

Abstract
Calpain is a family of calcium-dependent nonlysosomal neutral cysteine endopeptidases. Akt is a serine/threonine kinase that belongs to AGC kinases and plays important roles in cell survival, growth, proliferation, angiogenesis, and cell metabolism. Both calpain and Akt are the downstream signaling molecules of platelet-derived growth factor (PDGF) and mediate PDGF-induced collagen synthesis and proliferation of pulmonary artery smooth muscle cells (PASMCs) in pulmonary vascular remodeling. We found that inhibitions of calpain-2 by using calpain inhibitor MDL28170 and calpain-2 small interfering RNA attenuated Akt phosphorylations at serine-473 (S473) and threonine-308 (T308), as well as collagen synthesis and cell proliferation of PASMCs induced by PDGF. Overexpression of calpain-2 in PASMCs induced dramatic increases in Akt phosphorylations at S473 and T308. Moreover, knockout of calpain attenuated Akt phosphorylations at S473 and T308 in smooth muscle of pulmonary arterioles of mice with chronic hypoxic pulmonary hypertension. The cell-permeable-specific transforming growth factor (TGF)-β receptor inhibitor SB431542 attenuated Akt phosphorylations at both S473 and T308 induced by PDGF and by overexpressed calpain-2 in PASMCs. Furthermore, SB-431452 and knocking down activin receptor-like kinase-5 significantly reduced PDGF-induced collagen synthesis and cell proliferation of PASMCs. Nevertheless, neutralizing extracellular TGF-β1 using a cell-impermeable TGF-β1 neutralizing antibody did not affect PDGF-induced Akt phosphorylations at S473 and T308. Furthermore, inhibition of mammalian target of rapamycin complex 2 (mTORC2) by knocking down its component protein Rictor prevented Akt phosphorylations at S473 and T308 induced by PDGF and by overexpressed calpain-2. These data provide first evidence supporting that calpain-2 upregulates PDGF-induced Akt phosphorylation in pulmonary vascular remodeling via an intracrine TGF-β1/mTORC2 mechanism.
Keywords: PDGF, pulmonary hypertension, vascular smooth muscle cells, Rictor
pulmonary vascular remodeling is a key feature of pulmonary arterial hypertension (PAH) associated with accumulation of extracellular matrix, including collagen, and vascular smooth muscle cell proliferation and hypertrophy. These processes contribute to medial hypertrophy and muscularization, leading to obliteration of precapillary pulmonary arteries and sustained elevation of pulmonary arterial pressure (31, 32, 39). Several growth factors, including platelet-derived growth factor (PDGF) and transforming growth factor (TGF)-β1, participate in the process of pulmonary vascular remodeling in PAH patients and animal models (3, 11, 18, 27, 32). For example, PDGF and its receptor is upregulated in pulmonary arteries of patients with PAH (34, 35) and rodents exposed to chronic hypoxia and monocrotaline (MCT) (3, 28, 41). PDGF receptor antagonist prevents or reverses increased right ventricular pressure and pulmonary vascular changes induced by hypoxia, MCT, and hypoxia/SU5416 (8, 34). The TGF-β1/Smad pathway is a classical pathway regulating collagen synthesis. It has been reported that the TGF-β1/Smad pathway is activated in PAH animal models (3, 27, 28) and patients with PAH (38). Inhibition of TGF-β signaling attenuates pulmonary vascular remodeling and elevated right ventricular pressure in animal models (6, 27, 40). Despite these overwhelming data, approaches for intervention of these growth factors are limited, because the downstream signaling pathways of these growth factor receptors have not been clarified.
Calpain is a family of calcium-dependent nonlysosomal neutral cysteine endopeptidases (14). Calpain-1 and calpain-2 are two major typical calpains, which consist of a distinct large catalytic subunit (∼80 kDa) and a common small subunit (∼30 kDa, calpain-4) that helps regulate activity. Calpastatin functions as the major specific endogenous inhibitor for calpain-1 and calpain-2 (14). The mechanism for calpain activation involves calcium, phospholipid binding, autolysis, release of calpain from its inhibitor calpastatin, binding of activator proteins, and phosphorylation (12, 13). Binding of phospholipids may decrease the Ca2+ requirement for calpain-2 activation (4). Our laboratory has reported that calpain is a downstream signal molecule for PDGF and mediates PDGF-induced collagen synthesis and proliferation of pulmonary artery smooth muscle cells (PASMCs) (28). Inhibition of calpain by conditional knockout of calpain-4 and by using calpain inhibitor prevents pulmonary vascular remodeling in PAH animal models (28).
Akt is a serine/threonine kinase that belongs to AGC kinases. Akt plays important roles in cell survival, growth, proliferation, angiogenesis, and cell metabolism (1). It works as a downstream signaling molecule for several growth factors, including PDGF (10, 26, 30). Phosphorylation of Akt at serine-473 (S473) or threonine-308 (T308) causes activation of Akt (2). T308 residue is phosphorylated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) due to its association with phosphatidylinositol 3,4,5-trisphosphate, while mammalian target of rapamycin complex 2 (mTORC2) is responsible for the phosphorylation of S473 residue (15, 29). Akt plays an essential role in pulmonary vascular remodeling and the development of pulmonary hypertension (36). mTORC2 and phosphorylated (P)-Akt-S473 are increased in PASMCs of human lung with PAH and in PASMCs exposed to hypoxia and PDGF (15, 23). Knockout of Akt1 and mTOR attenuates pulmonary vascular remodeling of hypoxic PAH (36). Inhibition of mTOC using rapamycin prevents PAH in MCT rat model (20). Moreover, activations of Akt and mTOR are required for hypoxia-induced cell proliferation of vascular endothelial cells and adventitial fibroblasts of pulmonary arteries (9, 26). Nevertheless, the detail mechanism for the phosphorylation and activation of Akt in pulmonary vascular remodeling of PAH remains unclear.
Because both calpain and Akt work at the downstream signaling of PDGF, we hypothesize that there might be a connection between calpain activation and Akt in the collagen synthesis and cell proliferation of PASMCs caused by PDGF. In the present study, we demonstrate that inhibition of calpain-2 causes a reduction of PDGF-induced Akt phosphorylation, and that overexpression of calpain-2 induces an increase in Akt phosphorylation. We found, for the first time, that calpain-2 upregulates Akt phosphorylation via an intracrine TGF-β1/mTORC2 mechanism.
MATERIALS AND METHODS
Materials.
Human PASMCs and culture medium were obtained from Lonza (Walkersville, MD). MDL28170 was obtained from Calbiochem (San Diego, CA). SB431542 was from Tocris (Ellisville, MO). Anti-GAPDH, anti-P-Akt-S473, anti-P-Akt-T308, anti-total Akt, anti-GAPDH antibodies, Akt small interfering RNA (siRNA), LY294002, and bromodeoxyuridine (BrdU) cell proliferation kit were purchased from Cell Signaling Technology (Beverly, MA). Antibody against collagen I was from Novus Biologicals (Littleton, CO). Anti-calpain-1 and -2 antibodies were purchased from Triple Point Biologics (Forest Grove, OR). Anti-Rictor antibody was from Bethyl Laboratories (Montgomery, TX). Anti-activin receptor-like kinase (ALK)-5 antibody and ALK-5 siRNA were from Santa Cruz Biotechnology (Dallas, TX). PDGF-BB and all other regents were obtained from Sigma-Aldrich (St. Louis, MO).
Cell culture.
Human PASMCs were cultured according to the manufacturer's instructions. Before all experimentation, the third- to seventh-passage cells were equilibrated in growth factor-free medium for 24 h.
SDS-PAGE and Western blot analysis.
