

Abstract
Mechanistic target of rapamycin complex 1 (mTORC1) is a molecular node that couples extracellular cues to a wide range of cellular events controlling various physiological processes. Here, we identified mTORC1 signaling as a critical mediator of angiotensin II (Ang II) action in the brain. In neuronal GT1–7 cells, we show that Ang II stimulates neuronal mTORC1 signaling in an Ang II type 1 receptor-dependent manner. In mice, a single intracerebroventricular (ICV) injection or chronic sc infusion of Ang II activated mTORC1 signaling in the subfornical organ, a critical brain region in cardiovascular control and fluid balance. Moreover, transgenic sRA mice with brain-specific overproduction of Ang II displayed increased mTORC1 signaling in the subfornical organ. To test the functional role of brain mTORC1 in mediating the action of Ang II, we examined the consequence of mTORC1 inhibition with rapamycin on Ang II-induced increase in water intake and arterial pressure. ICV pretreatment with rapamycin blocked ICV Ang II-mediated increases in the frequency, duration, and amount of water intake but did not interfere with the pressor response evoked by Ang II. In addition, ICV delivery of rapamycin significantly reduced polydipsia, but not hypertension, of sRA mice. These results demonstrate that mTORC1 is a novel downstream pathway of Ang II type 1 receptor signaling in the brain and selectively mediates the effect of Ang II on drinking behavior.
The renin angiotensin system (RAS) plays a crucial role in the maintenance of cardiovascular and body fluid homeostasis. This is accomplished by the action of angiotensin II (Ang II), the main effector of the RAS, on its receptors present in various tissues including the brain. Circulating Ang II influences neuronal activity through Ang II type 1 receptor (AT1R) located in the brain regions, called the circumventricular organs, which are devoid of blood-brain barrier. The circumventricular organs include the subfornical organ (SFO) which is enriched in AT1R. Through projections to many other brain areas involved in blood pressure (BP) control and fluid balance, the SFO contributes to the long-term effects of systemic Ang II. In addition, all the components of the RAS have been identified in multiple brain nuclei, including the SFO, that are involved in the central control of body fluid and BP that allows local production and action of Ang II (1).
Dysregulation of the brain RAS has been implicated as a key mechanism in the development of hypertension and fluid imbalance. This is supported by substantial evidence including the ability of cerebroventricular administration of Ang II or brain-restricted amplification of Ang II production by expression of human renin and angiotensinogen genes to increase arterial pressure and water intake in rodents (2, 3). Conversely, pharmacological or genetic blockade of central Ang II production or action reverse the elevated arterial pressure, water intake, and salt appetite associated with various animal models of hypertension (4, 5). However, the molecular processes underlying central regulation of arterial pressure and fluid balance by brain action of Ang II are not well understood.
Mechanistic target of rapamycin (mTOR) is an evolutionarily conserved protein that belongs to the phosphatidylinositol kinase-related kinase family (6). mTOR interaction with several other proteins (regulator-associated protein of mTOR, mammalian lethal with Sec13 protein 8, proline-rich protein kinase B substrate 40 kDa, Dishevelled, Egl-10 and Pleckstrin-domain-containing mTOR-interacting protein [Deptor] and Ras homolog enriched in brain) leads to the formation of a large complex, termed mTOR complex 1 (mTORC1). Activity of mTORC1 is a critical determinant of a number of cellular processes, including gene transcription, protein synthesis, cell growth and proliferation, ribosome and mitochondria biogenesis, cytoskeleton organization, and autophagy. In response to various cues arising from growth factors, hormones, stress, nutrients, and energy status, mTORC1 modulate cellular processes through modulation of the activity of multiple downstream signaling pathways including p70 ribosomal protein S6 kinase (S6K) and its effector, the ribosomal protein S6, both of which are commonly used as markers of mTORC1 activity (6).
The mTORC1 pathway links Ang II and hypertrophy by promoting protein synthesis in vascular smooth muscle cells and cardiac myocytes (7–9). These studies raise a possible role for mTORC1 pathway in Ang II actions in the brain, particularly the SFO, where a key component of mTORC1, Deptor, is abundantly expressed along with the AT1R (10, 11). In the current study, we tested the hypothesis that mTORC1 signaling is involved in mediating neuronal actions of Ang II.
Methods and Methods
Animals
The current study used 8- to 20-week-old male and female mice. C57BL/6J mice were obtained from The Jackson Laboratory and bred in our own colony. Transgenic mice (termed sRA) with brain-specific active RAS were generated as described previously (12) by crossing mice expressing human renin (R) under transcriptional control of the neuron-specific synapsin (s) promoter with mice expressing human angiotensinogen (A) under transcriptional control by its own promoter. Nontransgenic littermate mice were used as controls. Mice were housed in air-conditioned room with constant temperature (22°C) and a 12-hour light, 12-hour dark cycle. Animals had ad libitum access to regular rodent chow diet and water. All animal studies were performed in accordance with accepted standards of humane animal care, as outlined in the Ethical Guidelines and approved by Institutional Animal Care and Use Committee of the University of Iowa.
