

Abstract
The critical regulation of the peripheral circadian gene implicated in osteoarthritis (OA) has been recently recognized; however, the causative role and clinical potential of the peripheral circadian rhythm attributable to such effects remain elusive. The purpose of this study was to elucidate the role of a circadian gene Bmal1 in human cartilage and pathophysiology of osteoarthritis. In our present study, the mRNA and protein levels of circadian rhythm genes, including nicotinamide adenine dinucleotide oxidase (NAD+) and sirtuin 1 (Sirt1), in human knee articular cartilage were determined. In OA cartilage, the levels of both Bmal1 and NAD+ decreased significantly, which resulted in the inhibition of nicotinamide phosphoribosyltransferase activity and Sirt1 expression. Furthermore, the knockdown of Bmal1 was sufficient to decrease the level of NAD+ and aggravate OA-like gene expression changes under the stimulation of IL-1β. The overexpression of Bmal1 relieved the alteration induced by IL-1β, which was consistent with the effect of the inhibition of Rev-Erbα (known as NR1D1, nuclear receptor subfamily 1, group D). On the other hand, the transfection of Sirt1 small interfering RNA not only resulted in a reduction of the protein expression of Bmal1 and a moderate increase of period 2 (per2) and Rev-Erbα but also further exacerbated the survival of cells and the expression of cartilage matrix-degrading enzymes induced by IL-1β. Overexpression of Sirt1 restored the metabolic imbalance of chondrocytes caused by IL-1β. These observations suggest that Bmal1 is a key clock gene to involve in cartilage homeostasis mediated through sirt1 and that manipulating circadian rhythm gene expression implicates an innovative strategy to develop novel therapeutic agents against cartilage diseases.
Osteoarthritis (OA) is the most common disorder in joint diseases characterized by loss of tissue cellularity and extracellular matrix (ECM) damage, causing joint pain and dysfunction in the affected patients, with currently no cure (1). OA primarily affects the articular cartilage lining the ends of long bones. In cartilage, chondrocytes are the only resident cells and are responsible for both synthesis and turnover of the abundant ECM (2). Therefore, maintaining the chondrocytes in a healthy condition seems to be a key factor for the maintenance of the entire cartilage and prevention of cartilage degeneration. Although age is considered a major risk factor, how aging contributes to OA is still not well understood.
It is known that during aging, circadian rhythms deteriorate in a wide range of species, including humans. The circadian (∼24 h) clock in mammals comprises a central component located in the hypothalamic suprachiasmatic nucleus and subordinate clocks in peripheral tissues. Both central and peripheral clocks are operated by positive- and negative-feedback loops of circadian genes (3–5), including those for transcriptional activators (Clock/Npas2 and Bmal1), transcriptional repressors (Per1/2 and Cry1/2), and nuclear hormone receptors (Rev-Erbα/β [known as NR1D1, nuclear receptor subfamily 1, group D] and Retinoic acid receptor related orphan receptor (Ror)a/Rorg) (3). This transcriptional-translational feedback loop controls the expression of downstream target genes (clock controlled genes) to maintain body homeostasis, regulating a wide variety of physiological and metabolic processes. Disruption of the circadian rhythm attributable to such effects as aging or shift work has been correlated with an increased risk of various human diseases, including obesity, diabetes, cardiovascular disease, and cancer (6).
Recent study showed an existence of clock genes in cartilage (7). Knockout mice with clock gene Bmal1 deficiency display an accelerated aging phenotype, including osteoporosis. Mice with mutations in clock genes also show altered regulation of bone volume (8), retarded long bone growth (9), and increased susceptibility to inflammatory arthritis (10); moreover, in vitro study also demonstrated that overexpression of Per1 suppressed chondrocyte differentiation by suppressing the functional E-box in the Indian hedgehog promoter (9). Together these studies provide both in vivo and in vitro evidence for the importance of the molecular clock in the skeletal system.
Furthermore, sirtuin 1 (Sirt1) is a nicotinamide adenine dinucleotide oxidase (NAD+)-dependent protein deacetylase that governs many physiological pathways, including circadian rhythm in peripheral tissues. An age-related decline in Sirt1 was implicated as a cause for circadian disruption (11). Some studies have also shown that Sirt1 interacts with the circadian system at multiple levels through the direct regulation of Bmal1 expression (11), and through Per2 and Bmal1 deacetylation (12–13). In mammalians, Sirt1 has been reported to play an important role in age-related diseases such as osteoporosis, diabetes, and cancer (14–16). Recently also shown was an involvement of Sirt1 in the pathogenesis of OA by modulating chondrocyte gene expression and hypertrophy (17), and its disruption exacerbates experimental OA (18–20). However, whether circadian change of Sirt1 is also involved in regulating cartilage gene expression is still unclear, and the underlying mechanisms for circadian expression of Sirt1 are yet to be elucidated.
Judging from the broad cell-regulating functions of circadian clocks in a variety of cells, circadian genes may also play a regulating role in human chondrocytes in the pathophysiological process of cartilage undergoing OA changes. In the present study, we investigated the roles of clock gene Bmal1 both in human cartilage tissue and chondrocytes and in the pathophysiology of OA. Our observations suggest that Bmal1 protects chondrocytes from stress-induced senescence and may prevent chondrocytes from undergoing OA changes.
Materials and Methods
OA and normal cartilage sample obtaining and processing
OA cartilage tissues were obtained from femoral condyles of six patients with medial knee OA during total knee joint replacement surgery (mean age 61.4 y). The cartilage samples were obtained from the distal part of both the lateral and medial femoral condyles of the knee joint. Articular cartilage tissues without OA were obtained from three age-matched patients undergoing surgery for femoral neck fracture (mean age 59.3 y). None of the three patients had a history of joint disease, and none of the samples showed macroscopically obvious progressed OA changes. All OA and normal cartilage samples were obtained in accordance with the World Medical Association Declaration of Helsinki ethical principles.
