

Abstract
During the absorptive state, the liver stores excess glucose as glycogen and synthesizes fatty acids for triglyceride synthesis for export as very low density lipoproteins. For de novo synthesis of fatty acids from glucose, the mitochondrial pyruvate dehydrogenase complex (PDC) is the gatekeeper for the generation of acetyl-CoA from glucose-derived pyruvate. Here, we tested the hypothesis that limiting the supply of PDC-generated acetyl-CoA from glucose would have an impact on expression of key genes in the lipogenic pathway. In the present study, although the postnatal growth of liver-specific PDC-deficient (L-PDCKO) male mice was largely unaltered, the mice developed hyperinsulinemia with lower blood glucose levels in the fed state. Serum and liver lipid triglyceride and cholesterol levels remained unaltered in L-PDCKO mice. Expression of several key genes (ACL, ACC1) in the lipogenic pathway and their upstream regulators (LXR, SREBP1, ChREBP) as well as several genes in glucose metabolism (Pklr, G6pd2, Pck1) and fatty acid oxidation (FAT, Cpt1a) was downregulated in livers from L-PDCKO mice. Interestingly, there was concomitant upregulation of lipogenic genes in adipose tissue from L-PDCKO mice. Although, the total hepatic acetyl-CoA content remained unaltered in L-PDCKO mice, modified acetylation profiles of proteins in the nuclear compartment suggested an important role for PDC-generated acetyl-CoA in gene expression in de novo fatty acid synthesis in the liver. This finding has important implications for the regulation of hepatic lipid synthesis in pathological states.
Keywords: liver-specific pyruvate dehydrogenase complex-deficient mice, acetyl-CoA, gene expression, lipogenic pathway, nuclear protein acetylation
glucose serves as the major fuel for liver metabolism only during the absorptive state after a balanced meal is consumed. Hepatic glycolysis, glycogenesis, and fatty acid (FA) biosynthesis are the major metabolic pathways for glucose utilization and fuel storage in the form of glycogen and export of triglycerides (TG) in the fed state. For TG synthesis, the liver converts excess glucose to acetyl-CoA (AcCoA) for the de novo synthesis of lipids (long-chain FAs and cholesterol) and also generates ATP from the oxidation of AcCoA in the tricarboxylic acid cycle to support biosynthetic processes (28). In the liver during the fed state, pyruvate generated from dietary glucose via glycolysis is converted to AcCoA by the mitochondrial pyruvate dehydrogenase complex (PDC), which in turn is converted to citrate and then transported into the cytosol, where citrate is reconverted to AcCoA by ATP-citrate lyase (ACL; Fig. 1) (29). The cytosolic pool of AcCoA serves as the precursor for synthesis of lipids and acetylation of specific cytosolic and nuclear proteins (2, 12).
Fig. 1.
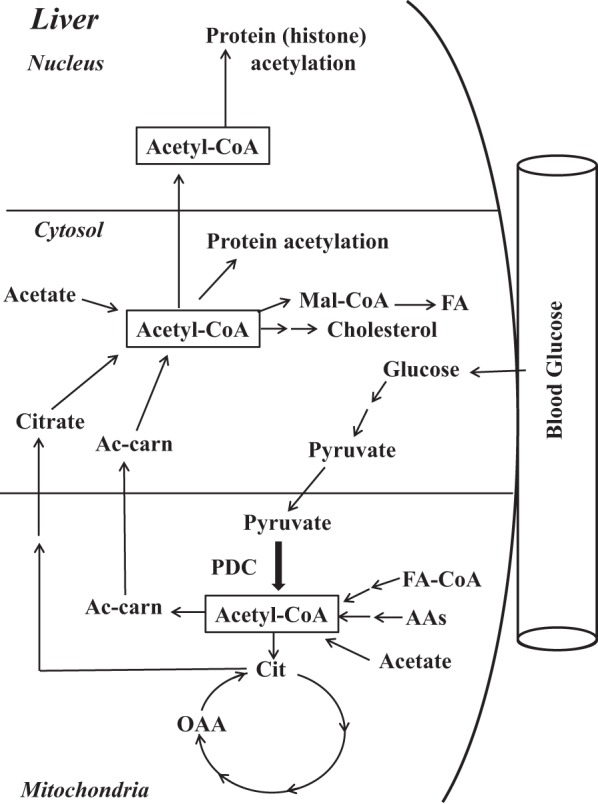
Schematic representation of the main cellular pathways involved in the generation of nuclear, cytosolic, and mitochondrial acetyl-CoA for lipid biosynthesis and protein acetylation. Mal-CoA, malonyl-CoA; FA, fatty acid; FA-CoA, fatty acid-CoA; AAs, amino acids; Cit, citrate; OAA, oxaloacetate; Ac-carn, acetyl-carnitine; PDC, pyruvate dehydrogenase complex.
The major contributors to the cytosolic AcCoA pool are the mitochondria-generated AcCoA from the catabolism of carbohydrates, FAs, and amino acid carbon skeletons (Fig. 1). In addition to the PDC reaction, there are other pathways by which AcCoA is generated in liver mitochondria. β-Oxidation of long-chain FAs as well as catabolism of amino acids and activation of acetate contribute to the AcCoA pool in the mitochondria. The contributions of these mitochondrial reactions to the cytosolic AcCoA pool in the fed and fasted states are not fully known. In the cytosolic compartment, AcCoA is generated by several reactions. The major pathway is mitochondria-derived citrate, which is cleaved by ACL (Fig. 1). Formation of AcCoA from acetate by acetyl-CoA synthetase-1 and conversion of mitochondria-derived acetyl-carnitine to AcCoA are other sources for AcCoA in the cytosol (Fig. 1).
The sources of lipids in the liver are 1) dietary lipids derived through chylomicron remnants, 2) de novo synthesis of FAs and cholesterol from excess of dietary carbohydrates and amino acids, and 3) free FAs transported from adipose tissues after lipolysis of stored TG. Relative contributions of these sources in hepatic lipid metabolism and their deposition have been investigated using several animal models in which a specific gene product was ablated or overexpressed. For examples, hepatic de novo synthesis of FAs was markedly diminished in mice with liver-specific deficiency of acetyl-CoA carboxylase 1 (ACC1KO) (27), whereas liver-specific overexpression of mature human SREBP-1c, a regulator of the FA biosynthetic pathway, resulted in upregulation of expression of key genes in the lipogenic pathway (23). In studies employing deletion mouse models for ACC1 (27) or fatty acid synthase (FAS) (5), there was, as expected, inhibition of FA synthesis due to the lack of a given enzyme activity in the pathway and not due to a lack of AcCoA availability as the substrate for FA synthesis. Furthermore, Wellen et al. (42) showed that RNAi knockdown of Acly, which generates cytosolic AcCoA from citrate, caused a reduction in global histone acetylation, decreasing the expression of several metabolic genes. Hence, variable effects are observed on the hepatic lipogenic pathway, depending on the sites being altered in the pathway.
We generated a line of mice with a conditional null mutation using the Cre-loxP system in the Pdha1 gene [encoding the α-subunit of the pyruvate dehydrogenase component (PDH) of PDC] localized on chromosome X (18). Generation of the systemic null mutation in male mice proved to be lethal at an early embryonic stage. However, tissue-specific (except the brain) deletion of the Pdha1 gene permitted both pre- and postnatal development and growth of male mice for metabolic studies (6, 30, 38, 39). Earlier, we generated liver-specific deletion of the Pdha1 gene in male mice (L-PDCKO) to investigate its impact on lipid biosynthesis from glucose in the liver (6). As expected, there was no incorporation in vitro of the [14C]glucose carbon into FAs by liver (6). Hence, it is of interest to investigate whether the absence of FA synthesis in the liver from L-PDCKO mice was due solely to the lack of availability of the precursor AcCoA for FA synthesis or to other possible factors contributing to this outcome in the liver.
