

Abstract
Mitochondrial Ca2+ homeostasis, the Ca2+ influx-efflux balance, is responsible for the control of numerous cellular functions, including energy metabolism, generation of reactive oxygen species, spatiotemporal dynamics of Ca2+ signaling, and cell growth and death. Recent discovery of the molecular identity of the mitochondrial Ca2+ uniporter (MCU) provides new possibilities for application of genetic approaches to study the mitochondrial Ca2+ influx mechanism in various cell types and tissues. In addition, the subsequent discovery of various auxiliary subunits associated with MCU suggests that mitochondrial Ca2+ uptake is not solely regulated by a single protein (MCU), but likely by a macromolecular protein complex, referred to as the MCU-protein complex (mtCUC). Moreover, recent reports have shown the potential role of MCU posttranslational modifications in the regulation of mitochondrial Ca2+ uptake through mtCUC. These observations indicate that mtCUCs form a local signaling complex at the inner mitochondrial membrane that could significantly regulate mitochondrial Ca2+ handling, as well as numerous mitochondrial and cellular functions. In this review we discuss the current literature on mitochondrial Ca2+ uptake mechanisms, with a particular focus on the structure and function of mtCUC, as well as its regulation by signal transduction pathways, highlighting current controversies and discrepancies.
Keywords: CCDC109A, MCUb, Ca2+/calmodulin-dependent protein kinase II, proline-rich tyrosine kinase 2, phosphorylation
mitochondria play an important role in Ca2+ homeostasis (82, 96), which is crucial for controlling cell survival and death (30, 36, 79). The first associations between mitochondria and Ca2+ were reported in the 1950s and early 1960s; several groups showed that isolated mitochondria from mammalian tissues accumulated Ca2+ (11, 27, 99, 112). These pioneering discoveries set in motion a series of investigations of the physiological and pharmacological features of mitochondrial Ca2+ uptake mechanisms that have been extensively characterized in various cell types (17, 41, 97, 98). From these observations, mitochondrial Ca2+ uptake was long considered as the result of a single transport mechanism mediated by the mitochondrial Ca2+ uniporter (MCU), a Ca2+-selective channel in the inner mitochondrial membrane (IMM), mainly because ruthenium red and lanthanides almost completely inhibited this channel (38). Although MCU has been widely recognized as the main mechanism for mitochondrial Ca2+ uptake, its molecular identity was not well characterized for several decades after its discovery. However, recent groundbreaking studies revealed that MCU is encoded by coiled-coil domain-containing protein 109A (CCDC109A) (3, 24). These findings open up the exciting possibility of investigating mitochondrial Ca2+ handling via genetic approaches. For instance, molecular identification of MCU laid the foundation for the discovery of several auxiliary subunits associated with this pore-forming protein [e.g., essential MCU regulator (EMRE), mitochondrial Ca2+ uptake (MICU) 1, 2, and 3, and MCU regulator 1 (MCUR1)] (3, 13, 16, 24, 70, 89, 91, 102). From this came a new concept in the field: mitochondrial Ca2+ uptake is not regulated by a single protein (MCU), but possibly by macromolecular protein complexes [mitochondrial Ca2+ uniporter protein complexes (mtCUCs)] (33, 57, 74, 78). Furthermore, several groups have shown that kinases in the cytosol can translocate into the mitochondria, interact with mtCUC, and directly modulate mtCUC function through posttranslational modifications (PTMs) of MCU (56, 64, 81). These reports also support the contention that MCU and its regulatory proteins form macromolecular local signaling complexes at the IMM that may significantly impact mitochondrial Ca2+ handling, as well as numerous mitochondrial and cellular functions, such as energy metabolism, reactive oxygen species (ROS) generation, spatiotemporal dynamics of Ca2+ signaling, and cell growth and death (30, 36, 82, 83). Here, concentrating on the function of each mtCUC subunit and the intracellular regulators of these microenvironments, we aim to summarize what is known about mitochondrial Ca2+ uptake mechanisms. We also highlight the current controversies and discrepancies in the field to provide a platform for future discussions and experiments to help close the gap between mtCUC functions and cellular functions.
THE MITOCHONDRIAL CALCIUM UNIPORTER: HISTORICAL OVERVIEW
As noted above, the study of mitochondria has captivated scientists, although the molecules responsible for mitochondrial Ca2+ uptake escaped discovery for half a century. Initial candidates for the molecular identity of MCU were uncoupling proteins (UCPs) 2 and 3 (107). However, Jiang and colleagues reported that knockdown of UCPs in cultured Drosophila cells did not alter mitochondrial Ca2+ or H+ homeostasis (55), suggesting that UCPs either do not form the MCU pore or only indirectly modulate MCU activities in mammalian cells (6, 22, 113, 114). Next, a combination of bioinformatics and siRNA screening was used to identify mitochondrial Ca2+ uptake 1 (MICU1), an important regulatory protein in the mitochondrial Ca2+ uptake mechanism (89). MICU1 possesses two Ca2+-binding EF-hand motifs but is predicted to have only one transmembrane domain (TMD), making it unlikely to form a MCU channel pore. Therefore, MICU1 was likely to be a modulator or auxiliary subunit of MCU (see Auxiliary Subunits). Lastly, two groups independently reported CCDC109A to be the molecular identity of the MCU pore subunit (3, 24) (see Pore-Forming Subunits). In addition, subsequent studies raised the possibility of additional Ca2+ uptake pathways, such as 1) the rapid mode of uptake (RaM), 2) the mitochondrial ryanodine receptor (mRyR), 3) leucine zipper EF-hand-containing transmembrane protein 1 (Letm1), 4) mCa1 and mCa2, and 5) coenzyme Q10, which exhibits pharmacological characteristics different from the original mtCUC theory (for review see Refs. 26 and 82). Indeed, recent reports using the mitoplast patch-clamp technique revealed the existence of MCU-independent single Ca2+ channel activity (5, 6, 100). Thus the mtCUC may not be the sole mechanism for transducing changes in Ca2+ concentration ([Ca2+]) in the cytosol ([Ca2+]c) into changes in mitochondrial matrix [Ca2+] ([Ca2+]mt). However, we focus here on the role of mtCUC, since remarkable progress has been made in the 5 years since the discovery of MCU.
MOLECULAR IDENTITIES OF THE MITOCHONDRIAL CALCIUM UNIPORTER PROTEIN COMPLEX
Pore-Forming Subunits
MCU (CCDC109A).
The MCU gene (known as CCDC109A) is highly conserved across all eukaryotes (with the exception of yeast) and encodes a 40-kDa protein containing a mitochondrial target sequence (MTS) at the NH2 terminus (3, 24). The characteristics of MCU reported in two original studies were confirmed by several other groups and are summarized as follows. 1) The MCU protein is composed of two coiled-coil domains, two TMDs, and a short motif of amino acids between the two TMDs, which allows a Ca2+ channel pore to form (3, 24, 64, 94) (Table 1, Fig. 1). 2) Knockdown of MCU dramatically reduces mitochondrial Ca2+ uptake in isolated mitochondria, in permeabilized cells and living cells (1, 3, 5, 24, 25, 34, 42, 44, 51, 61, 66, 69, 70, 72, 75, 85, 93, 102, 105, 106, 108, 115), an effect that can be rescued by overexpression of MCU (1, 3, 24, 34, 51, 64, 71, 106) (Table 2). 3) MCU down- or upregulation alone does not affect basal mitochondrial membrane potential (ΔΨmt) (3, 24, 34, 52, 93), oxygen consumption (3, 61, 69, 85), ATP synthesis (3, 61, 93, 106), or mitochondrial morphology (3, 24, 42, 80, 92) (Table 2). 4) Reconstituted MCU channels in planar lipid bilayers exhibit ruthenium red-sensitive Ca2+ current with 6- to 7-pS single-channel activity (24, 94). 5) The site-specific mutation of two negatively charged residues inside the motif of the pore region (D261/E264 in human MCU) showed loss-of-function or dominant-negative effects (3, 24, 42, 81, 95, 118) (Table 2).
Table 1.
Summary subunits that associate with the mitochondrial Ca2+ uniporter to form a complex
| Subunit Name | No. of TMDs | Location | Direct Binding to MCU? | Subunit Category |
|---|---|---|---|---|
| MCU | 2 (3, 24, 64) | IMM (3, 13, 24, 76) | Yes (3, 24, 94) | Pore (3, 13, 24) |
| MCUb | 2 (94) | IMM (94, 102) | Yes (94, 102) | Pore (94) |
| EMRE | 1 (102, 109) | IMM (102, 109) | Yes (102, 109) | Class 1 (102, 109) |
| MICU1 | 0 (86, 102, 109) | IMS (13, 16, 62, 86, 90, 102, 109) or Matrix (46, 71) | Yes (46, 86) or No (102, 109) | Class 2 (13, 16, 62, 86, 90, 102, 109) or 3 (46, 71) |
| MICU2 | 0 (86, 102, 109) | IMS (86, 102, 109) or Matrix? | Yes (86) or No (102) | Class 2 (86, 102, 109) or 3? |
| MICU3 | 0 (91) | IMS? or Matrix? | ? | Class 2? or 3? |
| MCUR1 | 2 (70) | IMM (70, 87) | Yes (70) or No (87) | Class 1 (70, 110) or not a subunit (87) |
| SLC25A23 | 6 (47) | IMS (47) | Yes (47) | Class 1 (47) |
For subunit classifications, see also Fig. 1.
TMD, transmembrane domain; IMM, inner mitochondrial membrane; IMS, intermembrane space. Numbers in parentheses are reference numbers.
Fig. 1.

Models of mitochondrial Ca2+ uniporter protein complexes. A: schematic drawing showing the subunit classification of an inner mitochondrial membrane (IMM)-localized ion channel. IMS, intermembrane space; OMM, outer mitochondrial membrane. B: model 1, in which mitochondrial Ca2+ uptake 1 and 2 (MICU1 and MICU2) are categorized as class 2 auxiliary subunits. Arrows show proposed interaction sites. MICU3, MCUR1, and SLC25A23 are omitted from this model. See also Table 1. EMRE, essential mitochondrial Ca2+ uniporter (MCU) regulator. C: model 2, in which MICU1 and MICU2 are grouped under class 3 auxiliary subunits. Arrow shows proposed interaction sites. MICU3, MCUR1, and SLC25A23 are omitted from this model. See also Table 1.