Chondrocyte culture and treatment
Normal human articular cartilage was harvested at the time of autopsy from the femoral condyles and the tibial plateaus of donors (age >55 y) without a history of joint disease and with macroscopically normal cartilage surfaces. Chondrocytes were isolated and cultured as described previously (21). First-passage chondrocytes were used in the experiments.
The chondrocytes were cultured in DMEM containing 10% fetal bovine serum. Once reaching 80% confluence, the chondrocytes were cultured for a further 24 hours and then stimulated by 50% fetal horse serum and 100 μM EX527 (Sigma-Aldrich) in serum-free DMEM, respectively, and cells were collected every 4 hours for 24 hours or 48 hours.
Small interfering RNA (siRNA) and plasmid against Bmal1 transfection under the simulation of IL-1β
siRNA targeted for Bmal1 (sc-38165) was purchased from Santa Cruz Biotechnology Inc. The siRNA for Sirt1, Rev-erbα, and overexpression plasmid of Bmal1 and Sirt1 were from Genechem Technology Inc. Primary chondrocytes were transfected with siRNA or plasmid by Lipofectamine2000 (Invitrogen) according to the manufacturer's instructions. After transfection, cells were seeded on a six-well plate and cultured for 2 days. The next day after transfection, cells were analyzed for green fluorescent protein using a microscope (Nicon). For catabolic stresses, cells were cultured with 10 ng/mL of IL-1β (R&D Systems) for another 24 hours after the transfection.
Cell proliferation assay
Cell proliferation was evaluated using the Cell Counting Kit-8 (CCK-8; Beyotime Institute of Biotechnology, Haimen, China) as described previously (22). Ten microliters of the CCK-8 solution was added to each well of 96-well plates, and cells were incubated at 37°C for 1 hour. Absorbance was determined at 450 nm using a microplate spectrophotometer.
Alcian blue stain
Alcian blue 8GX was purchased from Sigma-Aldrich. Primary cultured cells were plated at a density of 8–9 × 104 per well in six-well plates. At the end of indicated culture period, the cells were washed with ice-cold PBS, fixed at room temperature with 4% glutaraldehyde for 15 minutes, and rinsed with HCl 0.1 N. The cells were then stained at room temperature with 1% alcian blue in 3% glacial acetic acid and 1% HCl (pH 2.5). The cells were rinsed once with distilled water and twice with PBS. The amount of colorized dye was measured at an OD of 620 nm after extraction with 4 M guanidine-HCl (Sigma-Aldrich).
Assays of NAD production
Intracellular NAD+ levels were determined using a colorimetric NAD/nicotinamide adenine dinucleotide hydroxide (NADH) quantification kit (BioVision Inc). Briefly, chondrocytes were extracted in NAD/NADH extraction buffer. The reactions were prepared in 96-well plates and read at an OD of 450 nm. NAD+ concentration was determined by subtracting NADH concentration from total NAD concentration.
Immunohistochemistry
Cartilage tissues were fixed in 4% paraformaldehyde for 24 hours and embedded in paraffin wax. Each specimen was cut into 5-μm slices, and the sections were incubated with primary antibody at 4°C overnight. After this procedure, sections were incubated with horseradish peroxidase (HRP)-conjugated goat antirabbit IgG polyclonal antibody (Bioss Bioscience) at 37°C for 1 hour. The signal was developed as a brown reaction product using the peroxidase substrate 3,3-diaminobenzidine (Bioss Bioscience). Primary antibodies included the following: anti-Bmal1 (sc-48790, Santa Cruz Biotechnology); and anti-Sirt1 (number 9475L, Cell Signaling Technology; ab110304, abcam).
Immunofluorescence
Normal human knee articular chondrocytes were seeded on four-well chamber slides at a density of 5 × 104/well. Cells were cultured in 100 μM EX527 for 2 hours and fixed in 4% paraformaldehyde at 8 hours after EX527 was removed. The fixed cells were washed in PBS for 15 minutes and then incubated in blocking solution (3% BSA) for 1 hour. After incubation with a primary antibody against Bmal1 (sc-48790, Santa Cruz Biotechnology) overnight at 4°C, the cells were followed by an Alexa Fluor 488-conjugated secondary antibody (ab150077; Abcam) for 1 hour at room temperature. Images were obtained using a microscope (Nicon).
RNA extraction and quantitative real-time PCR
RNA was extracted using a QIAGEN RNeasy minikit (QIAGEN Inc). According to the manufacturer's instructions, quantification of PCR products was conducted by real-time PCR using a MJ Mini TM (Bio-Rad Laboratories) with an IQ SYBR Green supermix (Bio-Rad Laboratories). The PCR conditions were as follows: 95°C for 30 seconds followed by 40 cycles at 95°C for 5 seconds and 60°C for 1 minute. Values were quantified using the comparative cycle threshold method, and samples were normalized to GAPDH. The primer sequences are described in Supplemental Table 1.
Western blot
Tissues and chondrocytes under various treatments were lysed in buffer containing 25 mM Tris-Hcl (pH 6.8), 2% sodium dodecyl sulfate, 6% glycerol, 1% 2-mercaptoethanol, 2 mM phenylmethylsulfonylfuoride, 0.2% bromophenol blue, and a protease inhibitor cocktail for 20 minutes. Western blot was performed by using a standard protocol as described. In a brief, tissues and chondrocytes were harvested, and immunoblotting was performed using equal amounts of protein (50–100 μg) and the following primary antibodies: antiglyceraldehyde-3-phosphate dehydrogenase (GAPDH; ab181602; Abcam), anti-Bmal1 (sc-48790; Santa Cruz Biotechnology), anti-Per2 (ab180655; Abcam), anti-Rev-erbα (number 13418; Cell Signaling Technology), anti-Sirt1 (number 9475; Cell Signaling Technology); antinicotinamide phosphoribosyltransferase (NAMPT; ab45890; Abcam); rabbit polyclonal antibody against collagen 2a1 (col2a1), aggrecan, matrix metalloproteinase (MMP)-13, and A disintegrin and metalloproteinase with thrombospondin-like repeats 5 (ADAMTS5; ab34712, ab36861, ab39012, ab41037; Abcam).
