

Abstract
Low-dose administration of manganese chloride (MnCl2) causes release of hypothalamic LH-releasing hormone (LHRH) and advances puberty in rat. Recently, this element was shown to up-regulate mammalian target of rapamycin (mTOR), kisspeptin gene (KiSS-1), and LHRH gene expressions in the brain preoptic area (POA)/anteroventral periventricular (AVPV) nucleus. Because these genes are critical for puberty, this study was conducted to identify the upstream mechanism by which Mn activates the mTOR/KiSS-1 pathway. On day 12, immature female rats began receiving a daily supplemental dose of 10 mg/kg of MnCl2 or saline by gavage, and POA/AVPV tissues were collected on day 29 for specific protein assessments. Another experiment assessed in vitro IGF-1 release in response to Mn and assessed signal transduction pathways in the POA/AVPV region after Mn delivery into the third ventricle. Chronic Mn exposure increased (P < .05) basal expressions of mTOR and kisspeptin proteins. Mn increased protein kinase B (Akt) and Ras homolog enriched in brain, both capable of activating mTOR. Central Mn delivery increased expressions of phosphorylated IGF-1 receptor (IGF-1R) (P < .05) and Akt (P < .01) in the POA/AVPV region. The previous central delivery of JB1, an IGF-1R antagonist, blocked Mn-induced expressions of both phosphorylated IGF-1R and Akt. Downstream to Akt, centrally administered Mn increased tuberous sclerosis complex 2 (P < .05), Ras homolog enriched in brain (P < .01), mTOR (P < .05), and kisspeptin (P < .05). Finally, we observed that the early puberty induced by Mn was blocked by the administration of an mTOR inhibitor. These results suggest that Mn acts, at least in part, through the IGF-1/Akt/mTOR pathway to influence prepubertal kisspeptin and LHRH.
Timing of the onset of puberty depends on complex interactions within the hypothalamus involving metabolic and neurochemical signals, as well as genetic and environmental influences. These interactions ultimately result in the increased synthesis and release of hypothalamic LH-releasing hormone (LHRH). Manganese (Mn) is a naturally occurring environmental element that is required for normal mammalian physiological processes. Although exposure to high levels of this element can be toxic, Mn deficiencies are associated with impaired growth and reproduction (1, 2); thus, the latter suggesting that this nutrient plays a facilitative role in reproductive functions. We have shown previously that when prepubertal rats of both sexes were exposed to low but elevated levels of Mn, the element precociously stimulated the release of hypothalamic LHRH and resulted in early pubertal development (3, 4). Additionally, Mn causes increased LHRH secretion in adult male rats (5). Although the stimulation of LHRH is beneficial at the normal time of puberty, precocious development after exposure to low levels of Mn too early in life could cause problems. Early puberty is a serious disorder, and in females most cases have no identifiable cause (6). Thus, deriving a better understanding of the mechanism by which Mn activates the prepubertal LHRH releasing system is important. In this regard, low level Mn administration has been shown to up-regulate KiSS-1 (7), the gene responsible for the synthesis of kisspeptins (Kps), a family of peptides that are potent stimulators of LHRH (8–10) and critical for the onset of puberty (11–14). In addition to Mn, mammalian target of rapamycin (mTOR) has been shown to up-regulate KiSS-1 (15) and, thus, exerts a positive influence on LHRH neuronal activity. Recently, we observed that Mn stimulated the gene expressions of mTOR, KiSS-1, and LHRH (7), all from the same block of tissue which included the preoptic area (POA) and the anteroventral periventricular (AVPV) nucleus. The present study was conducted to determine whether the Mn induction of mTOR and KiSS-1 results in translation to their respective protein and then begin to determine the signal transduction pathway(s) by which Mn induces the mTOR protein.
Materials and Methods
Animals and surgery
Eighteen-day pregnant female rats of the Sprague Dawley line were purchased from Charles River and allowed to deliver pups normally in the Texas A&M University lab animal facility. Female pups were weaned at 21 days of age and housed 4 per cage under controlled conditions of light (lights on, 6 am; lights off, 6 pm) and temperature (23°C), with ad libitum access to food (Harland Teklad diet) and water. The diet was Harlan Teklad 2016, which contained 94.7-mg/kg Mn and 149.8-mg/kg iron, as analyzed by the Heavy Metal Analysis Laboratory, Department of Veterinary Integrative Biosciences, College of Veterinary Medicine, Texas A&M University. All procedures performed were approved by the University Animal Care and Use Committee and in accordance with the NAS-NRC Guidelines for the Care and Use of Laboratory Animals. Surgical anesthesia was an ip injection of 2.5% tribromoethanol (0.5-mL/60-g body weight; Sigma-Aldrich).
Chronic Mn experimental procedure
Rats were bred and delivered their pups normally. All litters were adjusted to 10–12 pups, with each having 4–6 females. Food and water was available to the mothers ad libitum, which was also available to the pups once they began eating solid food. When the animals were 12 days old, the females were numbered by tail markings and approximately half of the female pups in each litter were supplemented with a daily gastric gavage dose of 10-mg/kg manganese chloride (MnCl2) (0.25 mg in 0.2 mL/25 g of rat; Sigma-Aldrich). The other female pups in each litter received an equal volume of saline. When the pups were 21 days old, the mothers and the male pups were removed from the cage. One saline and 1 Mn-treated female pup were selected from each litter; hence, the final groupings of animals from which the tissues were assessed contained pups from different litters. The dosing continued for the female pups until they were terminated by decapitation when 29 days old. This 10-mg/kg dose causes a cumulative intake of 9.7 mg of Mn over the 17 days. Because oral Mn intake is absorbed at about 3%–10% and figuring 5% absorption, this dose results in a total of approximately 0.49 mg of Mn absorbed over the 17 days of dosing. This low dose is important because much higher doses of Mn have been used for many of the neurotoxicological studies (16, 17).