Table 2.
Summary genetic manipulations of mtCUC components and their impacts on mitochondrial Ca2+-handing profiles and cellular functions
| Gene Manipulation | Basal [Ca2+]mt | Mitochondrial Ca2+ Uptake | IMCU | In situ/In vivo Phenotype |
|---|---|---|---|---|
| MCU KO or KD | → (16, 61, 71) | ⇊ (1, 3, 5, 24, 25, 34, 42, 44, 51, 61, 64, 66, 69-72, 75, 85, 93, 102, 105, 106, 108, 115) | ⇊ (5, 13, 46, 69, 109) | In situ |
| →ΔΨmt (3, 24, 34, 52, 93) | ||||
| →Basal mitochondrial OCR (3, 61, 69, 85) | ||||
| ⇊Ca2+-dependent activation of the TCA cycle, OCR, and ATP production (3, 61, 69, 85, 93, 106) | ||||
| ⇊ (115) | ⇊Glucose-triggered ATP production (1) | |||
| →Mitochondrial morphology (3, 24, 42, 80, 92) | ||||
| ⇊Insulin secretion (93) | ||||
| ⇊Proinflammatory cytokine secretion (108) | ||||
| ⇊β-Hexosaminidase release in mast cells (34) | ||||
| ⇈Speed of synaptic vesicle endocytosis (75) | ||||
| →Basal mitochondrial ROS level (42, 69) | ||||
| ⇊Mitochondrial ROS level generation by stimuli (51, 69) | ||||
| ⇊Store-operated Ca2+ entry (25, 101, 105) | ||||
| →Proliferation or cell viability (19) | ||||
| ⇊Cell migration (105) | ||||
| ⇊Cell death rate by stimuli (19, 66) | ||||
| ⇈Cell death rate by stimuli (42) | ||||
| ⇊Replicative and oncogene-induced senescence (117) | ||||
| In vivo | ||||
| ⇊Exercise capacity and skeletal muscle performance (85) | ||||
| ⇊Skeletal muscle size (72) | ||||
| →I/R-mediated I/R-mediated heart injury (85) | ||||
| ⇊I/R-mediated I/R-mediated heart injury (61, 69) | ||||
| MCU-DN OX | ? | ⇊ (3, 24, 42, 81, 95, 118) | ? | In situ |
| →Basal mitochondrial OCR (95) | ||||
| ⇊Αpoptotic signaling activation by stimuli (81) | ||||
| In vivo | ||||
| →Resting heart rates (118) | ||||
| ⇊Fight-or-flight heart rate acceleration (118) | ||||
| ⇊Inotropic and lusitropic responses to stress (95) | ||||
| →Protection from I/R-mediated heart injury (109) | ||||
| MCU OX | ⇈ | ⇈ (24, 42, 81) | ⇈ (13) | In situ |
| ⇈Basal mitochondrial ROS level (81) | ||||
| ⇈Mitochondrial ROS generation by stimuli (81) | ||||
| ⇈Cell death rate by stimuli (24, 66, 81) | ||||
| ⇊ Speed of synaptic vesicle endocytosis (75) | ||||
| MCUb KD | ? | ⇈ (94) | ? | ? |
| MCUb OX | ? | ⇊ (81, 94) | ? | In situ |
| ⇊Basal mitochondrial superoxide levels (81) | ||||
| ⇊Mitochondrial superoxide generation by stimuli (81) | ||||
| ⇊Cell death rate by stimuli (81) | ||||
| EMRE KD | ⇊ (115) | ⇊ (102, 109, 115) | ⇊(102, 109) | ? |
| EMRE OX | ? | ? | ? | ? |
| MICU1 KO or KD | ⇈ (46, 71, 86, 109) | ⇈ (42, 77, 86) | ⇈ (46) | In situ |
| ⇊Basal ATP level (71) | ||||
| →(16, 21) | → (71) | →Basal respiration (89) | ||
| ⇊Glucose-triggered ATP production (1) | ||||
| ⇊(89) | ⇊ (1, 34, 50, 50, 51, 89, 91, 116) | |||
| ⇊Insulin secretion (1) | ||||
| ⇈at low [Ca2+]c elevation and ⇊at high [Ca2+]c elevation (2, 16, 21, 58) | →β-Hexosaminidase release in mast cells (34) | |||
| ⇈Basal mitochondrial ROS level (46, 71) | ||||
| →Basal mitochondrial ROS level (16, 42) | ||||
| ⇈Mitochondrial ROS generation during mitochondrial Ca2+ uptake(16) | ||||
| →Mitochondrial ROS generation by stimuli (42) | ||||
| ⇊Mitochondrial ROS generation by stimuli (50, 51) | ||||
| →Proliferation or cell viability (71) | ||||
| ⇈Cell death rate by stimuli (16, 71) | ||||
| ⇊Cell migration (46, 71) | ||||
| ⇈Oncogene-induced senescence (117) | ||||
| In vivo | ||||
| ⇊Liver regeneration and hepatocyte proliferation following partial hepatectomy (2) | ||||
| ⇈Proinflammatory response after partial hepatectomy (2) | ||||
| MICU1 OX | ? | → (58) | ? | ? |
| ⇊ (86) | ||||
| MICU2 KO or KD | ⇈ (77, 109) | ⇊ (91) | →(109) | ? |
| ⇈ (58, 77, 86) | ||||
| MICU2 OX | ? | → (58) | ? | ? |
| ⇊ (86) |
Because of the small number of publications available, MICU3, MCUR1, and SLC25A23 are omitted. All references are limited to reports using mammalian cells and tissues.
mtCUC, MCU-protein complex; [Ca2+]mt, mitochondrial Ca2+ concentration; MCU, mitochondrial Ca2+ uniporter; IMCU, MCU current; ΔΨmt, mitochondrial membrane potential; TCA, tricarboxylic acid cycle; ROS, reactive oxygen species; EMRE, essential MCU regulator; MICU, mitochondrial Ca2+ uptake; I/R, ischemia-reperfusion; KD, knockdown; KO, knockout; OX, overexpression; OCR, oxygen consumption rate.
After these two original reports, the significant effect of MCU on mitochondrial Ca2+ accumulation was confirmed by genetic manipulation of MCU expression (or activity) in various cell lines, as well as in primary cells, including cardiomyocytes (28, 81), pancreatic β-cells (106), neurons (92), and breast epithelial cells (42). Moreover, the attempt to knock down MCU in vivo also began to clarify the physiological and pathophysiological roles of MCU at the organ and tissue levels (3, 61, 69, 72, 85). Although there was originally some debate about MCU topology, it has been confirmed by several groups utilizing different approaches that both the NH2 and COOH termini of MCU face the mitochondrial matrix and a short motif of amino acids for the pore-forming region is exposed to the intermembrane space (IMS) (13, 16, 64, 76, 81). For instance, Ting and colleagues used the novel electron-microscopy tag “APEX” to show that both the NH2 and COOH termini of MCU face the matrix (76). Since a single MCU protein contains two TMDs, MCU needs to be oligomerized to form channels (3, 24, 29), analogous to some K+ channels. The crystal structure of the whole MCU protein has not been solved, but computational analysis using a combination of structural bioinformatics and molecular dynamics stimulation predicts that the MCU channel pore is formed by an MCU tetramer (94). The proposed structure was also confirmed by determination of the molecular weight of denatured proteins extracted from wheat germ or mammalian cells overexpressing MCU. In a native gel, immunoblotting of overexpressed MCU showed both a monomer and higher molecular weight compatible with a tetramer, indicating that MCU monomers oligomerized in situ to form higher-order complexes (81, 94).
MCUb (CCDC109B).
Rizzuto's group reported a novel protein that serves as an endogenous dominant-negative pore-forming subunit of mtCUC (94) (Table 1). MCUb, encoded by a gene related to CCDC109A, named CCDC109B, is a 33-kDa protein that has two predicted TMDs, shares 50% similarity to MCU with key amino acid substitutions (R251W and E256V) in the pore-forming region (94), and shows expression profiles different from MCU in various tissues. The biochemical and biophysical function of MCUb was characterized by several groups and is summarized as follows. 1) MCUb contains a MTS at its NH2 terminus and is expressed exclusively in the IMM (94, 102) (Table 1). 2) MCU and MCUb bind each other and form hetero-oligomers (94, 102) (Table 1). 3) MCUb overexpression in mammalian cells dramatically decreases mitochondrial Ca2+ uptake in response to increases in [Ca2+]c (81, 94) (Table 2). Interestingly, the MCU-to-MCUb mRNA expression ratio varies among tissue types (94), which might correlate to the size of mitochondrial Ca2+-selective currents recorded from mitoplasts isolated from a variety of mouse tissues (32). For instance, heart and skeletal muscle possess a high copy number of MCU mRNA (3, 24, 94), but MCUb expression is much higher in heart than skeletal muscle (94). Accordingly, the MCU current measured in whole mitoplast from hearts was 30-fold smaller than that from skeletal muscle (32). These observations suggest that the MCU-to-MCUb expression ratio may characterize different mitochondrial Ca2+ uptake profiles in different tissue types.
Auxiliary Subunits
Ion channels are composed of macromolecular protein complexes that open and close their pore subunit to regulate rapid ion fluxes across the cell membranes (9, 63). Generally, pore-forming subunits alone are sufficient for formation of a functional channel (121). However, without the coexistence of auxiliary subunits, ion channels frequently exhibit abnormal gating properties, as well as lower expression levels or lower efficiency of ion channel trafficking, in situ or in vivo (12, 121).