Given the major role of AcCoA generated from citrate formed from glucose carbon in the liver in acetylation of nucleocytosolic proteins for transcriptional regulation (2, 7, 13), we tested the hypothesis that limiting the generation of cytosolic AcCoA from glucose carbon in livers from L-PDCKO mice would have a significant impact on the expression of key genes in the lipid and glucose metabolic pathways as well as on expression of key upstream regulatory genes. The results presented here show that in the absence of hepatic mitochondrial PDC activity, which normally serves as the provider of the cytosolic AcCoA derived from glucose carbons during the fed state, there is downregulation of expression of key metabolic genes and several upstream regulatory genes involved in hepatic lipogenesis. Although surprisingly the total cellular AcCoA pool remained unaltered in livers of L-PDCKO mice, modified acetylation of proteins in the nuclear compartment most likely contributed to altered hepatic gene expression in L-PDCKO mice. These findings suggest a unique role of PDC-generated AcCoA in modulating the expression of genes that regulate its own utilization for hepatic lipid synthesis.
METHODS
Mouse model and animal care.
Animal colony maintenance and all experiments were performed in accordance with the Guide for the Use and Care of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the University at Buffalo. Mice harboring the Pdha1flox8 allele(s) were generated as described previously (18, 30). This mouse colony had a 129J genetic background. In our previously published paper (6), we bred PDH-floxed females with a 129J genetic background with liver-specific Cre transgenic mice (C57BL/6J genetic background), creating the progeny with a mixed genetic background (129J/B6). To overcome any effects of a mixed genotype on metabolic phenotype of L-PDCKO mice, the 129J PDH-floxed females were back-crossed with wild-type males (B6 genetic background) for 10 generations, as reported previously (30). The progeny of the last breeding were intrabred to derive a PDH-floxed colony with a B6 genetic background. In the present study, to generate deletion of exon 8 in the Pdha1 gene (L-PDCKO) in liver, homozygous Pdha1flox8/flox8 females with a B6 genetic background (30) were bred with males from the C57BL/6-TgN9AlbCre 21Mgn transgenic line from The Jackson Laboratory, which carried an autosomally integrated Cre gene with liver-specific albumin promoter (33). To generate control mice (L-PDCCT), wild-type B6 females were bred with transgenic liver-specific Cre B6 males. Only Cre-positive male progeny were used as the control. All animals had free access to a standard rodent diet and water.
Tail DNA from ∼15-day-old progeny were isolated using a kit (OmniprepTM I; Genotechnolgy) and genotyped (6). Mice were weaned on postnatal day 21 on a standard rodent diet and water ad libitum. Body weights were taken once/wk. Food intake was recorded in single-caged mice from days 49 to 56. Sixty-day-old mice in the fed state were deeply anesthetized, blood was collected by cardiac puncture, and serum was separated by centrifugation and stored at −20°C. Livers were quickly removed, frozen in liquid nitrogen, and stored at −80°C.
Insulin, glucose, and acetyl-CoA assays.
Serum insulin levels were measured using radioimmunoassay kits (Millipore). Blood glucose levels were determined using a glucometer (Abbott). Liver AcCoA content was assayed using an acetyl-CoA assay kit (Sigma-Aldrich). Pulverized liver (∼200 mg) was deproteinized in 1 N perchloric acid and centrifuged, and supernatants were neutralized with KHCO3 solution. Aliquots (50 μl) were used for AcCoA assays using the standards provided by the manufacturer.
Lipid analyses.
Serum total cholesterol was measured by an enzymatic kit (BioAssay Systems) according to the manufacturers' instructions. Serum TG was quantified by a commercial kit (Abcam). Hepatic TG was extracted by homogenization in aqueous Triton-X buffer (2%) and measured using a commercial kit (Abcam). For analysis of hepatic FAs, approximately 0.5 g of pulverized liver was spiked with heptadecanoic acid (C17:0) as an internal standard. Total lipids were isolated from liver tissue with a modified Dole mixture, followed by extraction with heptane, and saponified (36, 41). FA extracts were methylated with methanolic boron trifluoride (Sigma Aldrich). FA methyl esters were separated using a Supelcowax 10 column (30 m × 0.25 mm with 0.25-m film thickness; Supelco) in a Shimadzu GC-17A gas chromatograph fitted with a flame ionization detector. Relative hepatic FA content was calculated by using individual FA peak area relative to the total areas and expressed as the percentage of total FAs.
Hepatic cholesterol was extracted and analyzed as reported previously (36). Pulverized liver (∼500 mg) was spiked with α-cholestane as internal standard and saponified, extracted with petroleum diethyl ether, and dried under nitrogen gas. Sterol fractions were analyzed on the same gas chromatography system using a SAC-5 capillary column.
Quantitative real-time PCR for gene expression.
Liver and epididymal adipose tissue (∼100 mg) were homogenized in TRIzol reagent, and total RNA was extracted as per the manufacturer's instructions (Life Technologies). Total RNA (∼1 μg) was reverse transcribed into cDNA using an iScript cDNA kit (Bio-Rad). RT-PCR reactions were performed using appropriately diluted cDNA in triplicate with 18S rRNA serving as an internal control, and gene expression levels were quantified using a CFX96 Touch RT-PCR detection system (Bio-Rad) according to the manufacturer's recommendation. Relative quantification of amplified DNA was performed using the 2−ΔΔCT method (26, 30). Primers used for gene expression analysis are presented in Table 1.
Table 1.