Coimmunoprecipitation
Cytoplasmic lysate (200 μg) was incubated for 2 hours at 4°C with the corresponding antibodies coupled to 20 μL of packed protein A+G sepharose beads (number sc-2002; Santa Cruz Biotechnology). Immune complexes were resolved by means of SDS-PAGE and immunoblotted with the indicated antibodies.
Statistical analysis
The statistical analyses were performed using the GraphPad Prism software (GraphPad Software Inc). In case of data with normal distribution and/or equal variances, statistical differences between two and more than three groups were determined by a two-tailed Student's t test and a one-way ANOVA followed by Bonferroni's post hoc comparison test, respectively. A value of P < .05 was considered statistically significant. Results are presented as the mean ± SEM from at least three independent experiments.
Results
Expression of key genes of circadian rhythm in human articular cartilage tissues with or without OA
To explore the regulation of circadian genes in human articular cartilage, we examined the mRNA expression of key genes, Bmal1, Clock, per1/2, cry1/2, and Rev-erbα in human articular cartilage of OA patients. First, hematoxylin and eosin and Safranin O staining showed that chondrocytes and well-stained ECM in the non-OA cartilage and slightly less stained cartilage of the lateral condyle. The cartilage of the medial condyle was severely degenerated and was barely stained with Safranin O (Figure 1A). We then examined the mRNA expression of marker genes related to cartilage homeostasis, for example, col2a1, aggrecan, MMP13, and ADAMTS5, and the results showed that the expressions of the matrix catabolism genes increased significantly (Figure 1, B and C). Subsequently, the expressions of key circadian genes in patients with or without OA were determined, for example, Bmal1, Clock, per1/2, cry1/2, and Rev-erbα. Compared with normal articular cartilage, the expression of Bmal1 mRNA in OA patients' articular cartilage showed a weakening trend, whereas Per2 and Rev-erbα mRNA expression increased significantly (Figure 1D). There was no significance of other circadian genes.
Figure 1.
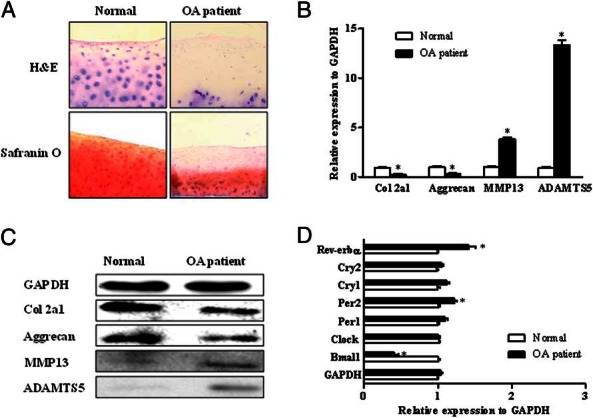
Expression of the key genes of circadian rhythm in human articular cartilage tissue in OA patients. OA cartilage tissues were obtained from the knee joint of patients with or without OA. A, The hematoxylin and eosin (H&E) and Safranin O stainings were performed on articular cartilage. B, The remarkable changes of cartilage genes related to synthetic metabolism and catabolism were examined by real-time PCR, including the col2a1, aggrecan, MMP13, and ADAMTS5 in the knee joint of the patients. C, The protein levels of col2a1, aggrecan, MMP13, and ADAMTS5 were examined by Western blot. D, The expression levels of the key circadian rhythm genes, for example, Bmal1, per1/2, cry1/2, and Rev-erbα were determined by real-time PCR. Bars represent a mean and SE of n = 3–6/group.
Reduction of Bmal1 expression was accompanied by the decrease of NAD+ and Sirt1 level in cartilage of OA patients' knee joint
We next examined the protein expression of Baml1 in the human articular cartilage of OA patients. The immunohistochemistry (IHC) results further showed fewer Bmal1-positive chondrocytes in OA knee joint cartilage, and the expression level of Bmal1 in OA cartilage tended to be lower than in normal cartilage confirmed by real-time PCR and Western blot (Figure 2, A and B).
Figure 2.

Bmal1 expression level was related with the Sirt1 activity, which is influenced by the level of NAD+. A, The representative IHC images of Sirt1 and Bmal1 in patients' knee joints. B, The expression levels of Bmal1 were determined by real-time PCR and Western blot. C, The level of NAD+ in knee joint tissue was detected by a colorimetric NAD/NADH quantification kit. D, The expression of NAMPT and Sirt1 was examined by Western blot and real-time PCR. Bars represent a mean and SE of n = 3–6/group.
Sirt1, an NAD+-dependent deacetylase involved in transcriptional silencing, genome stability, and longevity was reported to regulate circadian clock gene expression (12, 23, 24). Therefore, we next detected the level of NAD+ to investigate whether Bmal1 disruption interact with NAD+/NAMPT during the progress of osteoarthritis. The results indicated that there was a 40% decline in the level of NAD+ and a significant reduction of the expression of NAMPT (Figure 2, C and D). Meanwhile, the level of Sirt1 was significantly reduced in the OA articular cartilage (Figure 2, C and D), which is consistent with IHC images of Sirt1 (Figure 2A).
