiency. Genet Med. 1998;1:34–39. doi: 10.1097/00125817-199811000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Cooper DE, Young PA, Klett EL, Coleman RA. Physiological Consequences of Compartmentalized Acyl-CoA Metabolism. J Biol Chem. 2015;290:20023–20031. doi: 10.1074/jbc.R115.663260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berlanga A, Guiu-Jurado E, Porras JA, Auguet T. Molecular pathways in non-alcoholic fatty liver disease. Clin Exp Gastroenterol. 2014;7:221–239. doi: 10.2147/CEG.S62831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schrader M, Costello J, Godinho LF, Islinger M. Peroxisome-mitochondria interplay and disease. J Inherit Metab Dis. 2015;38:681–702. doi: 10.1007/s10545-015-9819-7. [DOI] [PubMed] [Google Scholar]
- 20.Anderson CM, Stahl A. SLC27 fatty acid transport proteins. Mol Aspects Med. 2013;34:516–528. doi: 10.1016/j.mam.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doege H, Stahl A. Protein-mediated fatty acid uptake: novel insights from in vivo models. Physiology (Bethesda) 2006;21:259–268. doi: 10.1152/physiol.00014.2006. [DOI] [PubMed] [Google Scholar]
- 22.Black PN, DiRusso CC. Yeast acyl-CoA synthetases at the crossroads of fatty acid metabolism and regulation. Biochim Biophys Acta. 2007;1771:286–298. doi: 10.1016/j.bbalip.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 23.Black PN, DiRusso CC. Vectorial acylation: linking fatty acid transport and activation to metabolic trafficking. Novartis Found Symp. 2007;286:127–138. doi: 10.1002/9780470985571.ch11. discussion 138–141, 162–123, 196–203. [DOI] [PubMed] [Google Scholar]
- 24.Krammer J, Digel M, Ehehalt F, Stremmel W, Fullekrug J, Ehehalt R. Overexpression of CD36 and acyl-CoA synthetases FATP2, FATP4 and ACSL1 increases fatty acid uptake in human hepatoma cells. Int J Med Sci. 2011;8:599–614. doi: 10.7150/ijms.8.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arias-Barrau E, Dirusso CC, Black PN. Methods to monitor Fatty Acid transport proceeding through vectorial acylation. Methods Mol Biol. 2009;580:233–249. doi: 10.1007/978-1-60761-325-1_13. [DOI] [PubMed] [Google Scholar]
- 26.Tong F, Black PN, Coleman RA, DiRusso CC. Fatty acid transport by vectorial acylation in mammals: roles played by different isoforms of rat long-chain acyl-CoA synthetases. Arch Biochem Biophys. 2006;447:46–52. doi: 10.1016/j.abb.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Zou Z, Tong F, Faergeman NJ, Borsting C, Black PN, DiRusso CC. Vectorial acylation in Saccharomyces cerevisiae. Fat1p and fatty acyl-CoA synthetase are interacting components of a fatty acid import complex. J Biol Chem. 2003;278:16414–16422. doi: 10.1074/jbc.M210557200. [DOI] [PubMed] [Google Scholar]
- 28.Binnert C, Koistinen HA, Martin G, Andreelli F, Ebeling P, Koivisto VA, Laville M, Auwerx J, Vidal H. Fatty acid transport protein-1 mRNA expression in skeletal muscle and in adipose tissue in humans. Am J Physiol Endocrinol Metab. 2000;279:E1072–1079. doi: 10.1152/ajpendo.2000.279.5.E1072. [DOI] [PubMed] [Google Scholar]
- 29.Melton EM, Cerny RL, Watkins PA, DiRusso CC, Black PN. Human fatty acid transport protein 2a/very long chain acyl-CoA synthetase 1 (FATP2a/Acsvl1) has a preference in mediating the channeling of exogenous n-3 fatty acids into phosphatidylinositol. J Biol Chem. 2011;286:30670–30679. doi: 10.1074/jbc.M111.226316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herrmann T, Buchkremer F, Gosch I, Hall AM, Bernlohr DA, Stremmel W. Mouse fatty acid transport protein 4 (FATP4): characterization of the gene and functional assessment as a very long chain acyl-CoA synthetase. Gene. 2001;270:31–40. doi: 10.1016/s0378-1119(01)00489-9. [DOI] [PubMed] [Google Scholar]
- 31.Herrmann T, van der Hoeven F, Grone HJ, Stewart AF, Langbein L, Kaiser I, Liebisch G, Gosch I, Buchkremer F, Drobnik W, Schmitz G, Stremmel W. Mice with targeted disruption of the fatty acid transport protein 4 (Fatp 4, Slc27a4) gene show features of lethal restrictive dermopathy. J Cell Biol. 2003;161:1105–1115. doi: 10.1083/jcb.200207080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klar J, Schweiger M, Zimmerman R, Zechner R, Li H, Torma H, Vahlquist A, Bouadjar B, Dahl N, Fischer J. Mutations in the fatty acid transport protein 4 gene cause the ichthyosis prematurity syndrome. Am J Hum Genet. 2009;85:248–253. doi: 10.1016/j.ajhg.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sobol PT, Boudreau JE, Stephenson K, Wan Y, Lichty BD, Mossman KL. Adaptive antiviral immunity is a determinant of the therapeutic success of oncolytic virotherapy. Mol Ther. 2011;19:335–344. doi: 10.1038/mt.2010.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hubbard B, Doege H, Punreddy S, Wu H, Huang X, Kaushik VK, Mozell RL, Byrnes JJ, Stricker-Krongrad A, Chou CJ, Tartaglia LA, Lodish HF, Stahl A, Gimeno RE. Mice deleted for fatty acid transport protein 5 have defective bile acid conjugation and are protected from obesity. Gastroenterology. 2006;130:1259–1269. doi: 10.1053/j.gastro.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 35.Mitsuyoshi H, Yasui K, Harano Y, Endo M, Tsuji K, Minami M, Itoh Y, Okanoue T, Yoshikawa T. Analysis of hepatic genes involved in the metabolism of fatty acids and iron in nonalcoholic fatty liver disease. Hepatol Res. 2009;39:366–373. doi: 10.1111/j.1872-034X.2008.00464.x. [DOI] [PubMed] [Google Scholar]