Intracerebroventricular (ICV) cannulation
Mice were anesthetized with ip administration of ketamine (91 mg/kg) and xylazine (9.1 mg/kg) and placed in a stereotactic device (Kopf Instruments). A cannula (9 mm) was implanted into the left lateral brain ventricle (0.3 mm posterior and 1 mm lateral relative to bregma, and 2 mm below the surface of the skull). Mice were allowed 7–10 days to recover from the surgery before experimentation.
Osmotic minipump implantation
C57BL/6J mice were anesthetized with isoflurane (5% for initial induction; 1.5%–2% for maintenance). An osmotic minipump loaded with Ang II (1000 ng/kg · min; Sigma-Aldrich) or vehicle (PBS) was implanted into a sc pocket in the back of each mouse.
Construction of AT1aR expression vector
Rat Ang II type 1a receptor (AT1aR) cDNA was first subcloned from rat brain using RT-PCR, and then inserted into TA TOPO cloning vector (Life Technologies), followed by cloning into the mammalian expression vector, pcDNA3.1.
Cell culture
Immortalized hypothalamic GT1–7 cells were used to study, in vitro, the ability of Ang II to activate neuronal mTORC1 signaling. The cells were authenticated through quantitative real-time PCR analysis of expression of agouti-related peptide as reported previously (13). GT1–7 cells were cultured in growth medium, DMEM containing high glucose (4.5 g/L; Gibco) supplemented with 10% (vol/vol) fetal bovine serum (Atlanta Biologicals) and 1mM sodium pyruvate (Gibco), at 37°C with 5% CO2. The rat AT1aR construct (3 μg) was transfected into 60%–70% confluent GT1–7 cells in 60-mm dish with Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. After incubation with the DNA/Lipofectamine mixture for 24 hours, the cells were recovered in the growth medium for 24 hours before Ang II stimulation.
After Ang II (100nM) treatment, GT1–7 cells were washed in ice-cold PBS and immediately lysed in 50mM Tris-Cl buffer (pH 7.5) containing 0.1mM EDTA, 0.1mM EGTA, 1% sodium deoxycholic acid (wt/vol), 1% Nonidet P-40 (vol/vol), 0.1% Sodium dodecyl sulfate (vol/vol), protease inhibitors, and phosphatase inhibitors for 30 minutes on ice. The extracts were centrifuged at 14 000g for 30 minutes at 4°C. Protein concentration of the obtained supernatant was determined by the Lowry method (14) with a spectrophotometer.
Western blotting
Whole cell lysate (20 μg) was resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to Polyvinylidene Difluoride membranes. The membranes were blocked in either 5% bovine serum albumin or nonfat dry milk in Tris-buffered saline (TBS) with 0.1% Tween 20 for 1 hour at room temperature, followed by incubation with phospho-S6K antibody (1:1000 dilution, 04–392; Millipore) or S6K antibody (1:1000 dilution, 2708; Cell Signaling) at 4°C overnight. The membranes were incubated with antirabbit IgG secondary antibody conjugated with horseradish peroxidase (1:5000 dilution, 7074S; Cell Signaling) for 1 hour at room temperature. Visualization was done with enhanced chemiluminescence (ECL plus; GE).
Immunohistochemistry
Mice anesthetized with ip ketamine and xylazine were perfused transcardially with PBS followed by ice-chilled 4% paraformaldehyde made in PBS (pH 7.4). Isolated brains were further fixed in paraformaldehyde at 4°C overnight (16–24 h). Brains were sliced into 20-μm-thick sections using a vibratome (Leica Biosystems). The sections were incubated in TBS with 0.005% Triton X-100 and 5% goat serum. After overnight incubation with a primary antibody (against phospho-S6 [5364; Cell Signaling] or c-Fos [sc-253; Santa Cruz Biotechnology, Inc]) at 4°C, the sections were incubated with biotinylated antirabbit IgG (Vector) at room temperature for 1 hour and then transferred to an horseradish peroxidase-conjugated avidin solution (ABC kit; Vector) for 30 minutes. The signal was developed using 3,3′-Diaminobenzidine substrate (Vector) and the sections were mounted on glass slides with Permount media (Fisher).
Effect of rapamycin on Ang II-evoked acute drinking response
A 60-minute adaptation period was given to single-housed C57BL/6J mice. The water bottle was temporally removed while the mouse was treated with ICV rapamycin (10 ng/μL) or vehicle (Dimethyl sulfoxide [DMSO], 1 μL) for 10 minutes. Ang II (200 ng/μL) or vehicle (PBS, 1 μL) was then slowly injected ICV before returning the animal to the home cage with the water bottle. The number of times the mouse reached the water bottle, the duration of each drinking episode and the total volume of water intake were recorded for 30 minutes.