Because Mn-treated rats attain puberty early, beginning about day 30, it was important to take the tissues before this time so that some of the rats would not be in first proestrus or estrus. This identical procedure was repeated 3 times to complete tissue collections for the various assessments (see below). On day 29, brains were removed and the POA/AVPV region was dissected out by a previously described method (18). The tissue block was then frozen on dry ice and stored at −80°C until subsequent protein analysis. All of the animals were confirmed to be in the late juvenile stage of puberty by confirming lack of vaginal opening (VO) and little to no uterine fluid detected when their tissues were collected (19).
Effect of mTOR inhibitor on Mn-induced precocious puberty
Another experiment was conducted and repeated as above with regard to the administration of saline and MnCl2. On day 25, however, half of the Mn-treated began receiving a daily oral dose via gastric gavage of Everolimus (EV) (5-mg/kg rat; Selleckchem), an mTOR inhibitor (20) that is known to cross the blood brain barrier in rodents (21). The saline and the other half of the Mn-treated animals received an equal volume of 30% polypropylene glycol in dH2O via daily gastric gavage. Rats were treated until VO occurred, and vaginal smears were preformed until first diestrus was recorded.
Effects of Mn on IGF-1 release in vitro
Thirty-day-old animals were killed by decapitation, and brains were removed. The block of tissue containing the POA/AVPV nucleus and the medial basal hypothalamus (MBH) was dissected under a stereomicroscope as described previously (18) Briefly, the brain tissue was cut approximately 1 mm rostral to the optic chiasm then cut caudally behind the mammillary bodies. The block was then formed by making cuts along the lateral borders of the optic chiasm and along the border of the thalamus, dorsally. This block of tissue contains both the AVPV nucleus and the arcuate nucleus. The tissue blocks were incubated as described previously (22) with minor modifications. Briefly, each tissue block was incubated in a vial containing 250 μL of Locke's buffer (2mM HEPES, 154mM NaCl, 5.6mM KCl, 1mM MgCl2, 6mM NaHCO3, 10mM glucose, and 1.25mM CaCl2; pH 7.4) inside a Dubnoff shaker (80 cycles/min) at 37°C in an atmosphere of 95% O2 and 5% CO2. After a 60-minute equilibration period, the medium was discarded and replaced with the following fresh medium; medium only, medium plus 1mM, 10mM, or 20mM MnCl2. Tissues were incubated for 4 hours, and then these media were removed and saved to determine IGF-1 release by an ELISA.
Acute Mn experimental procedure
Twenty-four-day-old female rats were implanted with third ventricular (3V) cannulae as described previously (23). After 4 days of recovery, the animals were divided into 2 groups. At 9 am, group 1 received a 3V injection over 1 minute of MnCl2 at a dose of 10-μg/3-μL saline, which we have used previously to stimulate LH release (3). Group 2 (control) was injected with an equal volume of saline. All animals were killed by decapitation at 1 pm, 4 hours after MnCl2 or saline. Brains were removed, placement of the 3V cannulae verified, and the animals were confirmed to be in the late juvenile stage of pubertal development (18). A block of tissue containing the POA/AVPV nucleus was dissected as described above. This experiment was repeated 4 times, and all tissues were stored frozen until assayed for total- and phosphorylated (p)-IGF-1 receptor (IGF-1R), total- and p-Akt protein, total- and p-tuberous sclerosis complex 2 (TSC2), Ras homolog enriched in brain (Rheb), total- and p-mTOR, and Kp expressions by Western blot analysis.
Effect of JB1, IGF-1R antagonist, on Mn-induced Akt expression
In a separate experiment, JB1, IGF-1R antagonist (18, 24), was administered before Mn in order to determine whether the Mn effect on Akt was mediated by IGF-1. The same dose of MnCl2 was administered as above, either in the presence or absence of a dose JB1 used previously (4-μg/4-μL saline; Sigma-Aldrich) (18). The JB1 was administered into the 3V at 5 pm, the day before experiment, and then again the following morning at both 8 and 9 am before the 3V administration of MnCl2 or saline at 9:30 am. All animals were killed by decapitation at 1:30 pm and the POA/AVPV tissue block was collected from 3 trials and stored as stated above until assessed for total- and p-IGF-1R and Akt protein expressions.