- 36.Auinger A, Helwig U, Pfeuffer M, Rubin D, Luedde M, Rausche T, Eddine El Mokhtari N, Folsch UR, Schreiber S, Frey N, Schrezenmeir J. A variant in the heart-specific fatty acid transport protein 6 is associated with lower fasting and postprandial TAG, blood pressure and left ventricular hypertrophy. Br J Nutr. 2012;107:1422–1428. doi: 10.1017/S0007114511004727. [DOI] [PubMed] [Google Scholar]
- 37.Oquendo P, Hundt E, Lawler J, Seed B. CD36 directly mediates cytoadherence of Plasmodium falciparum parasitized erythrocytes. Cell. 1989;58:95–101. doi: 10.1016/0092-8674(89)90406-6. [DOI] [PubMed] [Google Scholar]
- 38.Glatz JF, Bonen A, Ouwens DM, Luiken JJ. Regulation of sarcolemmal transport of substrates in the healthy and diseased heart. Cardiovasc Drugs Ther. 2006;20:471–476. doi: 10.1007/s10557-006-0582-8. [DOI] [PubMed] [Google Scholar]
- 39.Kiens B. Skeletal muscle lipid metabolism in exercise and insulin resistance. Physiol Rev. 2006;86:205–243. doi: 10.1152/physrev.00023.2004. [DOI] [PubMed] [Google Scholar]
- 40.Masuda D, Hirano K, Oku H, Sandoval JC, Kawase R, Yuasa-Kawase M, Yamashita Y, Takada M, Tsubakio-Yamamoto K, Tochino Y, Koseki M, Matsuura F, Nishida M, Kawamoto T, Ishigami M, Hori M, Shimomura I, Yamashita S. Chylomicron remnants are increased in the postprandial state in CD36 deficiency. J Lipid Res. 2009;50:999–1011. doi: 10.1194/jlr.P700032-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aitman TJ, Cooper LD, Norsworthy PJ, Wahid FN, Gray JK, Curtis BR, McKeigue PM, Kwiatkowski D, Greenwood BM, Snow RW, Hill AV, Scott J. Malaria susceptibility and CD36 mutation. Nature. 2000;405:1015–1016. doi: 10.1038/35016636. [DOI] [PubMed] [Google Scholar]
- 42.Mendez-Gimenez L, Rodriguez A, Balaguer I, Fruhbeck G. Role of aquaglyceroporins and caveolins in energy and metabolic homeostasis. Mol Cell Endocrinol. 2014;397:78–92. doi: 10.1016/j.mce.2014.06.017. [DOI] [PubMed] [Google Scholar]
- 43.Kim CA, Delepine M, Boutet E, El Mourabit H, Le Lay S, Meier M, Nemani M, Bridel E, Leite CC, Bertola DR, Semple RK, O'Rahilly S, Dugail I, Capeau J, Lathrop M, Magre J. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93:1129–1134. doi: 10.1210/jc.2007-1328. [DOI] [PubMed] [Google Scholar]
- 44.Razani B, Combs TP, Wang XB, Frank PG, Park DS, Russell RG, Li M, Tang B, Jelicks LA, Scherer PE, Lisanti MP. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J Biol Chem. 2002;277:8635–8647. doi: 10.1074/jbc.M110970200. [DOI] [PubMed] [Google Scholar]
- 45.Scherer PE, Okamoto T, Chun M, Nishimoto I, Lodish HF, Lisanti MP. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci U S A. 1996;93:131–135. doi: 10.1073/pnas.93.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Razani B, Wang XB, Engelman JA, Battista M, Lagaud G, Zhang XL, Kneitz B, Hou H, Jr, Christ GJ, Edelmann W, Lisanti MP. Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol Cell Biol. 2002;22:2329–2344. doi: 10.1128/MCB.22.7.2329-2344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang Z, Scherer PE, Okamoto T, Song K, Chu C, Kohtz DS, Nishimoto I, Lodish HF, Lisanti MP. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem. 1996;271:2255–2261. doi: 10.1074/jbc.271.4.2255. [DOI] [PubMed] [Google Scholar]
- 48.Gazzerro E, Sotgia F, Bruno C, Lisanti MP, Minetti C. Caveolinopathies: from the biology of caveolin-3 to human diseases. Eur J Hum Genet. 2010;18:137–145. doi: 10.1038/ejhg.2009.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008;7:489–503. doi: 10.1038/nrd2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grevengoed TJ, Klett EL, Coleman RA. Acyl-CoA metabolism and partitioning. Annu Rev Nutr. 2014;34:1–30. doi: 10.1146/annurev-nutr-071813-105541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin GG, Atshaves BP, McIntosh AL, Mackie JT, Kier AB, Schroeder F. Liver fatty acid binding protein gene ablation potentiates hepatic cholesterol accumulation in cholesterol-fed female mice. Am J Physiol Gastrointest Liver Physiol. 2006;290:G36–48. doi: 10.1152/ajpgi.00510.2004. [DOI] [PubMed] [Google Scholar]
- 52.Martin GG, Danneberg H, Kumar LS, Atshaves BP, Erol E, Bader M, Schroeder F, Binas B. Decreased liver fatty acid binding capacity and altered liver lipid distribution in mice lacking the liver fatty acid-binding protein gene. J Biol Chem. 2003;278:21429–21438. doi: 10.1074/jbc.M300287200. [DOI] [PubMed] [Google Scholar]
- 53.Newberry EP, Xie Y, Kennedy S, Han X, Buhman KK, Luo J, Gross RW, Davidson NO. Decreased hepatic triglyceride accumulation and altered fatty acid uptake in mice with deletion of the liver fatty acid-binding protein gene. J Biol Chem. 2003;278:51664–51672. doi: 10.1074/jbc.M309377200. [DOI] [PubMed] [Google Scholar]
- 54.Hotamisligil GS, Bernlohr DA. Metabolic functions of FABPs-mechanisms and therapeutic implications. Nat Rev Endocrinol. 2015;11:592–605. doi: 10.1038/nrendo.2015.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yeung DC, Wang Y, Xu A, Cheung SC, Wat NM, Fong DY, Fong CH, Chau MT, Sham PC, Lam KS. Epidermal fatty-acid-binding protein: a new circulating biomarker associated with cardio-metabolic risk factors and carotid atherosclerosis. Eur Heart J. 2008;29:2156–2163. doi: 10.1093/eurheartj/ehn295. [DOI] [PubMed] [Google Scholar]
- 56.Hotamisligil GS, Johnson RS, Distel RJ, Ellis R, Papaioannou VE, Spiegelman BM. Uncoupling of obesity from insulin resistance through a targeted mutation in aP2, the adipocyte fatty acid binding protein. Science. 1996;274:1377–1379. doi: 10.1126/science.274.5291.1377. [DOI] [PubMed] [Google Scholar]