Effect of rapamycin on the acute pressor response to Ang II
In anesthetized C57BL/6J mice (under ketamine and xylazine), mean arterial pressure (MAP) was recorded using a fluid-filled catheter inserted into the carotid artery and connected to a pressure transducer. Changes in hemodynamic parameters were monitored in real time using LabChart software (ADInstruments). ICV rapamycin (10 ng/μL) or vehicle (DMSO, 1 μL) was delivered 10 minutes before ICV Ang II (200 ng). The dose of ICV rapamycin was based on our previous studies (15, 16). MAP response to Ang II in the presence or absence of rapamycin was calculated by subtracting the maximum change in MAP (at ∼5 min after ICV injection of Ang II) from baseline MAP.
Effects of daily ICV rapamycin in sRA mice
The effect of daily ICV rapamycin on systolic BP (SBP) and drinking was compared between sRA mice and littermate controls. A noninvasive tail-cuff system (Visitech Systems BP-2000) was used to measure SBP due to the low survival rate of sRA mice after invasive implantation of a radiotelemetric BP probe.
Single-housed mice equipped with ICV cannula were acclimated to restraint, tail-cuff inflation, and handling by daily mock measurements for at least 1 week. A daily measurement session consisted of 30 cycles of mock recording for acclimation and 30 cycles of actual SBP recording. When less than 20 successful SBP recordings out of the 30 cycles were obtained due to errors caused by indistinct signals or excessive movement, the single session was not included in the dataset. SBP was determined by averaging the successful independent sessions during 7 days. After baseline SBP and water intake was determined, ICV rapamycin (10 ng/μL) or vehicle (1-μL DMSO) was administered once a day for 7 days just before the beginning of the dark cycle.
Data analysis
Results are presented as mean ± SEM. All data were analyzed by Student's t test, one- or two-way ANOVA with or without repeated measures, as appropriate. A Tukey test was employed post hoc when appropriate. P < .05 was considered statistically significant. Prism 6 for Macintosh (GraphPad Software, Inc) was used for statistical analysis.
Results
AT1aR-dependent mTORC1 activation by Ang II in neuronal GT1–7 cells
We began by determining the effect of Ang II on mTORC1 signaling in a neuronal cell line, GT1–7. Our screening revealed no expression of the 2 subtypes of rodent AT1R, AT1aR and AT1bR, in GT1–7 cells (Supplemental Figure 1) despite endogenous AT1R expression in the relevant cell types in the brain. Consistent with the absence of AT1R expression, Ang II treatment failed to increase the phosphorylated levels of S6K in nontransfected GT1–7 cells (Figure 1A, open columns). Thus, GT1–7 cells were transiently transfected with a rat AT1aR construct to assess the ability of Ang II to activate neuronal mTORC1 signaling.
Figure 1.

Effect of Ang II on mTORC1 activity in hypothalamic GT1–7 cells. A, Ang II (100nM) increased phosphorylated S6K (p-S6K) in a time-dependent manner in GT1–7 cells transfected with AT1aR (closed columns). Ang II had no effect on p-S6K at 15 minutes without the transfection (white columns). B, Effect of losartan (Los) on Ang II-induced activation of mTORC1 pathway. In AT1aR-transfected GT1–7 cells, losartan blocked the increase in phosphorylation of S6K by Ang II. Values are mean ± SEM, n = 3–4; *, P < .05 vs baseline (0 min) (A) and all other groups (B), one-way ANOVA.
Activation of mTORC1 pathway was determined by Western blot analysis of phosphorylated levels of S6K, a direct substrate of mTORC1 (17). Ang II treatment elicited gradual and time-dependent increase in phosphorylated levels of S6K. Ang II-induced activation of S6K reached a peak, approximately 2.3-fold increase compared with baseline, 10–20 minutes after the treatment (Figure 1A). To assess further the necessity of AT1R for the activation of mTORC1 evoked by Ang II, we used losartan, a selective antagonist of the AT1R (18, 19). In the presence of losartan (10μM, 15-min pretreatment), Ang II (100nM, 15 min) failed to induce S6K phosphorylation in the AT1aR-transfected GT1–7 cells (Figure 1B). Together, these findings show the AT1R-dependent activation of mTORC1 pathway by Ang II in neuronal GT1–7 cells.
Central Ang II stimulates mTORC1 signaling in the SFO and paraventricular nucleus of the hypothalamus (PVN) in mice
AT1R is expressed throughout the brain particularly in the nuclei involved in cardiovascular and body fluid regulation (11). To determine whether Ang II stimulates mTORC1 activity in the brain, we tested in C57BL/6J mice the effect of a single ICV injection of Ang II on immunoreactivity of phosphorylated S6, a downstream effector and surrogate marker of mTORC1 activity (15, 20). ICV Ang II caused a significant increase in the number of phospho-S6-positive cells in the SFO (32 ± 3 vs 13 ± 3 in vehicle group, P < .05) (Figure 2), a brain nucleus enriched in AT1R and where Ang II from the circulation or produced locally exerts its effects on arterial pressure and fluid balance (21). Mice treated with ICV Ang II also exhibited increased phospho-S6 immunoreactivity in the PVN, another Ang II-sensitive cardiovascular regulatory region (Supplemental Figure 2) (22). No effect of Ang II on mTORC1 activity was detected in any other brain region that expresses AT1R (data not shown). These data demonstrate the ability of Ang II to activate mTORC1 signaling in 2 brain nuclei (SFO and PVN) that are critically involved in cardiovascular and fluid homeostasis.