Effect of Bmal1 on chondrocyte survival and matrix proteoglycan synthesis
To explore the effect of Bmal1 on chondrocyte functions, we next determined whether the knockdown of Bmal1 had any impact on chondrocyte survival or matrix production. Our data demonstrated that knockdown of Bmal1 alone had no significant effect on chondrocyte survival as assessed by the CCK-8 (Figure 3A), whereas it significantly inhibited proteoglycan synthesis assessed by Alcian blue staining (Figure 3, B and C). Because it has been reported that the metabolic imbalance of chondrocytes is a cause of OA and IL-1β is a known metabolic imbalance inducer (25), we treated the chondrocyte with IL-1β after the transfection of Bmal1 siRNA. The results showed that transfection of Bmal1 siRNA or treatment of IL-1β decreased the expression of col2a1 and aggrecan mRNA both at the mRNA (Figure 3D) and protein levels (Figure 3F). Meanwhile, the expression of the cartilage-degrading enzyme MMP1, MMP3, MMP13, and ADAMTS5 was significantly increased by induction of IL-1β, with such effects being more pronounced with the transfection of Bmal1 siRNA (Figure 3, E and F).
Figure 3.

Knockdown of Bmal1 inhibited the matrix proteoglycan synthesis in chondrocytes, which was further aggravated by IL-1β. A, The proliferation of chondrocytes was determined using the CCK-8 assay after transfection with Bmal1 siRNA. B and C, Representative images of alcian blue staining and the levels of alcian blue-stained proteoglycans were detected by OD values at 620 nm. D, The chondrocytes were treated with IL-1β for 24 hour after the transfection of Bmal1 siRNA. Quantitative results for col2a1 and aggrecan in chondrocytes were measured by real-time PCR after transfection with Bmal1 siRNA under stimulation with IL-1β (10 ng/mL). E, The expression of MMP1, MMP3, MMP13, and ADAMT5 in chondrocytes was measured by real-time PCR after transfection with Bmal1 siRNA under stimulation of IL-1β (10 ng/mL). F, The representative Western blots of the col2a1 and aggrecan, MMP 13, and ADAMTS5 after transfection with Bmal1 siRNA under stimulation of IL-1β. Bars represent a mean and SE of n = 5/group. control, control siRNA; siBmal1, Bmal1siRNA.
On the other hand, we transfected the chondrocytes with Bmal1 plasmid to investigate whether Bmal1 overexpression may restore the changes of the chondrocytes functions induced by IL-1β. First, the transfection efficiency was validated by fluorescence microscope and reconfirmed by quantitative PCR and Western blot (Figure 4, A and B). The transfection of Bmal1 plasmid alone had no effect on the characteristic genes of the chondrocytes. However, it neutralized the effect of IL-1β on chondrocyte functions as assessed by expression of col2a1 and aggrecan mRNA (Figure 4C) as well as the expression of the cartilage-degrading enzyme MMP13 and ADAMTS5 (Figure 4D).
Figure 4.

Overexpression of Bmal1 neutralized the metabolic imbalance of chondrocytes induced by IL-1β. The chondrocytes were treated with IL-1β for 24 hours after transfection with the Bmal1 plasmid. A and B, The representative images of the chondrocyte transfection with the Bmal1 plasmid, and the transfection efficiency was evaluated by real-time PCR and Western blot. C, The quantitative results for col2a1 and aggrecan in chondrocytes transfected with Bmal1 under stimulation with IL-1β was determined by real-time PCR. D, Quantitative results for the expression levels of MMP13 and ADAMTS5 were measured by real-time PCR. Bars represent a mean and SE of n = 5/group. control, control plasmid; plaBmal1, Bmal1 plasmid.
The reciprocal interaction of Bmal1 and Sirt1 in cultured chondrocyte
Since an age related decline in Sirt1 has been implicated as a cause for circadian disruption, and our finding in current study also showed that Sirt1 expression decreased markedly in cartilage of OA patients compared with those of non-OA patients, we reasoned that Sirt1 could directly modulate Bmal1 function in chondrocyte. Endogenous Sirt1 and Bmal1 proteins interact as demonstrated by coimmunoprecipitation assays (Supplemental Figure 1A). To confirm whether Bmal1 regulates Sirt1 in chondrocytes, we knocked down Bmal1 with targeted siRNA and detected the transfection efficiency (Supplemental Figure 1B). After the knockdown of Bmal1, the protein expressions of Per2 and Rev-erbα were up-regulated (Supplemental Figure 1C). In addition, there was a significant reduction of NAD+ generation as well as the protein levels of NAMPT and Sirt1 in the cells transfected with Bmal1 siRNA (Supplemental Figure 1, D and E).
Inhibition of Rev-erbα resulted in the activation of Bmal1 and in turn protected the chondrocytes from the stimulation with IL-1β
Based on prior results of the expression level of Rev-erbα increase in the cartilage of the OA knee joint and the inhibition of Bmal1 worsening of the cartilage-degrading enzyme induced by IL-1β, we then treated the chondrocytes with siRNA-targeted Rev-erbα. The transfection efficiency was validated by Western blot (Supplemental Figure 2A). The results showed that the expression of Bmal1 and Sirt1 were significantly up-regulated by the inhibition of Rev-erbα (Supplemental Figure 2, B and C). The transfection of Rev-erbα siRNA alone had no effect on the characteristic genes of the chondrocytes. However, it partially nullified the effect of IL-1β as assessed by the expression of col2a1 and aggrecan mRNA and the cartilage-degrading enzymes MMP1, MMP3, MMP13, and ADAMTS5 (Supplemental Figure 2D).