- 57.Uysal KT, Scheja L, Wiesbrock SM, Bonner-Weir S, Hotamisligil GS. Improved glucose and lipid metabolism in genetically obese mice lacking aP2. Endocrinology. 2000;141:3388–3396. doi: 10.1210/endo.141.9.7637. [DOI] [PubMed] [Google Scholar]
- 58.Makowski L, Boord JB, Maeda K, Babaev VR, Uysal KT, Morgan MA, Parker RA, Suttles J, Fazio S, Hotamisligil GS, Linton MF. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med. 2001;7:699–705. doi: 10.1038/89076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boord JB, Maeda K, Makowski L, Babaev VR, Fazio S, Linton MF, Hotamisligil GS. Adipocyte fatty acid-binding protein, aP2, alters late atherosclerotic lesion formation in severe hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2002;22:1686–1691. doi: 10.1161/01.atv.0000033090.81345.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maeda K, Uysal KT, Makowski L, Gorgun CZ, Atsumi G, Parker RA, Bruning J, Hertzel AV, Bernlohr DA, Hotamisligil GS. Role of the fatty acid binding protein mal1 in obesity and insulin resistance. Diabetes. 2003;52:300–307. doi: 10.2337/diabetes.52.2.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Watkins PA, Maiguel D, Jia Z, Pevsner J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J Lipid Res. 2007;48:2736–2750. doi: 10.1194/jlr.M700378-JLR200. [DOI] [PubMed] [Google Scholar]
- 62.Hisanaga Y, Ago H, Nakagawa N, Hamada K, Ida K, Yamamoto M, Hori T, Arii Y, Sugahara M, Kuramitsu S, Yokoyama S, Miyano M. Structural basis of the substrate-specific two-step catalysis of long chain fatty acyl-CoA synthetase dimer. J Biol Chem. 2004;279:31717–31726. doi: 10.1074/jbc.M400100200. [DOI] [PubMed] [Google Scholar]
- 63.Neess D, Bek S, Engelsby H, Gallego SF, Faergeman NJ. Long-chain acyl-CoA esters in metabolism and signaling: Role of acyl-CoA binding proteins. Prog Lipid Res. 2015;59:1–25. doi: 10.1016/j.plipres.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 64.Wanders RJ. Peroxisomes in human health and disease: metabolic pathways, metabolite transport, interplay with other organelles and signal transduction. Subcell Biochem. 2013;69:23–44. doi: 10.1007/978-94-007-6889-5_2. [DOI] [PubMed] [Google Scholar]
- 65.Lee J, Wolfgang MJ. Metabolomic profiling reveals a role for CPT1c in neuronal oxidative metabolism. BMC Biochem. 2012;13:23. doi: 10.1186/1471-2091-13-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Violante S, Ijlst L, Te Brinke H, Koster J, Tavares de Almeida I, Wanders RJ, Ventura FV, Houten SM. Peroxisomes contribute to the acylcarnitine production when the carnitine shuttle is deficient. Biochim Biophys Acta. 2013;1831:1467–1474. doi: 10.1016/j.bbalip.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 67.Wanders RJ. Metabolic functions of peroxisomes in health and disease. Biochimie. 2014;98:36–44. doi: 10.1016/j.biochi.2013.08.022. [DOI] [PubMed] [Google Scholar]
- 68.Wanders RJ, Ferdinandusse S, Brites P, Kemp S. Peroxisomes, lipid metabolism and lipotoxicity. Biochim Biophys Acta. 2010;1801:272–280. doi: 10.1016/j.bbalip.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 69.De Marcos Lousa C, van Roermund CW, Postis VL, Dietrich D, Kerr ID, Wanders RJ, Baldwin SA, Baker A, Theodoulou FL. Intrinsic acyl-CoA thioesterase activity of a peroxisomal ATP binding cassette transporter is required for transport and metabolism of fatty acids. Proc Natl Acad Sci U S A. 2013;110:1279–1284. doi: 10.1073/pnas.1218034110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Roermund CW, Visser WF, Ijlst L, van Cruchten A, Boek M, Kulik W, Waterham HR, Wanders RJ. The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl-CoA esters. FASEB J. 2008;22:4201–4208. doi: 10.1096/fj.08-110866. [DOI] [PubMed] [Google Scholar]
- 71.van Roermund CW, Visser WF, Ijlst L, Waterham HR, Wanders RJ. Differential substrate specificities of human ABCD1 and ABCD2 in peroxisomal fatty acid beta-oxidation. Biochim Biophys Acta. 2011;1811:148–152. doi: 10.1016/j.bbalip.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 72.van Roermund CW, Ijlst L, Wagemans T, Wanders RJ, Waterham HR. A role for the human peroxisomal half-transporter ABCD3 in the oxidation of dicarboxylic acids. Biochim Biophys Acta. 2014;1841:563–568. doi: 10.1016/j.bbalip.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 73.Le Borgne F, Ben Mohamed A, Logerot M, Garnier E, Demarquoy J. Changes in carnitine octanoyltransferase activity induce alteration in fatty acid metabolism. Biochem Biophys Res Commun. 2011;409:699–704. doi: 10.1016/j.bbrc.2011.05.068. [DOI] [PubMed] [Google Scholar]
- 74.Wanders RJ, Komen J, Ferdinandusse S. Phytanic acid metabolism in health and disease. Biochim Biophys Acta. 2011;1811:498–507. doi: 10.1016/j.bbalip.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 75.Hardwick JP. Cytochrome P450 omega hydroxylase (CYP4) function in fatty acid metabolism and metabolic diseases. Biochem Pharmacol. 2008;75:2263–2275. doi: 10.1016/j.bcp.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 76.Sanders RJ, Ofman R, Dacremont G, Wanders RJ, Kemp S. Characterization of the human omega-oxidation pathway for omega-hydroxy-very-long-chain fatty acids. FASEB J. 2008;22:2064–2071. doi: 10.1096/fj.07-099150. [DOI] [PubMed] [Google Scholar]