Western blot analysis
Brain tissues were homogenized in 1% Igepal CA-630, 20mM Tris-Cl (pH 8.0), 137mM NaCl, 2mM EDTA, 10% glycerol, 10mM sodium pyruvate, 10mM sodium fluoride, 1mM sodium orthovanadate, 1mM phenylmethylsulfonyl fluoride, and 0.25% protease inhibitor cocktail (Sigma-Aldrich) at 4°C. The homogenates were incubated on ice for 30 minutes and centrifuged at 12 000g for 15 minutes. The concentration of total protein in the resulting supernatant was determined by the Pierce 660nm Protein assay kit (Thermo Scientific) using bovine serum albumin as standard. Immunoblot analysis was performed by solubilizing the proteins (100 μg) in a sample buffer containing 25mM Tris-Cl (pH 6.8), 1% sodium dodecyl sulfate, 5% β-mercaptoethanol, 1mM EDTA, 4% glycerol, and 0.01% bromophenol blue and electrophoresed through 8% SDS-PAGE for TSC2 and mTOR, 10% for IGF-1R and Akt, and 15% for Rheb and Kp under reducing conditions. The separated proteins were electrophoretically transblotted onto polyvinylidene difluoride membranes. After transfer, membranes were blocked with 5% nonfat dried milk/0.1% Tween 20 in PBS (pH 7.4) for 3 hours and subsequently incubated at 4°C overnight with rabbit antitotal-IGF-1R (1:1000) or anti-p-IGF-1R (1:250; Cell Signaling Technology), rabbit antitotal-Akt (1:1000) or anti-p-Akt (1:3000; Cell Signaling Technology), rabbit antitotal-TSC-2 (1:2000) or anti-p-TSC-2 (1:250; Cell Signaling Technology), rabbit anti-Rheb (1 μg/mL; Abcam, Inc), rabbit antitotal-mTOR or anti-p-mTOR (1:1000; Cell Signaling Tech), and rabbit anti-Kp (3 μg/mL; Novus Biologicals). Additional information regarding antisera is shown in Table 1. The specificity of each primary antibody was checked by preabsorbing at 23°C for 3 hours with an excess (4–5 fold) of specific peptide that is able to completely block the epitope recognized by the antibody being tested. After the incubation, membranes were washed in PBS/0.1% Tween 20 and then incubated with horseradish peroxidase-labeled goat antirabbit IgG (1:50 000; Santa Cruz Biotechnology, Inc) for 2 hours at room temperature. After washing, the specific signals were detected with the enhanced chemiluminescence (Western Lightning Plus-ECL; PerkinElmer) and quantified with NIH ImageJ software version 1.43 (National Institutes of Health). Subsequently, all membranes were also stripped using Re-Blot Plus kit (EMD Millipore) and reprobed with mouse monoclonal antibody to β-actin and goat antimouse secondary antibody, to normalize for the amount of sample loading when appropriate. After washing, the detection and quantitation of β-actin was conducted as described above.
Table 1.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Akt | Anti-Akt | Cell Signaling Technology, 4691 | Rabbit; monoclonal | 1:1000 | |
| p-Akt | Anti-p-Akt | Cell Signaling Technology, 4060 | Rabbit; monoclonal | 1:3000 | |
| β-Actin | DDDIAALVIDNGSGK | Anti-β-actin | Sigma-Aldrich, A1978 | Mouse; monoclonal | 1:50 000 |
| IGF-1Rβ | Anti-IGF-1Rβ | Cell Signaling Technology, 9750 | Rabbit; polyclonal | 1:1000 | |
| p-IGF-1Rβ | Anti-p-IGF-1Rβ | Cell Signaling Technology, 6113 | Rabbit; polyclonal | 1:250 | |
| Kp (KISS-1) | Anti-Kp (KISS-1) | Novus Biologicals, NBP1-45672 | Rabbit; polyclonal | 3 μg/mL | |
| mTOR | Anti-mTOR | Cell Signaling Technology, 2983 | Rabbit; monoclonal | 1:1000 | |
| p-mTOR | Anti-p-mTOR | Cell Signaling Technology, 5536 | Rabbit; monoclonal | 1:1000 | |
| Rheb | Anti-Rheb | Abcam, Ab25873 | Rabbit; polyclonal | 1 μg/mL | |
| Tuberin/TSC2 | Antituberin/TSC2 | Cell Signaling Technology, 4308 | Rabbit; monoclonal | 1:2000 | |
| p-Tuberin/TSC2 | Anti-p-tuberin/TSC2 | Cell Signaling Technology, 3617 | Rabbit; monoclonal | 1:250 |
IGF-1 analysis
IGF-1 released into the media was determined by an ELISA kit purchased from Ray Biotech, Inc. The sensitivity of this assay was 7.2 ng/mL.
Statistical analysis
Differences between 2 groups were analyzed by Student's t test. Multiple comparisons were performed using ANOVA with post hoc testing using the Student-Newman-Keuls multiple comparison test. These statistical tests were conducted with INSTAT and Prism software (GraphPad Software). P < .05 was considered to be statistically significant.
Results
Figure 1 demonstrates that the chronic Mn exposure stimulated an increase (P < .05) in the phosphorylation of mTOR and in the basal expression of Kp in the POA/AVPV region of Mn-treated animals compared with controls. Because Akt and Rheb are upstream components of pathways associated with the induction of mTOR (25), we then assessed, in the same tissues, whether these proteins were affected by Mn. Figure 2 shows that the expression of p-Akt (P < .05) and Rheb (P < .05) proteins were also increased in the Mn-treated animals; thus, further implicating an Akt/Rheb/mTOR pathway to Kp. Because Mn can induce an early puberty (3, 4), we tested whether the interruption of this pathway by EV, an mTOR inhibitor, would negate the advanced VO expected in the animals treated with Mn. Figure 3 demonstrates that MnCl2 advanced VO when compared with the saline-treated animals (P < .05) and that this action of Mn was blocked (P < .01) in the animals that received EV.
Figure 1.

Effect of chronic Mn exposure on mTOR and Kp protein expressions in the POA/AVPV region of prepubertal female rats. A, Representative Western blotting of p-mTOR and total mTOR proteins from saline (lanes 1–3) and Mn-treated (lanes 4–6) animals. B, Densitometric quantitation of all bands from 2 blots evaluating p-mTOR normalized to total mTOR protein. Note that the Mn exposure resulted in a marked increase in p-mTOR protein expression vs the saline-treated animals. C, Representative Western immunoblotting of Kp and β-actin proteins from saline (lanes 1–3) and Mn-treated (lanes 4–6) animals. D, Densitometric quantitation of all bands from 2 blots evaluating Kp protein expression normalized to β-actin protein. Note that the animals exposed to Mn showed a marked increase in Kp protein expression vs the saline-treated animals. The respective bars illustrate the mean (±SEM) of an n of 8 per group; *, P < .05 vs saline controls.