Rizzuto's group showed that recombinant MCU alone produces Ca2+ conductance in planar lipid bilayers (24, 86, 94) without the addition of other auxiliary subunits (see Pore-Forming Subunits). These reports strongly support the initial idea that MCU is a pore subunit and a sufficient minimal structure for the uniporter (3, 24). However, in lipid bilayers (in vitro setting) under symmetrical 100 mM Ca2+ conditions, the channel conductance from reconstituted channel by purified MCU proteins was only 6–7 pS (24, 86, 94), whereas an endogenous uniporter under mitoplast patch-clamp conditions exhibits a channel conductance of 14.3 pS (5, 54). The IMM has unique phospholipid compositions compared with the outer mitochondrial membrane (OMM), plasma membrane, and endoplasmic reticulum (ER)/sarcoplasmic reticulum (SR) membrane (104): the IMM does not possess cholesterol but does have a unique dimeric phospholipid cardiolipin that contributes to maintenance of the structure of cristae and has a unique ability to interact with proteins at the IMM, including the components in the electron transport chain (ETC), and supports their catalytic efficiencies (15, 104). Therefore, it is reasonable to speculate that mtCUC conductance may be smaller in the artificial planar lipid bilayers than in the natural IMM environment. Another factor may be the coexistence of auxiliary subunits with MCU pore subunits in the natural IMM environment.
Auxiliary subunits of the ion channel/transporter can be subdivided into three classes (Fig. 1, Table 1). Class 1 consists of subunits possessing at least one TMD and, in most cases, binds directly to the pore subunits [e.g., γ-subunit for the voltage-gated Ca2+ (Cav) channel (12)]. Class 2 consists of subunits that have no TMD, and the entire structure of class 2 subunits is on the extracellular side [for the channel expressed at the IMM, it should be at the IMS, e.g., the α2δ-subunit for Cav (12)]. Class 3 has no TMD, and the entire structure of class 3 subunits is at the cytoplasmic intracellular side [for the channel at the IMM, it should be at the matrix side, e.g., the β-subunit for the Cav channel (12)].
EMRE.
EMRE (known as C22ORF32) is the first identified class 1-type auxiliary subunit reported by Mootha and colleagues (102). EMRE is a 10-kDa protein with a MTS at its NH2 terminus and a highly conserved aspartate-rich COOH-terminal region (102). It contains a predicted single TMD and is located at the IMM (102); its COOH-terminal region may face the mitochondrial matrix (109) (Table 1, Fig. 1). Since EMRE knockdown in mammalian cells showed almost complete loss of uniporter activity in situ under the condition of MCU overexpression, Mootha and colleagues concluded that this protein is an essential auxiliary subunit (categorized as a class 1 auxiliary subunit) for production of mtCUC current in situ in mammalian cells (Table 1, Fig. 1). Recently, Graier's group achieved similar results: knockdown of EMRE strongly reduced mitochondrial Ca2+ uptake in HeLa cells (115). Foskett's group also confirmed the complete abrogation of MCU current (IMCU), originally known as IMiCa (59), in EMRE-knockdown cells (109). In addition, several other groups showed that, in cells lacking EMRE, the interaction between MCU and MICU1 and MICU2 was completely lost without affecting the submitochondrial localization, topology, or oligomerization of MCU (102, 109) (Fig. 1B). Moreover, using EMRE mutants carrying a COOH-terminal deletion or charge neutralization of its seven terminal acidic residues, Foskett's group proposed that the COOH-terminal region of EMRE may serve as a matrix Ca2+ sensor that is required for the gatekeeping function of the uniporter (see MICU1, MICU2, and MICU3).
Since the number of reports regarding EMRE remains limited, several important questions remain to be solved by additional experiments.
IS EMRE REQUIRED FOR MCU-MICU1 INTERACTIONS?
Mootha and colleagues proposed a model in which EMRE interacts with MICU1 and MICU2 at the IMS and with MCU at the IMM (possibly via their TMDs) (see MICU1, MICU2, and MICU3), thus acting as a bridge between MICUs and MCU and transmitting the function of MICUs to the pore subunits (60, 102) (Fig. 1B, Table 1). However, using a planar lipid bilayer system, Rizzuto's group clearly showed that recombinant MICU1 can activate the current derived from the reconstituted MCU channel, even when no other components are present (86). They also showed that mutations in a short protein loop of MCU facing the IMS (24, 94) nearly abolish the interactions between MCU and MICUs, suggesting that the pore structure of MCU might be important for MCU-MICU binding (86) (Fig. 1B). Therefore, this important question regarding our understanding of the whole picture of mtCUC stoichiometry remains open.
WHY IS EMRE REQUIRED FOR RECONSTITUTION OF THE UNIPORTER CURRENT IN SITU?
Mootha's group clearly demonstrated that mtCUC in a native gel became dramatically smaller after EMRE knockdown, suggesting that EMRE is an essential protein for efficient assembly and/or stabilization of mtCUC in mammalian cells (102). Moreover, they observed that when MCU is knocked down in mammalian cells, mitochondria also simultaneously lose EMRE protein, indicating that the protein's stability is impaired in the absence of MCU (102). It is conceivable that other mtCUC components (possibly unknown proteins) are required at the IMM for maintaining EMRE protein stability as well as its proper function in situ. Our understanding of the physiological role of EMRE is still very limited, because it is completely unknown how (or whether) EMRE itself affects the single-channel properties of the uniporter, although several groups have clearly shown that IMCU is almost abolished by EMRE knockdown under whole mitoplast patch-clamp conditions (102, 109) (Table 2). In a mitoplast patch-clamp study, Graier's group attempted to test the effect of double knockdown of MCU and EMRE on single Ca2+ channel properties (6), but the effect of EMRE alone on the single uniporter current remains unclear.
Another interesting observation is that, in yeast mitochondria, which do not endogenously possess both MCU and EMRE, EMRE is required for reconstitution of a functional uniporter when human MCU is introduced but is not necessary when a fungal (Dictyostelium discoideum) MCU homolog is expressed (60). Fungal MCU and human MCU have similar domain architectures but only ∼40% homology. Therefore, the comparison of the divergent sequences may give us clues in answering important questions: Which structures are important for MCU-EMRE associations? How are the two units functionally interdependent? Why is EMRE needed for reconstitution of the uniporter current in situ in mammalian mitochondria? Additional electrophysiological experiments to verify the role of EMRE in the uniporter current, hopefully, can answer these and other questions posed above.
MICU1, MICU2, and MICU3.
It is well recognized that mitochondrial Ca2+ stimulates oxidative phosphorylation and ETC activity, which results in stimulation of ATP synthesis (37). However, in early studies, high [Ca2+] (e.g., ≥10 μM) was required to efficiently activate Ca2+ uptake into isolated mitochondria (for reviews see Refs. 39 and 82). In addition, a mitoplast patch-clamp study revealed half-maximal activation (K0.5) of IMCU of ∼20 mM (59). However, in the intact cells, mitochondria take up Ca2+ during a global increase in [Ca2+]c (<10 μM). This discrepancy between isolated mitochondria/mitoplasts and intact cells was in part resolved by the discovery of high [Ca2+]c at the microdomains between mitochondria and the ER/SR during Ca2+ release from the ER/SR via inositol 1,4,5-trisphosphate receptors and/or ryanodine receptors (18) because of the juxtaposition of these two organelles via mitochondria-associated membranes (MAMs) (111). It is therefore conceivable that the transient increases in [Ca2+]mt in response to [Ca2+]c elevation at the MAM (e.g., ∼10 μM) (18) after cell signaling activation (e.g., Gq protein-coupled receptor stimulation) and/or after electrical stimulation (e.g., in cardiac and skeletal muscles) are important for triggering activation of Ca2+-sensitive dehydrogenases and acceleration of ATP production (82). However, if there is a scenario in which chronic and continuous Ca2+ accumulation into mitochondria occurs even in resting [Ca2+]c conditions, this might be an unfavorable situation for the cells to maintain their cellular functions, because mitochondrial Ca2+ overload causes ΔΨmt depolarization, ROS overproduction, and apoptotic cell death. Indeed, mitochondrial Ca2+ uptake rates are very slow near resting [Ca2+]c but rapidly increase if [Ca2+]c at the MAM reaches ∼10–20 μM (17, 18, 35, 97). To maintain this sigmoidal response to [Ca2+]c, mtCUC needs some sort of “gatekeeper” mechanism 1) to keep the channel activity low and the channel opening slow at resting [Ca2+]c, which does not increase basal [Ca2+]mt possibly via basal mitochondrial Ca2+ efflux and a Ca2+ buffering mechanism and 2) to facilitate mitochondrial Ca2+ uptake in response to [Ca2+]c elevation at the MAM (82). Indeed, MICU family proteins are the most likely candidates, but the detailed mechanisms are still under debate.
Historically, MICU1 (encoded by CBARA1/EFHA3) was the first regulatory element for the MCU pore, as discovered by Mootha's group in 2010 (see also the mitochondrial calcium uniporter: historical overview) just before the discovery of MCU (89). MICU1 is a 54-kDa protein with two highly conserved EF-hand Ca2+-binding motifs. Since MICU1 was originally predicted to have only one TMD (89), this protein was immediately believed unlikely to form the channel pore (Fig. 1, Table 1). In the original report (89) and a subsequent study from another group (1), knockdown of MICU1 was found to inhibit mitochondrial Ca2+ uptake; thus this protein was proposed as a key component for positive regulation of the uniporter (activator). Immediately after these reports, Madesh's group reported that MICU1-silenced cells possess mitochondria constitutively overloaded with Ca2+, suggesting that MICU1 is keeping the MCU pore closed under resting [Ca2+]c as a gatekeeper for the uniporter (46, 71). Indeed, some of the observations from Madesh's group were confirmed by several other groups: MICU1 knockdown increases basal [Ca2+]mt (86, 109), and MICU1-silenced cells increase mitochondrial Ca2+ uptake in response to low [Ca2+]c (range from nanomolar to a few micromolar) (16, 21, 42, 58, 77, 86) (Table 2). These reports offer clues to a partial understanding of the molecular gatekeeping function within mtCUC.
The remaining mystery was how the [Ca2+]c elevation [or proximity of mitochondria (18, 35)] overcomes the gatekeeping function of MICU1. Csordas and colleagues immediately proposed an answer: MICU1 not only determines the threshold of MCU pore opening but also works cooperatively in activating the channel open state in response to high [Ca2+]c, suggesting that MICU1 is the sole determinant of (or a main regulator for) the sigmoidal response of mitochondrial Ca2+ uptake (16). However, this model needs to be revisited and revised in light of the additional information that MICU1 has isoforms derived from its paralogous genes that also participate in mtCUC and modulate uniporter activities (91).