- 77.Wanders RJ, Komen J, Kemp S. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 2011;278:182–194. doi: 10.1111/j.1742-4658.2010.07947.x. [DOI] [PubMed] [Google Scholar]
- 78.Hoffmann L, Seibt A, Herebian D, Spiekerkoetter U. Monounsaturated 14:1n-9 and 16:1n-9 fatty acids but not 18:1n-9 induce apoptosis and necrosis in murine HL-1 cardiomyocytes. Lipids. 2014;49:25–37. doi: 10.1007/s11745-013-3865-4. [DOI] [PubMed] [Google Scholar]
- 79.Wong GK, Crawford MW. Carnitine deficiency increases susceptibility to bupivacaine-induced cardiotoxicity in rats. Anesthesiology. 2011;114:1417–1424. doi: 10.1097/ALN.0b013e31821a8d46. [DOI] [PubMed] [Google Scholar]
- 80.Saini-Chohan HK, Mitchell RW, Vaz FM, Zelinski T, Hatch GM. Delineating the role of alterations in lipid metabolism to the pathogenesis of inherited skeletal and cardiac muscle disorders: Thematic Review Series: Genetics of Human Lipid Diseases. J Lipid Res. 2012;53:4–27. doi: 10.1194/jlr.R012120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Anichini A, Fanin M, Vianey-Saban C, Cassandrini D, Fiorillo C, Bruno C, Angelini C. Genotype-phenotype correlations in a large series of patients with muscle type CPT II deficiency. Neurol Res. 2011;33:24–32. doi: 10.1179/016164110X12767786356390. [DOI] [PubMed] [Google Scholar]
- 82.Rose EC, di San Filippo CA, Ndukwe Erlingsson UC, Ardon O, Pasquali M, Longo N. Genotype-phenotype correlation in primary carnitine deficiency. Hum Mutat. 2012;33:118–123. doi: 10.1002/humu.21607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Koizumi A, Nozaki J, Ohura T, Kayo T, Wada Y, Nezu J, Ohashi R, Tamai I, Shoji Y, Takada G, Kibira S, Matsuishi T, Tsuji A. Genetic epidemiology of the carnitine transporter OCTN2 gene in a Japanese population and phenotypic characterization in Japanese pedigrees with primary systemic carnitine deficiency. Hum Mol Genet. 1999;8:2247–2254. doi: 10.1093/hmg/8.12.2247. [DOI] [PubMed] [Google Scholar]
- 84.Wilcken B, Wiley V, Sim KG, Carpenter K. Carnitine transporter defect diagnosed by newborn screening with electrospray tandem mass spectrometry. J Pediatr. 2001;138:581–584. doi: 10.1067/mpd.2001.111813. [DOI] [PubMed] [Google Scholar]
- 85.Therrell BL, Jr, Lloyd-Puryear MA, Camp KM, Mann MY. Inborn errors of metabolism identified via newborn screening: Ten-year incidence data and costs of nutritional interventions for research agenda planning. Mol Genet Metab. 2014;113:14–26. doi: 10.1016/j.ymgme.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lund AM, Joensen F, Hougaard DM, Jensen LK, Christensen E, Christensen M, Norgaard-Petersen B, Schwartz M, Skovby F. Carnitine transporter and holocarboxylase synthetase deficiencies in The Faroe Islands. J Inherit Metab Dis. 2007;30:341–349. doi: 10.1007/s10545-007-0527-9. [DOI] [PubMed] [Google Scholar]
- 87.Rasmussen J, Kober L, Lund AM, Nielsen OW. Primary Carnitine deficiency in the Faroe Islands: health and cardiac status in 76 adult patients diagnosed by screening. J Inherit Metab Dis. 2014;37:223–230. doi: 10.1007/s10545-013-9640-0. [DOI] [PubMed] [Google Scholar]
- 88.Rasmussen J, Nielsen OW, Janzen N, Duno M, Gislason H, Kober L, Steuerwald U, Lund AM. Carnitine levels in 26,462 individuals from the nationwide screening program for primary carnitine deficiency in the Faroe Islands. J Inherit Metab Dis. 2014;37:215–222. doi: 10.1007/s10545-013-9606-2. [DOI] [PubMed] [Google Scholar]
- 89.Wu X, Huang W, Prasad PD, Seth P, Rajan DP, Leibach FH, Chen J, Conway SJ, Ganapathy V. Functional characteristics and tissue distribution pattern of organic cation transporter 2 (OCTN2), an organic cation/carnitine transporter. J Pharmacol Exp Ther. 1999;290:1482–1492. [PubMed] [Google Scholar]
- 90.Amat di San Filippo C, Taylor MR, Mestroni L, Botto LD, Longo N. Cardiomyopathy and carnitine deficiency. Mol Genet Metab. 2008;94:162–166. doi: 10.1016/j.ymgme.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu X, Prasad PD, Leibach FH, Ganapathy V. cDNA sequence, transport function, and genomic organization of human OCTN2, a new member of the organic cation transporter family. Biochem Biophys Res Commun. 1998;246:589–595. doi: 10.1006/bbrc.1998.8669. [DOI] [PubMed] [Google Scholar]
- 92.Burckhardt G, Wolff NA. Structure of renal organic anion and cation transporters. Am J Physiol Renal Physiol. 2000;278:F853–866. doi: 10.1152/ajprenal.2000.278.6.F853. [DOI] [PubMed] [Google Scholar]
- 93.Filippo CA, Ardon O, Longo N. Glycosylation of the OCTN2 carnitine transporter: study of natural mutations identified in patients with primary carnitine deficiency. Biochim Biophys Acta. 2011;1812:312–320. doi: 10.1016/j.bbadis.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Amat di San Filippo C, Pasquali M, Longo N. Pharmacological rescue of carnitine transport in primary carnitine deficiency. Hum Mutat. 2006;27:513–523. doi: 10.1002/humu.20314. [DOI] [PubMed] [Google Scholar]
- 95.Amat di San Filippo C, Longo N. Tyrosine residues affecting sodium stimulation of carnitine transport in the OCTN2 carnitine/organic cation transporter. J Biol Chem. 2004;279:7247–7253. doi: 10.1074/jbc.M309171200. [DOI] [PubMed] [Google Scholar]
- 96.Longo N, Scaglia F, Wang Y. Insulin increases the turnover rate of Na+-K+-ATPase in human fibroblasts. Am J Physiol Cell Physiol. 2001;280:C912–919. doi: 10.1152/ajpcell.2001.280.4.C912. [DOI] [PubMed] [Google Scholar]