The inhibition of Sirt1 increased IL-1β-induced MMPs and ADAMTS5 expression along with the rhythm disorders of key clock genes
To clarify the effect of Sirt1 on human chondrocyte survival, the chondrocyte was transiently transfected with siRNA-targeted Sirt1. As shown in Figure 5A, Sirt1 was efficiently inhibited by day 2 after transfection as assessed by Western blot. We first evaluated the effect of Sirt1 siRNA on the clock genes, for example, Bmal1, per2, and Rev-erbα. The result indicated that the transfection of Sirt1 siRNA inhibited the protein expression of Bmal1 and a moderate increase of per2 and Rev-erbα (Figure 5B).We next examined the effect of Sirt1 inhibition on the IL-1β-induced cell survival and the expression cartilage matrix-degrading enzymes, for example, MMPs and ADAMTS5. Figure 5C showed that the proliferation drops 0.5-fold under the stimulation of IL-1β when Sirt1 was inhibited. The levels of MMP1, MMP3, MMP13, and ADAMTS5 caused by IL-1β was further up-regulated compared with the control, as determined by both real-time PCR and Western blot (Figure 5, D and E).
Figure 5.

Effects of Sirt1 siRNA on chondrocytes under stimulation with IL-1β. The chondrocytes were treated with IL-1β for 24 hours after the transfection with Sirt1 siRNA. A, Western blot for Sirt1 and representative data were from repeated experiments. B, Western blots for Bmal1, Per2, and Rev-erbα after Sirt1 siRNA treatment. C, The proliferation of chondrocytes was determined using the CCK-8 assay after the Sirt1 silence or IL-1β treatment. D, Real-time PCR for MMP1, MMP3, MMP13, and ADAMTS5 mRNA in chondrocytes. The values were at a relative expression level to the control expression. E, Western blots for MMP13 and ADAMTS 5 after Sirt1 siRNA treatment. Bars represent a mean and SE of n = 5/group. control, control siRNA; siBmal1, Sirt1 siRNA.
To further confirm the above results, we also examined the expression of clock genes in chondrocytes with treatment of the Sirt1 inhibitor EX527. Immunofluorescence indicated much less expression levels of Bmal1 in chondrocytes nucleus by treatment of EX527 as compared with the control (Figure 6A). Then we detected the effects of Sirt1 inhibitor on circadian rhythm as well as the clock gene expression in chondrocytes treated with EX527 for 2 hours. The result showed a rhythmic change of clock genes expression (Figure 6B). After the classic analysis of the rhythm, the curve showed that the amplitude of Bmal1 treated with EX527 was decreased throughout the 48 hours and the Per2 and Rev-erbα increased (Figure 6, C–E).
Figure 6.

EX527, a Sirt1 inhibitor, influenced the rhythmic expression levels of Bmal1, per2, and Rev-erbα. A, After 2 hours of EX527 (100 μM) treatment and then at the first 8 hours, the Bmal1 was stained by immunofluorescence. B, The protein expression of Bmal1, Per2, and Rev-erbα was detected after 2 hours of EX527 treatment. Samples were collected every 4 hours for 24 hours. C–E, The circadian rhythms of Bmal1, Per2, and Rev-erbα were detected after 2 hours of EX527 treatment. Samples were collected every 4 hours for 48 hours. Bars represent a mean and SE of n = 5/group.
In contrast, the Sirt1 plasmid was transfected into chondrocyte to confirm whether the recovery of Sirt1 expression may nullify the effect of IL-1β on chondrocyte functions. As shown in Figure 7A, plasmid transfection was validated by overexpressed Sirt1 protein assessed by Western blot. Figure 7B shows that the overexpression of Sirt1 significantly improved the chondrocyte proliferation in the presence of IL-1β. Addition of Sirt1 inhibitor EX527 in the culture medium abolished the protective effect of Sirt1 on cell proliferation. On the other hand, the overexpression of Sirt1 significantly inhibited the up-regulation of mRNA and protein expression of MMP1, MMP3, MMP13 and ADAMTS5 caused by IL-1β (Figure 7, C and D).
Figure 7.

Overexpression of Sirt1 restored the metabolic imbalance of chondrocytes caused by IL-1β. The chondrocytes were treated with IL-1β for 24 hours after the transfection with the Sirt1 plasmid. A, Western blot for Sirt1 and representative data from repeated experiments. B, The proliferation of chondrocytes was determined by the CCK-8 assay. C, The expression of MMP1, MMP3, MMP13, and ADAMTS5 mRNA in chondrocytes was measured by real-time PCR after transfection with the Sirt1 plasmid under stimulation with IL-1β (10 ng/mL). The values were at a relative expression level to the control expression. D, Western blots for MMP13 and ADAMTS5. Bars represent a mean and SE of n = 5/group. control, control plasmid; plaSirt1, Sirt1 plasmid.
Discussion
Although several studies have revealed an autonomous circadian clock in chondrocytes and that endochondral ossification is under the regulation of particular clock gene products expressed in chondrocytes during postnatal skeletogenesis, the effects of circadian disruption (eg, during aging) in cartilage homeostasis has not been addressed. The relationship between aging and OA is clinically and epidemiologically evident, and recent findings provide insight into mechanisms that lead to aging-related changes in cells and ECM (26), the characteristic progressive degeneration and loss of articular cartilage in OA. Articular cartilage appears to be highly susceptible to the accumulation of aging-related changes, in part due to the relatively low turnover of ECM and cells. In this study, we explored the regulation of clock gene Bmal1 on human articular cartilage of patients with OA and further elucidated the underlying mechanism. Our results implicated an important role of Bmal1 on the maintenance of cartilage homeostasis and pivotal interaction of Bmal1 and Sirt1 in the development of OA.