Subsequent analysis from Mootha's group after the discovery of MICU1 (89) revealed that MICU1 shares ∼25% sequence identity with two other human genes, EFHA1 and EFHA2, which were renamed MICU2 and MICU3, respectively (91) (Tables 1 and 2). Both of these proteins have an NH2-terminal MTS and two EF-hand motifs, similar to the structure of MICU1 (57). Importantly, a difference in tissue distribution patterns among the three isoforms was discovered: while MICU1 expression was detected in most tissues studied and seems ubiquitous (or at least broadly distributed) in all tissues, MICU2 was detectable in only half of the tissues tested, and MICU3 is expressed only in the central nervous system (with a very small amount in skeletal muscles) (91; for review see Refs. 40 and 78). Patron and colleagues clearly showed that MICU2 overexpression in situ decreases mitochondrial Ca2+ uptake elicited by [Ca2+]c elevation (86). In addition, the application of recombinant MICU2 to a planar lipid bilayer system decreased the open probability of the reconstituted MCU at lower [Ca2+] but had no effect at higher [Ca2+] (86). However, using a lipid bilayer system, they also showed that recombinant MICU1 did not affect or inhibit MCU channel activity at low [Ca2+] but did increase the MCU channel open probability under micromolar (∼1 μM) [Ca2+]; therefore, it serves as a cooperative activator. From these results, Patron and colleagues proposed that 1) MICU2 is the direct MCU gatekeeper in resting [Ca2+]c (gatekeeping effect) and 2) once [Ca2+]c elevation occurs, MICU2-dependent inhibition was achieved through some structural conformational changes, triggering MICU1-dependent activation of MCU channeling activity (cooperativity effect) (for review see Ref. 23). Several other groups also clarified some observations regarding MICU2 function (58, 77) (Table 2). However, there also exists an opposing contention, that MICU1 is more important than MICU2 for gatekeeping functions (40), because some tissue types express little (or no) MICU2 but are still capable of keeping the mitochondrial uniporter close to low [Ca2+]c (40, 78). In the parallel scenario, although MICU3 had been identified as a homolog protein of MICU1 and MICU2, MICU3 might play a minor role in this process, since it is expressed only in the central nervous system (91). Nevertheless, there are no reports regarding the exact functions of MICU3 protein; further investigations are required to understand its role in mtCUC regulation (Table 2).
We have reviewed recent reports regarding MICUs in chronological order since the discovery of their first member, MICU1. Below we summarize and discuss the important questions and discrepancies remaining in this research field.
WHY DOES MICU1 SILENCING YIELD VARYING RESULTS IN DIFFERENT STUDIES?
Genetic knockdown/knockout is a common procedure used to understand the functional role of specific proteins in situ or in vivo. As shown in Pore-Forming Subunits and Table 2, the effect of genetic knockdown/knockout of MCU on mitochondrial Ca2+ uptake has been extensively validated over the past 4 years by different groups achieving results quite similar to the original reports. However, for MICU1, that is not the case. For instance, while several groups have shown that MICU1 silencing decreases (or almost completely abolishes) mitochondrial Ca2+ uptake in response to strong agonist stimuli (1, 16, 21, 34, 50, 51, 89, 91, 116), other groups reported no change (71) or even the opposite: enhancement of mitochondrial Ca2+ uptake (42, 77, 86) (Table 2). For resting [Ca2+]mt, several groups reported significant increases in MICU1-silenced cells (46, 71, 86, 109), but others showed no change (16, 21) or significant decreases (89) (Table 2). One might argue that this is mainly due to the sensitivity of the experimental techniques (or, to some extent, the method of measurement of the resting [Ca2+]mt) and/or cell type differences [although HeLa cells were used in most of the tests (16, 71, 86, 89)]. However, we need to keep in mind the approaches used to knock down MICU1 (stable or transient) and the level of silencing achieved. It is generally accepted that some compensatory mechanism is frequently at work in stable and chronic knockdown compared with acute and transient silencing. For instance, Hajnoczky's group reported that mitochondrial buffering capacity was altered in their stable MICU1-silenced cells (16), but it is difficult to evaluate whether those results were produced via the direct effect of the MICU1 deletion or some compensatory mechanism (indirect effect) (71). Note that the lowered expression levels of MICU1 may also change the protein levels of other components within mtCUC. After MICU1 knockdown (stable or transient), MICU2 protein levels also significantly decreased without changes in the amount of MICU2 mRNA, suggesting that MICU1 protein plays a role in maintaining MICU2 protein stability in mitochondria (58, 86, 91). Therefore, further careful variations are required to completely determine whether the effects of the knockdown of MICU1 alone (16, 21, 34, 71, 89) are impacted by indirect MICU2 silencing. In addition, further research is needed to take into account cell and tissue types used because of the significant differences in expression levels and stoichiometry of MICU family proteins in various cell types/tissues (40, 91). Moreover, it is possible that the regulation and stoichiometry of MICU family proteins depend on the localization of the mitochondria in the cells and the interaction of mitochondria with the ER/SR membranes. For instance, in the cardiac and skeletal muscles, it is possible that perinuclear and interfibrillar mitochondria do not sense the similar [Ca2+]c during contraction, and, consequently, the role of MICUs might be different within the cell.
HOW DOES MICU SENSE THE CHANGE IN CYTOSOLIC OR MITOCHONDRIAL MATRIX CALCIUM CONCENTRATION TO SERVE AS A GATEKEEPER AND ACTIVATOR FOR THE UNIPORTER?
The initial contention was that MICUs are the ideal proteins to comprise the sigmoidal activation mechanism for mitochondrial Ca2+ uptake, since they have EF-hand motifs, structures that may induce Ca2+-dependent conformational changes in mtCUC (3, 58, 86, 91). However, which side of the changes in [Ca2+] can the MICUs sense: cytosol (IMS), mitochondrial matrix, or both? In other words, which categorization is suitable for MICUs: class 2 or class 3 auxiliary subunits? This important question is attracting a great deal of attention from researchers, since it is key for understanding the entire picture of mtCUC topology across the IMM.
Using a protease-sensitivity assay with isolated mitochondria or mitoplasts, several groups confirmed major submitochondrial localization of MICU1 in the IMS, but not the mitochondrial matrix (13, 16, 81, 86, 109) (Table 1, Fig. 1B). These observations are strongly supported by the findings of Mootha's group. Using a combination of proteomic and immunohistochemical approaches, they suggest that MICU1 and MICU2 exist at least as soluble proteins in the IMS (53, 62). Furthermore, biochemical (coimmunoprecipitation) (57, 58, 86, 91) and cell biological approaches, e.g., Förster resonance energy transfer (FRET)-based assay (86), suggest that MICU1 and MICU2 bind each other and form homo- or heteromultimers at the IMS. IMS localization of MICUs has also been validated by functional assay. 1) Patron and colleagues added recombinant MICU1 or MICU2 to the cis side of a planar lipid bilayer and observed a significant effect on recombinant MCU channels (activation or inhibition, respectively), suggesting that the MICU-MCU interaction at the IMS side may modulate MCU channels (86). 2) Using a live-cell FRET approach with the overexpression of CFP- and YFP-tagged MICU1, Waldeck-Weiermair and colleagues elegantly demonstrated that FRET changes (possibly the rearrangement of MICU1 multimers) follow [Ca2+]c elevation, but not [Ca2+]mt elevation (i.e., FRET changes are faster than [Ca2+]mt elevation) (115), indicating that MICU1 is capable of sensing changes in [Ca2+] at the extramitochondrial side (Fig. 1B).
Conversely, Madesh's group is proposing a different model (46, 71) (Table 1, Fig. 1C). Using a prepulse protocol for loading the mitochondrial matrix with Ca2+ in permeabilized cells, they found that MICU1 can specifically sense changes in [Ca2+]mt (71). Submitochondrial localization of MICU1 was investigated by observing the release profile of fluorescence-tagged MICU1 and other mitochondrial proteins when the OMM or both the OMM and IMM were selectively permeabilized. These researchers concluded that MICU1 behaved as a matrix protein (46). Moreover, using multiple different approaches, they identified the coiled-coil domains of MCU as strong candidate sites critical for MCU-MICU1 interaction, which is localized at the matrix side (46) (Fig. 1C).
Reasons behind the discrepancy in results reported by different groups remain unclear. However, the precise regulation of Ca2+ binding affinity at the EF-hand motifs of MICUs in the context of their homo- or heteromultimer and the careful observation of their conformational changes after Ca2+ binding may yield clear answers for the specific submitochondrial localization of MICUs. A recent study of the crystal structure of MICU1 confirmed that MICU1 has two EF-hand motifs, the Ca2+ binding affinities [dissociation constant (Kd)] of which are estimated to be ∼21.4 and 15.8 μM, suggesting that MICU1 is less likely to bind Ca2+ at resting [Ca2+]c or [Ca2+]mt (116). In addition, MICU1 forms a trimer of dimers (i.e., a hexamer) in the absence of Ca2+, which shows large conformational changes when Ca2+ is added. These results obtained using structural biological approaches also strongly support the contention that MICU1 serves as a Ca2+ sensor for mtCUC, possibly sensing the changes in [Ca2+]c. Further experiments to study the structural features of MICU1-MICU2 hetero-oligomers and their Ca2+ binding affinity will be helpful in revising this working model, since MICU1-MICU2 linking by the disulfide bonds is critical for maintenance of a normal mitochondrial Ca2+ uptake profile in response to [Ca2+]c elevation (90). The model proposed by Petrungaro and colleagues is that binding affinity of the MICU1-MICU2 heterodimer to MCU is dependent on [Ca2+]c (90); the MICU1-MICU2 heterodimer associates with MCU at low [Ca2+]c and inhibits uniporter activity at rest (gatekeeper) but dissociates upon high [Ca2+] (cooperative activator) possibly because of large conformational changes after Ca2+ binding to MICUs (116). Additional structural information, as well as electrophysiological approaches (lipid bilayer system and/or mitoplast patch) using MICU mutants (e.g., at EF-hand motifs), will strengthen this working model.