Figure 2.
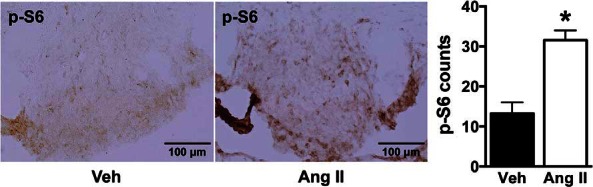
Effect of ICV Ang II on mTORC1 signaling in the SFO. Relative to vehicle, ICV administration of Ang II (1 μg, 30 min) significantly increased the positive cell number of phosphorylated S6 (p-S6) in the SFO of wild-type mice. Microphotographs (×20 magnification) of phospho-S6 staining in the SFO and quantification data are displayed. Values are mean ± SEM, n = 4–6; *, P < .05, t test.
Chronic systemic Ang II infusion activates mTORC1 in the SFO
We asked whether chronic systemic administration of Ang II can also activate brain mTORC1 signaling. In mice, sc infusion of Ang II significantly increased phospho-S6 immunoreactivity in the SFO after 1 week (27 ± 3 vs 11 ± 2 in vehicle, P < .05) or 2 weeks (35 ± 2 vs 18 ± 3 in vehicle, P < .05) of treatment (Figure 3). No change in phospho-S6 immunoreactivity was observed in the PVN or other brain regions (data not shown). Interestingly, induction of phospho-S6 in the SFO by Ang II was associated with a significant increase in nuclear c-Fos-positive cells (1 wk, 7 ± 1 vs 1 ± 0 in vehicle; and 2 wk, 13 ± 2 vs 3 ± 1 in vehicle) (Figure 4), indicative of neuronal activation in this nucleus. Of note, Ang II-induced increase in phospho-S6 and c-Fos-positive cells in SFO occurs in both male and female mice although the increase in c-Fos in males did not reach statistical significance (Supplemental Figure 3). Thus, chronic sc Ang II activated mTORC1 signaling selectively in the SFO in mice.
Figure 3.

Effect of systemic Ang II infusion on mTORC1 signaling in the SFO. Subcutaneous infusion of Ang II (1000 ng/kg · min) for 1 week (A) or 2 weeks (B) significantly increased the positive cell number of phosphorylated S6 (p-S6) in the SFO of wild-type mice. Microphotographs (×20 magnification) of phospho-S6 staining in the SFO and quantification data are displayed. Values are mean ± SEM, n = 5–8 per treatment; *, P < .05, t test.
Figure 4.

Effect of sc Ang II infusion on neuronal activity in the SFO of wild-type mice. Ang II infusion (1000 ng/kg · min) for 1 week (A) or 2 weeks (B) significantly increased the positive cell number of c-Fos in the SFO. Microphotographs (×20 magnification) of c-Fos immunoreactivity in the SFO and quantification data are displayed. Values are mean ± SEM, n = 5–8 per treatment; *, P < .05, t test.
Elevated mTORC1 activity in the SFO of sRA transgenic mice
To assess further the effect of Ang II on brain mTORC1 signaling, we employed transgenic mice (sRA model) with brain-specific overproduction of Ang II. Immunostaining for phospho-S6 revealed significantly elevated mTORC1 signaling in the SFO of sRA (54 ± 6) relative to control littermates (29 ± 5) (Figure 5A). The elevated phospho-S6 immunoreactivity in the SFO of sRA mice occurred in both males and females (Supplemental Figure 4). sRA mice also displayed higher nuclear c-Fos immunoreactivity in the SFO (21 ± 4 vs 2 ± 0 in controls) (Figure 5B), which again indicates neuronal activation in this nucleus. Assessment of the phospho-S6 immunoreactivity in other cardiovascular brain nuclei including the PVN revealed no significant difference between sRA and controls (data not shown). SFO-specific activation of mTORC1 signaling is consistent with the SFO-restricted Ang II overproduction in sRA mice (3).
Figure 5.

Effect of genetic overaction of brain RAS (sRA mice) on mTORC1 signaling and neuronal activity in the SFO. Increased immunoreactivities of phosphorylated S6 (A) and c-Fos (B) in the SFO of sRA mice. Microphotographs (×20 magnification) of p-S6 and c-Fos immunoreactivities in the SFO and quantification data are displayed. Values are mean ± SEM, n = 4–9; *, P < .05, t test.
mTORC1 pathway mediates Ang II-induced drinking but not pressor response
To study the functional role of mTORC1 in brain AT1R signaling, we tested the effect of rapamycin, a selective mTORC1 inhibitor (23), on the physiological responses evoked by Ang II. First, we examined the effect of rapamycin on the pressor effect of Ang II. ICV Ang II increased arterial pressure (+16 ± 3 mm Hg in MAP) in anesthetized mice, which was not altered by ICV pretreatment with rapamycin (10 ng, 10 min) (+17 ± 4 mm Hg) (Figure 6A).