Figure 2.

Effect of chronic Mn exposure on Akt and Rheb protein expressions in the POA/AVPV region of prepubertal female rats. A, Representative Western blotting of p-Akt and total Akt proteins from saline (lanes 1–3) and Mn-treated (lanes 4–6) animals. B, Densitometric quantitation of all bands from 2 blots evaluating p-Akt normalized to total Akt protein. Note that chronic exposure to Mn caused a marked increase in p-Akt protein expression vs the saline-treated animals. C, Representative Western blotting of Rheb and β-actin proteins from saline (lanes 1–3) and Mn-treated (lanes 4–6) animals. D, Densitometric quantitation of all bands from 2 blots evaluating Rheb protein expression normalized to β-actin protein. Chronic Mn exposure also increased Rheb protein expression vs the saline-treated animals. The respective bars illustrate the mean (±SEM) of an n of 8 per group; *, P < .05 vs saline controls.
Figure 3.

Effect of oral administration of EV, an mTOR inhibitor, on Mn-induced precocious puberty. Note that daily low level Mn exposure (open bar) advanced the age of VO when compared with saline-treated controls (solid bar) and the Mn+EV treated (hatched bar) animals. The respective bars illustrate the mean (±SEM) of an n of 16 per group; *, P < .05; +, P < .01 vs Mn only.
Because phosphorylation of Akt is involved in mTOR activation, we identified IGF-1, a peptide that acts through Akt signaling, as a candidate for mediating the Mn action on mTOR. In this regard, we first determined whether Mn could stimulate the secretion of IGF-1. In a block of tissue containing both the POA/AVPV nucleus and the MBH, Mn was shown to cause a dose-dependent increase in the release of IGF-1 in vitro (Figure 4). To test the influence of Mn in vivo, the element was injected into the brain 3V, and its ability to induce the phosphorylation of IGF-1R, as well as that of Akt, was assessed. Figure 5 demonstrates that the central delivery of Mn elicited an increase (P < .05) in the expression of p-IGF-1R in the POA/AVPV block at 4 hours after injection, an action associated with increased (P < .01) protein expression of p-Akt. In order to show that IGF-1R signaling was directly involved with Mn-induced stimulation of Akt, rats received a 3V injection of Mn in the absence or presence of JB1, an IGF-1R antagonist. Central delivery of JB1 blocked (P < .001) the Mn-induced phosphorylation of IGF-1R (Figure 6, A and B), resulting in the suppression (P < .05) of p-Akt protein (Figure 6, C and D).
Figure 4.

MnCl2 stimulates IGF-1 release from hypothalamic fragments of prepubertal female rats. Thirty-day-old female rats were killed, and a tissue block containing the POA/MBH region of each animal was excised and incubated in medium. Open bar represents tissue exposed to medium only, sold bars represent tissues exposed to the different concentrations of MnCl2. Note that the 1mM dose did not stimulate IGF-1 over medium only controls, but that the 10mM and the 20mM doses of MnCl2 stimulated increases in IGF-1 release over the controls and those exposed to 1mM. The respective bars illustrate the mean (±SEM) of an n of 8–10 animals/group; *, P < .05; **, P < .01 vs medium only.
Figure 5.

Effect of acute Mn exposure on IGF-1R and Akt protein expressions in the POA/AVPV region of prepubertal female rats. Animals were exposed via a 3V injection of MnCl2 (10 μg/3 μL) or saline and IGF-1R and Akt proteins were assessed in the AVPV tissue block 4 hours after injection. A, Representative Western blotting of p-IGF-1R and total IGF-1R proteins from saline (lanes 1–3) and Mn-treated (lanes 4–6) animals. B, Densitometric quantitation of all bands from 2 blots evaluating p-IGF-1R normalized to total IGF-1R protein. Note that 3V administration of Mn stimulated an increase in p-IGF-1R protein expression in the AVPV nucleus compared with the saline-treated animals. C, Representative Western blotting of p-Akt and total Akt proteins from saline (lanes 1–3) and Mn-treated (lanes 4–6) animals. D, Densitometric quantitation of all bands from 2 blots evaluating p-Akt normalized to total Akt protein. Note that Mn stimulated an increase in p-Akt when compared with the saline-treated animals. The respective bars illustrate the mean (±SEM) of an n of 8 per group; *, P < .05; **, P < .01 vs saline controls.
Figure 6.

Effect of IGF-1R inhibition on Mn-induced IGF-1R and Akt protein expression in the POA/AVPV region of prepubertal female rats. The IGF-1R antagonist, JB1, was injected into the 3V the day before and 2 hours before the administration of MnCl2 (10 μg/3 μL). IGF-1R and Akt protein levels were assessed by Western blotting at 4 hour after Mn injection. A, Representative Western blotting of p-IGF-1R and total IGF-1R proteins from saline (lanes 1–3), Mn (lanes 4–6) and Mn+JB1-treated (lanes 7–9) animals. B, Densitometric quantitation of all bands from 2 blots evaluating p-IGF-1R normalized to total IGF-1R protein. Note that the Mn-induced increase in p-IGF-1R was blocked in the animals that received the JB1. C, Representative Western blotting of p-Akt and total Akt proteins from saline (lanes 1–3), Mn (lanes 4–6), and Mn+JB1-treated (lanes 7–9) animals. D, Densitometric quantitation of all bands from 2 blots evaluating p-Akt normalized to total Akt protein. Note that the Mn-induced increase in p-Akt was blocked in the animals that received the JB1. The respective bars illustrate the mean (±SEM) of an n of 5–7 per group; ***, P < .001; *, P < .05 vs saline controls and Mn+JB1.