Recently, Foskett's group reported that the COOH-terminal end of EMRE contains a conserved acidic motif similar to the Ca2+-binding “Ca2+ bowl” structure in the Ca2+-activated large-conductance K+ channel and that this structure makes EMRE a sensor for [Ca2+]mt that governs the inhibition of uniporter activity under basal [Ca2+]mt and protects the mitochondria from Ca2+ overload under resting [Ca2+]c. However, they also proposed that this novel function of EMRE requires IMS-localized MICU1 and/or MICU2 binding to EMRE, indicating that the gatekeeping or Ca2+-sensing function of the uniporter may be governed not by the single-class subunits (class 2 or 3) but, instead, is elicited by the macrocomplex structure of mtCUC, including the multiclass subunits from both sides of the IMM. In addition, this observation from Foskett's group is quite reasonable, because mtCUC seems to possess Ca2+ sensors on both sides of the IMM (IMS and matrix) similar to the case of Cav at the plasma membrane (12).
Other proposed auxiliary subunits.
MCUR1, encoded by CCDC90A, was proposed as a possible class 1 auxiliary subunit for MCU by the Foskett-and-Madesh group (70) (Table 1). MCUR1 is a 40-kDa protein that has a MTS at the NH2 terminus, two TMDs, and one coiled-coil region. Its NH2 and COOH termini face the IMS, and the major part of the protein is exposed to the mitochondrial matrix (70). The Foskett-and-Madesh group demonstrated that knockdown of MCUR1 not only inhibits agonist-induced mitochondrial Ca2+ uptake, but also decreases resting [Ca2+]mt. Furthermore, overexpression of MCUR1 increases mitochondrial Ca2+ uptake, but only when MCU is expressed. Since the protein interaction between MCUR1 and MCU was observed in this study, they concluded that MCUR1 is a component of mtCUC and that direct interaction of MCU and MCUR1 is required for efficient Ca2+ uptake. On the other hand, Mootha's group failed to detect any interaction of MCU and MCUR1 (102). In addition, Shoubridge and colleagues recently raised a question about the direct modulation of uniporter activity by MCUR1 (87). They demonstrated that MCUR1 knockdown causes inhibition of ETC activity via a decrease in cytochrome c oxidase (complex IV) assembly and activity, indicating that the involvement of MCUR1 in MCU activity may be an indirect effect due to a drop in ΔΨmt. However, using a mitoplast patch, the Foskett-and-Madesh group directly measured IMCU under the precise control of ΔΨmt and confirmed that IMCU was indeed significantly reduced in cells with stable knockdown of MCUR1 (110). These electrophysiological data suggest that ΔΨmt reduction might not be the sole mechanism by which MCUR1 knockdown reduces mitochondrial Ca2+ uptake.
In addition to MCUR1, Madesh's group proposed another protein, SLC25A23, as a class 1 auxiliary subunit (47) (Table 1). This protein was originally known as a phosphate transporter across the IMM. They reported that SLC25A23 participates in mitochondrial Ca2+ uptake probably via its interaction with MCU and MICU1.
In summary, since reports regarding the protein functions of MCUR1 and SLC25A23 remain limited, further investigations by multiple laboratories are required to revisit the published data and also to understand the detailed mechanisms of how MCUR1 or SLC25A23 modulates uniporter activity.
REGULATION OF MCU BY POSTTRANSLATIONAL MODIFICATIONS
As summarized above, advancements in genetic approaches have uncovered the molecular identity of mtCUC components, including the MCU pore and its auxiliary subunits. Generally, ion channels/transporters located at the plasma membrane receive multiple levels of regulation, from gene expression of each subunit to PTMs that influence single-channel properties. It is reasonable to suggest that mitochondria-localized channels/transporters, including the uniporter, are also subject to such regulation. However, research efforts to identify the PTMs in mtCUC and their impact on uniporter activities have only just begun. Possible PTMs that regulate ion channel/transporter functions include modifications of 1) Ser, Thr, and/or Tyr residues by phosphorylation (20, 65) and 2) Cys residues by S-nitrosylation, S-oxidation, and S-palmitoylation (83, 103). For instance, basal Thr phosphorylation (at the NH2-terminal region) and Tyr phosphorylation (at the COOH-terminal region), but not Ser phosphorylation (see online database PhosphoSitePlus), of MCU were detected through mass spectroscopy analyses of human samples (http://www.phosphosite.org/homeAction.do) (48, 49) (Fig. 2, A and B). Thus, here we specifically focus on recent publications regarding the effect of MCU phosphorylation on mitochondrial Ca2+ uptake.
Fig. 2.
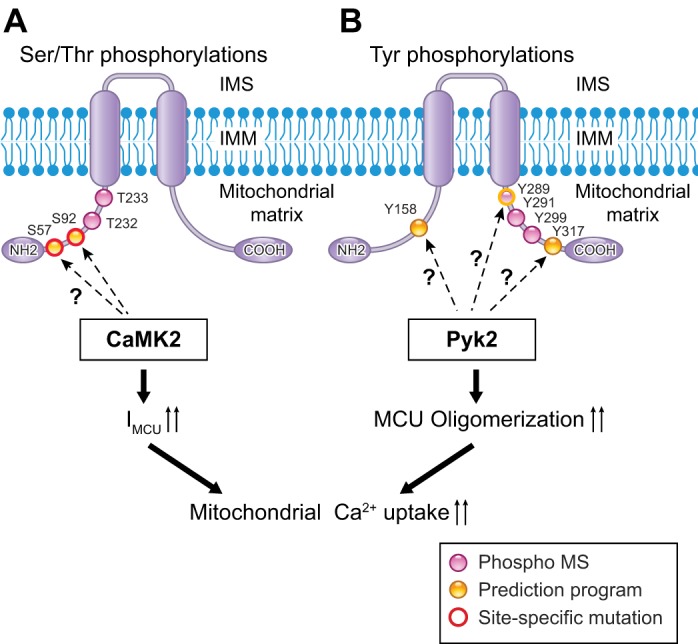
Phosphorylation candidate sites on MCU. A: Ser/Thr phosphorylation candidate sites on MCU. Predicted Ca2+/calmodulin-dependent protein kinase II (CaMKII) target sites are indicated with arrows. Purple dots, phosphorylation sites detected by mass spectroscopy (phospho MS; PhosphoSitePlus); yellow dots, phosphorylation sites predicted by putative phosphorylation motifs and computational programs; red circles, sites whose functions were investigated using site-specific mutagenesis. B: Tyr phosphorylation candidate sites on MCU. Predicted proline-rich tyrosine kinase 2 (Pyk2) target sites are indicated with arrows. Purple dots, phosphorylation sites detected by mass spectroscopy; yellow dots, phosphorylation sites predicted by putative phosphorylation motifs for general protein tyrosine kinase (PTK) and computational programs.
Anderson's group provided the first report that candidate cellular signaling (kinases) can phosphorylate MCU (56) (Fig. 2A). Their findings are summarized as follows. 1) Ca2+ and Ca2+/calmodulin-dependent protein kinase II (CaMKII) exist endogenously in the cardiac mitochondrial matrix, and matrix-localized CaMKII is capable of becoming activated during pathophysiological conditions such as ischemia-reperfusion (I/R), myocardial infarction, and neurohumoral injury, common causes of myocardial death and heart failure. 2) IMCU, measured using the patch-clamp technique on a whole mitoplast, was significantly increased by addition of a constitutively active monomeric CaMKII mutant (T287D) to the pipette solution (i.e., to the mitochondrial matrix), and IMCU became significantly reduced when CaMKII-inhibitory peptide was expressed at the mitochondrial matrix. 3) S57 and S92 at the NH2 terminus of the human MCU are putative CaMKII target sites (Fig. 2A), and overexpression of a dephosphorylated mimetic MCU mutant (S57A/S92A) abolished the CaMKII effect on IMCU. Recently, Lee and colleagues also tested the effect of MCU-S92A on mitochondrial Ca2+ uptake in response to [Ca2+] elevation (64). They found that MCU-S92A overexpression failed to restore the Ca2+ uptake profile in MCU-silenced cells. Moreover, they succeeded in determining the crystal structure of the NH2-terminal domain of MCU, including the S92 site, which turned out to be an essential domain for the modulation of MCU function. They concluded that S92A mutation induced a significant conformational change within this region, which subsequently impaired mitochondrial Ca2+ uptake activity. This report from Anderson's group was exciting, but several questions remain. 1) Since the NH2 terminus of MCU (amino acids 1–56 in human MCU) is predicted to be a MTS (80) and undergoes cleavage after insertion into the IMM, it is unlikely to form a putative CaMKII target sequence around S57 at the IMM. 2) Because Ser/Thr phosphorylation levels in MCU elicited by activated CaMKII or by cellular signaling (e.g., Gq protein-coupled receptor stimulation) were not investigated in these studies, it remains unclear whether this phosphorylation occurs in situ or in vivo. 3) Basal phosphorylation of neither S57A nor S92A was detected by analysis of phosphorylation sites by mass spectroscopy using human and mouse samples (Fig. 2A). Recently, using mouse cardiac mitoplasts, Fieni and colleagues (31) revisited the effect of CaMKII on IMCU, but they were unable to reproduce the findings of Anderson's group. Further investigations are required to determine whether CaMKII is a direct modulator of MCU through direct phosphorylation.
Another PTM of MCU reported is Tyr phosphorylation. Our group reported that a Ca2+- and ROS-dependent protein tyrosine kinase (PTK), called proline-rich tyrosine kinase 2 (Pyk2), which is activated by α1-adrenergic signaling, directly phosphorylates MCU and enhances mitochondrial Ca2+ uptake by promoting MCU channel oligomerization and formation of tetrameric channels (81) (Fig. 2B). We detected Pyk2 translocation from the cytosol to the mitochondrial matrix, as well as Pyk2 binding to MCU concomitant with MCU Tyr phosphorylation. Human MCU contains 15 Tyr residues, five in the NH2 terminus, six in the COOH terminus, and four in TMDs, which are conserved across all eukaryotic species. No Tyr residues are located in the pore-forming region. Since the consensus substrate motifs for Pyk2 have not been established (7), potential general PTK phosphorylation sites were identified using several phosphorylation prediction programs (4, 119). However, only three Tyr residues were predicted to be potential PTK phosphorylation sites (Fig. 2B). Among them, basal Y289 phosphorylation was reported in mass spectroscopy of human tissue (PhosphoSitePlus) (48, 49) (Fig. 2B). Future studies are needed to definitively identify Pyk2-specific phosphorylation site(s) in MCU and to test the functional importance of these sites using nonphosphorylation and phosphorylation mimetic MCU mutants.