Proteins (12–25 μg) were resolved in 7.5% or 4–15% criterion SDS-polyacrylamide gel electrophoresis (Bio-Rad). Proteins were then transferred to nitrocellulose membrane using the criterion transfer system. P-Akt-S473, P-Akt-T308, total Akt, collagen I, ALK-5, and Rictor were detected by immunoblotting the membrane using primary antibodies against calpain-1 (1:5,000), calpain-2 (1:5,000), collagen I (1:1,000), P-Akt-S473 (1:2,000), P-Akt-T308 (1:2,000), total Akt (1:1,000), ALK-5 (1:1,000), Rictor (1:1,000), and GAPDH (1:1,000) overnight in Tris-buffered saline with Tween 20 with 1% BSA. Bound antibodies were detected using a goat anti-rabbit (or mouse) IgG-AP conjugate (Bio-Rad) at 1:5,000 dilution using Tris-buffered saline with Tween 20 with 1% BSA. Proteins were detected using an Immun-Star AP chemiluminescence kit (Bio-Rad). Densitometric analysis of the protein bands were performed using Quantity One 1-D Analysis Software.
RNA interference in PASMCs.
The expressions of calpain-1, calpain-2, Akt, ALK-5, and Rictor were silenced using siRNA technology. Calpain-1 siRNA (target sequence: 5′-AAGCTAGTGTTCGTGCACTCT-3′), calpain-2 siRNA (target sequence: 5′-CTGGAACACTATAGACCCAGA-3′), and the nontargeting siRNA (target sequence: 5′- AACGTACGCGGAATACTTCGA-3′) were synthesized by Qiagen. The Akt siRNA (no. 6211) is from Cell Signaling Technology. ALK-5 siRNA (no. SC40222) is from Santa Cruz Biotechnology. Rictor siRNA is a mix of two Rictor siRNAs (no. 184233638 and no. 184233637) from Qiagen. The sequences of siRNA were not disclosed by the companies. The siRNAs were transfected into PASMCs with siPORT amine transfection reagent, according to the manufacturer's instruction. Three days after transfection, the medium was changed to serum-free medium for 24 h, followed by the treatments of PASMCs.
Overexpression of calpain-2 in PASMCs.
Total RNA was isolated from PASMCs with RNeasy RNA Isolation Kit (Qiagen, Valencia, CA), and the mRNA was reverse-transcribed with High-Capacity cDNA Reverse Transcription Kit (Life Technologies, Grand Island, NY). The cDNA was amplified by PCR using Expand High Fidelity PCR System (Roche Applied Science, Indianapolis, IN), as well as calpain-2 forward 5′-AAAAAAAGCTTGGATGGCGGGCATCGCGGC-3′ and calpain-2 reverse 5′-AAAAATCTAGAAAGTACTGAGAAACAGAGCCAAGAGATAAGGTCG-3′ primers (Integrated DNA Technologies, Coralville, IA) containing HindIII and XbaI restrictions sites, respectively. Primers were designed according to the homo sapiens calpain-2 sequence (National Center for Biotechnology Information database accession number: NM_001748.4). Then the cDNA was cloned into a pcDNA3.1/V5-His (Life Technologies, Grand Island, NY) mammalian expression vectors to create the pcDNA3.1/V5-His-human calpain-2 wild-type plasmid. The vector construct was confirmed by DNA sequencing. PASMCs were transfected with pcDNA3.1/V5-His-human calpain-2 wild-type construct using X-tremeGENE HP transfection reagent (Roche Applied Science, Indianapolis, IN), according to the manufacturer's protocol. Forty-eight hours after the transfection, the medium was changed to serum-free medium for 24 h, followed by the treatments of cells.
Calpain activity assay.
Calpain activity in PASMCs was measured with a SpectraMax M2e microplate reader (Molecular Devices, Sunnyvale, CA), as described previously (28). Briefly, after the treatments, the cells were washed with PBS, and the fluorogenic Suc-Leu-Leu-Val-Tyr-AMC peptide substrate was added to a final concentration of 80 μM in the presence and absence of MDL28170. Fluorescence was immediately recorded at 2-min intervals for 20 min. Calpain activity was calculated as fluorescence units in the absence of MDL28170, subtracted by those in the presence of MDL28170.
Cell proliferation assay.
Cells were cultured in 96-well plates for 24 h and serum-starved for 12 h. A colorimetric BrdU ELISA kit was used. After the treatments, BrdU incorporation was detected by using anti-BrdU peroxidase conjugate antibody, according to the manufacturer's instructions. Absorbance at 450 nm was measured by using a Spectra-Max M2e microplate reader (Molecular Devices, Sunnyvale, CA).
Determination of Akt phosphorylation in pulmonary arteries from calpain knockout mice and control mice with chronic hypoxic PAH.
To investigate the role of calpain in Akt phosphorylation in pulmonary vascular remodeling in vivo, calpain activity was inhibited in a mouse line of inducible global knock out of calpain-4 (ER-Cre+/−Capn4flox/flox), as described previously (28). The animal experiments were performed in accordance with the guiding principles of the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Augusta University. Because calpain-4 is required for the activity of both calpain-1 and -2, deletion of calpain-4 prevents activation of calpain-1 and -2. Deletion of calpain-4 was induced by administration of tamoxifen (20 mg·kg−1·day−1 ip) for 5 days. Control mice were littermate Capn4flox/flox or Capn4flox/+ mice treated with the same tamoxifen regimen. Mice between 12 and 16 wk old were used for the hypoxic PAH model, as described previously (28). To determine the extent of phosphorylated Akt in pulmonary arterioles, double immunostaining of P-Akt/α-actin was performed on lung tissue slides. The slides were incubated first with a rabbit polyclonal antibody against P-Akt-S473, P-Akt-T308, or total Akt and mouse monoclonal antibody against α-actin overnight and then with goat anti-rabbit IgG Alexa Fuor 488 and goat anti-mouse IgG Alexa Fuor 594. The slides were sealed with mounting solution containing anti-fade reagent and were examined using a Zeiss LSM 510 laser scanning confocal microscope. The fluorescence intensities in the vascular smooth muscle layer of pulmonary arterioles were measured using ImageJ.
Statistical analysis.
Within each experiment, cells were matched for number of passages to avoid differences related to tissue culture variables. Results are shown as means ± SE for n experiments. One-way ANOVA and t-test analysis (2-tailed) were used to determine the significance of differences between the means of experimental and control groups. A value of P < 0.05 was considered significant.
RESULTS
Inhibition of Akt attenuates PDGF-induced collagen synthesis and proliferation of PASMCs.
Incubation of PASMCs with PDGF-BB (10 ng/ml) for 0.5–24 h induces a rapid increase in Akt activation and phosphorylations at S473 and T308 (Fig. 1A). The increases in P-Akt-S473 and P-Akt-T308 were at the highest levels at 0.5- and 1-h incubation and last for at least 24 h. To investigate the role of Akt activation in PDGF-induced collagen synthesis and cell proliferation of PASMCs, Akt was inhibited by the specific Akt inhibitor triciribine and by using Akt siRNA gene silencing. As shown in Fig. 1, B–G, incubation of PASMCs with PDGF-BB (10 ng/ml) for 24 h caused increases in collagen I protein level and cell proliferation. Triciribine (10 μM) and Akt knockdown significantly attenuated PDGF-induced increases in collagen I protein level and cell proliferation. These data confirm that Akt plays an important role in PDGF-induced collagen synthesis and cell proliferation of PASMCs.
Fig. 1.