Primers for gene expression analyses
| Gene Symbol | Gene ID | Primer Sequence (5′→3′) | PCR Product (bp) |
|---|---|---|---|
| ChREBP (Mlxipl) | NM 021455.4 | Forward: 5′-GCATCCTCATCCGACCTTTA-3′ | 170 |
| Reverse: 5′-GATGCTTGTGGAAGTGCTGA-3′ | |||
| SREBP-1c (Srebf1) | AF286470 | Forward: 5′-GGAGCCATGGATTGCACATT-3′ | 104 |
| Reverse: 5′-CCTGTCTCACCCCCAGCATA-3′ | |||
| SREBP-2 (Srebf2) | NM 033218.1 | Forward: 5′-CAAAATCATAGAGTTGAAGGACTTAGTCA-3′ | 86 |
| Reverse: 5′-TGTAATCAATGGCCTTCCTCAGA-3′ | |||
| Pgc-1α (Ppargc1a) | NM 008904.2 | Forward: 5′-GACTCAGTGTCACCACCGAAATC-3′ | 86 |
| Reverse: 5′-GACCTGTGTCGAGAAAAGGATCTT-3′ | |||
| Foxo1 | NM 019739.3 | Forward: 5′-TGTTACTTAGCTCTCCCCTCG-3′ | 144 |
| Reverse: 5′-AGACGAGCAGTGGCTCAAT-3′ | |||
| LXRα (Nr1 h3) | AF085745 | Forward: 5′-GCTCTGCTCATTGCCATCAG-3′ | 80 |
| Reverse: 5′-TGTTGCAGCCTCTCTACTTGGA-3′ | |||
| Sirt6 | NM 181586.3 | Forward: 5′-GGCTACGTGGATGAGGTGAT-3′ | 131 |
| Reverse: 5′-GGCTCAGCCTTGAGTGCTAC-3′ | |||
| Gck | NM 010292.5 | Forward: 5′-CTGGATGACAGAGCCAGGATG-3′ | 247 |
| Reverse: 5′-AGTTGGTTCCTCCCAGGTCT-3′ | |||
| Pklr | NM 013631.2 | Forward: 5′-CTGGATGGGGCTGACTGTAT-3′ | 142 |
| Reverse: 5′-GGCGTAGCTCCTCAAACAAC-3′ | |||
| Acc1 (Acaca) | NM 133360.2 | Forward: 5′-GCAGCCCTGGGCACAG-3′ | 67 |
| Reverse: 5′-AACTACTCCCACGGGTATTCCC-3′ | |||
| Acc2 (Acacb) | NM 133904 | Forward: 5′-TGTTCTCGGCCTCTCTTCAC-3′ | 95 |
| Reverse: 5′-GAGGCTGCATTGAACACAAG-3′ | |||
| G6pd2 | NM 019468.2 | Forward: 5′-GCTGGATCTAACTTATGGCAACA-3′ | 104 |
| Reverse: 5′-CGGACAAAGTGCATCTGGC-3′ | |||
| Pck1 | NM 011044.2 | Forward: 5′-CAGCTGCTGCAGAACACAAGG-3′ | 237 |
| Reverse: 5′-GCTAACTGCTACAGCTAACGTG-3′ | |||
| G6Pase (G6pc) | NM 008061.3 | Forward: 5′-CCTGAGGAACGCCTTCTATG-3′ | 214 |
| Reverse: 5′-TCACAGGTGACAGGGAACTG-3′ | |||
| FAT (Cd36) | NM 001159558.1 | Forward: 5′-TGGCCTTACTTGGGATTGG-3′ | 111 |
| Reverse: 5′-CCAGTGTATATGTAGGCTCATCCA-3′ | |||
| Cpt1a | NM013495 | Forward: 5′-CCTGGGCATGATTGCAAAG-3′ | 84 |
| Reverse: 5′-ACGCCACTCACGATGTTCTTC-3′ | |||
| Ppara | NM 001113418.1 | Forward: 5′-GGGCAAGAGAATCCACGAAG-3′ | 91 |
| Reverse: 5′-GTTGTTGCTGGTCTTTCCCG-3′ | |||
| Hmgcr | NM008255 | Forward: 5′-TGACCTTTCTAGAGCGAGTGCAT −3′ | 83 |
| Reverse: 5′-CACGAGCTATATTTTCCCTTACTTCA-3′ | |||
| Acat1 | NM 144784 | Forward: 5′-TGCATAACTTCGTTCCAGGC-3′ | 109 |
| Reverse: 5′-AGCCTTTCGCGTCTCCAT-3′ | |||
| Acat2 | NM 009338 | Forward: 5′-CACCTCACCCATCCTGACTC-3′ | 92 |
| Reverse: 5′-ACTCCACCATTGTGGTAGCTG-3′ | |||
| AceCS1 (Acss2) | NM 019811 | Forward: 5′-TGGAGATGATCCTGTCACCA-3′ | 167 |
| Reverse: 5′-GCATATGGCCACCTGTTTCT-3′ | |||
| AceCS2 (Acss1) | NM 080575 | Forward: 5′-CTCGGTGCACTCCATGTCT-3′ | 104 |
| Reverse: 5′-GGAGAGAGATGAACCTGGGA-3′ | |||
| Pdha1 | NM 008810.2 | Forward: 5′-TGGCAGCACTGTGGAAATTA-3′ | 154 |
| Reverse: 5′-CGCACAAGATATCCATTCCA-3′ | |||
| Acly | NM 134037.3 | Forward: 5′-AGGAAGTGCCACCTCCAACAGT-3′ | 103 |
| Reverse: 5′-CGCTCATCACAGATGCTGGTCA-3′ | |||
| β-Actin | NM 007393.5 | Forward: 5′-GCTCTTTTCCAGCCTTCCTT-3′ | 168 |
| Reverse: 5′-CTTCTGCATCCTGTCAGCAA-3′ |
ChREBP, carbohydrate response element-binding protein; SREBP-1c, sterol regulatory element-binding protein-1c; peroxisome proliferator-activated receptor-γ coactivator-1α; Foxo1, forkhead box O1; LXRα, liver X receptor-α; Sirt6, sirtuin 6; Gck, glucokinase; Pklr, liver-pyruvate kinase; Acc1 and 2, acetyl-CoA carboxylase 1 and 2, respectively; Pck1, PEP carboxykinase; G6Pase, glucose-6-phosphatase; FAT, fatty acid translocase; Cpt1a, carnitine palmitoyltransferase 1α; Ppara, peroxisome proliferator-activated receptor-α; Hmgcr, hydroxy-methylglutaryl-CoA reductase; Acat1 and -2, acetyl-CoA:acetyltransferase 1 and 2, respectively; AceCS1 and -2, acetyl-CoA synthetase 1 and 2, respectively.
Subcellular fractionation of liver and Western blotting.
Liver homogenates were fractionated according to the procedure described earlier (8). Briefly, liver (∼100 mg) was homogenized in buffered sucrose containing a protease inhibitor cocktail (Sigma-Aldrich), kept on ice for 30 min, and centrifuged at 800 g for 15 min. Following differential centrifugation, nuclear, mitochondrial, and cytosolic fractions were prepared and stored at −80°C. Nuclei fractions were washed in the same buffer and resuspended in nuclei resuspension buffer containing a protease inhibitor cocktail (Sigma-Aldrich). The nuclei were sonicated for 15 s, the lysate was centrifuged at 9,000 g for 30 min at 4°C, and the supernatant was stored at −80°C. Mitochondrial fractions were solubilized in resuspension buffer containing protease inhibitor cocktail (Sigma-Aldrich) and sonicated for 15 s and stored −80°C. For whole cell lysate preparation, liver tissues were homogenized in RIPA buffer (Abcam) containing protease inhibitor cocktail, agitated for 2 h at 4°C, and centrifuged at 13,000 g for 20 min. The supernatant was saved and stored at −80°C. The protein content of all tissue preparations was determined using Bio-Rad protein assay. Proteins were separated and immunodetected using the Western blotting technique as described (6). Equal amounts of protein (50 μg) were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred on the nitrocellulose membrane, and detected using specified antibodies as listed: anti-acetyl-CoA carboxylase 1 (04–322; EMD Millipore), SREBP1 antibody (NB600-582; Novus Biologicals), anti-acetyl-CoA synthetase (ab133664; Abcam), anti-histone H3 (acetyl K9) antibody (ab10812; Abcam), SIRT6 antibody (12486; Cell Signaling Technology), acetylated-lysine antibody (9441; Cell Signaling Technology), and anti-PDH antibody (6) to detect pyruvate dehydrogenase component of PDC. β-Actin used as a loading control was detected using β-actin (D6A8) antibody (8457; Cell Signaling Technology), and gels were stained with Ponceau S stain, followed by densitometry analysis (for nuclear protein analysis only). Protein bands were visualized using an Enhanced Chemiluminescence kit (Perkin-Elmer) and analyzed using Bio-Rad ChemiDoc MP image analyzer.
Data analysis.
Results are presented as means ± SE of eight animals unless otherwise indicated. Differences between the means of the L-PDCCT and L-PDCKO groups were performed using Students' t-test. For postnatal growth of mice, body weight data were analyzed using the analysis of variance (one-way ANOVA), followed by the Holm-Sidak method for two groups of mice at different age periods. Significance was assigned when the P value was ≤0.05.
RESULTS
Analysis of liver-specific PDC-deficient mice.