PHYSIOLOGICAL AND PATHOPHYSIOLOGICAL ROLE OF THE MITOCHONDRIAL CALCIUM UNIPORTER PROTEIN COMPLEX
Before discovery of mtCUC, mitochondrial Ca2+ uptake had been suggested as an important regulator of fundamental cellular processes, including 1) cellular metabolism, 2) cytosolic Ca2+ buffering, 3) secretory functions, 4) cell survival and proliferation/migration, and 5) cell death. Taking advantage of the various methods of genetic manipulation of mtCUC activity summarized above, we have gained a better understanding of the physiological and pathophysiological roles of mitochondrial Ca2+ uptake mechanisms in mammalian cells (Table 2). Several groups have demonstrated that MCU-dependent Ca2+ accumulation may activate the tricarboxylic acid cycle and oxidative phosphorylation (1, 3, 61, 69, 85, 93, 106) to meet the cellular ATP demands for such purposes as secretory functions (1, 34, 93, 106, 108) and cell migration (105). These results are possibly due to the [Ca2+]mt-dependent activation profiles of several intramitochondrial dehydrogenases, as well as the ATPase (complex V) and adenosine nucleotide translocase (8, 37, 37a, 83). However, the precise molecular mechanisms underlying how a gradual increase in [Ca2+]mt via mtCUC efficiently activates these enzymes, stimulates ETC activity, and results in higher mitochondrial ATP output remain unclear, since [Ca2+]mt is regulated not only by mitochondrial Ca2+ influx, but also by the balance between Ca2+ buffering at the mitochondrial matrix and mitochondrial Ca2+ efflux (10). Moreover, further studies are required to determine whether mtCUC activity is involved in the maintenance of basal mitochondrial ATP levels. In regard to cytosolic Ca2+ buffering, several groups using cultured cell lines have shown that MCU expression levels modulate the magnitude of [Ca2+]c elevation (24, 64, 81) or store-operated Ca2+ entry (25, 101, 105) in various cultured cell lines (24, 64, 81) (Table 2). Using primary neonatal cardiomyocytes, Pozzan's group showed that [Ca2+]c peaks under spontaneous contractions are reduced or enhanced by MCU overexpression and siRNA silencing, respectively (28); however, further studies are required to understand the relative contribution of mitochondrial Ca2+ uptake to [Ca2+]c regulation in native cells or in vivo conditions.
In addition to energy metabolism, recent reports suggest that mtCUC activity may participate in the regulatory mechanisms underlying cell death, especially apoptotic signaling. First, genetic manipulation of MCU activities/expression levels is capable of changing the amount of ROS production following [Ca2+]c elevation induced by various stimuli (Table 2). Several groups showed that silencing MCU or inhibiting MCU activity through the introduction of dominant-negative MCU or MCUb decreases mitochondrial ROS generation by various stimuli (51, 69, 81). Conversely, overexpression of MCU enhances mitochondrial ROS generation (81). One of the possible mechanisms underlying MCU-dependent mitochondrial ROS production is the acceleration of oxidative phosphorylation by [Ca2+]mt elevation, as shown above. Another possible mechanism is that prolonged mitochondrial Ca2+ overload via the uniporter may eventually depolarize ΔΨmt, which decreases the electron flow efficiency at the ETC and generates more ROS. Both [Ca2+]mt elevation and mitochondrial ROS accumulation via MCU activity during various apoptotic challenges (e.g., treatment with H2O2) may directly or indirectly induce mitochondrial permeability transition pore (mPTP) opening and/or increase OMM permeability (51, 81), which initiates cell death (19, 24, 66, 67, 81, 92, 120). Moreover, inhibition of the cellular signaling pathway that phosphorylates MCU and, thus, activates the uniporter (e.g., CaMKII and Pyk2) can also prevent mitochondrial Ca2+ overload, ROS production, and cell death (56, 81) (see REGULATION OF MCU BY PTMs).
Genetic manipulation of MICU1 expression levels also modulates ROS production and cell death rate under stress (Table 2). MICU1 knockdown increases basal ROS levels (46, 71) or increases ROS during mitochondrial Ca2+ uptake (16) compared with control cells, and MICU1-knockdown (or knockout) cells are highly subject to apoptotic cell death during stress, probably due to the loss of gatekeeping effects mediated by MICU1 (protecting mitochondria from Ca2+ overload at resting [Ca2+]c and/or during moderate [Ca2+]c elevation) (see Auxiliary Subunits). In a wound-healing assay, Medesh's group also showed that MICU1 knockdown impairs cell migration without changing the basal proliferation rate (47, 71), implying that MICU1 knockdown sensitizes cells to apoptotic cell death in response to stress. However, it should be noted that several other groups, when using different cell types or different ROS measurement systems, reported no effect or even opposite results regarding the effect of MICU1 silencing on mitochondrial ROS (42, 50, 51). In summary, studies employing genetic approaches in situ confirmed the effects of mitochondrial Ca2+ on key cellular functions, which, interestingly, were proposed before discovery of MCU.
After these in situ experiments, several groups tested the role of mtCUC in vivo. The Finkel-and-Murphy group generated whole body MCU-knockout mice by using genetic ablation (85), which, very surprisingly, resulted in only mild phenotypes. Mitochondria from these MCU-knockout mice lack Ca2+ uptake, and these mice display only a low tolerance to exercise but live normally. In addition, although [Ca2+]mt-dependent mPTP opening was ablated in cardiomyocytes from MCU-knockout mice, their hearts were not significantly protected from I/R injury. These results raise the possibility that some unexpected adaptations must take place in these animals (for review see Refs. 23, 43, 45, and 88). Indeed, Anderson's group also encountered unexpected compensatory changes that affected cytoplasmic Ca2+ homeostasis when cardiac MCU was chronically inhibited by cardiac-specific overexpression of dominant-negative MCU, thereby preventing anticipated therapeutic responses to I/R injury (95). To avoid any compensatory effect produced by chronic mtCUC inhibition, conditional and inducible knockout animals or in vivo gene delivery by virus might serve as a suitable alternative approach to understand the physiological and pathophysiological role of mitochondrial Ca2+ uptake in tissue and organs. Two groups recently reported the cardiac phenotype of conditional and cardiomyocyte-specific MCU-knockout mice (61, 69). Hearts from these mice clearly showed the anticipated therapeutic responses to I/R injury, which were completely different from results in either the original MCU-knockout mice or the dominant-negative MCU overexpression mice. Several groups have also tested viral-based gene delivery systems in vivo to manipulate MCU (3, 14, 72) and MICU1 expression levels in specific organs (2, 91).
Finally, the possible roles of mtCUC in human disease have also been reported. Pinton's group showed that microRNA-25 expression can decrease MCU gene expression and function (73). Specifically, microRNA-25 is upregulated in human colon and prostate cancers, which leads to decreased MCU levels, followed by reduced mitochondrial Ca2+ uptake and resistance to Ca2+-dependent apoptotic challenges (73). Logan et al. (68) identified human pedigrees carrying MICU1 mutations with proximal myopathy, learning difficulties, and a progressive extrapyramidal movement disorder. The mutations discovered in this family produce truncated MICU1, which leads to complete loss of MICU1 expression. These reports are important for understanding the transcriptional and posttranscriptional mechanisms for regulating mtCUC activity in vivo, particularly within the context of human disease.
CONCLUSIONS
As summarized in this review, studies of the uniporter subunits have expanded our view of MCU channels from a single protein to macromolecular signaling complexes, which encompasses the properties and functions of mitochondrial Ca2+ uptake in a cell type- and tissue-specific manner. In addition, recent reports have gradually revealed the complex control of mtCUC at the transcriptional, posttranscriptional, and posttranslational levels under physiological and pathophysiological conditions. Future research must comprehensively analyze the role of mtCUC from the cellular to the organ level and how these interact systematically; this will allow us a broader understanding of mitochondrial biology, as well as human disease and how mtCUCs play a crucial part.
GRANTS
This work was partly supported by American Heart Association Grants 14BGIA18830032 and 16SDG27260248 (to J. O-Uchi), W. W. Smith Charitable Trust Medical Research Grant H1403 (to J. O-Uchi), and National Heart, Lung, and Blood Institute Grants 2R01 HL-093671 and 1R01 HL-122124 (to S. S. Sheu). J. O-Uchi is a recipient of 2015 New Investigator Award from American Physiological Society, Cell and Molecular Physiology Section.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.S.J., J.M., S.-S.S., and J.O.-U. developed the concept and designed the research; B.S.J., S.-S.S., and J.O.-U. prepared the figures; B.S.J., J.M., S.-S.S., and J.O.-U. drafted the manuscript; B.S.J., J.M., S.M., D.F., W.J., S.-S.S., and J.O.-U. edited and revised the manuscript; B.S.J., J.M., S.M., D.F., W.J., S.-S.S., and J.O.-U. approved the final version of the manuscript. This work was originally done in S.-S.S. lab.