Figure 6.

Effect of rapamycin (Rap) on Ang II-induced acute dipsogenic and pressor responses. Rapamycin pretreatment (10 ng, 10 min, ICV) inhibited ICV Ang II (200 ng, 30 min)-evoked increase in the frequency (A), duration (B), and amount (C) of water intake. ICV Ang II-induced increase in MAP was not affected by rapamycin pretreatment (D). n = 4–8; *, P < .05, one-way ANOVA or t test.
Next, we determined how ICV pretreatment with rapamycin influences the drinking response evoked by ICV Ang II in conscious C57BL/6J mice. ICV injection of Ang II substantially increased the drinking episodes (Figure 6B) and the time spent drinking (Figure 6C) leading to higher total water intake (Figure 6D). Notably, ICV pretreatment with rapamycin significantly reduced Ang II-induced dipsogenic response (Figure 6, B–D). In contrast, ICV rapamycin alone caused no change in drinking behavior in wild-type mice (Figure 6, B–D). These results indicate that the mTORC1 pathway is required for acute drinking, but not the pressor response produced by stimulation of brain AT1R.
ICV rapamycin reduced polydipsia, but not hypertension, of sRA mice
We also assessed the effect of ICV administration of rapamycin on the elevated arterial pressure and water intake associated with sRA transgenic mice. Arterial pressure was significantly (P < .05) elevated in sRA mice (SBP of 128 ± 4 mm Hg) compared with littermate controls (109 ± 2 mm Hg) (Figure 7A). Daily ICV administration of rapamycin for 7 days failed to decrease SBP in sRA mice (123 ± 4 mm Hg) relative to the vehicle-treated sRA group (124 ± 8 mm Hg) (Figure 7A). Absence of effect of rapamycin on SBP in sRA mice was noted in males and females (data not shown). In addition, ICV rapamycin had no effect on SBP in control mice (110 ± 2 mm Hg vs 107 ± 3 mm Hg in vehicle). Thus, the brain mTORC1 pathway appears not to be involved in the maintenance of the hypertension associated with the sRA mouse model with overactive brain RAS.
Figure 7.

Effect of rapamycin on SBP and water intake in sRA mice. A, Daily ICV treatment with rapamycin (10 ng/d for 7 d) failed to decrease SBP in sRA mice. B, Daily ICV administration of rapamycin (10 ng/d for 3 d) reduced 24-hour water intake in sRA mice. C, Schematic summarizing the selective role of mTORC1 signaling in the SFO in underlying Ang II-mediated control of water intake vs BP. Values are mean ± SEM, n = 6–20 per group; *, P < .05 vs control groups; †, P < .05 vs all other groups, one-way ANOVA.
Finally, we determined the contribution of mTORC1 to the polydipsia of sRA mice. sRA mice consumed a substantial amount of water (9.5 ± 0.6 mL) relative to control littermates (3.2 ± 0.2 mL, P < .05). Interestingly, daily ICV rapamycin administration significantly (P < .05) reduced water intake of sRA mice (−3.4 ± 0.6 mL), whereas vehicle treatment had no effect (−0.4 ± 0.4 mL) (Figure 7B). ICV rapamycin decreased water intake of both male (−2.6 ± 1.0 mL) and female (−4.1 ± 0.7 mL) sRA mice. In control mice, water intake was not affected by either ICV rapamycin (−0.3 ± 0.1 mL) or vehicle (−0.2 ± 0.1 mL). This finding indicates that mTORC1 activation underlies the increased drinking behavior of sRA mice.
Discussion
This study demonstrates that mTORC1 is an important downstream pathway for neuronal action of Ang II and AT1R signaling. In neuronal GT1–7 cells, Ang II elicited a time-dependent increase in activity of mTORC1 as indicated by the phosphorylated levels of S6K. Moreover, AT1R blockade with losartan abolished the activation of mTORC1 signaling. We also show that mice treated with Ang II, ICV or sc, exhibit elevated neuronal mTORC1 signaling as indicated by the elevated phospho-S6 immunoreactivity. Similarly, transgenic sRA mice with brain-specific overactive RAS display higher mTORC1 activity in the SFO. The ability of rapamycin to interfere with the drinking response evoked by exogenous Ang II and polydipsia of sRA mice highlights the physiological significance of mTORC1 signaling in mediating brain action of Ang II. Together, these results implicate neuronal mTORC1 as a novel mechanism underlying selective regulation of fluid homeostasis by Ang II (Figure 7C).