Progeny from the breeding of Pdha1-targeted homozygous females with liver-specific Cre males were found to be normal in average litter size with no embryonic lethality. In the present study, only 2 mo-old male progeny were analyzed unless otherwise indicated. Genomic DNA analysis of liver (L), skeletal muscle (SM), heart (H) and adipose tissue (AT) from 2-mo-old control (L-PDCCT) mice by PCR showed a 700-bp band corresponding to the wild-type allele (Pdha1wt) (Fig. 2A). Tissue DNA analysis of L-PDCKO mice detected the 800-bp Pdha1flox allele in skeletal muscle, heart, and adipose tissue, whereas liver showed the 400-bp Pdha1Δex8 deleted allele, indicating that the Pdha1 gene was deleted in the liver only (Fig. 2A). As expected, a 240-bp Cre allele was present in all tissues analyzed (Fig. 2A), indicating that the Cre transgene was ubiquitously present in all of the tissues. Liver Pdha1 mRNA analysis by qRT-PCR using one primer in the region of the deleted exon 8 showed the level at 1.3% in L-PDCKO mice compared with control (L-PDCCT) mice (Fig. 2B). Western blot analyses showed the complete absence of the α- and β-subunits of PDH in livers of three L-PDCKO mice (L1, L2, and L3) compared with three L-PDCCT mice (Fig. 2C). As expected, two PDH subunits were present in other tissues analyzed in mice (Fig. 2C). Previously, it was shown that in the absence of the α-subunit of PDH, the β-subunit of PDH was found to be absent due to its instability (17).
Fig. 2.
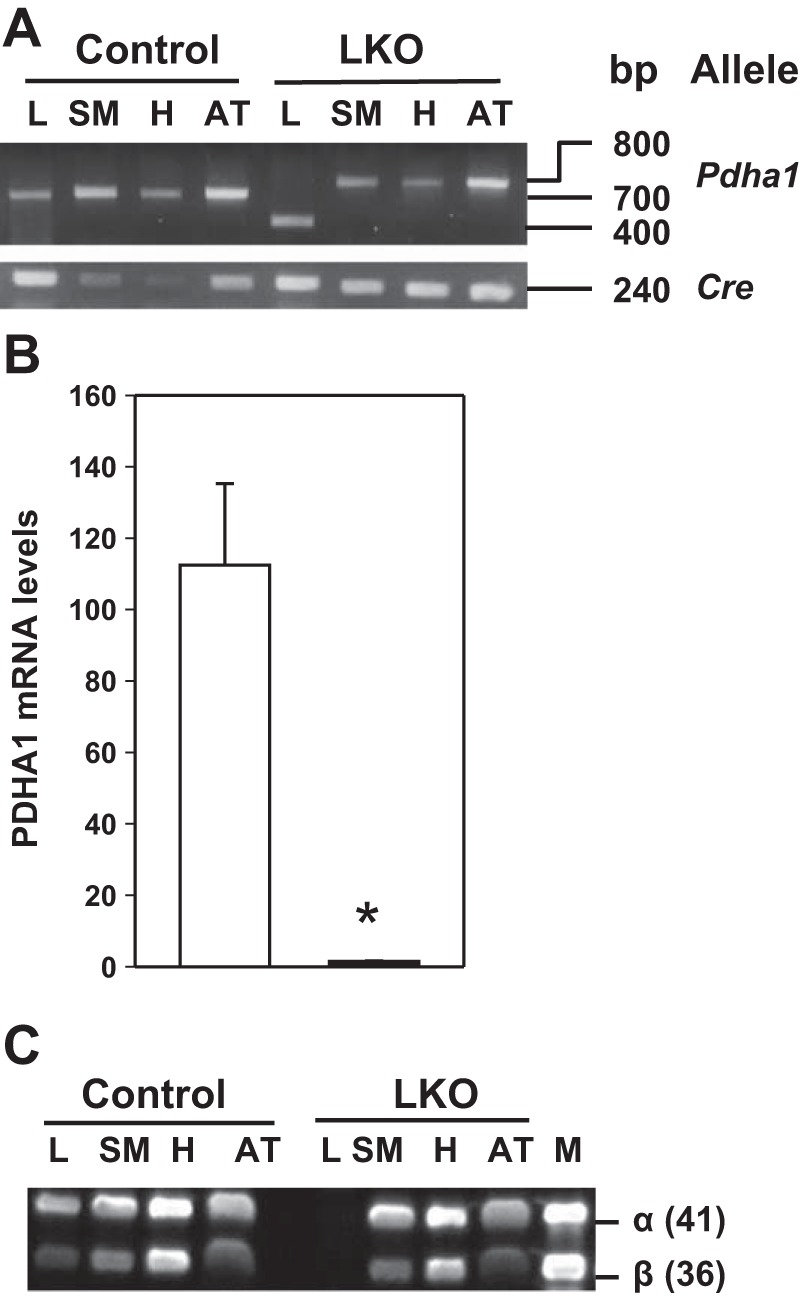
Genetic and protein analyses of control (L-PDCCT) and liver-specific pyruvate dehydrogenase complex-deficient (L-PDCKO) mice. A: PCR amplification of 3 different alleles in liver (L), skeletal muscle (SM), heart (H), and adipose tissue (AT). B: quantitative RT-PCR (qRT-PCR) analysis of Pdha1 mRNA in L-PDCKO liver (black bar) compared with L-PDCCT liver (open bar). Results are means ± SE (n = 6). *P < 0.05. C: Western blot analyses of PDH proteins (both the 41-kDa α- and 36-kDa β-subunits) in tissue lysates from L-PDCCT and L-PDCKO mice.
Metabolic characteristics of L-PDCCT and L-PDCKO male mice.
Body weights were not significantly different between L-PDCCT and L-PDCKO mice, except at one time point (postnatal day 49) from days 10 to 120 (Fig. 3A). There was a significant increase (14%) in food intake by L-PDCKO mice compared with L-PDCCT mice from postnatal days 49 to 56 (Fig. 3B). Liver weight did not differ significantly between two groups of mice on day 60; however, there was a significant reduction (12%) in the weight of epididymal adipose tissue in 2-mo-old L-PDCKO (0.29 ± 0.01; n = 10) vs. L-PDCCT (0.33 ± 0.007; n = 10) mice.
Fig. 3.

Body weights, food intake, blood glucose levels, serum insulin levels, and liver lipid analyses in L-PDCCT and L-PDCKO male mice in the fed state. A: body weights of mice from days 10 to 120. Body weights were recorded every 7 days. B: food intake of mice (single caged) was determined for 1 wk from days 49 to 56. C: tail blood glucose levels in randomly fed mice were recorded on day 60. D and E: serum insulin and serum triglyceride (TG) levels of mice were measured on day 60. F–H: levels of liver cholesterol and TG and fatty acid composition of TG of mice on day 60. Results are expressed as means ± SE (n = 8–12). *P < 0.05.
In the fed state, blood glucose levels were decreased by 24% in L-PDCKO mice (Fig. 3C), whereas serum insulin levels were increased by 63% in L-PDCKO mice (Fig. 3D). Analysis of serum TG from fed mice showed no significant change in L-PDCKO compared with L-PDCCT mice (Fig. 3E). The levels of liver cholesterol and TG were not significantly different between the two groups of mice (Fig. 3, F and G). However, the FA composition of hepatic TG was variable in L-PDCKO mice as follows: linoleic acid (C18:2) increased by 8.7% in L-PDCKO mice, whereas oleic acid (C18:1) and vaccinic acid (C18:1) decreased by 19.7 and 23.4%, respectively, in L-PDCKO mice (Fig. 3H). The latter finding most likely reflects changes due to elongation and desaturation and not de novo FA synthesis. It should be noted that there are differences in body weights, blood glucose levels, and serum insulin levels in L-PDCKO mice used in the present study and that of L-PDCKO mice reported previously (6). These phenotypic differences are due to differences in the genetic background of these two strains of mice. Mice used in the present study had a B6 genetic background, whereas mice used in our previously reported paper (6) had a mixed (129J/B6) genetic background. Earlier, we also reported some differences for β-cell structure and function between two pancreatic β-cell-specific PDCKO mouse strains (30, 39). So it is not surprising that we have observed some variations in the levels of serum insulin and blood glucose of L-PDCKO mice (B6 genetic background) in the present study compared with that of the L-PDCKO mice (129J/B6 mixed genetic background), as reported previously (6).