To determine effects of Mn downstream to Akt, we assessed the effect of centrally administered Mn on the protein expression of TSC2, an upstream signal that controls the actions of Rheb, a protein that is important for the activation of mTOR. Figure 7 demonstrates that Mn caused an increase in the phosphorylation of TSC2 (P < .05). The increase in phosphorylation of TSC2 deactivates the inhibitory tone that TSC2 has on Rheb, and as a result, Rheb levels increased (P < .01) in the POA/AVPV tissue block from the Mn-treated animals. Figure 8 shows that the increase in Rheb was associated with concomitant increases in p-mTOR (P < .05) and Kp (P < .05) proteins in the same tissues of the Mn-treated animals.
Figure 7.
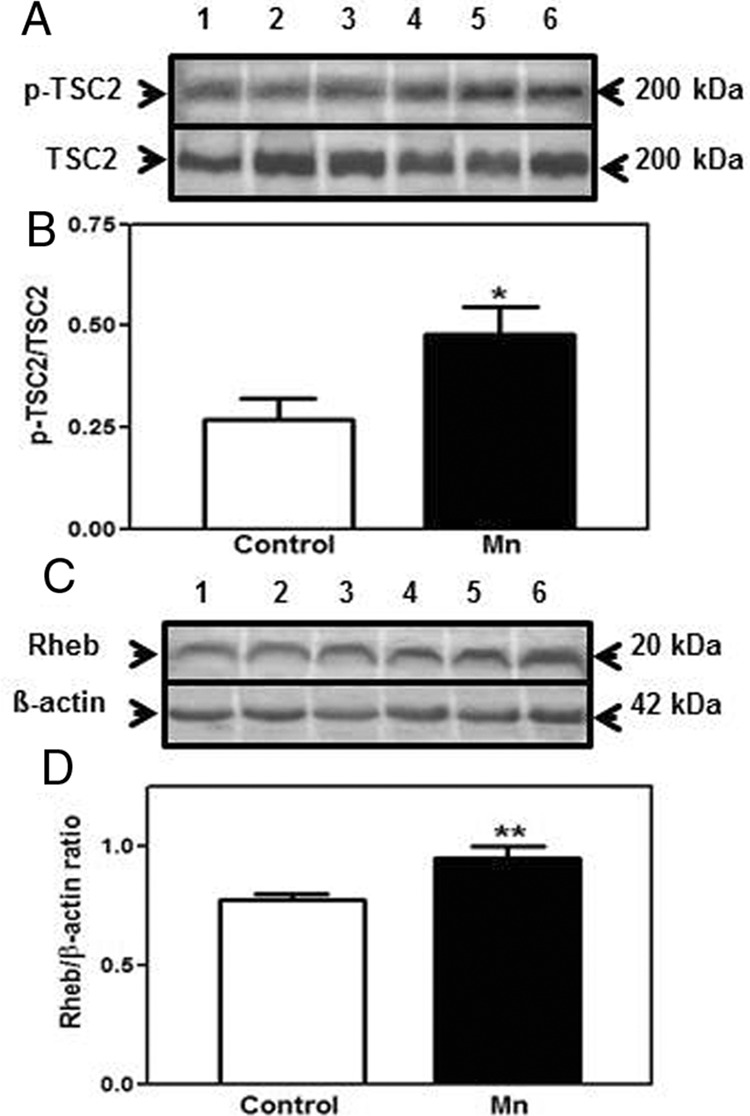
Effect of acute Mn exposure on TSC2 and Rheb protein expressions in the POA/AVPV region of prepubertal female rats. TSC2 and Rheb protein levels were assessed from the same AVPV tissue samples as in Figure 4. A, Representative Western blotting of p-TSC2 and total TSC2 proteins from saline (lanes 1–3) and Mn-treated (lanes 4–6) animals. B, Densitometric quantitation of all bands from 2 blots evaluating p-TSC2 normalized to total TSC2 protein. Central administration of Mn induced a marked increase in p-TSC2 protein expression when compared with the saline-treated animals. C, Representative Western blotting of Rheb and β-actin proteins from saline (lanes 1–3) and Mn-treated (lanes 4–6) animals. D, Densitometric quantitation of all bands from 2 blots evaluating Rheb protein expression normalized to β-actin protein. Rheb protein expression was also markedly increased after central administration of Mn when compared with the animals that received saline in the 3V. The respective bars illustrate the mean (±SEM) of an n of 8 per group; *, P < .05; **, P < .01 vs saline controls.
Figure 8.

Effect of acute Mn exposure on mTOR and Kp protein expressions in the POA/AVPV region of prepubertal female rats. Protein levels of mTOR and Kp were assessed from the same AVPV tissue samples as in Figure 4. A, Representative Western blotting of p-mTOR and total mTOR proteins from saline (lanes 1–3) and Mn-treated (lanes 4–6) animals. B, Densitometric quantitation of all bands from 2 blots evaluating p-mTOR normalized to total mTOR protein. Note that phosphorylation of mTOR protein was significantly increased after central administration of Mn when compared with the animals that received saline. C, Representative Western blotting of Kp and β-actin proteins from saline (lanes 1–3) and Mn-treated (lanes 4–6) animals. D, Densitometric quantitation of all bands from 2 blots evaluating Kp protein expression normalized to β-actin protein. Central Mn administration also increased Kp protein expression when compared with saline-treated animals. The respective bars illustrate the mean (±SEM) of an n of 8 per group; *, P < .05 vs saline controls.