Previous reports showed autonomous circadian clock existing in mice cartilage dysregulates with aging and chronic inflammation (7, 9, 27, 28). Circadian amplitude of cartilage oscillation was significantly reduced in aged tissue (7). Furthermore, environmental disruption of circadian rhythms in mice (mimicking chronic jet lag) predisposes knee cartilage to OA-like damage (29), supporting the notion of an involvement of circadian rhythm disruption in OA development. In the present study, our results demonstrated that clock genes for transcriptional activators such as Bmal1 displayed a significant decrease in cartilage of patients with OA, whereas in contrast, clock genes for transcriptional repressors such as Per2, increased markedly, indicating an important regulating role of Bmal1 in human articular cartilage degeneration. More importantly, our finding that knockdown of Bmal1 exacerbated the effects on chondrocytes induced by IL-1β in vitro implies a protective role of Bmal1 in cartilage homeostasis under such stress. Consistently, a recent study demonstrated a significant decrease of Bmal1 expression level in human OA cartilage; moreover, their finding in the same literature also showed a reduction of Bmal1-positive chondrocyte number in aged mouse knee cartilage (22–24 mo) as compared with young (2–3 mo) mouse knee cartilage, indicating an age-related change of an endogenous clock factor (30). Similarly, it was demonstrated that in cultured costal chondrocytes from Bmal−/− mice, a temporal increase of matrix proteoglycan as assessed by Alcian blue staining was significantly suppressed throughout the culture period from 7 to 21 days. A Bmal1 deficit led to the disruption of the rhythmic expression profiles of both Per1 and Indian hedgehog in the growth plate (9). Although Takarada et al (9) also demonstrated a discouraging finding in unchanged collagen 2a1 expression in Bmal1-deficient mice, this discrepancy may be attributed to the different model used. In their study, the unaltered collagen 2a1 attributed to its preferential expression at an early differentiation stage in Bmal1-deficient mice. Furthermore, another in vitro study identified the clock genes as being down-regulated by mechanic stress (31). Along with these evidences, our results indicated a regulating role of Bmal1 in chondrocyte function.
Although the report regarding the existence of circadian variation in cartilage dated back to early 1990 (32), it was only until recently that evidence for a functional circadian clock in cartilage tissue capable of driving downstream clock controlled genes has been largely circumstantial (7). In cartilage, several physiological processes, including the processes of matrix synthesis, the growth rate in the growth plate, and mineralization exhibit diurnal variation (32–33). Evidence demonstrated mice with mutant clock genes exhibited altered regulation of bone volume (8) and increasing susceptibility to inflammatory arthritis (10). A previous study showed expression of clock protein or gene in synovial tissue and cells from OA and rheumatoid arthritis patients (34); however, no studies have addressed how clock genes changed in articular cartilage of patients with OA.
Aging is well known to affect the circadian clock in the central nervous system, and sleep disorder is a main concern in aging (35). It is noteworthy that the average life span of Bmal1-deficient mice is 8 months, much less than those of wild-type mice, whose average life span is 26 months (36). These knockout mice display an accelerating aging phenotype, including osteoporosis. On the other hand, reduction of Bmal1 protein expression and attenuation of its circadian rhythm in aged articular cartilage could lead to increased cell senescence, as evidenced by the aged chondrocyte with higher levels of stress-induced senescence (37). Given the evidence that an age-related decline of Sirt1 has also implied a cause of circadian disruption, it is plausible to hypothesize that Bmal1 regulated cartilage homeostasis through Sirt1.
Our coimmunoprecipitation result confirmed a direct interaction of endogenous Bmal1-Sirt1 protein. Further findings showed that blockade of Sirt1 either by siRNA transfection or by Sirt1 inhibitor significantly decreased Bmal1 expression in cultured chondrocytes, coupled with a significant increase of Rev-erbα and per2. It was known that Bmal1 expression is negatively regulated by Rev-Erbα (38), controlling the formation of the Clock/Bmal1 complex (39). This complex then switches the NAMPT gene expression and NAD+ synthesis and thus the NAD+-dependent deacetylase activity of Sirt1. In turn, Sirt1 interacts with the Clock/Bmal1 complex closing the clock circuit of Sirt1 control (13). In our study, transfection of Rev-erbα siRNA caused a marked increase of Bmal1 and Sirt1 expression. Silence of Rev-erbα alleviated the effects of IL-1β on chondrocytes as assessed by gene expressions such as col2a, aggreacan, ADAMTS5, and MMPs. Taken together, our study implicates a reciprocal interaction of Bmal1 and Sirt1 in chondrocytes and at least partially via Rev-erbα regulation.
Consistent with our findings, there are some literatures that suggested a reciprocal interaction of circadian clock and Sirt1. For example, it was reported that Sirt1 disruption could lead to a change of the molecular clock and its downstream targets, or reciprocally changes to the circadian clock could disrupt Sirt1 activity through NAMPT expression (40). Another study reported that Clock and Bmal1 are under the transcriptional regulation of RORa and Rev-Erbα (41), posing a loop that ultimately regulates the innate immune system via Sirt1 (42). Moreover, a recent study described Sirt1 in the brain governs central circadian control by activating transcription of the two major circadian regulators, Bmal1 and Clock (43).
In summary, our findings demonstrated a direct circadian regulation of Bmal1 in cartilage homeostasis mediated by NAD+-dependent deacetylase Sirt1. Further studies, using genetically engineered mice with inducible tissue-specific target gene deletion/overexpression (ie, cartilage tissue-specific Bmal1 gene knockout mice) in combination with aging or environmental circadian rhythm disruption, are warranted to better understand the multiple catabolic and antianabolic effects mediated by circadian rhythm disturbances on joint tissue. Such studies may open up for new insights regarding the role of circadian genes in cartilage homeostasis as well as in the complex pathophysiology of OA and potentially develop novel therapeutic and intervention strategies for disease prevention and/or treatment.