Hepatic gene expression analyses in L-PDCKO mice.
Using quantitative RT-PCR analysis, we carried out gene expression analyses of several key enzymes as well as their upstream regulatory transcriptional factors in the liver of L-PDCKO mice. Significant reductions in the expression of key lipogenic genes [40% Acly and 60% Acc1 for FA synthesis, 60% hydroxy-methylglutaryl-CoA reductase (Hmgcr) for cholesterol biosynthesis, and 62% glucose-6-phosphate dehydrogenase (G6pd2) for both of these pathways] were observed in livers of L-PDCKO mice compared with L-PDCCT mice (Fig. 4A). Furthermore, for three key enzymes involved in FA oxidation, namely fatty acid translocase (FAT; a.k.a. Cd36), carnitine-palmitoyl-CoA transferase-1 (Cpt1a), and Acc2, their mRNA levels were significantly reduced in L-PDCKO livers by 56, 51, and 29%, respectively (Fig. 4A). Interestingly, for two key enzymes in the glycolytic pathway, the level of hepatic glucokinase (Gck) mRNA was not significantly altered in L-PDCKO mice; whereas the mRNA level of liver-pyruvate kinase (Pklr) was significantly decreased (65%) in L-PDCKO mice (Fig. 4A). In the gluconeogenic pathway, the level of PEP-carboxykinase (Pck1) mRNA was significantly decreased (62%), whereas the level of glucose-6-phosphatase [G6Pase (G6pc)] mRNA was not significantly affected (Fig. 4A).
Fig. 4.

Expression of metabolic genes and their transcription factors in the livers from L-PDCCT and L-PDCKO male mice. A: hepatic expression of glycolytic (Glyco), lipogenic (Lipo), fatty acid oxidation (FA oxid), and gluconeogenic (Gluconeo) genes. B: hepatic expression of transcription factors and regulators of metabolic genes. C: gene expression of acetyl-CoA:acetyl transferases (Acat1 and Acat2) and acetyl-CoA synthetases (AceCS1 and AceCS2) in the livers of mice. D: expression of lipogenic and FA oxidation genes in epididymal adipose tissue. E: expression of transcription factors and regulators of metabolic genes in adipose tissue. Expression was measured by qRT-PCR, with β-actin mRNA as an internal control. Tissues from 60-day-old L-PDCCT (CONT; open bars) and L-PDCKO (LKO; black bars) male mice fed a rodent diet were used for analyses. Results are expressed as means ± SE (n = 6–8/group; *P < 0.05).
Hepatic lipid metabolism is controlled by transcription factors such as liver X receptors (LXRs), sterol regulatory element-binding protein (SREBP)-1c, SREBP2, and carbohydrate response element-binding protein (ChREBP) that regulate the expression of critical enzymes involved in the lipogenic and glycolytic pathways (11). Furthermore, several other key transcription factors and coactivators such as peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α (PGC-1α), PPARα, forkhead box O1 (FOXO1), and Sirt6 are known to play important roles in the gene expression of several key enzymes in carbohydrate and lipid metabolism (14, 22, 31, 32). Interestingly, mRNA levels of all these transcription factor modifiers were decreased significantly in the liver from L-PDCKO mice compared with L-PDCCT mice [%reduction: 52 LXRα (Nr1h3), 61 SREBP1c (Srebf1), 40 SREBP2 (Srebf2), 53 ChREBP (Mlxipl), 65 Pgc-1α (Ppargc1a), 64 Ppara, 76 Foxo1, and 63Sirt6; Fig. 4B].
The mRNA levels of the cytosolic acetyl-CoA synthetase gene (AceCS1) and the mitochondrial acetyl-CoA synthetase gene (AceCS2) were determined. The mRNA levels of AceCS1 were significantly decreased (45%) in livers from L-PDCKO mice, whereas the mRNA levels AceCS2 were similar between the two groups of animals (Fig. 4C). The mRNA levels of acetyl-CoA:acetyltransferase 1 (Acat1) and acetyl-CoA:acetyltransferase 2 (Acat2) were also similar between the two groups of mice (Fig. 4C).
Earlier we reported a compensatory increase in FA biosynthesis in epididymal adipose tissue from male L-PDHKO mice (6). In epididymal adipose tissue the level of Acc1 mRNA was significantly increased (145%), whereas the level of Hmgcr mRNA was decreased by 60%, and the mRNA level of G6pd2 remained unaltered in L-PDHKO mice (Fig. 4D). The levels of mRNAs of FAT (Cd36; 67%) and Cpt1a (30%) were significantly reduced in adipose tissue from L-PDHKO mice, whereas the Acc2 mRNA level was significantly increased (58%) in L-PDHKO mice (Fig. 4D). The mRNA levels of SREBP1c, ChREBP, LXRα, and Sirt6 were not altered in adipose tissue from L-PDHKO mice, but the mRNA levels of Pgc-1α, PPARα, and Foxo1 were significantly increased by 132, 124, and 95%, respectively, in L-PDHKO mice (Fig. 4E). Interestingly, the expression of SREBP2 was decreased by 41% in adipose tissue from L-PDHKO mice (Fig. 4E), which is consistent with the observed reduction in Hmgcr mRNA levels (Fig. 4D).
Protein analyses in livers from L-PDCKO mice.
In light of alterations in expression of several genes in livers of L-PDCKO mice, we investigated the amounts of four hepatic proteins. The level of cytosolic ACC1 was significantly decreased (36%) in livers of L-PDCKO mice (Fig. 5A). The levels of SREBP-1c were significantly decreased in both the nuclear (33%) (Fig. 5B) and the cytosolic (43%) (Fig. 5C) fractions of L-PDCKO liver. Also, the protein level of AceCS1 that converts acetate to AcCoA in the cytosol was decreased (42%) in L-PDCKO livers (Fig. 5D). In contrast, the nuclear level of SIRT6 protein was significantly increased (57%) in L-PDCKO livers (Fig. 5E), although its mRNA level in L-PDCKO livers was not significantly altered (Fig. 4B). The increase in SIRT6 protein levels is most likely due to its stabilization, as there was no change in Sirt6 gene transcription (Fig. 5E) (21).
Fig. 5.

Western blot analyses of liver proteins from L-PDCCT and L-PDCKO mice. A: acetyl-CoA carboxylase 1 (ACC1) in liver cytosol. B: sterol regulatory element-binding protein (SREBP-1) in liver nuclei. C: SREBP-1 in liver cytosol. D: acetyl-CoA synthetase gene (AceCS1) in liver cytosol. E: sirtuin 6 (SIRT6) in liver nuclei. Top (blots A–E): Western blots for listed proteins. Bottom (graphs A–E): quantitation of Western blot proteins by densitometry analysis. Results were normalized with β-actin and expressed as means ± SE (n = 6). *P < 0.05. LKO, L-PDCKO.
AcCoA content and protein acetylation in livers from L-PDCKO mice.
We reasoned that PDC deficiency in the liver could impact the steady-state levels of AcCoA in ad libitum-fed L-PDCKO mice. Surprisingly, there was no significant difference in total liver AcCoA content between L-PDCCT and L-PDCKO livers (Fig. 6A). However, measuring the total hepatic amount of AcCoA does not allow the discrimination of any changes in the subcellular AcCoA pools in mitochondria and nucleocytosolic compartments. Since histone acetylation has been shown to be associated with changes in the cellular AcCoA levels (2, 42), we investigated possible changes in these pools by measuring their impact on the level of protein acetylation in liver nuclear fraction from L-PDCKO mice. Interestingly, there was a marked increase (2.1-fold) in histone H3K9 acetylation in liver nuclei from L-PDCKO mice compared with L-PDCCT mice (Fig. 6, B and C). When the SDS-PAGE-separated nuclear proteins were immunodetected using antiacetylated lysine antibodies, at least three major protein bands were significantly altered in L-PDCKO mice, namely 1) a 2.1-fold increase in the ∼22-kDa band representing histone H3K9, 2) a 24% reduction in the ∼35-kDa protein band, and 3) a 44% reduction in the ∼45-kDa band (Fig. 6, D and E).