Discussion
Previously we showed that prepubertal exposure to low levels of Mn induced increases in hypothalamic mTOR and KiSS-1 gene expressions (7) and now have shown that these increases are associated with increased translation of their respective proteins. Because the mTOR/KiSS-1/Kp pathway is critical for the central control of prepubertal LHRH (8–15), we began to focus our efforts on discerning upstream mechanism(s) by which Mn activates mTOR, a serine/threonine protein kinase known to be regulated by growth factors, amino acids, and cellular energy levels (25, 26). Furthermore, Akt is a known transduction signal capable of regulating mTOR (27–29); therefore, we identified IGF-1 as a potential upstream effector that could be influenced by Mn, because it is known that IGF-1 is capable of activating Akt in the basal hypothalamus (30, 31). The present study has revealed that Mn stimulated a dose-dependent release of hypothalamic IGF-1 in vitro and induced increases in the hypothalamic expressions of p-IGF-1R and Akt within the POA/AVPV region when injected directly into the brain 3V. This data further demonstrates that IGF-1 acts via its own type 1 receptor located on neurons and glia throughout the brain, including the POA and MBH (32, 33) to stimulate Akt. Importantly, we showed that this Mn induction of p-IGF-1R and Akt was blocked by JB1, an IGF-1R antagonist. JB1 can inhibit cell proliferation and prevents autophosphorylation of IGF-1R (24). We also showed previously that JB1 inhibits the IGF-1-induced increase in KiSS-1 gene expression (18) in the AVPV region of the brain. Taken together, these results demonstrate for the first time that Mn is capable of inducing IGF-1R-activated Akt; hence, revealing an upstream action of Mn in the regulation of Akt.
Because we observed that Mn increased Rheb protein expression after chronic exposure, we assessed whether Mn could be acting through the Akt/TSC2 pathway leading to the activation of Rheb and subsequently, mTOR and Kp. This rational was supported by several studies showing that Akt directly phosphorylates TSC2 and that this phosphorylation inhibits TSC2 activity, leading to the activation of Rheb and mTOR (27, 28, 34, 35). Additionally, the IGF-1/Akt pathway has been shown to up-regulate Rheb/mTOR through direct Akt phosphorylation of TSC2 (27, 36). Therefore, in the present study we demonstrated that the central administration of Mn caused increased phosphorylation of TSC2 and activation of Rheb, actions that coincided with concomitant increases in mTOR and Kp protein expressions. These results, along with the action of Mn to induce IGF-1R-activated Akt, strongly suggest that the IGF-1/Akt/Rheb pathway is intimately involved in the Mn activation of mTOR.
Our previous studies have shown that Mn is capable of acting downstream from the above mentioned events by directly stimulating prepubertal LHRH release from the neuron terminals in the MBH. In this regard, Mn activated soluble guanylyl cyclase, which then induced the cGMP-protein kinase G pathway to induce LHRH secretion (22). Importantly, to drive the pubertal process LHRH secretion must be sustained, indicating an action to promote LHRH synthesis to keep up with its release. Our present results relate to this, because they reveal an upstream action of Mn within the POA/AVPV region. The Kp neurons are localized within the AVPV nucleus, an area of the anterior hypothalamus that is critical to providing necessary inputs to most LHRH neurons located in the adjacent POA (37). In this regard, several points are worthy of further discussion. We demonstrated the action of Mn to activate the IGF-1 system, which is a contributor to the control of LHRH at puberty (38–41). As a result of IGF-1R activation by Mn, Akt was induced and its downstream effect was to activate Rheb, which acts as a mediator of the nutrient signaling input to mTOR (42). It is known that Rheb binds directly to mTOR (26), an interaction that is necessary for the activation of mTOR. This is a nutrient-responsive complex that mediates cell growth (25, 26) and plays a key role in the central activation of puberty through its regulation of KiSS-1 (15). The KiSS-1/Kp system is an important regulator of prepubertal LHRH synthesis and critical for the onset of puberty (8–15). Thus, our present results clearly demonstrate an upstream pathway by which Mn can activate the prepubertal mTOR/Kp/LHRH system, and that blockade of the pathway with an mTOR inhibitor negates Mn-induced early pubertal development.
It is important to note that the supplemental dose of Mn used is within the upper end of the estimated safe range for children (16, 43) and is 10–20 times below those amounts used for studying some of the neurotoxic effects of this element (17, 44–46). The present results show that Mn is among a unique category of a limited number of substances that can act at the hypothalamic level to advance the timing of puberty. As a result of this central action, we suggest that Mn, a peripherally derived environmental nutrient, is capable of acting along with other metabolic signals, as well as with genetic factors, to influence normal pubertal development; however, the possibility also exists that individuals exposed to low but elevated levels of the element during their juvenile or early adolescent years could be at risk for precocious puberty. This possibility is supported by several lines of evidence. We have shown that after prepubertal administration of Mn, the element accumulates in key areas of the hypothalamus that are responsible for both the synthesis and secretion of the LHRH peptide (3). The present study demonstrates that Mn is capable of activating an upstream pathway that ultimately contributes to the regulation of LHRH neuronal activity. These and other effects of Mn on the LHRH releasing system are consistent with the elevated serum levels of LH, FSH, and gonadal steroids noted in prepubertal rats of both sexes after low-dose exposure to the element. Importantly, these elevated puberty-related hormones have been shown to be associated with advanced puberty, with females appearing to be more sensitive to Mn than males (3, 4). Of potential importance is that in females, compared with males, 95% of precocious puberty cases have no identifiable cause and puberty appears normal, other than being too early (6). Interestingly, gender differences in Mn metabolism have also been reported, with male rats clearing the element over 2 times faster than females (47). Other studies describing Mn effects in the young also support the above noted actions. Infants and children are considered more sensitive to Mn (16, 43), because it can more efficiently cross the blood brain barrier in the young (48), which do not yet have the full capacity to eliminate the element (49). Mn is abundant in the environment, with some regions having greater amounts than others, and with some cultures of people consuming more in their diets. The cumulative information presented suggests that environmental Mn may contribute to the normal onset of puberty, but that a potential also exists for an increased risk of precocious puberty if an individual is exposed to a modestly elevated level of element too early in life. Epidemiological studies in children are needed to further address the potential relationship between early life Mn exposure and precocious puberty in humans.