The SFO located at the ventral side of the fornix in the dorsal third ventricle is a key nucleus in mediating the brain actions of Ang II (21). Lack of blood-brain barrier allows SFO neurons to sense circulating endocrine Ang II. In addition, locally produced Ang II can act in a paracrine/autocrine manner within the SFO. In agreement with these facts, our data show stimulation of mTORC1 signaling in the SFO by ICV or systemic administration or transgenic overproduction of Ang II. The relevance of the AT1R signaling in the SFO to the neural control of BP and fluid intake through efferent projections to other cardiovascular nuclei has been well documented (24). For instance, SFO-specific deletion of AT1R is sufficient to block the increase in arterial pressure and fluid intake in DOCA-salt hypertensive mice, a model of active brain RAS (25). Conversely, overproduction of Ang II in the SFO such as in sRA mice leads to hypertension and polydipsia (3). Although components of mTORC1 such as Deptor are present throughout the brain, certain nuclei, particularly the SFO display high expression (10). This raises the possibility that high density of both AT1R and mTORC1 subunits in the SFO may have contributed to Ang II-mediated activation of mTORC1 signaling in this nucleus. Activation of mTORC1 signaling in the PVN after ICV, but not sc, administration of Ang II may relate to the ability of this hormone to access this nucleus when injected directly into the brain ventricle as compared with systemic administration.
Notably, ICV injection of rapamycin interfered with the effect of Ang II and brain RAS activation on water intake but not arterial pressure indicating a selective involvement of mTORC1 in Ang II control of drinking behavior. These findings point to divergent signaling pathways in Ang II control of water intake vs arterial pressure (Figure 7C). This concept is further supported by our previous report that blockade of endoplasmic reticulum (ER) stress in the SFO corrects the polydipsia but not the hypertension of mouse models with active brain RAS (26). The requirement of both mTORC1 and ER stress for Ang II control of water intake points to a possible interaction between these pathways in neuronal AT1R signaling. This is further supported by the recent evidence demonstrating a reciprocal cross-talk between mTORC1 signaling and ER stress (27). In neurons, activation of the mTORC1 pathway promotes ER stress through a mechanism that involves the peroxisome proliferator-activated receptor-γ coactivator-1β (28). In turn, neuronal ER stress modulates mTOR activity through the tuberous sclerosis complex (29). Further investigations are required to tease out the potential relationship between ER stress and mTORC1 in neuronal AT1R signaling.
Recent studies have implicated protein kinase C (PKC) in the SFO in the control of fluid intake and the polydipsic response evoked by Ang II (30). In vascular smooth muscle cells, Ang II activation of PKC was found to lead to stimulation of the mTORC1 pathway through early endosomal antigen 1 (31). In addition to PKC axis, phosphoinositol-3 kinase and ERK are key downstream pathways of AT1R in neurons as Ang II-induced activation of these 2 pathways augments excitatory neuronal activity (32, 33). In peripheral tissues, Ang II induces hypertrophy by promoting S6K-dependent protein synthesis in a phosphoinositol-3 kinase- and ERK-dependent manner (7–9). However, in the brain inhibition of PKC signaling, but not ERK signaling, reversed the elevated water intake of sRA mice, mimicking the effects of mTORC1 blockade (30). Thus, we speculate that neuronal AT1R regulation of mTORC1 activity may implicate PKC signaling through intracellular calcium. This is further supported by the fact that Ang II binding to neuronal AT1R triggers activation of PKC via phospholipase C, resulting in an influx of calcium (29, 34), a potent activator of mTORC1 signaling (35).
Table 1.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used (Application) |
|---|---|---|---|---|---|
| S6K | N/A | p70 S6K (49D7) | Cell Signaling, 2708 | Rabbit; monoclonal | 1:1000 (WB) |
| Phospho-S6K (T389) | N/A | Antiphospho-p70 S6K (Thr389) | Millipore, 04-392 | Rabbit; monoclonal | 1:1000 (WB) |
| Phospho-S6 (S240/244) | N/A | Phospho-S6 ribosomal protein (Ser240/244) (D68F8) | Cell Signaling, 5364 | Rabbit; monoclonal | 1:1000 (IHC) |
| c-Fos | N/A | c-Fos antibody (K-25) | Santa Cruz Biotechnology, Inc, sc-253 | Rabbit; polyclonal | 1:2000 (IHC) |
| Rabbit IgG | N/A | Antirabbit IgG, HRP-linked antibody | Cell Signaling, 7074 | Goat | 1:5000 (WB) |
| Rabbit IgG | N/A | Biotinylated goat antirabbit IgG antibody | Vector, BA-1000 | Goat | 1:200 (IHC) |
Ang II-induced hypertension appears to depend on processes in the SFO that involve predominantly nicotinamide adenine dinucleotide phosphate oxidase (NOX)-derived superoxide, cyclooxygenase-derived prostanoids, and inducible transcription factors, nuclear factor-κB, and activator protein 1 (36). For instance, small interfering RNA-mediated silencing of either NOX2 or NOX4 in the SFO attenuates the pressor effect of central Ang II. It should be noted, however, that silencing NOX2 also interfered with the water drinking response evoked by central Ang II (37). These studies highlight the complexity of the signaling pathways associated with AT1R in the SFO that mediates Ang II actions.