Inhibition of Akt attenuates PDGF-induced collagen synthesis and proliferation of PASMCs. A: PASMCs with PDGF-BB (10 ng/ml) for 0.5–24 h after which P-Akt-S473, P-Akt-T308, and total Akt were measured by Western blot. The image shown is representative of immunoblots from 4 experiments. B–D: PASMCs were incubated with PDGF-BB (10 ng/ml) in the absence and presence of triciribine (10 μM) for 24 h, after which collagen I protein level and cell proliferation were determined. E–G: PASMCs were transfected with Akt siRNA or control siRNA and then incubated with PDGF-BB (10 ng/ml) for 24 h, after which collagen I protein level and cell proliferation were determined. B and E: representative immunoblots of collagen I from 4 experiments. C and F: bar graphs showing the changes in protein levels of collagen I quantified by scanning densitometry. D and G: bar graphs showing the changes in cell proliferation. Values are means ± SE; n = 4. *P < 0.05 vs. control. **P < 0.05 vs. PDGF only. #P < 0.05 vs. control siRNA + PDGF.
Inhibition of calpain-2 prevents PDGF-induced collagen synthesis and proliferation of PASMCs.
We have previously shown that PDGF causes calpain activation, and that calpain-2 rather than calpain-1 is responsible for increases in calpain activities induced by PDGF in PASMCs (22). Here we showed that pan calpain inhibitor MDL28170 (20 μM) prevented PDGF-induced collagen synthesis and cell proliferation of PASMCs (Fig. 2, A–C). To investigate whether calpain-mediated collagen synthesis and cell proliferation are caused by activation calpain-1 or calpain-2, calpain-1 and calpain-2 were specifically knocked down using their siRNA. As shown in Fig. 2, D–F, knockdown of calpain-2, but not calpain-1, prevented PDGF-induced collagen synthesis and cell proliferation of PASMCs. These data indicate that calpain-2 rather than calpain-1 is responsible for increases in collagen synthesis and cell proliferation induced by PDGF.
Fig. 2.

Inhibition of calpain-2 prevents PDGF-induced collagen synthesis and proliferation of PASMCs. A–C: PASMCs were incubated with PDGF-BB (10 ng/ml) in the absence and presence of MDL28170 (MDL; 20 μM) for 24 h, after which collagen I and cell proliferation were measured. D–F: PASMCs were transfected with calpain-1 or calpain-2 siRNA or control siRNA and then incubated with PDGF-BB (10 ng/ml) for 24 h, after which collagen I and cell proliferation were measured. A and D: representative immunoblots of collagen I. B and D: bar graphs showing the changes in protein levels of collagen I. C and F: bar graphs showing the changes in cell proliferation. Values are means ± SE; n = 4. *P < 0.05 vs. control. #P < 0.05 vs. PDGF only. ##P < 0.05 vs. control siRNA + PDGF.
Inhibition of calpain attenuates Akt phosphorylation in PASMCs.
As shown in Fig. 3, A and B, incubation of PASMCs with calpain inhibitor MDL28170 prevented PDGF-induced increases in the levels of both P-Akt-S473 and P-Akt-T308. The reduction of PDGF-induced increase in P-Akt-S473 was much greater than those in P-Akt-T308 in MDL28170-treated PASMCs, suggesting that calpain may primarily impact Akt phosphorylation at S473. Consistent with the importance of calpain-2 in PDGF-induced collagen synthesis and cell proliferation, knockdown of calpain-2, but not calpain-1, prevented PDGF-induced increases in the levels of P-Akt-S473 and P-Akt-T308 (Fig. 3, C and D). These results provide first evidence that it is calpain-2 rather than calpain-1 that leads to Akt activation and phosphorylation induced by PDGF in PASMCs.
Fig. 3.

Inhibition of calpain attenuates Akt phosphorylation. A and B: PASMCs were incubated with PDGF-BB (10 ng/ml) in the absence and presence of MDL28170 (20 μM) for 30 min, after which P-Akt-S473, P-Akt-T308, and total Akt were measured by Western blot. C and D: PASMCs were transfected with calpain-1 or calpain-2 siRNA or control siRNA and then incubated with PDGF-BB (10 ng/ml) for 30 min, after which P-Akt-S473, P-Akt-T308, and total Akt were measured. A and C: representative immunoblots of P-Akt-S473, P-Akt-T308, and total Akt. B and D: bar graphs showing the changes in protein levels of P-Akt-S473 and P-Akt-T308. Values are means ± SE; n = 4. *P < 0.05 vs. control. **P < 0.05 vs. PDGF only. #P < 0.05 vs. control siRNA. ##P < 0.05 vs. control siRNA + PDGF.
Overexpression of calpain-2 increases the levels of P-Akt-S473 and P-Akt-T308.
To further investigate the role of calpain-2 in Akt phosphorylation, calpain-2 was overexpressed in PASMCs by using a plasmid containing human calpain-2 gene. As shown in Fig. 4, the levels of P-Akt-S473 and P-Akt-T308 were much higher in PASMCs transfected with plasmids containing calpain-2 gene than those with empty plasmids. The increase in phosphorylation at S473 is much higher than those at T308. These data further support that calpain-2 signaling leads to Akt phosphorylation in PASMCs.
Fig. 4.
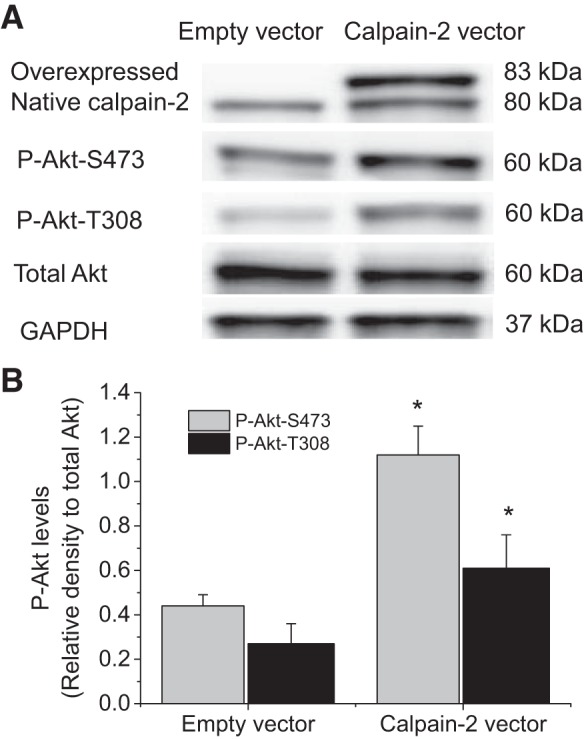
Overexpression of calpain-2 increases the levels of P-Akt-S473 and P-Akt-T308. PASMCs were transfected with plasmids containing human calpain-2 gene or empty plasmids for 24 h after which P-Akt-S473, P-Akt-T308, and total Akt were measured by Western blot. A: representative immunoblots of P-Akt-S473, P-Akt-T308, and total Akt. B: bar graph showing the changes in protein levels of P-Akt-S473 and P-Akt-T308. Values are means ± SE; n = 4. *P < 0.05 vs. empty vector.
Calpain knockout inhibits Akt phosphorylation in smooth muscle of pulmonary arteries in mice with chronic hypoxic PAH.
We have reported that knockout of calpain-4 (small unit) attenuates hypoxic pulmonary hypertension and vascular remodeling (22, 28). To investigate the role of calpain in Akt phosphorylation in pulmonary vascular remodeling in vivo, we determined the levels of P-Akt-S473 and P-Akt-T308 in smooth muscle of pulmonary arteries from calpain knockout mice and control mice with chronic hypoxic PAH. As shown in Fig. 5, the levels of P-Akt-S473 and P-Akt-T308 in smooth muscle of pulmonary arteries were much higher in control mice exposed to hypoxia than those exposed to normoxia. However, the levels of P-Akt-S473 and P-Akt-T308 in smooth muscle of pulmonary arteries were comparable between calpain knockout mice exposed to normoxia and those exposed to hypoxia. These results indicate that inhibition of calpain by conditional knockout of calpain-4 inhibits Akt phosphorylation in hypoxia-induced pulmonary vascular remodeling in vivo.