- 97.Scaglia F, Wang Y, Singh RH, Dembure PP, Pasquali M, Fernhoff PM, Longo N. Defective urinary carnitine transport in heterozygotes for primary carnitine deficiency. Genet Med. 1998;1:34–39. doi: 10.1097/00125817-199811000-00008. [DOI] [PubMed] [Google Scholar]
- 98.Amat di San Filippo C, Wang Y, Longo N. Functional domains in the carnitine transporter OCTN2, defective in primary carnitine deficiency. J Biol Chem. 2003;278:47776–47784. doi: 10.1074/jbc.M307911200. [DOI] [PubMed] [Google Scholar]
- 99.Wang Y, Meadows TA, Longo N. Abnormal sodium stimulation of carnitine transport in primary carnitine deficiency. J Biol Chem. 2000;275:20782–20786. doi: 10.1074/jbc.M000194200. [DOI] [PubMed] [Google Scholar]
- 100.Wang Y, Kelly MA, Cowan TM, Longo N. A missense mutation in the OCTN2 gene associated with residual carnitine transport activity. Hum Mutat. 2000;15:238–245. doi: 10.1002/(SICI)1098-1004(200003)15:3<238::AID-HUMU4>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 101.Amat di San Filippo C, Longo N. Tyrosine residues affecting sodium stimulation of carnitine transport in the OCTN2 carnitine/organic cation transporter. J Biol Chem. 2004;279:7247–7253. doi: 10.1074/jbc.M309171200. [DOI] [PubMed] [Google Scholar]
- 102.Wang Y, Ye J, Ganapathy V, Longo N. Mutations in the organic cation/carnitine transporter OCTN2 in primary carnitine deficiency. Proc Natl Acad Sci U S A. 1999;96:2356–2360. doi: 10.1073/pnas.96.5.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang Y, Korman SH, Ye J, Gargus JJ, Gutman A, Taroni F, Garavaglia B, Longo N. Phenotype and genotype variation in primary carnitine deficiency. Genet Med. 2001;3:387–392. doi: 10.1097/00125817-200111000-00002. [DOI] [PubMed] [Google Scholar]
- 104.Spiekerkoetter U, Huener G, Baykal T, Demirkol M, Duran M, Wanders R, Nezu J, Mayatepek E. Silent and symptomatic primary carnitine deficiency within the same family due to identical mutations in the organic cation/carnitine transporter OCTN2. J Inherit Metab Dis. 2003;26:613–615. doi: 10.1023/a:1025968502527. [DOI] [PubMed] [Google Scholar]
- 105.Saheki T, Li MX, Kobayashi K. Antagonizing effect of AP-1 on glucocorticoid induction of urea cycle enzymes: a study of hyperammonemia in carnitine-deficient, juvenile visceral steatosis mice. Mol Genet Metab. 2000;71:545–551. doi: 10.1006/mgme.2000.3093. [DOI] [PubMed] [Google Scholar]
- 106.Shibbani K, Fahed AC, Al-Shaar L, Arabi M, Nemer G, Bitar F, Majdalani M. Primary carnitine deficiency: novel mutations and insights into the cardiac phenotype. Clin Genet. 2014;85:127–137. doi: 10.1111/cge.12112. [DOI] [PubMed] [Google Scholar]
- 107.Fu L, Huang M, Chen S. Primary carnitine deficiency and cardiomyopathy. Korean Circ J. 2013;43:785–792. doi: 10.4070/kcj.2013.43.12.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fu LJ, Chen SB, Han LS, Guo Y, Zhao PJ, Zhu M, Li F, Huang MR. Clinical presentation and therapeutic outcomes of carnitine deficiency-induced cardiomyopathy. Zhonghua Er Ke Za Zhi. 2012;50:929–934. [PubMed] [Google Scholar]
- 109.De Biase I, Champaigne NL, Schroer R, Pollard LM, Longo N, Wood T. Primary Carnitine Deficiency Presents Atypically with Long QT Syndrome: A Case Report. JIMD Rep. 2012;2:87–90. doi: 10.1007/8904_2011_52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.El-Hattab AW, Li FY, Shen J, Powell BR, Bawle EV, Adams DJ, Wahl E, Kobori JA, Graham B, Scaglia F, Wong LJ. Maternal systemic primary carnitine deficiency uncovered by newborn screening: clinical, biochemical, and molecular aspects. Genet Med. 2010;12:19–24. doi: 10.1097/GIM.0b013e3181c5e6f7. [DOI] [PubMed] [Google Scholar]
- 111.Lee NC, Tang NL, Chien YH, Chen CA, Lin SJ, Chiu PC, Huang AC, Hwu WL. Diagnoses of newborns and mothers with carnitine uptake defects through newborn screening. Mol Genet Metab. 2010;100:46–50. doi: 10.1016/j.ymgme.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 112.Li FY, El-Hattab AW, Bawle EV, Boles RG, Schmitt ES, Scaglia F, Wong LJ. Molecular spectrum of SLC22A5 (OCTN2) gene mutations detected in 143 subjects evaluated for systemic carnitine deficiency. Hum Mutat. 2010;31:E1632–1651. doi: 10.1002/humu.21311. [DOI] [PubMed] [Google Scholar]
- 113.Schimmenti LA, Crombez EA, Schwahn BC, Heese BA, Wood TC, Schroer RJ, Bentler K, Cederbaum S, Sarafoglou K, McCann M, Rinaldo P, Matern D, di San Filippo CA, Pasquali M, Berry SA, Longo N. Expanded newborn screening identifies maternal primary carnitine deficiency. Mol Genet Metab. 2007;90:441–445. doi: 10.1016/j.ymgme.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 114.Wang Y, Taroni F, Garavaglia B, Longo N. Functional analysis of mutations in the OCTN2 transporter causing primary carnitine deficiency: lack of genotype-phenotype correlation. Hum Mutat. 2000;16:401–407. doi: 10.1002/1098-1004(200011)16:5<401::AID-HUMU4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 115.Lamhonwah AM, Olpin SE, Pollitt RJ, Vianey-Saban C, Divry P, Guffon N, Besley GT, Onizuka R, De Meirleir LJ, Cvitanovic-Sojat L, Baric I, Dionisi-Vici C, Fumic K, Maradin M, Tein I. Novel OCTN2 mutations: no genotype-phenotype correlations: early carnitine therapy prevents cardiomyopathy. Am J Med Genet. 2002;111:271–284. doi: 10.1002/ajmg.10585. [DOI] [PubMed] [Google Scholar]