Table 1.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised (Monoclonal or Polyclonal) | Dilution Used |
|---|---|---|---|---|---|
| Bmal1 | BMAL1 antibody (H-170) | Santa Cruz Biotechnology, sc-48790 | Rabbit; polyclonal | 1:200 | |
| Sirt1 | SirT1 (D1D7) | Cell Signaling Technology, number 9475 | Rabbit; mAb | 1:1000 | |
| Anti-SIRT1 antibody | Abcam, ab110304 | Mouse; mAb | 1:200 | ||
| Rev-erbα | Rev-Erbα (E1Y6D) | Cell Signaling Technology, number 13418 | Rabbit; mAb | 1:1000 | |
| GAPDH | Anti-GAPDH antibody | Abcam, ab181602 | Rabbit; monoclonal | 1:10 000 | |
| per2 | Anti-PER2 antibody | Abcam, ab180655 | Rabbit; polyclonal | 1:100 | |
| NAMPT | Antivisfatin antibody | Abcam, ab45890 | Rabbit polyclonal | 1:1000 | |
| col2a1 | Anticollagen II antibody | Abcam, ab34712 | Rabbit; polyclonal | 1:5000 | |
| aggrecan | Antiaggrecan antibody | Abcam, ab36861 | Rabbit; polyclonal | 1:1000 | |
| MMP13 | Anti-MMP13 antibody | Abcam, ab39012 | Rabbit; polyclonal | 1:3000 | |
| ADAMTS5 | Anti-ADAMTS5 antibody | Abcam, ab41037 | Rabbit; polyclonal | 1:250 | |
| Rabbit IgG H&L | Antirabbit IgG H&L (Alexa Fluor 488) | Abcam, ab150077 | Goat; polyclonal | 1:200 |
Abbreviations: H&L, heavy and light chains; mAb, monoclonal antibody.
Acknowledgments
Author contributions included the following: W.Y. and X.K. wrote the manuscript and researched the data. J.L., H.L., Z.M., X.J., Q.Z., T.X., D.F., F.L., W.P., and Q.C. researched the data and contributed to the discussion. S.W. and H.S. contributed to the experimental designs and reviewed and edited the manuscript. S.W. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
This work was supported by programs from the National Natural Science Foundation of China (Grants 81472038, 81370899, and 81170741); National Excellent Young Scientist Program Award 81222026; the New Century Excellent Talents from the Ministry of Education of China Award NCET-11-0437; and National Institutes of Health Grant P20GM104937.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ADAMTS5
- A disintegrin and metalloproteinase with thrombospondin-like repeats 5
- CCK-8
- Cell Counting Kit-8
- col2a1
- collagen 2a1
- ECM
- extracellular matrix
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- HRP
- horseradish peroxidase
- IHC
- immunohistochemistry
- MMP
- matrix metalloproteinase
- NAD+
- nicotinamide adenine dinucleotide oxidase
- NADH
- nicotinamide adenine dinucleotide hydroxide
- NAMPT
- nicotinamide phosphoribosyltransferase
- OA
- osteoarthritis
- Per2
- period 2
- siRNA
- small interfering RNA
- Sirt1
- sirtuin 1.
References
- 1. Vignon E, Arlot M, Meunier P, Vignon G. Quantitative histological changes in osteoarthritic hip cartilage. Morphometric analysis of 29 osteoarthritic and 26 normal human femoral heads. Clin Orthop Relat Res. 1974(103):269–278. [PubMed] [Google Scholar]
- 2. Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423(6937):332–336. [DOI] [PubMed] [Google Scholar]
- 3. Reppert SM, Weaver DR. Weaver. Coordination of circadian timing in mammals. Nature. 2002;418(6901):935–941. [DOI] [PubMed] [Google Scholar]
- 4. Schibler U, Sassone-Corsi P. A web of circadian pacemakers. Cell. 2002;111(7):919–922. [DOI] [PubMed] [Google Scholar]
- 5. Fu L, Lee CC. The circadian clock: pacemaker and tumour suppressor. Nat Rev Cancer. 2003;3(5):350–361. [DOI] [PubMed] [Google Scholar]
- 6. Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9(10):764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gossan N, Zeef L, Hensman J, et al. The circadian clock in murine chondrocytes regulates genes controlling key aspects of cartilage homeostasis. Arthritis Rheum. 2013;65(9):2334–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maronde E, Schilling AF, Seitz S, et al. The clock genes Period 2 and Cryptochrome 2 differentially balance bone formation. PLoS One. 2010;5(7):e11527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takarada T, Kodama A, Hotta S, et al. Clock genes influence gene expression in growth plate and endochondral ossification in mice. J Biol Chem. 2012;287(43):36081–36095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hashiramoto A, Yamane T, Tsumiyama K, et al. Mammalian clock gene Cryptochrome regulates arthritis via proinflammatory cytokine TNF-α. J Immunol. 2010;184(3):1560–1565. [DOI] [PubMed] [Google Scholar]
- 11. Chang HC, Guarente L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell. 2013;153(7):1448–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Asher G, Gatfield D, Stratmann M, et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134(2):317–328. [DOI] [PubMed] [Google Scholar]
- 13. Nakahata Y, Kaluzova M, Grimaldi B, et al. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134(2):329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Michan S, Sinclair D. Sinclair, Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bäckesjö CM, Li Y, Lindgren U, Haldosén LA. Activation of Sirt1 decreases adipocyte formation during osteoblast differentiation of mesenchymal stem cells. J Bone Miner Res. 2006;21(7):993–1002. [DOI] [PubMed] [Google Scholar]
- 16. Zeng L, Chen R, Liang F, et al. Silent information regulator, Sirtuin 1, and age-related diseases. Geriatr Gerontol Int. 2009;9(1):7–15. [DOI] [PubMed] [Google Scholar]
- 17. Fujita N, Matsushita T, Ishida K, et al. Potential involvement of SIRT1 in the pathogenesis of osteoarthritis through the modulation of chondrocyte gene expressions. J Orthop Res. 2011;29(4):511–515. [DOI] [PubMed] [Google Scholar]