Fig. 5.

Calpain knockout (KO) inhibits Akt phosphorylation in smooth muscle of pulmonary arteries in mice with chronic hypoxic PAH. Five days after regimen of tamoxifen administration, control mice and ER-Cre+/−Capn4flox/flox mutant mice were exposed to room air (normoxia) or 10% oxygen (hypoxia) for 3 wk. Lung slides from ER-Cre+/−Capn4flox/flox mutant and control mice exposed to normoxia or hypoxia were double-stained for α-actin (red) and P-Akt-S473, P-Akt-T308, or total Akt (green). A: representative images of 8 independent experiments. B: bar graph depicting the changes of P-Akt-S473 and P-Akt-T308 in smooth muscle of pulmonary arteries. Values are means ± SE; n = 8. *P < 0.05 vs. normoxia in control mice. #P < 0.05 vs. chronic hypoxia in control mice.
Inhibition of TGF-β receptor prevents calpain-2-induced increases in the levels of P-Akt-S473 and P-Akt-T308.
We have previously reported that calpain cleaves and activates latent TGF-β in vitro and in PASMCs (28). To investigate whether calpain-2 increases the levels of P-Akt-S473 and P-Akt-T308 through TGF-β pathway, PASMCs with and without calpain-2 overexpression were incubated with and without specific TGF-β receptor blocker SB431542 (10 μM). As shown in Fig. 6, SB431542 attenuated the increases in P-Akt-S473 level and to a less extent in P-Akt-T308 level in calpain-2-overexpressed PASMCs. These results indicate that calpain-2 increases the levels of P-Akt-S473 and P-Akt-T308 through TGF-β pathway.
Fig. 6.
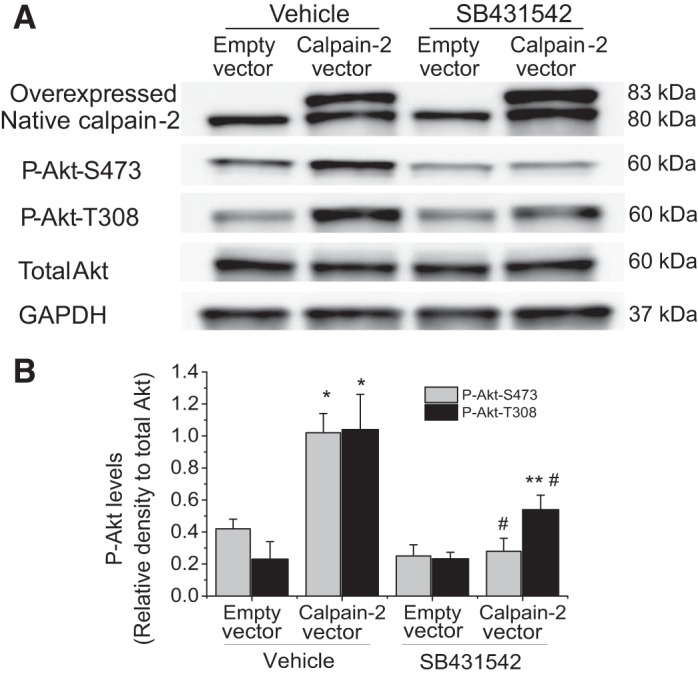
Inhibition of TGF-β receptor prevents calpain-2-induced increases in the levels of P-Akt-S473 and P-Akt-T308. PASMCs were transfected with plasmids containing human calpain-2 gene or empty plasmids and then incubated with and without SB431542 (10 μM) for 24 h, after which P-Akt-S473, P-Akt-T308, and total Akt were measured by Western blot. A: representative immunoblots of P-Akt-S473, P-Akt-T308, and total Akt. B: bar graph showing the changes in protein levels of P-Akt-S473 and P-Akt-T308. Values are means ± SE; n = 5. *P < 0.05 vs. empty vector in vehicle. **P < 0.05 vs. empty vector in SB431542. #P < 0.05 vs. calpain-2 vector in vehicle.
Inhibition of TGF-β receptor, but not neutralizing extracellular TGF-β, reduces PDGF-induced Akt phosphorylation, collagen synthesis, and cell proliferation.
We have previously reported that PDGF does not activate extracellular TGF-β, and that calpain activates an intracrine TGF-β pathway induced by PDGF in PASMCs (28). To examine whether this intracrine TGF-β pathway is involved in the calpain-mediated Akt phosphorylation, we first assessed the effect of a cell-impermeable TGF-β neutralizing antibody (2 μg/ml) on Akt phosphorylation. This antibody has been previously shown to be able to block the effect of extracellular TGF-β1 on PASMCs (28). We found that neutralizing extracellular TGF-β did not affect PDGF-induced increases in P-Akt-S473 and P-Akt-T308 (Fig. 7). Nevertheless, the cell-permeable SB-431452 (10 μM) significantly reduced PDGF-induced increases in P-Akt-S473, P-Akt-T308, collagen I, and cell proliferation of PASMCs (Fig. 8). Moreover, knock down of ALK-5 prevented PDGF-induced increases in collagen I protein level and cell proliferation of PASMCs (Fig. 9). These results indicate that the intracrine TGF-β pathway is involved in the calpain-mediated Akt phosphorylation in PASMCs.
Fig. 7.
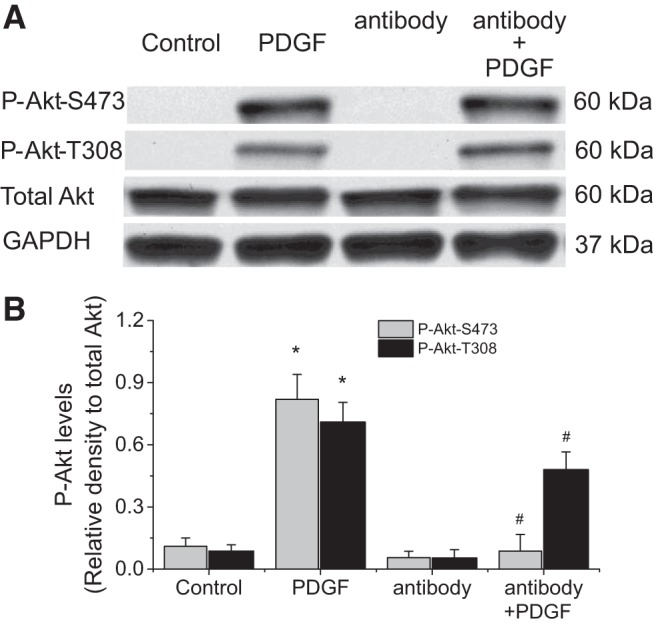
TGF-β neutralizing antibody does not affect PDGF-induced Akt phosphorylation. PASMCs were incubated with PDGF-BB (10 ng/ml) in the absence and presence of a cell-impermeable TGF-β neutralizing antibody (2 μg/ml) for 30 min, after which protein levels of P-Akt-S473, P-Akt-T308, and total Akt collagen I were measured. A: representative immunoblots from 4 experiments. B: bar graph showing the changes in protein levels of P-Akt-S473 and P-Akt-T308 quantified by scanning densitometry. Values are means ± SE; n = 4. *P < 0.05 vs. control. #P < 0.05 vs. antibody only.
Fig. 8.