Fig. 6.

Hepatic acetyl-CoA (AcCoA) content and analyses of acetylated proteins in liver nuclei from L-PDCCT (control) and L-PDCKO (LKO) mice. A: hepatic AcCoA content in mice. B: Western blot analysis of histone H3 acetyl K9 protein (H3K9) in liver nuclei from mice. C: densitometry analysis of histone H3 acetyl K9 protein detected in B. D: Western blot analysis of acetyl-lysine proteins in liver nuclei. E: densitometry analysis of 22-, 35-, and 45-kDa acetyl-lysine proteins bands detected in D. Protein loading was analyzed by staining of gels with Ponceau S stain, followed by densitometry analysis (results not shown). Results are expressed as means ± SE (n = 6–8). *P < 0.05.
DISCUSSION
During the absorptive period, the liver utilizes glucose as the primary substrate for the synthesis of glycogen and fatty acids/cholesterol. During this period, dephosphorylation/activation of PDC by insulin-mediated activation of PDH phosphatase allows the formation of AcCoA from pyruvate in the mitochondria (3, 19). The transfer of the acetyl moiety as citrate to the cytosol and regeneration of AcCoA from citrate serve as the major sources for lipid biosynthesis and protein acetylation in the nucleocytosolic compartments (Fig. 7). The null mutation in the Pdha1 gene resulting in the complete loss of hepatic PDC activity eliminates the availability of glucose-derived AcCoA for FA synthesis in the cytosol (6). The results of the present study extends this finding showing downregulation of several key genes involved in lipid synthesis and glucose metabolism in livers of L-PDCKO mice. This is a novel finding for glucose-derived AcCoA, which is not only the precursor for lipid biosynthesis but also serves as a modulator of gene expression of several key enzymes in de novo lipogenesis as well as their upstream regulators.
Fig. 7.
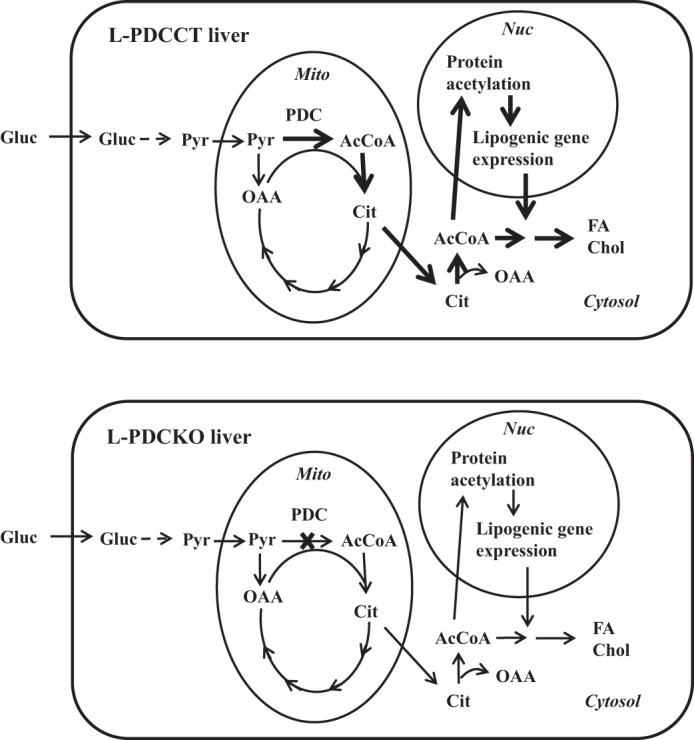
Schematic representation of the main cellular reactions involved in the generation and utilization of acetyl-CoA in L-PDCCT and L-PDCKO livers in the fed state. Corresponding arrows in L-PDCCT (top) and L-PDCKO (bottom) indicate carbon flux, protein modification, or gene expression. Thicker arrows in L-PDCCT liver indicate increased carbon flux, protein modification, or gene expression; arrows similar in thickness indicate no change between the 2 livers. Gluc, glucose; Pyr, pyruvate; AcCoA, acetyl-CoA; Cit, citrate; OAA, oxaloacetate; FA, fatty acid; Chol, cholesterol; Nuc, nucleus; Mito, mitochondria; PDC, pyruvate dehydrogenase complex.
Since PDC-generated AcCoA is the primary source of cytosolic citrate in the fed state, mitochondria-generated citrate derived from pyruvate metabolism via PDC cannot be the source for the cytosolic AcCoA pool in the liver of L-PDCKO mice (Fig. 7). However, there are other pathways such as β-oxidation of long-chain FAs, amino acid catabolism, and acetate activation by which AcCoA is generated in liver mitochondria. The acetyl moiety of mitochondrial AcCoA is transported out into the cytosol in the forms of citrate and acetyl-carnitine. As shown, when the contribution of the mitochondria-generated AcCoA via the deletion of PDC was completely eliminated in the liver, there was no effect on the level of total AcCoA in the liver of L-PDCKO mice, suggesting either increased generation by the alternate pathways or/and reduced utilization of AcCoA for lipid biosynthesis, resulting in the maintenance of the steady-state levels of AcCoA in the liver of L-PDCKO mice. The latter possibly appears to be a major contributor to this outcome. The levels of mRNA of FAT and Cpt1a were reduced in livers of L-PDCKO mice (Fig. 4A), suggesting that FA oxidation is not a source of the cytosolic AcCoA in the fed state. Another source of mitochondrial AcCoA is acetyl-CoA synthetase 2 (AceCS2) utilizing acetate as the substrate, whose mRNA levels were not altered in livers of L-PDCKO livers (Fig. 4C). The availability of acetate in the mitochondria of L-PDCKO mice is not known, and hence, its contribution to the cytosolic AcCoA pool cannot be evaluated. Formation of AcCoA from acetate by acetyl-CoA synthetase-1 and conversion of acetyl-carnitine are other potential sources of cytosolic AcCoA. Interestingly, the level of AceCS1 mRNA was markedly decreased in livers of L-PDCKO mice (Fig. 4C), suggesting a possible decrease in its contribution to the cytosolic AcCoA pool. It is possible that amino acid catabolism could serve as an alternate source of mitochondrial AcCoA in livers of L-PDCKO mice. This, however, remains to be further investigated.
Liver X receptors (LXRs), SREBPs, and ChREBP are key transcription factors controlling glycolysis and lipid biosynthesis (20, 24, 32, 37). SREBP-1c is involved in transcriptional regulation of several genes in de novo biosynthesis of fatty acids, whereas SREBP-2 regulates cholesterol synthesis in the liver (9, 14, 20). In liver, LXR stimulates the expression of both SREBP-1c and ChREBP by binding to their cognitive promoters (4, 35). Both SREBPs and ChREBP regulate the expression of glycolytic genes (Pklr and Gck) and lipogenic genes (Acc1, Acc2, and Hmgcr). Although L-PDCKO mice were hyperinsulinemic with enhanced insulin sensitivity (6), both lipogenic genes (Acly, Acc1, Hmgcr, and G6pd2) and glycolytic genes (Gck and Pklr) and several genes of upstream transcription factors (LXR, SREBP-1c, SREBP-2, and ChREBP) were downregulated in livers of these mice (Fig. 4, A and B). This finding is contrary to the expected outcome for hepatic lipogenic gene expression and associated regulators in the fed state. The suppression of LXR expression in L-PDCKO liver appears to be a key regulatory event controlling glycolytic and lipogenic responses, although the mechanism(s) by which this is accomplished in liver from L-PDCKO mice remains to be investigated. One possibility is that the hyperinsulinemic state of these mice causes enhanced lipogenesis in epididymal adipose tissue. It should be noted that the gene expression profile of the lipogenic pathway in adipose tissue was selective (an increase in the expression of Acc1 and Acc2 genes only and not of the LXR, SREBP1c, or ChREBP genes), suggesting that other mechanism(s) contribute to this outcome.