REFERENCES
- 1.Alam MR, Groschner LN, Parichatikanond W, Kuo L, Bondarenko AI, Rost R, Waldeck-Weiermair M, Malli R, Graier WF. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial Ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic β-cells. J Biol Chem 287: 34445–34454, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antony AN, Paillard M, Moffat C, Juskeviciute E, Correnti J, Bolon B, Rubin E, Csordas G, Seifert EL, Hoek JB, Hajnoczky G. MICU1 regulation of mitochondrial Ca2+ uptake dictates survival and tissue regeneration. Nat Commun 7: 10955, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341–345, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294: 1351–1362, 1999. [DOI] [PubMed] [Google Scholar]
- 5.Bondarenko AI, Jean-Quartier C, Parichatikanond W, Alam MR, Waldeck-Weiermair M, Malli R, Graier WF. Mitochondrial Ca2+ uniporter (MCU)-dependent and MCU-independent Ca2+ channels coexist in the inner mitochondrial membrane. Pflügers Arch 466: 1411–1420, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bondarenko AI, Parichatikanond W, Madreiter CT, Rost R, Waldeck-Weiermair M, Malli R, Graier WF. UCP2 modulates single-channel properties of a MCU-dependent Ca inward current in mitochondria. Pflügers Arch 467: 2509–2518, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonnette PC, Robinson BS, Silva JC, Stokes MP, Brosius AD, Baumann A, Buckbinder L. Phosphoproteomic characterization of PYK2 signaling pathways involved in osteogenesis. J Proteomics 73: 1306–1320, 2010. [DOI] [PubMed] [Google Scholar]
- 8.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 287: C817–C833, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Cai X. Subunit stoichiometry and channel pore structure of ion channels: all for one, or one for one? J Physiol 586: 925–926, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camara AK, Bienengraeber M, Stowe DF. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front Physiol 2: 13, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carafol IE, Rossi CS, Lehninger AL. Cation and anion balance during active accumulation of Ca2+ and Mg2+ by isolated mitochondria. J Biol Chem 239: 3055–3061, 1964. [PubMed] [Google Scholar]
- 12.Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 3: a003947, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaudhuri D, Sancak Y, Mootha VK, Clapham DE. MCU encodes the pore conducting mitochondrial calcium currents. Elife 2: e00704, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chemello F, Mammucari C, Gherardi G, Rizzuto R, Lanfranchi G, Cagnin S. Gene expression changes of single skeletal muscle fibers in response to modulation of the mitochondrial calcium uniporter (MCU). Genom Data 5: 64–67, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol 292: C33–C44, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, Hajnoczky G. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab 17: 976–987, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Csordas G, Thomas AP, Hajnoczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J 18: 96–108, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, Balla T, Hajnoczky G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell 39: 121–132, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Curry MC, Peters AA, Kenny PA, Roberts-Thomson SJ, Monteith GR. Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun 434: 695–700, 2013. [DOI] [PubMed] [Google Scholar]
- 20.Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Gui P, Hill MA, Wilson E. Regulation of ion channels by protein tyrosine phosphorylation. Am J Physiol Heart Circ Physiol 281: H1835–H1862, 2001. [DOI] [PubMed] [Google Scholar]
- 21.de la Fuente S, Matesanz-Isabel J, Fonteriz RI, Montero M, Alvarez J. Dynamics of mitochondrial Ca2+ uptake in MICU1-knockdown cells. Biochem J 458: 33–40, 2014. [DOI] [PubMed] [Google Scholar]
- 22.De Marchi U, Castelbou C, Demaurex N. Uncoupling protein 3 (UCP3) modulates the activity of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) by decreasing mitochondrial ATP production. J Biol Chem 286: 32533–32541, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Stefani D, Patron M, Rizzuto R. Structure and function of the mitochondrial calcium uniporter complex. Biochim Biophys Acta 1853: 2006–2011, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336–340, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deak AT, Blass S, Khan MJ, Groschner LN, Waldeck-Weiermair M, Hallstrom S, Graier WF, Malli R. IP3-mediated STIM1 oligomerization requires intact mitochondrial Ca2+ uptake. J Cell Sci 127: 2944–2955, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dedkova EN, Blatter LA. Calcium signaling in cardiac mitochondria. J Mol Cell Cardiol 58: 125–133, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deluca HF, Engstrom GW. Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci USA 47: 1744–1750, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drago I, De Stefani D, Rizzuto R, Pozzan T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci USA 109: 12986–12991, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drago I, Pizzo P, Pozzan T. After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J 30: 4119–4125, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duchen MR, Verkhratsky A, Muallem S. Mitochondria and calcium in health and disease. Cell Calcium 44: 1–5, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Fieni F, Johnson DE, Hudmon A, Kirichok Y. Mitochondrial Ca2+ uniporter and CaMKII in heart. Nature 513: E1–E2, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fieni F, Kirichok Y. Patch-clamp analysis of mitochondrial Ca2+ uniporter in different tissues (Abstract). J Gen Physiol 138: 56A, 2011. [Google Scholar]
- 33.Foskett JK, Philipson B. The mitochondrial Ca2+ uniporter complex. J Mol Cell Cardiol 78: 3–8, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furuno T, Shinkai N, Inoh Y, Nakanishi M. Impaired expression of the mitochondrial calcium uniporter suppresses mast cell degranulation. Mol Cell Biochem 410: 215–221, 2015. [DOI] [PubMed] [Google Scholar]
- 35.Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, Pozzan T. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell 38: 280–290, 2010. [DOI] [PubMed] [Google Scholar]
- 36.Giacomello M, Drago I, Pizzo P, Pozzan T. Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ 14: 1267–1274, 2007. [DOI] [PubMed] [Google Scholar]
- 37.Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51: 2959–2973, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37a.Griffiths EJ, Rutter GA. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim Biophys Acta 1787: 1324–1333, 2009. [DOI] [PubMed] [Google Scholar]
- 38.Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol Cell Physiol 258: C755–C786, 1990. [DOI] [PubMed] [Google Scholar]
- 39.Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca2+ transport mechanisms. Biochim Biophys Acta 1787: 1291–1308, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hajnoczky G, Booth D, Csordas G, Debattisti V, Golenar T, Naghdi S, Niknejad N, Paillard M, Seifert EL, Weaver D. Reliance of ER-mitochondrial calcium signaling on mitochondrial EF-hand Ca2+ binding proteins: Miros, MICUs, LETM1 and solute carriers. Curr Opin Cell Biol 29: 133–141, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82: 415–424, 1995. [DOI] [PubMed] [Google Scholar]
- 42.Hall DD, Wu Y, Domann FE, Spitz DR, Anderson ME. Mitochondrial calcium uniporter activity is dispensable for MDA-MB-231 breast carcinoma cell survival. PLos One 9: e96866, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harrington JL, Murphy E. The mitochondrial calcium uniporter: mice can live and die without it. J Mol Cell Cardiol 78: 46–53, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He J, Shi W, Guo Y, Chai Z. ERp57 modulates mitochondrial calcium uptake through the MCU. FEBS Lett 588: 2087–2094, 2014. [DOI] [PubMed] [Google Scholar]
- 45.Herzig S, Maundrell K, Martinou JC. Life without the mitochondrial calcium uniporter. Nat Cell Biol 15: 1398–1400, 2013. [DOI] [PubMed] [Google Scholar]
- 46.Hoffman NE, Chandramoorthy HC, Shamugapriya S, Zhang X, Rajan S, Mallilankaraman K, Gandhirajan RK, Vagnozzi RJ, Ferrer LM, Sreekrishnanilayam K, Natarajaseenivasan K, Vallem S, Force T, Choi ET, Cheung JY, Madesh M. MICU1 motifs define mitochondrial calcium uniporter binding and activity. Cell Rep 5: 1576–1588, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoffman NE, Chandramoorthy HC, Shanmughapriya S, Zhang XQ, Vallem S, Doonan PJ, Malliankaraman K, Guo S, Rajan S, Elrod JW, Koch WJ, Cheung JY, Madesh M. SLC25A23 augments mitochondrial Ca2+ uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol Biol Cell 25: 936–947, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B. PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics 4: 1551–1561, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 40: D261–D270, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hou T, Jian C, Xu J, Huang AY, Xi J, Hu K, Wei L, Cheng H, Wang X. Identification of EFHD1 as a novel Ca sensor for mitoflash activation. Cell Calcium 59: 262–270, 2016. [DOI] [PubMed] [Google Scholar]
- 51.Hou T, Zhang X, Xu J, Jian C, Huang Z, Ye T, Hu K, Zheng M, Gao F, Wang X, Cheng H. Synergistic triggering of superoxide flashes by mitochondrial Ca2+ uniport and basal reactive oxygen species elevation. J Biol Chem 288: 4602–4612, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang G, Vercesi AE, Docampo R. Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nat Commun 4: 2865, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hung V, Zou P, Rhee HW, Udeshi ND, Cracan V, Svinkina T, Carr SA, Mootha VK, Ting AY. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol Cell 55: 332–341, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jean-Quartier C, Bondarenko AI, Alam MR, Trenker M, Waldeck-Weiermair M, Malli R, Graier WF. Studying mitochondrial Ca2+ uptake—a revisit. Mol Cell Endocrinol 353: 114–127, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 326: 144–147, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS, Anderson ME. CaMKII determines mitochondrial stress responses in heart. Nature 491: 269–273, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kamer KJ, Mootha VK. The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol 16: 545–553, 2015. [DOI] [PubMed] [Google Scholar]
- 58.Kamer KJ, Mootha VK. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep 15: 299–307, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427: 360–364, 2004. [DOI] [PubMed] [Google Scholar]
- 60.Kovacs-Bogdan E, Sancak Y, Kamer KJ, Plovanich M, Jambhekar A, Huber RJ, Myre MA, Blower MD, Mootha VK. Reconstitution of the mitochondrial calcium uniporter in yeast. Proc Natl Acad Sci USA 111: 8985–8990, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep 12: 15–22, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK, Ting AY. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12: 51–54, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee A, Fakler B, Kaczmarek LK, Isom LL. More than a pore: ion channel signaling complexes. J Neurosci 34: 15159–15169, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee Y, Min CK, Kim TG, Song HK, Lim Y, Kim D, Shin K, Kang M, Kang JY, Youn HS, Lee JG, An JY, Park KR, Lim JJ, Kim JH, Kim JH, Park ZY, Kim YS, Wang J, Kim do H, Eom SH. Structure and function of the N-terminal domain of the human mitochondrial calcium uniporter. EMBO Rep 16: 1318–1333, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Levitan IB. Modulation of ion channels by protein phosphorylation and dephosphorylation. Annu Rev Physiol 56: 193–212, 1994. [DOI] [PubMed] [Google Scholar]