- 18. Matsushita T, Sasaki H, Takayama K, et al. The overexpression of SIRT1 inhibited osteoarthritic gene expression changes induced by interleukin-1β in human chondrocytes. J Orthop Res. 2013;31(4):531–537. [DOI] [PubMed] [Google Scholar]
- 19. Gabay O, Oppenhiemer H, Meir H, Zaal K, Sanchez C, Dvir-Ginzberg M. Increased apoptotic chondrocytes in articular cartilage from adult heterozygous SirT1 mice. Ann Rheum Dis. 2012;71(4):613–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gabay O, Sanchez C, Dvir-Ginzberg M, et al. Sirtuin 1 enzymatic activity is required for cartilage homeostasis in vivo in a mouse model. Arthritis Rheum. 2013;65(1):159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takayama K, Ishida K, Matsushita T, et al. SIRT1 regulation of apoptosis of human chondrocytes. Arthritis Rheum. 2009;60(9):2731–2740. [DOI] [PubMed] [Google Scholar]
- 22. Ishiyama M, Miyazono Y, Sasamoto K, Ohkura Y, Ueno K. A highly water-soluble disulfonated tetrazolium salt as a chromogenic indicator for NADH as well as cell viability. Talanta. 1997;44:1299–1305. [DOI] [PubMed] [Google Scholar]
- 23. Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J. Sirtuins: the 'magnificent seven': function, metabolism and longevity. Ann Med. 2007;39(5):335–345. [DOI] [PubMed] [Google Scholar]
- 24. Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol. 2010;72:517–549. [DOI] [PubMed] [Google Scholar]
- 25. Goldring MB, Birkhead J, Sandell LJ, Kimura T, Krane SM. Interleukin 1 suppresses expression of cartilage-specific types II and IX collagens and increases types I and III collagens in human chondrocytes. J Clin Invest. 1988;82(6):2026–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aigner T, Soder S, Gebhard PM, McAlinden A, Haag J. Mechanisms of disease: role of chondrocytes in the pathogenesis of osteoarthritis-structure, chaos and senescence. Nat Clin Pract Rheumatol. 2007;3:391–399. [DOI] [PubMed] [Google Scholar]
- 27. Honda KK, Kawamoto T, Ueda HR, et al. Different circadian expression of major matrix-related genes in various types of cartilage: modulation by light-dark conditions. J Biol Chem. 2013;154(4):373–381. [DOI] [PubMed] [Google Scholar]
- 28. Guo B, Yang N, Borysiewicz E, et al. Catabolic cytokines disrupt the circadian clock and the expression of clock-controlled genes in cartilage via an NFκB-dependent pathway. Osteoarthritis Cartilage. 2015;23(11):1981–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kc R, Li X, Voigt RM, et al. Environmental disruption of circadian rhythm predisposes mice to osteoarthritis-like changes in knee joint. J Cell Physiol. 2015;230(9):2174–2183. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30. Dudek M, Gossan N, Yang N, et al. The chondrocyte clock gene Bmal1 controls cartilage homeostasis and integrity. J Clin Invest. 2016;126(1):365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kanbe K, Inoue K, Xiang C, Chen Q. Identification of clock as a mechanosensitive gene by large-scale DNA microarray analysis: downregulation in osteoarthritic cartilage. Mod Rheumatol. 2006;16(3):131–136. [DOI] [PubMed] [Google Scholar]
- 32. Kaoru I, Shuichi S, Hisashi S. Diurnal rhythms in the incorporation and secretion of 3H-proline and 3H-galactose by cartilage cells and osteoblasts in various bone-forming sites in growing rats. Orthodontic Waves. 2013;72(1):11–15. [Google Scholar]
- 33. Russell JE, Grazman B, Simmons DJ. Mineralization in rat metaphyseal bone exhibits a circadian stage dependency. Proc Soc Exp Biol Med. 1984;176(4):342–345. [DOI] [PubMed] [Google Scholar]
- 34. Haas S, Straub RH. Disruption of rhythms of molecular clocks in primary synovial fibroblasts of patients with osteoarthritis and rheumatoid arthritis, role of IL-1beta/TNF. Arthritis Res Ther. 2012;14(3):R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khapre RV, Samsa WE, Kondratov RV. Circadian regulation of cell cycle: molecular connections between aging and the circadian clock. Ann Med. 2010;42(6):404–415. [DOI] [PubMed] [Google Scholar]
- 36. Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 2006;20(14):1868–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Loeser R. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the extracellular cartilage matrix. Osteoarthritis Cartilage. 2009;17:971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Preitner N, Damiola F, Lopez-Molina L, et al. The orphan nuclear receptor REV-ERBα controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251–260. [DOI] [PubMed] [Google Scholar]
- 39. Sato TK, Panda S, Miraglia LJ, Hogenesch JB, et al. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron. 2004;43:527–537. [DOI] [PubMed] [Google Scholar]
- 40. Gossan N, Boot-Handford R, Meng QJ. Ageing and osteoarthritis: circadian rhythm connection. Biogerontology. 2015;16(2):209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ueda HR, Hayashi S, Chen W, et al. System-level identification of transcriptional circuits underlying mammalian circadian clocks. Nat Genet. 2005;37:187–192. [DOI] [PubMed] [Google Scholar]
- 42. Kong S, McBurney MW, Fang D. Sirtuin 1 in immune regulation and autoimmunity. Immunol Cell Biol. 2012;90:6–13. [DOI] [PubMed] [Google Scholar]
- 43. Chang HC, Guarente L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell. 2013;153(7):1448–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]