TGF-β receptor inhibitor SB431542 reduces PDGF-induced Akt phosphorylation, collagen synthesis, and cell proliferation. PASMCs were incubated with PDGF-BB (10 ng/ml) in the absence and presence of SB431542 (10 μM) for 0.5–24 h, after which protein levels of P-Akt-S473, P-Akt-T308, total Akt, collagen I, and cell proliferation were measured. A: representative immunoblots from 4 experiments. B and C: bar graph showing the changes in protein levels of P-Akt-S473, P-Akt-T308, and collagen I quantified by scanning densitometry. D: bar graph showing the changes in cell proliferation. Values are means ± SE; n = 4. *P < 0.05 vs. control. #P < 0.05 vs. PDGF.
Fig. 9.

Knocking down ALK-5 attenuates PDGF-induced collagen synthesis and cell proliferation. PASMCs were transfected with ALK-5 siRNA or control siRNA and then incubated with PDGF-BB (10 ng/ml) for 24 h, after which collagen I protein level and cell proliferation were determined. A: representative immunoblots of collagen I from 4 experiments. B: bar graph showing the changes in protein levels of collagen I quantified by scanning densitometry. C: bar graph showing the changes in cell proliferation. Values are means ± SE; n = 4. *P < 0.05 vs. control. #P < 0.05 vs. control siRNA + PDGF.
Knock down of Rictor attenuates PDGF- and calpain-2-induced Akt phosphorylation in PASMCs.
More recent studies revealed that the phosphorylation of Akt at S473 is primarily mediated by mTORC2 (15, 29). To investigate whether mTORC2 contributes PDGF- and calpain-2-induced Akt phosphorylation, mTORC2 was inhibited by knocking down Rictor protein in this complex. As shown in Fig. 10, knock down of Rictor attenuates PDGF-induced Akt phosphorylation at S473 and to a less extent at T308 in PASMCs. Moreover, knock down of Rictor prevented Akt phosphorylations at S473 and T308 in calpain-2-overexpressed PASMCs (Fig. 11). These data suggest that mTORC2 contributes to calpain-2-mediated Akt phosphorylation induced by PDGF.
Fig. 10.
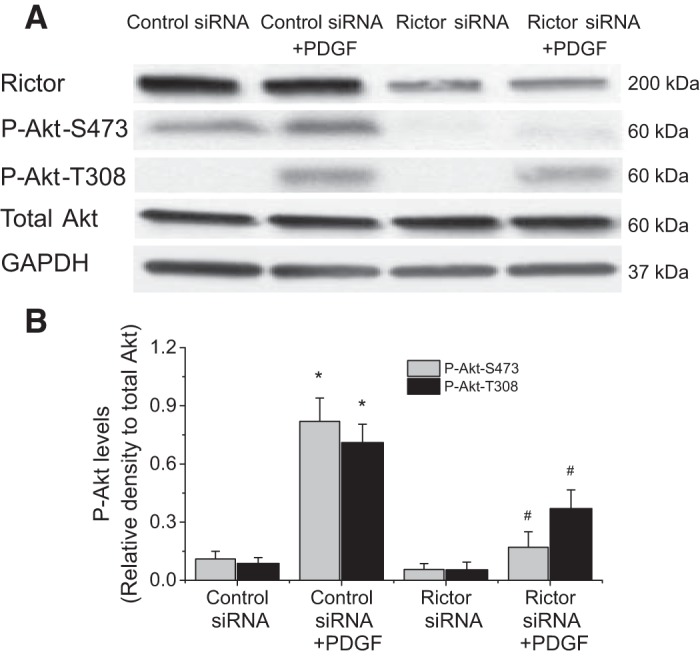
Knocking down Rictor attenuates PDGF-induced Akt phosphorylation in PASMCs. PASMCs were transfected with Rictor siRNA or control siRNA and then incubated with PDGF-BB (10 ng/ml) for 30 min, after which protein levels of P-Akt-S473, P-Akt-T308, and total Akt were measured. A: representative immunoblots from 4 experiments. B: bar graph showing the changes in protein levels of P-Akt-S473 and P-Akt-T308. Values are means ± SE; n = 4. *P < 0.05 vs. control siRNA. #P < 0.05 vs. control siRNA + PDGF.
Fig. 11.
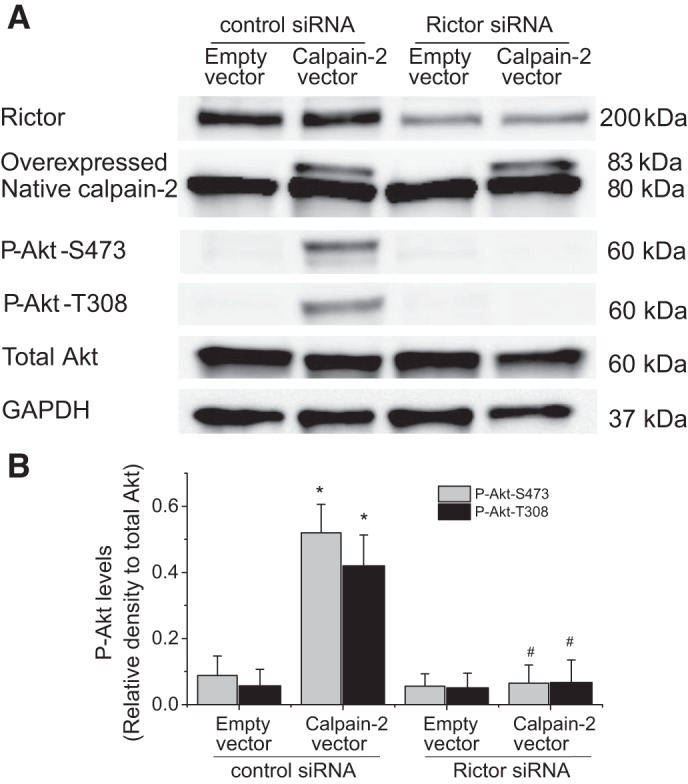
Knocking down Rictor attenuates calpain-2-induced Akt phosphorylation in PASMCs. PASMCs were transfected with Rictor siRNA or control siRNA and then transfected plasmids containing human calpain-2 gene or empty plasmids for 24 h, after which protein levels of P-Akt-S473, P-Akt-T308, and total Akt were measured. A: representative immunoblots from 3 experiments. B: bar graph showing the changes in protein levels of P-Akt-S473 and P-Akt-T308. Values are means ± SE; n = 4. *P < 0.05 vs. empty vector in control siRNA. #P < 0.05 vs. calpain-2 vector in control siRNA.
Phosphatidylinositol 3-kinase inhibitor LY294002 does not affect PDGF-induced calpain activation and calpain-2-induced Akt phosphorylation.
To investigate whether phosphatidylinositol 3 (PI3)-kinase is involved in PDGF-induced calpain activation and calpain-mediated Akt phosphorylation, PI3-kinase was inhibited by using LY294002. As shown in Fig. 12A, the PI3-kinase inhibitor LY294002 did not affect PDGF-induced increase in calpain activity. Similarly, LY294002 did not inhibit calpain-2-induced Akt phosphorylations at S473 and T308 (Fig. 12, B and C). These results suggest that PI3-kinase is not involved in PDGF-induced calpain activation and calpain-mediated Akt phosphorylation.
Fig. 12.