In a study in which the use of cytosolic AcCoA for FA biosynthesis was restricted by liver-specific deletion of Acc1, the mRNA expression of several genes (Acly, Acc2) involved in de novo FA synthesis was significantly increased in livers from L-ACC1KO mice (27). These investigators suggested that reduction in FA biosynthesis in the liver triggered increased expression of the lipogenic genes. In contrast, the levels of these genes remained unaffected in the epididymal adipose tissue from L-ACC1KO mice (27). In another study using the liver-specific ACC1 null mutation, expression of the hepatic ACC2 gene was enhanced to compensate for the formation of malonyl-CoA and lipogenic capacity (15). Mice with Acc2 deletion demonstrated a significant upregulation of lipogenic enzymes [Acly, Acc1, FAS (Fasn), and Hmgcr] and their upstream transcription factor genes (such as SREBP1, SREBP2, and ChREBP) (1). Interestingly, no significant effect was observed in the expression of other regulatory genes such as SREBP-1c, Pgc-1α, and Pgc-1β in double-KO (deletion of Acc1 and Acc2) mice (7).
In yet another study, liver-specific deletion of fatty acid synthase (L-FASKO), allowing the conversion of cytosolic AcCoA to malonyl-CoA by ACC1 but restricting the use of malonyl-CoA for FA synthesis, resulted in reduction in SREBP-1c expression in chow-fed L-FASKO mice with low circulating insulin levels (5). The levels of mRNAs of Gck, Pck1, PPARα, Pgc-1α, Cpt1a, and LXRα were not altered in livers from chow-fed L-FASKO mice (5). In contrast, our L-PDCKO mice with ablation of AcCoA formation from pyruvate exhibited completely opposite effects on expression of lipogenic genes in the liver. The mRNA expression of several lipogenic genes (Acly, Acc1, Acc2, Hmgcr, and G6pd2) and their upstream regulators (LXR, SREBP-1c, and ChREBP as well as Pgc-1α, PPARα, and Foxo1) were significantly downregulated in livers of L-PDCKO mice (Fig. 4, A and B). These results suggest that complete inhibition in the ability to generate AcCoA from glucose carbons as the precursor for lipogenesis in the liver triggers a downregulation in the expression of upstream regulators of hepatic lipogenic genes.
PGC-1α enhances gluconeogenesis as well as the uptake of FAs and their β-oxidation by coactivating hepatic transcription factors such as PPARα and FOXO1 (25, 32, 43). Insulin indirectly regulates PGC-1α by lowering cyclic AMP levels and also by inactivating FOXO1 (14, 16, 34). In livers of L-PDCKO mice, the expression of coactivator PGC-1α and its transcription factors (e.g., PPARα and FOXO1) was significantly reduced. Expression of their target genes such as Pck1 (gluconeogenesis), Cpt1a (FA oxidation), and FAT (FA uptake) was also significantly reduced in livers from L-PDCKO mice (Fig. 3A). Given the hyperinsulinemic state of L-PDCKO mice in the fed state, this is an expected outcome for downregulation of key gene expression in gluconeogenesis and FA oxidation in the livers of L-PDCKO mice.
A link between intracellular AcCoA levels and protein acetylation was shown in budding yeast-metabolizing glucose (2, 12, 40). Studies in yeast showed that increased availability of AcCoA due to Acc1 gene deletion was sufficient to enhance histone acetylation (13, 45). Similarly, activation of ACC1 by inhibition of AMPK in yeast resulted in decreased histone acetylation (44). In budding yeast, AcCoA production by the PDC in the mitochondria was indirectly altered by deletion of mitochondrial pyruvate carrier-1 (MPC1), resulting in accumulation of acetate in the cytosol, triggering upregulation of the cytosolic AceCS1 for AcCoA production, increased histone acetylation, and repression of autophagy genes (10). When mitochondrial AcCoA generation was eliminated by combined deletion of ACH1 (acetyl-CoA hydrolase/acetyl-CoA-CoAtransferase-1) and MPC1 in yeast, an upregulation of the Acs2p to synthesize AcCoA in the nucleocytosolic pathway was reported. In the present study, elimination of PDC activity in liver mitochondria resulted in a significant decrease in cytosolic AceCS1 gene expression but no change in AceCS2 gene expression, suggesting that AceCS1 is not able to compensate for reduced AcCoA supply due to PDC deficiency.
Protein acetylation of intermediary metabolic enzymes is highly prevalent and plays a major role in metabolic regulation (45). For example, liver-specific double-Acc1 and -Acc2 knockout mice had no effect on the mitochondrial and cytosolic AcCoA levels in the liver. However, an increase in the acetylation of proteins in the nucleocytosolic space and hypoacetylation of mitochondrial proteins was observed in these mice (7). Interestingly, many enzymes in glucose metabolism and FA synthesis were altered in livers of double (Acc1/Acc2) KO mice (7). We have observed modifications in acetylation status of several proteins in the nuclear compartment (Fig. 6, B–E). Since lipogenic gene expression is downregulated in the liver of PDCKO mice, altered acetylation profile of specific transcription involved in the regulation of lipogenic genes appears to be a contributing factor for this outcome.
In summary, AcCoA generated in liver mitochondria from glucose-derived pyruvate not only is necessary as the building block for de novo lipid biosynthesis in mice during the fed state (6) but also serves as a modulator of gene expression involved in de novo lipid biosynthesis, most likely via acetylation of histones and other specific proteins in the nuclear compartment (present study). It is suggested that altered acetylation of a key transcriptional regulatory sensor(s) (yet unidentified) plays a key role in influencing the altered metabolic state of the liver of L-PDCKO mice. Furthermore, alternate pathways for the generation of AcCoA in the liver appear not to be sufficient to compensate for gene expression in L-PDCKO mice during the fed state. These findings have important implications for regulation of hepatic fatty acid synthesis in obesity, type 2 diabetes, and nonalcoholic fatty liver disease.
GRANTS
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-20478 (M. S. Patel).
DISCLOSURES
The authors declare that there are no conflicts of interest, financial or otherwise, for this research.
AUTHOR CONTRIBUTIONS
S.M., B.B., T.C.R., and M.S.P. conception and design of research; S.M., B.B., and T.C.R. performed experiments; S.M., B.B., T.C.R., and M.S.P. analyzed data; S.M., B.B., T.C.R., and M.S.P. interpreted results of experiments; S.M. and T.C.R. prepared figures; S.M., B.B., T.C.R., and M.S.P. drafted manuscript; S.M., B.B., T.C.R., and M.S.P. edited and revised manuscript; S.M., B.B., T.C.R., and M.S.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Amy Raslawsky for performing analyses of lipids. We thank Dr. Murray Ettinger and Dr. Suzanne Laychock of the University at Buffalo for critical reading of the manuscript.