Acknowledgments
This work was supported by National Institutes of Health/National Institute of Environmental Health Sciences Grant ES013143 (to W.L.D.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Akt
- protein kinase B
- AVPV
- anteroventral periventricular
- EV
- Everolimus
- IGF-1R
- IGF-1 receptor
- JB-1
- IGF-1 receptor antagonist
- KiSS-1
- kisspeptin gene
- Kp
- kisspeptin
- LHRH
- LH-releasing hormone
- MBH
- medial basal hypothalamus
- mTOR
- mammalian target of rapamycin
- p
- phosphorylated
- POA
- preoptic area
- Rheb
- Ras homolog enriched in brain
- TSC2
- tuberous sclerosis complex 2
- 3V
- third ventricle
- VO
- vaginal opening.
References
- 1. Boyer PH, Shaw JH, Phillips PH. Studies on manganese deficiency in the rat. J Biol Chem. 1942;143:417–425. [Google Scholar]
- 2. Smith SE, Medlicott M, Ellis GH. Manganese deficiency in the rabbit. Arch Biochem Biophys. 1944;4:281–289. [Google Scholar]
- 3. Pine M, Lee B, Dearth RK, Hiney JK, Dees WL. Manganese acts centrally to stimulate LH secretion in immature female rats: a potential influence on female pubertal development. Toxicol Sci. 2005;85:880–885. [DOI] [PubMed] [Google Scholar]
- 4. Lee B, Pine M, Johnson L, Rettori V, Hiney JK, Dees WL. Manganese acts centrally to activate reproductive hormone secretion and pubertal development in male rats. Reprod Toxicol. 2006;22:580–585. [DOI] [PubMed] [Google Scholar]
- 5. Prestifilippo JP, Fernández-Solari J, Mohn C, et al. Effects of manganese on lutenizing hormone-releasing hormone secretion in adult male rats. Toxicol Sci. 2007;97:75–80. [DOI] [PubMed] [Google Scholar]
- 6. Rosenfield RL. Puberty in the female and its disorders. In: Sperling MA, ed. Pediatric Endocrinology. 2nd ed Philadelphia, PA: Saunders; 2002:495–511. [Google Scholar]
- 7. Srivastava VK, Hiney JK, Dees WL. Early life manganese exposure upregulates tumor-associated genes in the hypothalamus of female rats: relationship to manganese-induced precocious puberty. Toxicol Sci. 2013;136:373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Navarro VM, Fernández-Fernández R, Castellano JM, et al. Advanced vaginal opening and precocious activation of the reproductive axis by KiSS-1 peptide, the endogenous ligand of GRP54. J Physiol. 2004;561:379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thompson EL, Patterson M, Murphy KG, et al. Central and peripheral administration of kisspeptin-10 stimulates the hypothalamic-pituitary-gonadal axis. J Neuroendocrinol. 2004;16:850–858. [DOI] [PubMed] [Google Scholar]
- 10. Keen KL, Wegner FH, Bloom SR, Ghatei MA, Terasawa E. An increase in kisspeptin-54 release occurs with the pubertal increase in luteinizing hormone-releasing hormone-1 release in the stalk-median eminence of female rhesus monkeys in vivo. Endocrinology. 2008;149:4151–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GRP54. Proc Natl Acad Sci USA. 2003;100:10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seminara SB, Messager S, Chatzidaki E, et al. The GPR54 gene as a regulator of puberty. New Engl J Med. 2003;349:1614–1627. [DOI] [PubMed] [Google Scholar]
- 13. Navarro VM, Castellano JM, Fernández-Fernández R, et al. Developmental and hormonally regulated messenger ribonucleic acid expression of KiSS-1 and its putative receptor, GPR54, in rat hypothalamus and potent luteinizing hormone-releasing activity of KiSS-1 peptide. Endocrinology. 2004;145:4565–4574. [DOI] [PubMed] [Google Scholar]
- 14. Shahab M, Mastronardi C, Seminara SB, Crowley WF, Ojeda SR, Plant TM. Increased hypothalamic GPR54 signaling: a potential mechanism for initiation of puberty in primates. Proc Natl Acad Sci USA. 2005;102:2129–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roa J, Garcia-Galiano D, Varela L, et al. The mammalian target of rapamycin as novel central regulator of puberty onset via modulation of hypothalamic Kiss1 system. Endocrinology. 2009;150:5016–5026. [DOI] [PubMed] [Google Scholar]
- 16. Environmental Protection Agency. Health Effects Support Document for Manganese. EPA Report 02-029 Washington, DC: United States Environmental Protection Agency; 2002. [Google Scholar]
- 17. Newland MC. Animal models of manganese neurotoxicity. Neurotoxicology. 1999;20:415–432. [PubMed] [Google Scholar]
- 18. Hiney JK, Srivastava VK, Pine MD, Les Dees W. Insulin-like growth factor-I activates KiSS-1 gene expression in the brain of the prepubertal female rat. Endocrinology. 2009;150:376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dees WL, Skelley CW. Effects of ethanol during the onset of puberty. Neuroendocrinology. 1990;51:64–69. [DOI] [PubMed] [Google Scholar]
- 20. Fox JH, Connor T, Chopra V, et al. The mTOR kinase inhibitor Everolimus decreases S6 kinase phosphorylation but fails to reduce mutant huntingtin levels in brain and is not neuroprotective in the R6/2 mouse model of Huntington's disease. Mol Neurodegener. 2010;5:26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pawaskar DK, Straubinger RM, Fetterly GJ, et al. Physiologically based pharmacokinetic models for everolimus and sorafenib in mice. Cancer Chemother Pharmacol. 2013;71:1219–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee B, Hiney JK, Pine MD, Srivastava VK, Dees WL. Manganese stimulates luteinizing hormone releasing hormone secretion in prepubertal female rats: hypothalamic site and mechanism of action. J Physiol. 2007;578:765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dees WL, Srivastava VK, Hiney JK. Alcohol alters insulin-like growth factor-1 induced oct 2 POU homeodomain genes in the prepubertal hypothalamus. J Stud Alcohol. 2005;66:35–45. [DOI] [PubMed] [Google Scholar]