- 66.Liao Y, Hao Y, Chen H, He Q, Yuan Z, Cheng J. Mitochondrial calcium uniporter protein MCU is involved in oxidative stress-induced cell death. Protein Cell 6: 434–442, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu X, Xu S, Wang P, Wang W. Transient mitochondrial permeability transition mediates excitotoxicity in glutamate-sensitive NSC34D motor neuron-like cells. Exp Neurol 271: 122–130, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, Kriek M, Phadke R, Johnson CA, Roberts NY, Bonthron DT, Pysden KA, Whyte T, Munteanu I, Foley AR, Wheway G, Szymanska K, Natarajan S, Abdelhamed ZA, Morgan JE, Roper H, Santen GW, Niks EH, van der Pol WL, Lindhout D, Raffaello A, De Stefani D, den Dunnen JT, Sun Y, Ginjaar I, Sewry CA, Hurles M, Rizzuto R, UK 10K Consortium, Duchen MR, Muntoni F, Sheridan E. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet 46: 188–193, 2014. [DOI] [PubMed] [Google Scholar]
- 69.Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M, Elrod JW. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep 12: 23–34, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, Miller R, Kolesar JE, Molgo J, Kaufman B, Hajnoczky G, Foskett JK, Madesh M. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 14: 1336–1343, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 151: 630–644, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mammucari C, Gherardi G, Zamparo I, Raffaello A, Boncompagni S, Chemello F, Cagnin S, Braga A, Zanin S, Pallafacchina G, Zentilin L, Sandri M, De Stefani D, Protasi F, Lanfranchi G, Rizzuto R. The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep 10: 1269–1279, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, Bononi A, Corra F, Giorgi C, De Marchi E, Poletti F, Gafa R, Lanza G, Negrini M, Rizzuto R, Pinton P. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol 23: 58–63, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol 592: 829–839, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marland JR, Hasel P, Bonnycastle K, Cousin MA. Mitochondrial calcium uptake modulates synaptic vesicle endocytosis in central nerve terminals. J Biol Chem 291: 2080–2086, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, Ting AY. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol 30: 1143–1148, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matesanz-Isabel J, Arias-Del-Val J, Alvarez-Illera P, Fonteriz RI, Montero M, Alvarez J. Functional roles of MICU1 and MICU2 in mitochondrial Ca uptake. Biochim Biophys Acta 1858: 1110–1117, 2016. [DOI] [PubMed] [Google Scholar]
- 78.Murgia M, Rizzuto R. Molecular diversity and pleiotropic role of the mitochondrial calcium uniporter. Cell Calcium 58: 11–17, 2015. [DOI] [PubMed] [Google Scholar]
- 79.Orrenius S, Gogvadze V, Zhivotovsky B. Calcium and mitochondria in the regulation of cell death. Biochem Biophys Res Commun 460: 72–81, 2015. [DOI] [PubMed] [Google Scholar]
- 80.O-Uchi J, Jhun BS, Hurst S, Bisetto S, Gross P, Chen M, Kettlewell S, Park J, Oyamada H, Smith GL, Murayama T, Sheu SS. Overexpression of ryanodine receptor type 1 enhances mitochondrial fragmentation and Ca2+-induced ATP production in cardiac H9c2 myoblasts. Am J Physiol Heart Circ Physiol 305: H1736–H1751, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.O-Uchi J, Jhun BS, Xu S, Hurst S, Raffaello A, Liu X, Yi B, Zhang H, Gross P, Mishra J, Ainbinder A, Kettlewell S, Smith GL, Dirksen RT, Wang W, Rizzuto R, Sheu SS. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxid Redox Signal 21: 863–879, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.O-Uchi J, Pan S, Sheu SS. Molecular identities of mitochondrial Ca2+ influx mechanism: updated passwords for accessing mitochondrial Ca2+-linked health and disease. J Gen Physiol 139: 435–443, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.O-Uchi J, Ryu SY, Jhun BS, Hurst S, Sheu SS. Mitochondrial ion channels/transporters as sensors and regulators of cellular redox signaling. Antioxid Redox Signal 21: 987–1006, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 15: 1464–1472, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Stefani D, Rizzuto R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53: 726–737, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Paupe V, Prudent J, Dassa EP, Rendon OZ, Shoubridge EA. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab 21: 109–116, 2015. [DOI] [PubMed] [Google Scholar]
- 88.Pendin D, Greotti E, Pozzan T. The elusive importance of being a mitochondrial Ca2+ uniporter. Cell Calcium 55: 139–145, 2014. [DOI] [PubMed] [Google Scholar]
- 89.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467: 291–296, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Petrungaro C, Zimmermann KM, Kuttner V, Fischer M, Dengjel J, Bogeski I, Riemer J. The Ca2+-dependent release of the Mia40-induced MICU1-MICU2 dimer from MCU regulates mitochondrial Ca2+ uptake. Cell Metab 22: 721–733, 2015. [DOI] [PubMed] [Google Scholar]
- 91.Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLos One 8: e55785, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, Wyllie DJ, Bading H, Hardingham GE. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun 4: 2034, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Quan X, Nguyen TT, Choi SK, Xu S, Das R, Cha SK, Kim N, Han J, Wiederkehr A, Wollheim CB, Park KS. Essential role of mitochondrial Ca2+ uniporter in the generation of mitochondrial pH gradient and metabolism-secretion coupling in insulin-releasing cells. J Biol Chem 290: 4086–4096, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabo I, Rizzuto R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J 32: 2362–2376, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, Wagner BA, Soto J, McCormick ML, Kutschke W, Weiss RM, Yu L, Boudreau RL, Abel ED, Zhan F, Spitz DR, Buettner GR, Song LS, Zingman LV, Anderson ME. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci USA 112: 9129–9134, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13: 566–578, 2012. [DOI] [PubMed] [Google Scholar]
- 97.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763–1766, 1998. [DOI] [PubMed] [Google Scholar]
- 98.Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358: 325–327, 1992. [DOI] [PubMed] [Google Scholar]
- 99.Rossi CS, Lehninger AL. Stoichiometry of respiratory stimulation, accumulation of Ca++ and phosphate, and oxidative phosphorylation in rat liver mitochondria. J Biol Chem 239: 3971–3980, 1964. [PubMed] [Google Scholar]
- 100.Ryu SY, Beutner G, Kinnally KW, Dirksen RT, Sheu SS. Single channel characterization of the mitochondrial ryanodine receptor in heart mitoplasts. J Biol Chem 286: 21324–21329, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Samanta K, Douglas S, Parekh AB. Mitochondrial calcium uniporter MCU supports cytoplasmic Ca2+ oscillations, store-operated Ca2+ entry and Ca2+-dependent gene expression in response to receptor stimulation. PLos One 9: e101188, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342: 1379–1382, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shipston MJ. Ion channel regulation by protein palmitoylation. J Biol Chem 286: 8709–8716, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sparagna GC, Lesnefsky EJ. Cardiolipin remodeling in the heart. J Cardiovasc Pharmacol 53: 290–301, 2009. [DOI] [PubMed] [Google Scholar]
- 105.Tang S, Wang X, Shen Q, Yang X, Yu C, Cai C, Cai G, Meng X, Zou F. Mitochondrial Ca2+ uniporter is critical for store-operated Ca2+ entry-dependent breast cancer cell migration. Biochem Biophys Res Commun 458: 186–193, 2015. [DOI] [PubMed] [Google Scholar]
- 106.Tarasov AI, Semplici F, Ravier MA, Bellomo EA, Pullen TJ, Gilon P, Sekler I, Rizzuto R, Rutter GA. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells. PLos One 7: e39722, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol 9: 445–452, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Triantafilou K, Hughes TR, Triantafilou M, Morgan BP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci 126: 2903–2913, 2013. [DOI] [PubMed] [Google Scholar]
- 109.Vais H, Mallilankaraman K, Mak DO, Hoff H, Payne R, Tanis JE, Foskett JK. EMRE is a matrix Ca2+ sensor that governs gatekeeping of the mitochondrial Ca2+ uniporter. Cell Rep 14: 403–410, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vais H, Tanis JE, Muller M, Payne R, Mallilankaraman K, Foskett JK. MCUR1, CCDC90A, is a regulator of the mitochondrial calcium uniporter. Cell Metab 22: 533–535, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vance JE. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta 1841: 595–609, 2014. [DOI] [PubMed] [Google Scholar]
- 112.Vasington FD, Murphy JV. Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J Biol Chem 237: 2670–2677, 1962. [PubMed] [Google Scholar]
- 113.Waldeck-Weiermair M, Jean-Quartier C, Rost R, Khan MJ, Vishnu N, Bondarenko AI, Imamura H, Malli R, Graier WF. Leucine zipper EF hand-containing transmembrane protein 1 (Letm1) and uncoupling proteins 2 and 3 (UCP2/3) contribute to two distinct mitochondrial Ca2+ uptake pathways. J Biol Chem 286: 28444–28455, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Waldeck-Weiermair M, Malli R, Naghdi S, Trenker M, Kahn MJ, Graier WF. The contribution of UCP2 and UCP3 to mitochondrial Ca2+ uptake is differentially determined by the source of supplied Ca2+. Cell Calcium 47: 433–440, 2010. [DOI] [PubMed] [Google Scholar]
- 115.Waldeck-Weiermair M, Malli R, Parichatikanond W, Gottschalk B, Madreiter-Sokolowski CT, Klec C, Rost R, Graier WF. Rearrangement of MICU1 multimers for activation of MCU is solely controlled by cytosolic Ca2+. Sci Rep 5: 15602, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang L, Yang X, Li S, Wang Z, Liu Y, Feng J, Zhu Y, Shen Y. Structural and mechanistic insights into MICU1 regulation of mitochondrial calcium uptake. EMBO J 33: 594–604, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wiel C, Lallet-Daher H, Gitenay D, Gras B, Le Calve B, Augert A, Ferrand M, Prevarskaya N, Simonnet H, Vindrieux D, Bernard D. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun 5: 3792, 2014. [DOI] [PubMed] [Google Scholar]
- 118.Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, Luczak ED, Wang Q, Rokita AG, Wehrens XH, Song LS, Anderson ME. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun 6: 6081, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol Cell Proteomics 7: 1598–1608, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yoon MJ, Lee AR, Jeong SA, Kim YS, Kim JY, Kwon YJ, Choi KS. Release of Ca2+ from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget 5: 6816–6831, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yu FH, Yarov-Yarovoy V, Gutman GA, Catterall WA. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol Rev 57: 387–395, 2005. [DOI] [PubMed] [Google Scholar]