- 116.de Sain-van der Velden MG, Diekman EF, Jans JJ, van der Ham M, Prinsen BH, Visser G, Verhoeven-Duif NM. Differences between acylcarnitine profiles in plasma and bloodspots. Mol Genet Metab. 2013;110:116–121. doi: 10.1016/j.ymgme.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 117.Rasmussen J, Nielsen OW, Lund AM, Kober L, Djurhuus H. Primary carnitine deficiency and pivalic acid exposure causing encephalopathy and fatal cardiac events. J Inherit Metab Dis. 2013;36:35–41. doi: 10.1007/s10545-012-9488-8. [DOI] [PubMed] [Google Scholar]
- 118.Rijlaarsdam RS, van Spronsen FJ, Bink-Boelkens MT, Reijngoud DJ, Wanders RJ, Niezen-Koning KE, Van der Sluijs FH, Dorland B, Beaufort-Krol GC. Ventricular fibrillation without overt cardiomyopathy as first presentation of organic cation transporter 2-deficiency in adolescence. Pacing Clin Electrophysiol. 2004;27:675–676. doi: 10.1111/j.1540-8159.2004.00507.x. [DOI] [PubMed] [Google Scholar]
- 119.Mazzini M, Tadros T, Siwik D, Joseph L, Bristow M, Qin F, Cohen R, Monahan K, Klein M, Colucci W. Primary carnitine deficiency and sudden death: in vivo evidence of myocardial lipid peroxidation and sulfonylation of sarcoendoplasmic reticulum calcium ATPase 2. Cardiology. 2011;120:52–58. doi: 10.1159/000333127. [DOI] [PubMed] [Google Scholar]
- 120.Vijay S, Patterson A, Olpin S, Henderson MJ, Clark S, Day C, Savill G, Walter JH. Carnitine transporter defect: diagnosis in asymptomatic adult women following analysis of acylcarnitines in their newborn infants. J Inherit Metab Dis. 2006;29:627–630. doi: 10.1007/s10545-006-0376-y. [DOI] [PubMed] [Google Scholar]
- 121.Sarafoglou K, Tridgell AH, Bentler K, Redlinger-Grosse K, Berry SA, Schimmenti LA. Cardiac conduction improvement in two heterozygotes for primary carnitine deficiency on L-carnitine supplementation. Clin Genet. 2010;78:191–194. doi: 10.1111/j.1399-0004.2009.01368.x. [DOI] [PubMed] [Google Scholar]
- 122.Crombez EA, Cederbaum SD, Spector E, Chan E, Salazar D, Neidich J, Goodman S. Maternal glutaric acidemia, type I identified by newborn screening. Mol Genet Metab. 2008;94:132–134. doi: 10.1016/j.ymgme.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Leydiker KB, Neidich JA, Lorey F, Barr EM, Puckett RL, Lobo RM, Abdenur JE. Maternal medium-chain acyl-CoA dehydrogenase deficiency identified by newborn screening. Mol Genet Metab. 2011;103:92–95. doi: 10.1016/j.ymgme.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 124.Kor D, Mungan NO, Yilmaz BS, Oktem M. An asymptomatic mother diagnosed with 3-methylcrotonyl-CoA carboxylase deficiency after newborn screening. Journal of pediatric endocrinology & metabolism : JPEM. 2015;28:669–671. doi: 10.1515/jpem-2014-0302. [DOI] [PubMed] [Google Scholar]
- 125.Frigeni M, Yin X, Pasquali M, Longo N. Functional and molecular evaluation of patients with primary carnitine deficiency. J Inherit Metab Dis. 2015;38(Suppl 1):S199, Abs P-356. [Google Scholar]
- 126.Nezu J, Tamai I, Oku A, Ohashi R, Yabuuchi H, Hashimoto N, Nikaido H, Sai Y, Koizumi A, Shoji Y, Takada G, Matsuishi T, Yoshino M, Kato H, Ohura T, Tsujimoto G, Hayakawa J, Shimane M, Tsuji A. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet. 1999;21:91–94. doi: 10.1038/5030. [DOI] [PubMed] [Google Scholar]
- 127.Dobrowolski SF, McKinney JT, Amat di San Filippo C, Giak Sim K, Wilcken B, Longo N. Validation of dye-binding/high-resolution thermal denaturation for the identification of mutations in the SLC22A5 gene. Hum Mutat. 2005;25:306–313. doi: 10.1002/humu.20137. [DOI] [PubMed] [Google Scholar]
- 128.Urban TJ, Gallagher RC, Brown C, Castro RA, Lagpacan LL, Brett CM, Taylor TR, Carlson EJ, Ferrin TE, Burchard EG, Packman S, Giacomini KM. Functional genetic diversity in the high-affinity carnitine transporter OCTN2 (SLC22A5) Mol Pharmacol. 2006;70:1602–1611. doi: 10.1124/mol.106.028126. [DOI] [PubMed] [Google Scholar]
- 129.Rahbeeni Z, Vaz FM, Al-Hussein K, Bucknall MP, Ruiter J, Wanders RJ, Rashed MS. Identification of two novel mutations in OCTN2 from two Saudi patients with systemic carnitine deficiency. J Inherit Metab Dis. 2002;25:363–369. doi: 10.1023/a:1020143632011. [DOI] [PubMed] [Google Scholar]
- 130.Makhseed N, Vallance HD, Potter M, Waters PJ, Wong LT, Lillquist Y, Pasquali M, Amat di San Filippo C, Longo N. Carnitine transporter defect due to a novel mutation in the SLC22A5 gene presenting with peripheral neuropathy. J Inherit Metab Dis. 2004;27:778–780. doi: 10.1023/b:boli.0000045837.23328.f4. [DOI] [PubMed] [Google Scholar]
- 131.Han L, Wang F, Wang Y, Ye J, Qiu W, Zhang H, Gao X, Gong Z, Gu X. Analysis of genetic mutations in Chinese patients with systemic primary carnitine deficiency. Eur J Med Genet. 2014;57:571–575. doi: 10.1016/j.ejmg.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 132.Tang NL, Ganapathy V, Wu X, Hui J, Seth P, Yuen PM, Wanders RJ, Fok TF, Hjelm NM. Mutations of OCTN2, an organic cation/carnitine transporter, lead to deficient cellular carnitine uptake in primary carnitine deficiency. Hum Mol Genet. 1999;8:655–660. doi: 10.1093/hmg/8.4.655. [DOI] [PubMed] [Google Scholar]
- 133.Kilic M, Ozgul RK, Coskun T, Yucel D, Karaca M, Sivri HS, Tokatli A, Sahin M, Karagoz T, Dursun A. Identification of mutations and evaluation of cardiomyopathy in Turkish patients with primary carnitine deficiency. JIMD Rep. 2012;3:17–23. doi: 10.1007/8904_2011_36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Burwinkel B, Kreuder J, Schweitzer S, Vorgerd M, Gempel K, Gerbitz KD, Kilimann MW. Carnitine transporter OCTN2 mutations in systemic primary carnitine deficiency: a novel Arg169Gln mutation and a recurrent Arg282ter mutation associated with an unconventional splicing abnormality. Biochem Biophys Res Commun. 1999;261:484–487. doi: 10.1006/bbrc.1999.1060. [DOI] [PubMed] [Google Scholar]