- 24. Pietrzkowski Z, Wernicke D, Porcu P, Jameson BA, Baserga R. Inhibition of cellular proliferation by peptide analogues of insulin-like growth factor 1. Cancer Res. 1992;52:6447–6451. [PubMed] [Google Scholar]
- 25. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. [DOI] [PubMed] [Google Scholar]
- 26. Avruch J, Long X, Ortiz-Vega S, Rapley J, Papgeorgiou A, Dai N. Amino acid regulation of TOR complex 1. Am J Physiol Endocrinol Metab. 2009;296:592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nature Cell Biol. 2002;4:648–657. [DOI] [PubMed] [Google Scholar]
- 28. Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinistide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. [DOI] [PubMed] [Google Scholar]
- 29. Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004;167:399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cardona-Gomez GP, Mendez P, Garcia-Segura LM. Synergistic interaction of estradiol and insulin-like growth factor-1 in the activation of P13K/Akt signaling in the adult rat hypothalamus. Mol Brain Res. 2002;107:80–88. [DOI] [PubMed] [Google Scholar]
- 31. Hiney JK, Srivastava VK, Dees WL. Insulin-like growth factor-1 stimulation of hypothalamic KiSS-1 gene expression is mediated by Akt: effect of alcohol. Neuroscience. 2010;166:625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lesniak MA, Hill JM, Kiess W, Rojeski M, Pert CB, Roth J. Receptors for insulin-like growth factors I and II: autoradiographic localization in rat brain and comparison to receptors for insulin. Endocrinology. 1988;123:2089–2099. [DOI] [PubMed] [Google Scholar]
- 33. Bondy C, Werner H, Roberts CT, Jr, LeRoith D. Cellular pattern of type-I insulin-like growth factor receptor gene expression during maturation of the rat brain: comparison with insulin-like growth factors I and II. Neuroscience. 1992;46:909–923. [DOI] [PubMed] [Google Scholar]
- 34. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. [DOI] [PubMed] [Google Scholar]
- 35. Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–713. [DOI] [PubMed] [Google Scholar]
- 36. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. [DOI] [PubMed] [Google Scholar]
- 37. Lehman MN, Merkley CM, Coolen LM, Goodman RL. Anatomy of the kisspeptin neural network in mammals. Brain Res. 2010;1364:90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hiney JK, Srivastava V, Nyberg CL, Ojeda SR, Dees WL. Insulin-like growth factor I of peripheral origin acts centrally to accelerate the initiation of female puberty. Endocrinology. 1996;137:3717–3728. [DOI] [PubMed] [Google Scholar]
- 39. Wilson ME. Premature elevation in serum insulin-like growth factor-I advances first ovulation in rhesus monkeys. J Endocrinol. 1998;158:247–257. [DOI] [PubMed] [Google Scholar]
- 40. Daftary SS, Gore AC. Developmental changes in hypothalamic insulin-like growth factor-1: relationship to gonadotropin-releasing hormone neurons. Endocrinology. 2003;144:2034–2045. [DOI] [PubMed] [Google Scholar]
- 41. Wolfe A, Divall S, Wu S. The regulation of reproductive neuroendocrine function by insulin and insulin-like growth factor-1 (IGF-1). Front Neuroendocrinol. 2014;35:558–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;12:1259–1268. [DOI] [PubMed] [Google Scholar]
- 43. Food and Nutrition Board, Institute of Medicine. Manganese. Dietary Reference Intakes for Vitamin A, Vitamin K, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium and Zinc. Washington, DC: National Academy Press; 2001:394–419. [PubMed] [Google Scholar]
- 44. Gray Le, Jr, Laskey JW. Multivariate analysis of the effects of manganese on the reproductive physiology and behavior of the male house mouse. J Toxicol Environ Health. 1980;6:861–867. [DOI] [PubMed] [Google Scholar]
- 45. Laskey JW, Rehnberg GL, Hein JF, Carter SD. Effects of chronic manganese (Mn3O4) exposure on selected reproductive parameters in rats. J Toxicol Environ Health. 1982;9:677–687. [DOI] [PubMed] [Google Scholar]
- 46. Moreno JA, Streifel KM, Sullivan KA, Legare ME, Tjalkens RB. Developmental exposure to manganese increases adult susceptibility to inflammatory activation of glia and neuronal protein nitration. Toxicol Sci. 2009;112:405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zheng W, Kim H, Zhao Q. Comparative toxicokinetics of manganese chloride and methylcyclopentadienyl manganese tricarbonyl (MMT) in Sprague-Dawley rats. Toxicol Sci. 2000;54:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mena I. The role of manganese in human disease. Ann Clin Lab Sci. 1974;4:487–491. [PubMed] [Google Scholar]
- 49. Fechter LD. Distribution of manganese in development. Neurotoxicology. 1999;20:197–201. [PubMed] [Google Scholar]