Dysregulation of the mTORC1 pathway has been implicated in a variety of human diseases (6). Our study extends the pathophysiological effects of mTORC1 signaling by implicating this pathway in the polydipsia evoked by brain Ang II. However, mTORC1 signaling does not appear to contribute to brain Ang II-evoked hypertension. Unraveling the signaling mechanisms underlying the differential control of fluid intake and BP by brain Ang II can help develop specific treatments for polydipsia and hypertension.
In summary, our data demonstrate that mTORC1 is a novel signaling pathway downstream to AT1R signaling in the brain. Our findings also highlight the critical role of mTORC1 signaling in mediating selectively the effect of Ang II on drinking behavior.
Acknowledgments
We thank Dr Deng-Fu Guo for assistance with GT1–7 cell line authentication and Paul Casella for editorial feedback.
This work was supported by the United States National Institutes of Health Grant HL084207, the American Heart Association Grant 14EIA18860041, the University of Iowa Fraternal Order of Eagles Diabetes Research Center, and the University of Iowa Healthcare Center for Hypertension Research.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Ang II
- angiotensin II
- AT1R
- Ang II type 1 receptor
- AT1aR
- Ang II type 1a receptor
- BP
- blood pressure
- Deptor
- DEP-domain-containing mTOR-interacting protein
- DMSO
- Dimethyl sulfoxide
- ER
- endoplasmic reticulum
- ICV
- intracerebroventricular
- MAP
- mean arterial pressure
- mTOR
- mechanistic target of rapamycin
- mTORC1
- mTOR complex 1
- NOX
- nicotinamide adenine dinucleotide phosphate oxidase
- PKC
- protein kinase C
- PVN
- paraventricular nucleus of the hypothalamus
- RAS
- renin angiotensin system
- SBP
- systolic BP
- SFO
- subfornical organ
- S6K
- S6 kinase
- TBS
- Tris-buffered saline.
References
- 1. O'Callaghan EL, Choong YT, Jancovski N, Allen AM. Central angiotensinergic mechanisms associated with hypertension. Auton Neurosci. 2013;175:85–92. [DOI] [PubMed] [Google Scholar]
- 2. Davisson RL, Oliverio MI, Coffman TM, Sigmund CD. Divergent functions of angiotensin II receptor isoforms in the brain. J Clin Invest. 2000;106:103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sakai K, Agassandian K, Morimoto S, et al. Local production of angiotensin II in the subfornical organ causes elevated drinking. J Clin Invest. 2007;117:1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Okuno T, Nagahama S, Lindheimer MD, Oparil S. Attenuation of the development of spontaneous hypertension in rats by chronic central administration of captopril. Hypertension. 1983;5:653–662. [DOI] [PubMed] [Google Scholar]
- 5. Evered MD, Robinson MM, Richardson MA. Captopril given intracerebroventricularly, subcutaneously or by gavage inhibits angiotensin-converting enzyme activity in the rat brain. Eur J Pharmacol. 1980;68:443–449. [DOI] [PubMed] [Google Scholar]
- 6. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hafizi S, Wang X, Chester AH, Yacoub MH, Proud CG. ANG II activates effectors of mTOR via PI3-K signaling in human coronary smooth muscle cells. Am J Physiol Heart Circ Physiol. 2004;287:H1232–H1238. [DOI] [PubMed] [Google Scholar]
- 8. Eguchi S, Iwasaki H, Ueno H, et al. Intracellular signaling of angiotensin II-induced p70 S6 kinase phosphorylation at Ser(411) in vascular smooth muscle cells. Possible requirement of epidermal growth factor receptor, Ras, extracellular signal-regulated kinase, and Akt. J Biol Chem. 1999;274:36843–36851. [DOI] [PubMed] [Google Scholar]
- 9. Sadoshima J, Izumo S. Rapamycin selectively inhibits angiotensin II-induced increase in protein synthesis in cardiac myocytes in vitro. Potential role of 70-kD S6 kinase in angiotensin II-induced cardiac hypertrophy. Circ Res. 1995;77:1040–1052. [DOI] [PubMed] [Google Scholar]
- 10. Caron A, Baraboi ED, Laplante M, Richard D. DEP domain-containing mTOR-interacting protein in the rat brain: distribution of expression and potential implication. J Comp Neurol. 2015;523:93–107. [DOI] [PubMed] [Google Scholar]
- 11. Gonzalez AD, Wang G, Waters EM, et al. Distribution of angiotensin type 1a receptor-containing cells in the brains of bacterial artificial chromosome transgenic mice. Neuroscience. 2012;226:489–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morimoto S, Cassell MD, Sigmund CD. Glia- and neuron-specific expression of the renin-angiotensin system in brain alters blood pressure, water intake, and salt preference. J Biol Chem. 2002;277:33235–33241. [DOI] [PubMed] [Google Scholar]
- 13. Li B, Lee K, Martin RJ. Overexpression of glucose transporter 2 in GT1–7 cells inhibits AMP-activated protein kinase and agouti-related peptide expression. Brain Res. 2006;1118:1–5. [DOI] [PubMed] [Google Scholar]