- 135.Vaz FM, Scholte HR, Ruiter J, Hussaarts-Odijk LM, Pereira RR, Schweitzer S, de Klerk JB, Waterham HR, Wanders RJ. Identification of two novel mutations in OCTN2 of three patients with systemic carnitine deficiency. Hum Genet. 1999;105:157–161. doi: 10.1007/s004399900105. [DOI] [PubMed] [Google Scholar]
- 136.Tang NL, Hwu WL, Chan RT, Law LK, Fung LM, Zhang WM. A founder mutation (R254X) of SLC22A5 (OCTN2) in Chinese primary carnitine deficiency patients. Hum Mutat. 2002;20:232. doi: 10.1002/humu.9053. [DOI] [PubMed] [Google Scholar]
- 137.Cederbaum SD, Koo-McCoy S, Tein I, Hsu BY, Ganguly A, Vilain E, Dipple K, Cvitanovic-Sojat L, Stanley C. Carnitine membrane transporter deficiency: a long-term follow up and OCTN2 mutation in the first documented case of primary carnitine deficiency. Mol Genet Metab. 2002;77:195–201. doi: 10.1016/s1096-7192(02)00169-5. [DOI] [PubMed] [Google Scholar]
- 138.Komlosi K, Magyari L, Talian GC, Nemes E, Kaposzta R, Mogyorosy G, Mehes K, Melegh B. Plasma carnitine ester profile in homozygous and heterozygous OCTN2 deficiency. J Inherit Metab Dis. 2009;32(Suppl 1):S15–19. doi: 10.1007/s10545-009-0926-1. [DOI] [PubMed] [Google Scholar]
- 139.Mayatepek E, Nezu J, Tamai I, Oku A, Katsura M, Shimane M, Tsuji A. Two novel missense mutations of the OCTN2 gene (W283R and V446F) in a patient with primary systemic carnitine deficiency. Hum Mutat. 2000;15:118. doi: 10.1002/(SICI)1098-1004(200001)15:1<118::AID-HUMU28>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 140.Akpinar B, Coteli C, Yucel D, Ozgul RK, Dursun A, Demirkol M, Baykal T, Gokcay G. A NOVEL MUTATION (L363P) IN THE OCTN2 GENE AND ITS EFFECT ON SECONDARY PROTEIN STRUCTURE. Clinical genetics. 2010;78:89, Abs J06. [Google Scholar]
- 141.Mutlu-Albayrak H, Bene J, Oflaz MB, Tanyalcin T, Caksen H, Melegh B. Identification of SLC22A5 Gene Mutation in a Family with Carnitine Uptake Defect. Case Rep Genet. 2015;2015:259627. doi: 10.1155/2015/259627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rasmussen J, Lund AM, Risom L, Wibrand F, Gislason H, Nielsen OW, Kober L, Duno M. Residual OCTN2 transporter activity carnitine levels and symptoms correlate in patients with primary carnitine deficiency. Molecular Genetics and Metabolism Reports. 2014;1:241–248. doi: 10.1016/j.ymgmr.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rasmussen J, Thomsen JA, Olesen JH, Lund TM, Mohr M, Clementsen J, Nielsen OW, Lund AM. Carnitine levels in skeletal muscle, blood, and urine in patients with primary carnitine deficiency during intermission of L-carnitine supplementation. JIMD Rep. 2015;20:103–111. doi: 10.1007/8904_2014_398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang Y, Ye J, Ganapathy V, Longo N. Mutations in the organic cation/carnitine transporter OCTN2 in primary carnitine deficiency. Proc Natl Acad Sci U S A. 1999;96:2356–2360. doi: 10.1073/pnas.96.5.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schimmenti LA, Crombez EA, Schwahn BC, Heese BA, Wood TC, Schroer RJ, Bentler K, Cederbaum S, Sarafoglou K, McCann M, Rinaldo P, Matern D, di San Filippo CA, Pasquali M, Berry SA, Longo N. Expanded newborn screening identifies maternal primary carnitine deficiency. Mol Genet Metab. 2007;90:441–445. doi: 10.1016/j.ymgme.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 146.Filippo CA, Ardon O, Longo N. Glycosylation of the OCTN2 carnitine transporter: study of natural mutations identified in patients with primary carnitine deficiency. Biochim Biophys Acta. 2011;1812:312–320. doi: 10.1016/j.bbadis.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rose EC, di San Filippo CA, Ndukwe Erlingsson UC, Ardon O, Pasquali M, Longo N. Genotype-phenotype correlation in primary carnitine deficiency. Hum Mutat. 2012;33:118–123. doi: 10.1002/humu.21607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Amat di San Filippo C, Pasquali M, Longo N. Pharmacological rescue of carnitine transport in primary carnitine deficiency. Hum Mutat. 2006;27:513–523. doi: 10.1002/humu.20314. [DOI] [PubMed] [Google Scholar]
- 149.Wang Y, Taroni F, Garavaglia B, Longo N. Functional analysis of mutations in the OCTN2 transporter causing primary carnitine deficiency: lack of genotype-phenotype correlation. Hum Mutat. 2000;16:401–407. doi: 10.1002/1098-1004(200011)16:5<401::AID-HUMU4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 150.Nakanishi T, Hatanaka T, Huang W, Prasad PD, Leibach FH, Ganapathy ME, Ganapathy V. Na+- and Cl--coupled active transport of carnitine by the amino acid transporter ATB(0,+) from mouse colon expressed in HRPE cells and Xenopus oocytes. J Physiol. 2001;532:297–304. doi: 10.1111/j.1469-7793.2001.0297f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Stanley CA. Carnitine disorders. Adv Pediatr. 1995;42:209–242. [PubMed] [Google Scholar]
- 152.Pochini L, Scalise M, Galluccio M, Indiveri C. OCTN cation transporters in health and disease: role as drug targets and assay development. J Biomol Screen. 2013;18:851–867. doi: 10.1177/1087057113493006. [DOI] [PubMed] [Google Scholar]