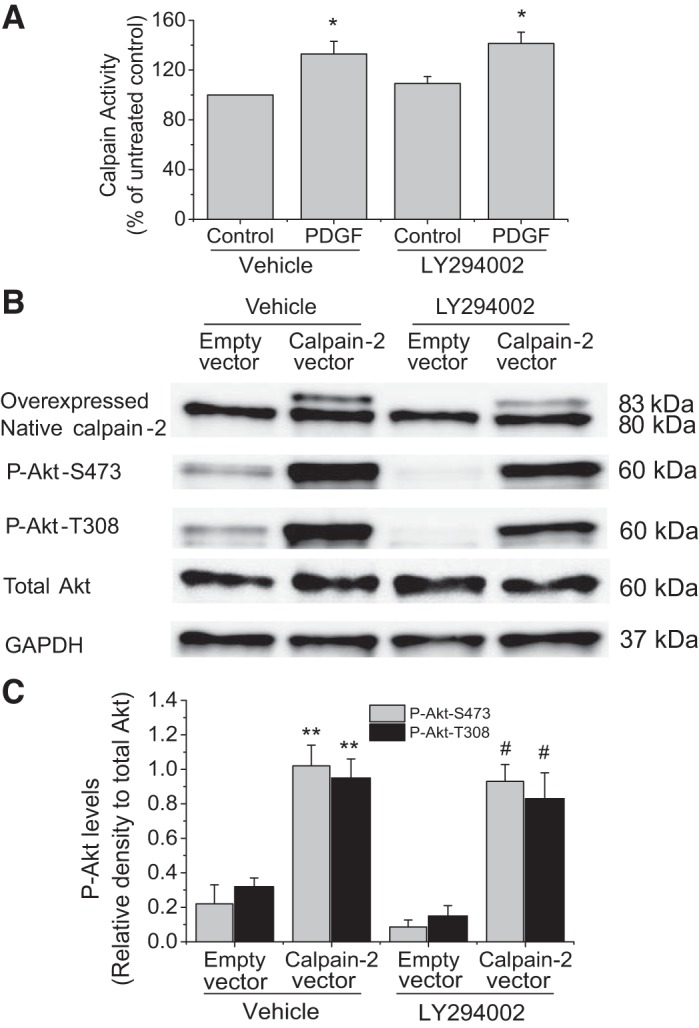
PI3-kinase inhibitor LY294002 does not affect PDGF-induced calpain activation and calpain-2-induced Akt phosphorylation. A: PASMCs were incubated with PDGF-BB (10 ng/ml) in the presence and absence of LY294002 (50 μM) for 30 min, after which calpain activity was measured, as described in materials and methods. B and C: PASMCs were transfected with plasmids containing human calpain-2 gene or empty plasmids and then incubated with and without LY294002 (50 μM) for 24 h, after which P-Akt-S473, P-Akt-T308, and total Akt were measured by Western blot. B: representative immunoblots of P-Akt-S473, P-Akt-T308, and total Akt. C: bar graph showing the changes in protein levels of P-Akt-S473 and P-Akt-T308. Values are means ± SE; n = 3. *P < 0.05 vs. control in vehicle. **P < 0.05 vs. empty vector in vehicle. #P < 0.05 vs. empty vector in LY294002.
DISCUSSION
Akt is the downstream signaling molecule of PDGF and mediates PDGF-induced collagen synthesis and proliferation of PASMCs, the most critical process in the medial layer thickening in pulmonary vascular remodeling (15, 30). Knocking out Akt1 attenuates the development and progression of pulmonary vascular remodeling and pulmonary hypertension (36). The activation of Akt depends on the phosphorylation of the most important residues at T308 or S473. T308 residue is phosphorylated by PI3-kinase activated PDK1 due to its association with phosphatidylinositol 3,4,5-trisphosphate, while mTORC2 is responsible for the phosphorylation of S473 residue (29). PDGF induces phosphorylation of both T308 and S473 in PASMCs. Akt phosphorylation is increased in PASMCs of human lung with PAH and in PASMCs exposed to hypoxia and PDGF (15, 23). Moreover, activation of Akt is required for hypoxia-induced cell proliferation of vascular endothelial cells and adventitial fibroblasts of pulmonary arteries (9, 26). In the present study, inhibition of Akt significantly attenuates PDGF-induced increases in collagen I protein level and cell proliferation, confirming that Akt plays an important role in PDGF-induced collagen synthesis and cell proliferation of PASMCs in pulmonary vascular remodeling (30, 36).
Our laboratory has previously reported that calpain is a downstream signal molecule for PDGF (28). PDGF-induced calpain activation is through ERK-mediated calpain phosphorylation (22). We have shown that calpain mediates PDGF-induced collagen synthesis and proliferation of PASMCs (28). Inhibition of calpain by conditional knockout of calpain and by using calpain inhibitor prevents pulmonary vascular remodeling in PAH animal models (22, 28). Our present work here identified a novel link between calpain and Akt phosphorylation in PASMCs. Inhibition of calpain prevents PDGF-induced Akt phosphorylation at S473 and partially blocks PDGF-induced Akt phosphorylation at T308. By separately knocking down calpain-1 and calpain-2 using siRNA technology, we found that it is calpain-2 rather than calpain-1 that leads to phosphorylation and activation of Akt, collagen synthesis, and cell proliferation of PASMCs induced by PDGF. This is consistent with our previous finding that calpain-2 rather than calpain-1 is responsible for increases in calpain activity induced by PDGF (22). Interestingly, Ho et al. (19) have also shown that mouse mammary carcinoma AC2M2 cells deficient in calpain-2 have lower levels of Akt phosphorylation at S473. We have shown that overexpression of calpain-2 in PASMCs induces dramatic increases in Akt phosphorylations at S473 and T308. Thus our work provides solid evidence, for the first time, showing that calpain-2 upregulates Akt phosphorylation.
We have reported that calpain activation and calpain phosphorylation at S50 are much higher in smooth muscle of pulmonary arteries in rodents of PAH models and patients with PAH (22, 28). Consistently, in the present study, Akt phosphorylations at S473 and T308 are also increased in smooth muscle of pulmonary arteries in mice with hypoxic PAH. Interestingly, inhibition of calpain activity by using knockout of calpain-4 dramatically reduces Akt phosphorylations at S473 and T308 in smooth muscle of pulmonary arteries in mice with hypoxic PAH. These findings provide further evidence supporting that calpain-2 upregulates Akt phosphorylation in pulmonary vascular remodeling of PAH.
PDGF activates PI3-kinase, leading to the interaction of Akt and PDK1 that causes Akt phosphorylation (1). Inhibition of PI3-kinase using LY294002 attenuates PDGF-induced Akt phosphorylation and activation (7, 26). We found here that inhibition of PI3-kinase using LY294002 did not affect PDGF-induced calpain activation. Similarly, LY294002 did not inhibit calpain-2-induced Akt phosphorylation. Therefore, the calpain-mediated Akt phosphorylation is independent of PI3-kinase. On the other hand, PDGF-induced collagen synthesis and proliferation are through an intracellular activation of TGF-β1 in PASMCs (28). Calpain cleaves and activates intracellular latent TGF-β1 and initiates this intracrine TGF-β1 pathway (5, 16, 28). It has been reported that TGF-β activates Akt in many types of cells (21, 25, 37). We examined whether this intracrine TGF-β pathway is involved in the calpain-mediated Akt phosphorylation. We found that neutralizing extracellular TGF-β1 using a cell-impermeable TGF-β1 neutralizing antibody did not affect PDGF-induced Akt phosphorylations at S473 and T308, suggesting that the paracrine or autocrine TGF-β pathways do not play a role in calpain-mediated Akt phosphorylation. Nevertheless, the cell-permeable-specific TGF-β receptor blocker SB431542 attenuated Akt phosphorylations at both S473 and T308 induced by overexpressed calpain-2 and PDGF in PASMCs. Moreover, SB-431452 and knocking down ALK-5 significantly reduced PDGF-induced collagen synthesis and cell proliferation of PASMCs. These results indicate that calpain-2 upregulates Akt phosphorylation via an intracrine TGF-β1 pathway.