- 14. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 15. Muta K, Morgan DA, Rahmouni K. The role of hypothalamic mTORC1 signaling in insulin regulation of food intake, body weight, and sympathetic nerve activity in male mice. Endocrinology. 2015;156:1398–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harlan SM, Guo DF, Morgan DA, Fernandes-Santos C, Rahmouni K. Hypothalamic mTORC1 signaling controls sympathetic nerve activity and arterial pressure and mediates leptin effects. Cell Metab. 2013;17:599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J. 2012;441:1–21. [DOI] [PubMed] [Google Scholar]
- 18. Yano N, Suzuki D, Endoh M, Zhao TC, Padbury JF, Tseng YT. A novel phosphoinositide 3-kinase-dependent pathway for angiotensin II/AT-1 receptor-mediated induction of collagen synthesis in MES-13 mesangial cells. J Biol Chem. 2007;282:18819–18830. [DOI] [PubMed] [Google Scholar]
- 19. Shibata S, Arroyo JP, Castañeda-Bueno M, et al. Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc Natl Acad Sci USA. 2014;111:15556–15561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liao YM, Sy A, Yen Y. Markers for efficacy of mammalian target of rapamycin inhibitor. Anticancer Res. 2012;32:4235–4244. [PubMed] [Google Scholar]
- 21. Coble JP, Grobe JL, Johnson AK, Sigmund CD. Mechanisms of brain renin angiotensin system-induced drinking and blood pressure: importance of the subfornical organ. Am J Physiol Regul Integr Comp Physiol. 2015;308:R238–R249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Northcott CA, Watts S, Chen Y, Morris M, Chen A, Haywood JR. Adenoviral inhibition of AT1a receptors in the paraventricular nucleus inhibits acute increases in mean arterial blood pressure in the rat. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1202–R1211. [DOI] [PubMed] [Google Scholar]
- 23. Ballou LM, Lin RZ. Rapamycin and mTOR kinase inhibitors. J Chem Biol. 2008;1:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Llewellyn T, Zheng H, Liu X, Xu B, Patel KP. Median preoptic nucleus and subfornical organ drive renal sympathetic nerve activity via a glutamatergic mechanism within the paraventricular nucleus. Am J Physiol Regul Integr Comp Physiol. 2012;302:R424–R432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hilzendeger AM, Cassell MD, Davis DR, et al. Angiotensin type 1a receptors in the subfornical organ are required for deoxycorticosterone acetate-salt hypertension. Hypertension. 2013;61:716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jo F, Jo H, Hilzendeger AM, et al. Brain endoplasmic reticulum stress mechanistically distinguishes the saline-intake and hypertensive response to deoxycorticosterone acetate-salt. Hypertension. 2015;65:1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Appenzeller-Herzog C, Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012;22:274–282. [DOI] [PubMed] [Google Scholar]
- 28. Camacho A, Rodriguez-Cuenca S, Blount M, et al. Ablation of PGC1 β prevents mTOR dependent endoplasmic reticulum stress response. Exp Neurol. 2012;237:396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Di Nardo A, Kramvis I, Cho N, et al. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J Neurosci. 2009;29:5926–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Coble JP, Johnson RF, Cassell MD, Johnson AK, Grobe JL, Sigmund CD. Activity of protein kinase C-α within the subfornical organ is necessary for fluid intake in response to brain angiotensin. Hypertension. 2014;64:141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nazarewicz RR, Salazar G, Patrushev N, et al. Early endosomal antigen 1 (EEA1) is an obligate scaffold for angiotensin II-induced, PKC-α-dependent Akt activation in endosomes. J Biol Chem. 2011;286:2886–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Veerasingham SJ, Raizada MK. Brain renin-angiotensin system dysfunction in hypertension: recent advances and perspectives. Br J Pharmacol. 2003;139:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wei SG, Yu Y, Zhang ZH, Felder RB. Angiotensin II upregulates hypothalamic AT1 receptor expression in rats via the mitogen-activated protein kinase pathway. Am J Physiol Heart Circ Physiol. 2009;296:H1425–H1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Makadia HK, Anderson WD, Fey D, Sauter T, Schwaber JS, Vadigepalli R. Multiscale model of dynamic neuromodulation integrating neuropeptide-induced signaling pathway activity with membrane electrophysiology. Biophys J. 2015;108:211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gulati P, Gaspers LD, Dann SG, et al. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008;7:456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Young CN, Davisson RL. Angiotensin-II, the brain, and hypertension: an update. Hypertension. 2015;66:920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peterson JR, Burmeister MA, Tian X, et al. Genetic silencing of Nox2 and Nox4 reveals differential roles of these NADPH oxidase homologues in the vasopressor and dipsogenic effects of brain angiotensin II. Hypertension. 2009;54:1106–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]