- 153.Hirano T, Yasuda S, Osaka Y, Kobayashi M, Itagaki S, Iseki K. Mechanism of the inhibitory effect of zwitterionic drugs (levofloxacin and grepafloxacin) on carnitine transporter (OCTN2) in Caco-2 cells. Biochim Biophys Acta. 2006;1758:1743–1750. doi: 10.1016/j.bbamem.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 154.Hu C, Lancaster CS, Zuo Z, Hu S, Chen Z, Rubnitz JE, Baker SD, Sparreboom A. Inhibition of OCTN2-mediated transport of carnitine by etoposide. Mol Cancer Ther. 2012;11:921–929. doi: 10.1158/1535-7163.MCT-11-0980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pochini L, Scalise M, Indiveri C. Inactivation by omeprazole of the carnitine transporter (OCTN2) reconstituted in liposomes. Chem Biol Interact. 2009;179:394–401. doi: 10.1016/j.cbi.2008.10.052. [DOI] [PubMed] [Google Scholar]
- 156.Lombard KA, Olson AL, Nelson SE, Rebouche CJ. Carnitine status of lactoovovegetarians and strict vegetarian adults and children. Am J Clin Nutr. 1989;50:301–306. doi: 10.1093/ajcn/50.2.301. [DOI] [PubMed] [Google Scholar]
- 157.Slonim AE, Borum PR, Tanaka K, Stanley CA, Kasselberg AG, Greene HL, Burr IM. Dietary-dependent carnitine deficiency as a cause of nonketotic hypoglycemia in an infant. J Pediatr. 1981;99:551–555. doi: 10.1016/s0022-3476(81)80252-1. [DOI] [PubMed] [Google Scholar]
- 158.Schmidt-Sommerfeld E, Penn D. Carnitine and total parenteral nutrition of the neonate. Biol Neonate. 1990;58(Suppl 1):81–88. doi: 10.1159/000243302. [DOI] [PubMed] [Google Scholar]
- 159.Carter AL, Abney TO, Lapp DF. Biosynthesis and metabolism of carnitine. J Child Neurol. 1995;10(Suppl 2):S3–7. [PubMed] [Google Scholar]
- 160.Paik WK, Kim S. Protein methylation. Science. 1971;174:114–119. doi: 10.1126/science.174.4005.114. [DOI] [PubMed] [Google Scholar]
- 161.Rebouche CJ, Engel AG. Tissue distribution of carnitine biosynthetic enzymes in man. Biochim Biophys Acta. 1980;630:22–29. doi: 10.1016/0304-4165(80)90133-6. [DOI] [PubMed] [Google Scholar]
- 162.Olson AL, Rebouche CJ. gamma-Butyrobetaine hydroxylase activity is not rate limiting for carnitine biosynthesis in the human infant. J Nutr. 1987;117:1024–1031. doi: 10.1093/jn/117.6.1024. [DOI] [PubMed] [Google Scholar]
- 163.Celestino-Soper PB, Shaw CA, Sanders SJ, Li J, Murtha MT, Ercan-Sencicek AG, Davis L, Thomson S, Gambin T, Chinault AC, Ou Z, German JR, Milosavljevic A, Sutcliffe JS, Cook EH, Jr, Stankiewicz P, State MW, Beaudet AL. Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum Mol Genet. 2011;20:4360–4370. doi: 10.1093/hmg/ddr363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Celestino-Soper PB, Violante S, Crawford EL, Luo R, Lionel AC, Delaby E, Cai G, Sadikovic B, Lee K, Lo C, Gao K, Person RE, Moss TJ, German JR, Huang N, Shinawi M, Treadwell-Deering D, Szatmari P, Roberts W, Fernandez B, Schroer RJ, Stevenson RE, Buxbaum JD, Betancur C, Scherer SW, Sanders SJ, Geschwind DH, Sutcliffe JS, Hurles ME, Wanders RJ, Shaw CA, Leal SM, Cook EH, Jr, Goin-Kochel RP, Vaz FM, Beaudet AL. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc Natl Acad Sci U S A. 2012;109:7974–7981. doi: 10.1073/pnas.1120210109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nava C, Lamari F, Heron D, Mignot C, Rastetter A, Keren B, Cohen D, Faudet A, Bouteiller D, Gilleron M, Jacquette A, Whalen S, Afenjar A, Perisse D, Laurent C, Dupuits C, Gautier C, Gerard M, Huguet G, Caillet S, Leheup B, Leboyer M, Gillberg C, Delorme R, Bourgeron T, Brice A, Depienne C. Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Transl Psychiatry. 2012;2:e179. doi: 10.1038/tp.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ziats MN, Comeaux MS, Yang Y, Scaglia F, Elsea SH, Sun Q, Beaudet AL, Schaaf CP. Corrigendum to “Improvement of regressive autism symptoms in a child with TMLHE deficiency following carnitine supplementation”. Am J Med Genet A. 2015;167A:2496. doi: 10.1002/ajmg.a.37192. [DOI] [PubMed] [Google Scholar]
- 167.Ziats MN, Comeaux MS, Yang Y, Scaglia F, Elsea SH, Sun Q, Beaudet AL, Schaaf CP. Improvement of regressive autism symptoms in a child with TMLHE deficiency following carnitine supplementation. Am J Med Genet A. 2015;167:2162–2167. doi: 10.1002/ajmg.a.37144. [DOI] [PubMed] [Google Scholar]
- 168.Filipek PA, Juranek J, Nguyen MT, Cummings C, Gargus JJ. Relative carnitine deficiency in autism. Journal of autism and developmental disorders. 2004;34:615–623. doi: 10.1007/s10803-004-5283-1. [DOI] [PubMed] [Google Scholar]
- 169.Wu ZM, Lu X, Wang YW, Sun J, Tao JW, Yin FH, Cheng HJ. Short-term medication of L-carnitine before intracytoplasmic sperm injection for infertile men with oligoasthenozoospermia. Zhonghua Nan Ke Xue. 2012;18:253–256. [PubMed] [Google Scholar]
- 170.Rashidi-Nezhad A, Talebi S, Saebnouri H, Akrami SM, Reymond A. The effect of homozygous deletion of the BBOX1 and Fibin genes on carnitine level and acyl carnitine profile. BMC Med Genet. 2014;15:75. doi: 10.1186/1471-2350-15-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wakahara T, Kusu N, Yamauchi H, Kimura I, Konishi M, Miyake A, Itoh N. Fibin, a novel secreted lateral plate mesoderm signal, is essential for pectoral fin bud initiation in zebrafish. Dev Biol. 2007;303:527–535. doi: 10.1016/j.ydbio.2006.11.041. [DOI] [PubMed] [Google Scholar]
- 172.Rigault C, Le Borgne F, Demarquoy J. Genomic structure, alternative maturation and tissue expression of the human BBOX1 gene. Biochim Biophys Acta. 2006;1761:1469–1481. doi: 10.1016/j.bbalip.2006.09.014. [DOI] [PubMed] [Google Scholar]