Hart and Vogt (17) discovered that mimicking phosphorylation at S473 by mutating S473 for aspartate leads to an enhancement of phosphorylation at T308. We noted that the increase in phosphorylation at S473 is much higher than those at T308 in calpain-2-overexpressed cells. Moreover, the reduction of PDGF-induced Akt phosphorylation caused by calpain inhibition is much more remarkable at S473 than those at T308. Similarly, the reduction of PDGF- and calpain-2-induced Akt phosphorylation caused by TGF-β inhibition is much greater at S473 than those at T308. These results suggest that phosphorylation of Akt at S473 might be a primary event. The TGF-β1 signaling may first phosphorylate Akt at S473, which is followed by Akt binding to PDK1, and phosphorylation at T308 ensues. Akt phosphorylation at S473 is mediated by mTORC2 (15, 29). Indeed, mTORC2 is at the downstream of TGF-β receptor and mediates the fibrotic effects of TGF-β1 (24, 33). mTORC2 is increased in PASMCs of human lung with PAH and in PASMCs exposed to hypoxia and PDGF (15, 23). Activations of mTOR and Akt are required for hypoxia-induced cell proliferation of vascular endothelial cells and adventitial fibroblasts of pulmonary arteries (9, 26). Our studies show that inhibition of mTORC2 by knocking down its component protein Rictor prevents PDGF- and calpain-2-induced Akt phosphorylation at S473 and attenuates in less magnitude Akt phosphorylation at T308. Taken together, our data provide the first evidence supporting that calpain-2 upregulates Akt phosphorylation via mTORC2.
In summary, we have demonstrated that overexpression of calpain-2 induces an increase in Akt phosphorylation, and that inhibition of calpain-2 causes a reduction of Akt phosphorylation in PASMCs induced by PDGF and in hypoxic PAH mice. Inhibition of TGF-β signaling attenuates Akt phosphorylation induced by PDGF and by overexpressed calpain-2 in PASMCs. Neutralizing extracellular TGF-β1 using a cell-impermeable TGF-β1 neutralizing antibody does not affect PDGF-induced Akt phosphorylation. Thus we have identified a novel pathway of calpain-2-upregulated Akt phosphorylation via an intracrine TGF-β1/mTORC2 mechanism that is independent of PI3-kinase (Fig. 13).
Fig. 13.
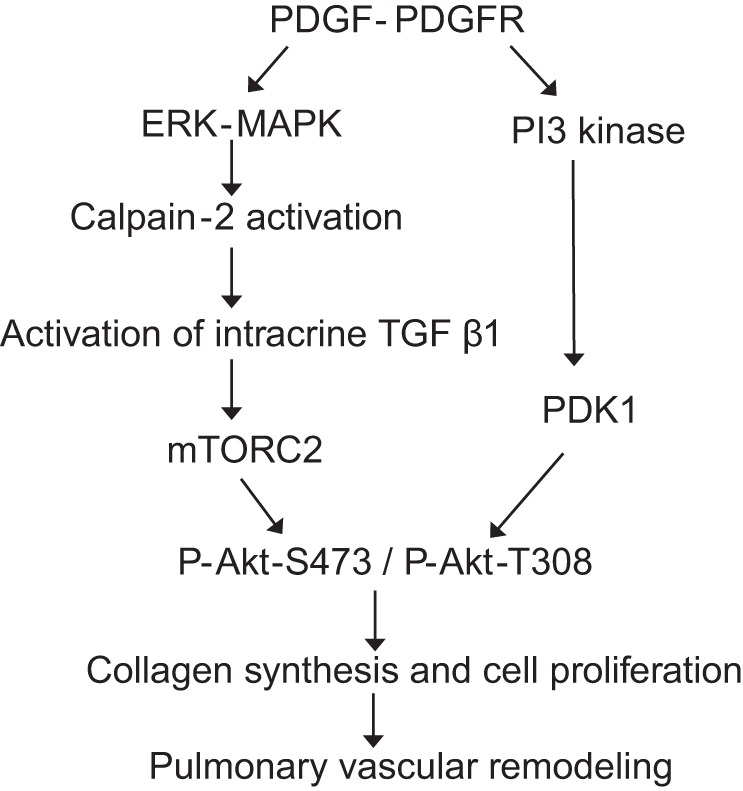
Schematic pathway illustrating the role of calpain-2 in PDGF-induced Akt phosphorylation in PASMCs of pulmonary vascular remodeling.
GRANTS
This work was supported, in whole or in part, by National Heart, Lung, and Blood Institute Grants HL-088261 and HL-115078, Flight Attendants Medical Research Institute Grants 113018_CIA and 140083_CIA, and by the Department of Veterans Affairs.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
P.A., L.K., and W.H. performed experiments; P.A., L.K., and Y.S. analyzed data; P.A., L.K., and Y.S. interpreted results of experiments; P.A. and L.K. prepared figures; P.A. and Y.S. drafted manuscript; P.A. and Y.S. edited and revised manuscript; P.A., L.K., W.H., and Y.S. approved final version of manuscript; Y.S. conception and design of research.
REFERENCES
- 1.Abeyrathna P, Su Y. The critical role of Akt in cardiovascular function. Vascul Pharmacol 74: 38–48, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541–6551, 1996. [PMC free article] [PubMed] [Google Scholar]
- 3.Arcot SS, Lipke DW, Gillespie MN, Olson JW. Alterations of growth factor transcripts in rat lungs during development of monocrotaline-induced pulmonary hypertension. Biochem Pharmacol 46: 1086–1091, 1993. [DOI] [PubMed] [Google Scholar]
- 4.Arora AS, de Groen PC, Croall DE, Emori Y, Gores GJ. Hepatocellular carcinoma cells resist necrosis during anoxia by preventing phospholipase-mediated calpain activation. J Cell Physiol 167: 434–442, 1996. [DOI] [PubMed] [Google Scholar]
- 5.Caver TE, O'Sullivan FX, Gold LI, Gresham HD. Intracellular demonstration of active TGFbeta1 in B cells and plasma cells of autoimmune mice. IgG-bound TGFbeta1 suppresses neutrophil function and host defense against Staphylococcus aureus infection. J Clin Invest 98: 2496–2506, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen YF, Feng JA, Li P, Xing D, Zhang Y, Serra R, Ambalavanan N, Majid-Hassan E, Oparil S. Dominant negative mutation of the TGF-beta receptor blocks hypoxia-induced pulmonary vascular remodeling. J Appl Physiol 100: 564–571, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Choi KH, Kim JE, Song NR, Son JE, Hwang MK, Byun S, Kim JH, Lee KW, Lee HJ. Phosphoinositide 3-kinase is a novel target of piceatannol for inhibiting PDGF-BB-induced proliferation and migration in human aortic smooth muscle cells. Cardiovasc Res 85: 836–844, 2010. [DOI] [PubMed] [Google Scholar]
- 8.Ciuclan L, Hussey MJ, Burton V, Good R, Duggan N, Beach S, Jones P, Fox R, Clay I, Bonneau O, Konstantinova I, Pearce A, Rowlands DJ, Jarai G, Westwick J, Maclean MR, Thomas M. Imatinib attenuates hypoxia-induced pulmonary arterial hypertension pathology via reduction in 5-hydroxytryptamine through inhibition of tryptophan hydroxylase 1 expression. Am J Respir Crit Care Med 187: 78–89, 2013. [DOI] [PubMed] [Google Scholar]
- 9.Gerasimovskaya EV, Tucker DA, Stenmark KR. Activation of phosphatidylinositol 3-kinase, Akt, and mammalian target of rapamycin is necessary for hypoxia-induced pulmonary artery adventitial fibroblast proliferation. J Appl Physiol (1985) 98: 722–731, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh CG, Lenin M, Calhaun C, Zhang JH, Abboud HE. PDGF inactivates forkhead family transcription factor by activation of Akt in glomerular mesangial cells. Cell Signal 15: 161–170, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Gillespie MN, Rippetoe PE, Haven CA, Shiao RT, Orlinska U, Maley BE, Olson JW. Polyamines and epidermal growth factor in monocrotaline-induced pulmonary hypertension. Am Rev Respir Dis 140: 1463–1466, 1989. [DOI] [PubMed] [Google Scholar]