REFERENCES
- 1.Abu-Elheiga L, Wu H, Gu Z, Bressler R, Wakil SJ. Acetyl-CoA carboxylase 2−/− mutant mice are protected against fatty liver under high-fat, high-carbohydrate dietary and de novo lipogenic conditions. J Biol Chem 287: 12578–12588, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai L, Tu BP. On acetyl-CoA as a gauge of cellular metabolic state. Cold Spring Harbor Symp Quant Biol 76: 195–202, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Caruso M, Maitan MA, Bifulco G, Miele C, Vigliotta G, Oriente F, Formisano P, Beguinot F. Activation and mitochondrial translocation of protein kinase Cdelta are necessary for insulin stimulation of pyruvate dehydrogenase complex activity in muscle and liver cells. J Biol Chem 276: 45088–45097, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Cha JY, Repa JJ. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J Biol Chem 282: 743–751, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Chakravarthy MV, Pan Z, Zhu Y, Tordjman K, Schneider JG, Coleman T, Turk J, Semenkovich CF. “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab 1: 309–322, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Choi CS, Ghoshal P, Srinivasan M, Kim S, Cline G, Patel MS. Liver-specific pyruvate dehydrogenase complex deficiency upregulates lipogenesis in adipose tissue and improves peripheral insulin sensitivity. Lipids 45: 987–995, 2010. [DOI] [PubMed] [Google Scholar]
- 7.Chow JD, Lawrence RT, Healy ME, Dominy JE, Liao JA, Breen DS, Byrne FL, Kenwood BM, Lackner C, Okutsu S, Mas VR, Caldwell SH, Tomsig JL, Cooney GJ, Puigserver PB, Turner N, James DE, Villen J, Hoehn KL. Genetic inhibition of hepatic acetyl-CoA carboxylase activity increases liver fat and alters global protein acetylation. Mol Metab 3: 419–431, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dimauro I, Pearson T, Caporossi D, Jackson MJ. A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res Notes 5: 513, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eberle D, Hegarty B, Bossard P, Ferre P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86: 839–848, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Eisenberg T, Schroeder S, Andryushkova A, Pendl T, Kuttner V, Bhukel A, Marino G, Pietrocola F, Harger A, Zimmermann A, Moustafa T, Sprenger A, Jany E, Buttner S, Carmona-Gutierrez D, Ruckenstuhl C, Ring J, Reichelt W, Schimmel K, Leeb T, Moser C, Schatz S, Kamolz LP, Magnes C, Sinner F, Sedej S, Frohlich KU, Juhasz G, Pieber TR, Dengjel J, Sigrist SJ, Kroemer G, Madeo F. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab 19: 431–444, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foufelle F, Ferre P. New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1c. Biochem J 366: 377–391, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friis RM, Wu BP, Reinke SN, Hockman DJ, Sykes BD, Schultz MC. A glycolytic burst drives glucose induction of global histone acetylation by picNuA4 and SAGA. Nucleic Acids Res 37: 3969–3980, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galdieri L, Vancura A. Acetyl-CoA carboxylase regulates global histone acetylation. J Biol Chem 287: 23865–23876, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene 27: 2320–2336, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Harada N, Oda Z, Hara Y, Fujinami K, Okawa M, Ohbuchi K, Yonemoto M, Ikeda Y, Ohwaki K, Aragane K, Tamai Y, Kusunoki J. Hepatic de novo lipogenesis is present in liver-specific ACC1-deficient mice. Mol Cell Biol 27: 1881–1888, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413: 179–183, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Ho L, Hu CW, Packman S, Patel MS. Deficiency of the pyruvate dehydrogenase component in pyruvate dehydrogenase complex-deficient human fibroblasts. Immunological identification. J Clin Invest 78: 844–847, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson MT, Mahmood S, Hyatt SL, Yang HS, Soloway PD, Hanson RW, Patel MS. Inactivation of the murine pyruvate dehydrogenase (Pdha1) gene and its effect on early embryonic development. Mol Genet Metab 74: 293–302, 2001. [DOI] [PubMed] [Google Scholar]
- 19.Johnson SA, Denton RM. Insulin stimulation of pyruvate dehydrogenase in adipocytes involves two distinct signalling pathways. Biochem J 369: 351–356, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jump DB. N-3 polyunsaturated fatty acid regulation of hepatic gene transcription. Curr Opin Lipidol 19: 242–247, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanfi Y, Shalman R, Peshti V, Pilosof SN, Gozlan YM, Pearson KJ, Lerrer B, Moazed D, Marine JC, de Cabo R, Cohen HY. Regulation of SIRT6 protein levels by nutrient availability. FEBS Lett 582: 543–548, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A, Vazquez-Ortiz G, Jeong WI, Park O, Ki SH, Gao B, Deng CX. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab 12: 224–236, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knebel B, Haas J, Hartwig S, Jacob S, Kollmer C, Nitzgen U, Muller-Wieland D, Kotzka J. Liver-specific expression of transcriptionally active SREBP-1c is associated with fatty liver and increased visceral fat mass. PLoS One 7: e31812, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem 277: 9520–9528, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab 1: 361–370, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 27.Mao J, DeMayo FJ, Li H, Abu-Elheiga L, Gu Z, Shaikenov TE, Kordari P, Chirala SS, Heird WC, Wakil SJ. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc Natl Acad Sci USA 103: 8552–8557, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel MS, Korotchkina LG. The biochemistry of the pyruvate dehydrogenase complex. Biochem Mol Biol Ed 31: 5–15, 2003. [Google Scholar]
- 29.Patel MS, Jomain-Baum M, Ballard FJ, Hanson RW. Pathway of carbon flow during fatty acid synthesis from lactate and pyruvate in rat adipose tissue. J Lipid Res 12: 179–191, 1971. [PubMed] [Google Scholar]
- 30.Patel MS, Srinivasan M, Strutt B, Mahmood S, Hill DJ. Featured Article: Beta cell specific pyruvate dehydrogenase alpha gene deletion results in a reduced islet number and β-cell mass postnatally. Exp Biol Med (Maywood) 239: 975–985, 2014. [DOI] [PubMed] [Google Scholar]
- 31.Pei L, Waki H, Vaitheesvaran B, Wilpitz DC, Kurland IJ, Tontonoz P. NR4A orphan nuclear receptors are transcriptional regulators of hepatic glucose metabolism. Nat Med 12: 1048–1055, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest 118: 829–838, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274: 305–315, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92: 829–839, 1998. [DOI] [PubMed] [Google Scholar]
- 35.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 14: 2819–2830, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rideout TC, Harding SV, Jones PJ. Consumption of plant sterols reduces plasma and hepatic triglycerides and modulates the expression of lipid regulatory genes and de novo lipogenesis in C57BL/6J mice. Mol Nutr Food Res 54, Suppl 1: S7–S13, 2010. [DOI] [PubMed] [Google Scholar]
- 37.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B. Role of LXRs in control of lipogenesis. Genes Dev 14: 2831–2838, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sidhu S, Gangasani A, Korotchkina LG, Suzuki G, Fallavollita JA, Canty JM Jr, Patel MS. Tissue-specific pyruvate dehydrogenase complex deficiency causes cardiac hypertrophy and sudden death of weaned male mice. Am J Physiol Heart Circ Physiol 295: H946–H952, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Srinivasan M, Choi CS, Ghoshal P, Pliss L, Pandya JD, Hill D, Cline G, Patel MS. β-Cell-specific pyruvate dehydrogenase deficiency impairs glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab 299: E910–E917, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell 23: 207–217, 2006. [DOI] [PubMed] [Google Scholar]
- 41.van der Vusse GJ, Roemen TH, Reneman RS. The content of non-esterified fatty acids in rat myocardial tissue. A comparison between the Dole and Folch extraction procedures. J Mol Cell Cardiol 17: 527–531, 1985. [DOI] [PubMed] [Google Scholar]
- 42.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324: 1076–1080, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413: 131–138, 2001. [DOI] [PubMed] [Google Scholar]
- 44.Zhang M, Galdieri L, Vancura A. The yeast AMPK homolog SNF1 regulates acetyl coenzyme A homeostasis and histone acetylation. Mol Cell Biol 33: 4701–4717, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science 327: 1000–1004, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]