- 12.Glading A, Bodnar RJ, Reynolds IJ, Shiraha H, Satish L, Potter DA, Blair HC, Wells A. Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol Cell Biol 24: 2499–2512, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glading A, Lauffenburger DA, Wells A. Cutting to the chase: calpain proteases in cell motility. Trends Cell Biol 12: 46–54, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev 83: 731–801, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Goncharov DA, Kudryashova TV, Ziai H, Ihida-Stansbury K, DeLisser H, Krymskaya VP, Tuder RM, Kawut SM, Goncharova EA. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 129: 864–874, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gressner OA, Lahme B, Siluschek M, Rehbein K, Herrmann J, Weiskirchen R, Gressner AM. Activation of TGF-beta within cultured hepatocytes and in liver injury leads to intracrine signaling with expression of connective tissue growth factor. J Cell Mol Med 12: 2717–2730, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hart JR, Vogt PK. Phosphorylation of AKT: a mutational analysis. Oncotarget 2: 467–476, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hassoun PM, Mouthon L, Barbera JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, Newman JH, Rabinovitch M, Schermuly R, Stenmark KR, Voelkel NF, Yuan JX, Humbert M. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol 54: S10–S19, 2009. [DOI] [PubMed] [Google Scholar]
- 19.Ho WC, Pikor L, Gao Y, Elliott BE, Greer PA. Calpain 2 regulates Akt-FoxO-p27(Kip1) protein signaling pathway in mammary carcinoma. J Biol Chem 287: 15458–15465, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Houssaini A, Abid S, Mouraret N, Wan F, Rideau D, Saker M, Marcos E, Tissot CM, Dubois-Rande JL, Amsellem V, Adnot S. Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am J Respir Cell Mol Biol 48: 568–577, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kattla JJ, Carew RM, Heljic M, Godson C, Brazil DP. Protein kinase B/Akt activity is involved in renal TGF-beta1-driven epithelial-mesenchymal transition in vitro and in vivo. Am J Physiol Renal Physiol 295: F215–F225, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kovacs L, Han W, Rafikov R, Bagi Z, Offermanns S, Saido TC, Black SM, Su Y. Activation of calpain-2 by mediators in pulmonary vascular remodeling of pulmonary arterial hypertension. Am J Respir Cell Mol Biol 54: 384–393, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krymskaya VP, Snow J, Cesarone G, Khavin I, Goncharov DA, Lim PN, Veasey SC, Ihida-Stansbury K, Jones PL, Goncharova EA. mTOR is required for pulmonary arterial vascular smooth muscle cell proliferation under chronic hypoxia. FASEB J 25: 1922–1933, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamouille S, Connolly E, Smyth JW, Akhurst RJ, Derynck R. TGF-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J Cell Sci 125: 1259–1273, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C, Wang Q, Wang JF. Transforming growth factor-beta (TGF-beta) induces the expression of chondrogenesis-related genes through TGF-beta receptor II (TGFRII)-AKT-mTOR signaling in primary cultured mouse precartilaginous stem cells. Biochem Biophys Res Commun 450: 646–651, 2014. [DOI] [PubMed] [Google Scholar]
- 26.Li L, Xu M, Li X, Lv C, Zhang X, Yu H, Zhang M, Fu Y, Meng H, Zhou J. Platelet-derived growth factor-B (PDGF-B) induced by hypoxia promotes the survival of pulmonary arterial endothelial cells through the PI3K/Akt/Stat3 pathway. Cell Physiol Biochem 35: 441–451, 2015. [DOI] [PubMed] [Google Scholar]
- 27.Long L, Crosby A, Yang X, Southwood M, Upton PD, Kim DK, Morrell NW. Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation 119: 566–576, 2009. [DOI] [PubMed] [Google Scholar]
- 28.Ma W, Han W, Greer PA, Tuder RM, Toque HA, Wang KKW, Caldwell RW, Su Y. Calpain mediates pulmonary vascular remodeling in rodent models of pulmonary hypertension and its inhibition attenuates pathologic features of disease. J Clin Invest 121: 4548–4566, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moschella PC, McKillop J, Pleasant DL, Harston RK, Balasubramanian S, Kuppuswamy D. mTOR complex 2 mediates Akt phosphorylation that requires PKCepsilon in adult cardiac muscle cells. Cell Signal 25: 1904–1912, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogawa A, Firth AL, Smith KA, Maliakal MV, Yuan JX. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 302: C405–C411, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rabinovitch M. Pathobiology of pulmonary hypertension. Annu Rev Pathol 2: 369–399, 2007. [DOI] [PubMed] [Google Scholar]
- 32.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 118: 2372–2379, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rahimi RA, Andrianifahanana M, Wilkes MC, Edens M, Kottom TJ, Blenis J, Leof EB. Distinct roles for mammalian target of rapamycin complexes in the fibroblast response to transforming growth factor-beta. Cancer Res 69: 84–93, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest 115: 2811–2821, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Selimovic N, Bergh CH, Andersson B, Sakiniene E, Carlsten H, Rundqvist B. Growth factors and interleukin-6 across the lung circulation in pulmonary hypertension. Eur Respir J 34: 662–668, 2009. [DOI] [PubMed] [Google Scholar]
- 36.Tang H, Chen J, Fraidenburg DR, Song S, Sysol JR, Drennan AR, Offermanns S, Ye RD, Bonini MG, Minshall RD, Garcia JG, Machado RF, Makino A, Yuan JX. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L208–L220, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Terme JM, Lhermitte L, Asnafi V, Jalinot P. TGF-beta induces degradation of TAL1/SCL by the ubiquitin-proteasome pathway through AKT-mediated phosphorylation. Blood 113: 6695–6698, 2009. [DOI] [PubMed] [Google Scholar]
- 38.Thomas M, Docx C, Holmes AM, Beach S, Duggan N, England K, Leblanc C, Lebret C, Schindler F, Raza F, Walker C, Crosby A, Davies RJ, Morrell NW, Budd DC. Activin-like kinase 5 (ALK5) mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrotaline. Am J Pathol 174: 380–389, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tuder RM. Pathology of pulmonary arterial hypertension. Semin Respir Crit Care Med 30: 376–385, 2009. [DOI] [PubMed] [Google Scholar]
- 40.Zaiman AL, Podowski M, Medicherla S, Gordy K, Xu F, Zhen L, Shimoda LA, Neptune E, Higgins L, Murphy A, Chakravarty S, Protter A, Sehgal PB, Champion HC, Tuder RM. Role of the TGF-beta/Alk5 signaling pathway in monocrotaline-induced pulmonary hypertension. Am J Respir Crit Care Med 177: 896–905, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ziino AJ, Ivanovska J, Belcastro R, Kantores C, Xu EZ, Lau M, McNamara PJ, Tanswell AK, Jankov RP. Effects of rho-kinase inhibition on pulmonary hypertension, lung growth, and structure in neonatal rats chronically exposed to hypoxia. Pediatr Res 67: 177–182, 2010. [DOI] [PubMed] [Google Scholar]